
Abstract
Various polymeric nanoparticles have been used as drug carriers in drug delivery systems (DDSs). Most of them were constructed from dynamic self-assembly systems formed via hydrophobic interactions and from structures that are unstable in an in vivo environment owing to their relatively weak formation forces. As a solution to this issue, physically stabilized core-crosslinked particles (CP) with chemically crosslinked cores have received attention as alternatives to the dynamic nanoparticles. This focused review summarizes recent advances in the construction, structural characterization, and in vivo behavior of polymeric CPs. First, we introduce a nanoemulsion-mediated method to create polyethylene glycol (PEG)-bearing CPs and their structural characterization. The relationship between the PEG chain conformations in the particle shell and the in vivo fate of the CPs is also discussed. After that, the development and advantages of zwitterionic amino acid-based polymer (ZAP)-bearing CPs are presented to address the poor penetration and the internalization of PEG-based CPs into tumor tissues and cells, respectively. Finally, we conclude and discuss prospects for application of polymeric CPs in the DDS field.
Similar content being viewed by others


Introduction
Considerable effort has been devoted to developing anticancer drugs with low molecular weights [1,2,3]. Owing to the relatively small sizes of those drugs, they can easily enter the target cells and reach the target organelles or proteins inside the cells via diffusion. However, their specific accumulation levels in cancer tissues in actual in vivo systems are extremely low owing to fast clearance of those small drugs in the kidneys, so overdoses are required to obtain the desired therapeutic effects. This raises medical costs and concerns about side effects because of undesired accumulation in healthy organs. For instance, hydrophobic anticancer drugs, such as doxorubicin (dox), are immediately captured by serum proteins after administration and accumulate in all healthy organs at high levels (especially in the liver), causing severe side effects [4].
To avoid undesired biodistribution of anticancer drugs and enhance their therapeutic efficacies at the tumor site, self-assembled nanoparticles, such as liposomes, polymer micelles, and polymersomes, were developed decades ago as carriers for such anticancer drugs [5,6,7,8,9,10,11,12,13]. In this strategy, specific drug delivery systems (DDSs), such as anticancer drugs, are encapsulated with biocompatible polymer-coated nanoparticles that are not recognized by serum proteins in vivo and avoid capture by the reticuloendothelial system. If the nanoparticle size is controlled within an appropriate range, renal clearance is inhibited, and this leads to prolonged blood circulation of the drug to be delivered [6, 14]. Furthermore, the nanoparticles are believed to specifically accumulate in the tumor tissues by passing through the gaps between underdeveloped endothelial cells [15,16,17] or by using the intracellular pathway based on the transcytosis of the cells around cancer tissues [18, 19]. The accumulated nanoparticles are thought to be retained relatively longer at the tumor site due to the immature lymphatic systems in solid tumors. These phenomena are known as the enhanced permeability and retention (EPR) effect and have been used as a standard strategy in most nanoparticle-based cancer chemotherapies [20, 21].
Although there are various debates over this EPR effect, the impact of nanoparticle sizes on the in vivo fate is unavoidable and is based on both pharmacokinetics (PK) and pharmacodynamics (PD). Renal clearance of relatively small nanoparticles (<5 nm) cannot be avoided [22]. Particles with relatively large sizes (>150 nm) might be trapped in the lung capillary network or filtered in the liver and spleen [22]. Because of these concerns, nanoparticle sizes are designed to fall within the range 20−150 nm. In addition to the size issue, it is essential to impart biocompatible polymers onto the nanoparticle surfaces to control blood retention by avoiding protein recognition during blood circulation. Polyethylene glycol (PEG) and zwitterionic polymers are frequently used on DDS nanoparticles to prevent recognition by proteins and the resulting immune responses [23,24,25,26]. Doxil, a commercially available liposomal formulation encapsulating dox, and liposomal vaccines encapsulating COVID-19 mRNA also have PEGs on their particle surfaces to enhance biocompatibility [27, 28].
However, although various self-assembly based nanoparticle systems have been developed thus far, their therapeutic effects have not been as beneficial as anticipated [29]; this is due to low selectivity in tumor delivery owing to poor retention in the blood, which has been an issue even for conventional small-molecule anticancer drugs. Since these nanoparticles are formed in aqueous solutions by self-assembly of amphiphilic unimers via relatively weak hydrophobic interactions, the solution condition impacts the equilibrium between the unimers and the nanoparticles. Because of this equilibrium, the administered nanoparticles always suffer from various effects in the in vivo environment, including dilution, protein interactions, and high shear stress in the blood, which shift the equilibrium toward dissociation into the unimers even though their surfaces are protected with biocompatible polymers [30,31,32,33,34]. Even in polymeric micelles with a polystyrene core whose glass transition temperature is ~100 °C, over 80% of the micelles are cleared from the blood after intravenous administration owing to these equilibrium issues [35]; this limits the tumor delivery efficiency as long as self-assembled nanoparticles are employed.
As a solution for this issue, physically stabilized core-crosslinked particles (CPs) with polymer chains on the particle surfaces have been proposed as next-generation drug carriers [36,37,38,39]. Since those nanoparticles do not establish dynamic equilibria, they are retained in the blood longer than self-assembled systems; this enhances accumulation in tumors by reducing undesired biodistribution into healthy organs. Based on our studies, this review is focused on recent progress in developing polymeric CPs as drug carriers.
Polymeric CPs prepared via the formation of nanoemulsions
Preparation and structural characterization
Polymeric CPs are primarily conducted via crosslinking of micellar cores. The resulting particles are often called “core-crosslinked micelles (CCMs).” In terms of the micellar concept, this expression is not quite appropriate; the “CCMs” cannot be categorized as micelles because these nanoparticles are not in dynamic structures [40, 41], and there is no equilibrium between the unimers and micelles [42, 43]. The traditional strategy for preparing CCMs involves introducing polymerizable groups (e.g., methyl acrylate) into the hydrophobic blocks of amphiphilic block copolymers [44, 45]. After micelle formation, the hydrophobic polymer chains are linked via polymerization reactions to produce the CCMs. Rijcken et al. prepared a block copolymer composed of PEG-b-poly N-(2-hydroxyethyl)methacrylamide with lactate side chains (pHEMAm-Lacn), and the hydroxyl groups were partially modified with methacryloyl chloride to introduce polymerizable methyl methacrylate groups [46]. UV-induced polymerization converted the micelles prepared from the block copolymer into CCMs. Those CCMs showed relatively prolonged blood circulation times compared to those of noncrosslinked micelles, which demonstrated the advantage of the core-crosslinked strategy in improving the PK of the nanoparticles.
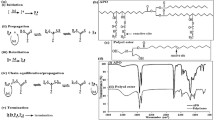
In contrast to the previous study, our approach to producing polymeric CPs was based on forming nanoemulsions composed of amphiphilic molecules with crosslinkable oil agents [47, 48]. As shown in Fig. 1, we first prepared PEGylated surfactants with vinyl groups at the ends of the hydrophobic chains. The surfactant was named PxDU, where x indicates the molecular weight of the PEG. We added crosslinkable oil agents, such as D4H and D4V, into the solution of PxDU micelles containing a Pt catalyst and produced nanoemulsions. By vigorously stirring with probe-type ultrasonication at 60 °C, a hydrosilylation reaction formed the D4H/D4V network structures [49] in the nanoemulsion core. The vinyl groups in the surfactant were also involved in the reaction and developed PEGylated CP (PEGx@CP). The advantage of this method is that various CPs can be prepared from the surfactant by simply changing the type and amount of crosslinker [47].

Schematic illustration of the synthesis of PEGylated core-crosslinked particles (PEGx@CP) from nanoemulsions composed of D4H/D4V with hydrosilylation reactions. Reprinted from [48], Copyright 2020, with permission from Elsevier
The structure of PEGx@CPs was elucidated with light and X-ray scattering measurements. Small-angle X-ray scattering (SAXS) determined the particle morphology, accurate core, and particle sizes with a model-based analysis. In addition, multiangle light scattering (MALS) measurements coupled with an asymmetrical flow field-flow fractionation (AF4) system indicated the accurate molar masses and size distributions of nanoparticles [50,51,52]. AF4 systems separate nanoparticles based on their sizes by combining vertical and horizontal flows without any stationary phase, which enables fractionation of the self-assembled systems, including micelles and liposomes that would otherwise be adsorbed on stationary phases, such as those in gel permeation columns. As a typical example, the AF4-MALS fractograms of P2kDU micelles and PEG2k@CP are displayed in Fig. 2. Both nanoparticles showed only one peak in the fractogram, indicating their narrow sizes and molar mass distributions and lack of secondary aggregation in solution. The concentrations of whole particles and PxDU components were determined with RI and UV detectors, respectively. We also know the molar mass from the MALS detector. Therefore, based on those parameters, the PEG chain number (NPEG) was estimated for each nanoparticle by using a measurement, which estimated the PEG chain density on the particle shell, as discussed later.

AF4 fractograms of P2kDU micelles (a) and 150 mM PEG2k@CP (b) in aqueous NaCl at 25 °C measured with UV (red lines), LS (gray points), and RI (blue lines) detectors. The particle molar masses (red points) and the aggregation number for P2kDU in the nanoparticles (Nagg, green points) were determined for each fraction. Reprinted from [48], Copyright 2020, with permission from Elsevier
Impact of PEG conformations on the in vivo performance of PEGylated CPs
The PEG chain conformation is an important structural factor that determines the in vivo fates of PEGylated nanoparticles [53,54,55]. Cao et al. reported that a denser brush conformation in PEGylated micelles (PEG-b-poly(D,L-lactide) (PLA)) suppressed protein adsorption and improved their blood retention in vivo [56]. However, other studies reported that the PEG density was not dependent on the in vivo PK of the PEGylated nanoparticles. Wang et al. found that rather than PEG, a higher molecular weight PEG in a micelle (PEG-b-poly(ε-caprolactone)) improved blood retention in vivo [57]. Another study on the in vivo fates of PEGylated gold nanoparticles demonstrated a nonsignificant difference in the effects of the PEG brush and mushroom conformations on blood retention [58]. Because of these inconsistent reports, the relationship between the PEG chain conformations and the in vivo fates of the particles remains unclear. Herein, we attempt to clarify the relationship by using PEGx@CP as a model PEGylated nanoparticle.
The PEG chain density can be calculated from NPEG, and the particle core size is determined using SAXS measurements, which thus elucidates the PEG chain conformation for each nanoparticle. We found that the PEG chain density could be controlled by changing the PEG molecular weight of the surfactant PxDU. In PEGx@CP composed of a D4H/decadiene (DD) crosslinked structure in the core, the PEG density decreased with increasing the PEG molecular weight (from 1k to 2k and then to 5k Da), which changed the PEG chain conformation from dense brush, to brush, to mushroom conformations, as shown in Fig. 3 [59]. The changes in PEG chain density could depend on the micelle or nanoemulsion structures that serve as the precursors. These self-assembled nanoparticle morphologies can be determined by using packing parameter theory, which is based on the geometric parameters of the surfactant [60]. According to this theory, the aggregation number of the nanoparticles decreases as the volume of the surfactant headgroup increases while keeping the structure of the hydrophobic moiety the same. As the hydrophilic volume of the head group of PxDU increases with the PEG molecular weight, NPEG decreases in the PEGx@CPs, resulting in a lower PEG chain density.

Schematic illustration of the PEG conformations on PEGx@CPs: mushroom, brush, and dense brush. Reprinted (adapted) with permission from [59]. Copyright 2022 American Chemical Society
We monitored the in vivo PK of intravenously administered PEGx@CPs, by incorporating a fluorescence dye Cy5 via chemical bonding, in tumor-bearing BALB/c mice (Fig. 4a). Since the number concentration of the nanoparticles impacted their blood retention [29], we fixed the particle concentration at one quadrillion nanoparticles, which is beyond the proposed dose threshold of 1 trillion, to eliminate the effect of the particle concentration. As shown in their PK profiles (Fig. 4a), no significant difference was observed for those PEGx@CPs even though their PEG conformations differed from each other. These results suggested that the PEGylated nanoparticles exhibited high blood retention as long as the PEG chains covered the nanoparticles to the extent that the hydrophobic interface was not exposed and induced aggregation.

a In vivo pharmacokinetics of PEGx@CPCy5s (red: PEG1k@CPCy5, blue: PEG2k@CPCy5, green: PEG5k@CPCy5) and the P2kDUNBD micelle (black) after intravenous (IV) administration into mice with a 1.0 quadrillion nanoparticle dosage. b Biodistribution of PEGx@CPCy5s (red: PEG1k@CPCy5, blue: PEG2k@CPCy5, green: PEG5k@CPCy5) after 24 h of IV administration into mice. In vivo drug release kinetics of (c) sCy5 and (d) nCy5 included in PEGx@CPs (red: PEG1k@CP, blue: PEG2k@CP, green: PEG5k@CP) after intravenous administration into mice with a 1.0 quadrillion nanoparticle dosage. The chemical structures of sCy5 and nCy5 are displayed, where the R group is an alkyl chain with an azide, and the details are displayed in Fig. S15. All data are represented as the mean ± standard deviation (n = 5). n.s.: not significant. *P < 0.05 and **P < 0.01 (one-way ANOVA with Tukey’s multiple comparison test). The right images are schematic illustrations describing the relative release kinetics for each dye from PEGx@CPs with various PEG conformations. Reprinted (adapted) with permission from [59]. Copyright 2022 American Chemical Society
We also examined the in vivo release kinetics of physically trapped model drugs, including sulfo-Cy5 (sCy5) and native-Cy5 (nCy5), into PEGx@CP (Fig. 4c, d, respectively). These drugs were encapsulated by the particle during the core-crosslinking reaction, and then the free drug was removed using spin dialysis. To maintain the particle structure, we controlled the loading contents of those drugs at <1.0 wt%. In fact, it was difficult to increase the loading content of this system by >3.0 wt% owing to the absence of strong interactions between those drugs and the particle core, which did not affect the particle structure. This depended significantly on the difference in their PEG conformations. Interestingly, the different polarities of the hydrophilic sCy5 (Fig. 4c) and hydrophobic nCy5 (Fig. 4d) model drugs might be important in determining the in vivo release kinetics of the model drugs. PEG5kCP with a mushroom PEG conformation released sCy5 relatively faster than the other PEGx@CPs. However, the order for the nCy5 release kinetics was the opposite, and PEG5k@CP held the dye relatively longer than the others. This might be related to diffusion of the dyes from the particle core to the water-swollen PEG shell.
The hydrophobic character of nCy5 was similar to those of the hydrophobic components in PEGx@CPs according to the logarithmic values of the predicted octanol/water partition coefficients (miLog P), which were positive and in the range 5−7 [59]. This indicated that nCy5 was homogeneously dispersed in the particle cores. However, the miLog P value of sCy5 was negative, suggesting that it could not be uniformly distributed in the particle core and was probably unevenly distributed at the core–shell interface. Furthermore, water molecules were expected to easily penetrate the particle shell due to it relatively low PEG density and mushroom conformation, which might allow easy removal of the hydrophilic drug from the particle core to the water-swollen shell and the water interface. In other words, the brush and dense brush conformations suppressed diffusion of the hydrophilic dye from the particle core to the water phase owing to chain crowding. In contrast, in hydrophobic drugs, drug diffusion is most likely to be suppressed by the mushroom conformation, which should be swollen with water molecules, since drugs are averse to contact with water. From these perspectives, we concluded that the PEG chain conformation did not significantly affect the PK of PEGylated nanoparticles, which is important in controlling the in vivo drug release kinetics.
The results described above also indicated that in vivo drug release would depend on the particle core composition and the drug polarity. For example, when encapsulating a hydrophilic drug into a hydrophilic particle core, the drug is expected to be uniformly dispersed in the core. In that system, the drug release behavior depends only on drug diffusion from the core to the water interface, which would rely strongly on the polymer chain morphology of the particle surface, as in the experiments described above. However, this drug release behavior may also depend on the polymer on the particle shell and the drug. If the encapsulated drug in the particle core has the potential to interact with the polymer chain, the drug would remain in the particle shell and exhibit drug release behavior that differed from those of the systems described above. In other words, to precisely control drug release behavior in vivo, the molecular design must consider not only the morphology of the polymer chain in the particle shell but also drug–particle core and drug–polymer interactions.
We also need to address the issue of physically trapped drugs in the particle core and their PD. As shown in Fig. 4b, PEGx@CPs were found in all organs, including the liver, at reasonably low levels, whereas the model drugs nCy5 and sCy5 accumulated in all organs at relatively high levels (Fig. 5a, b). If these model drugs were anticancer drugs, the high-level accumulations in healthy organs would cause serious side effects; this indicates that drug encapsulation through physical interactions is not sufficient to retain the drug during blood circulation until it reaches the target sites.

Biodistribution of (a) nCy5 and (b) sCy5 contained in PEGx@CP (red: PEG1k@CP, blue: PEG2k@CP, green: PEG5k@CP) after 24 h of IV administration in mice. All data are represented as the mean ± SD (n = 5). n.s.: not significant. *P < 0.05 (one-way ANOVA with Tukey’s multiple comparison test). Reprinted (adapted) with permission from [59]. Copyright 2022 American Chemical Society
One solution to these undesired PD is to chemically bind the drug into the particle, which also requires a strategy for detaching the drug at the target site. Some strategies utilizing the unique physiological environments of cancer cells have been proposed to address this demand. One of those strategies is to use disulfide bonds to crosslink the cores of the CPs. Disulfide bonds are readily cleaved by glutathione (GSH), which is a tripeptide comprising cysteine, glutamic acid, and glycine and exists in the cytosol. Since the concentration of GSH in cancer cells is higher than that in normal cells, CPs with disulfide-based crosslinked structures internalized into a target cancer cell can be destabilized with GSH, which facilitates diffusion of the physically encapsulated drug in the CPs into the cell. In this strategy, the particle core should have a composition that swells in water; otherwise, the hydrophilic GSH will not reach the particle core and cleave the disulfide-based network. In previous studies, anticancer drugs were physically trapped in disulfide-based network structures using this strategy [61,62,63], which might have caused undesired drug PD, as discussed above. If the drug is also chemically bonded with the disulfide in the particle core, the drug will be stably delivered to the target cancer cell and then released as the particle collapses, which might be an ideal DDS using polymeric CPs.
Zwitterionic amino acid polymer-grafted CPs
Synthesis of glutamine polymer-grafted CPs
As shown above, physical stability in nanoparticles is important for tumor-targeting polymer-grafted nanoparticles because it improves blood circulation and tumor accumulation while covering the particle surfaces with biocompatible polymers such as PEG. Although PEGylated CPs work well until the stage of ambulation into tumor tissues, the therapeutic efficacy might be affected by their limited ability to enter the target cancer cells due to the inertness of PEG [64]. To address this issue, pilot molecules, such as sugars [65], peptides [66], and aptamers [67], have been introduced at the ends of the PEG chains on the nanoparticles, and these can be recognized by specific proteins on the target cancer cells. Unlike these approaches, we utilized zwitterionic amino acid-based polymers (ZAPs), which are as biocompatible as other conventional biocompatible polymers, such as PEG, and exist as zwitterions at neutral pH [68, 69]. In addition to biocompatibility, we took advantage of their cancer-targeting ability. Cancer cells generally require more external nutrients than normal cells to maintain rapid cell proliferation and enhanced intracellular metabolism, so they overexpress amino acid transporters (AATs) [70, 71]. Several studies have found that phenylalanine-, lysine-, and glutamine-based ZAPs are recognized by the AATs on cancer cells and enter the cells via the AAT-mediated endocytosis pathway [72,73,74]. This cancer-targeting function and the zwitterionic natures of the ZAPs allows them to serve as “pilot polymers” in CP-based DDSs to improve tumor delivery.
Herein, we synthesized a poly(glutamine methacrylate) (pGlnMA)-grafted CP (pGlnMA@CP) as shown in Fig. 6 [75]. The hydrophilic pGlnMA in the amphiphilic polymer was prepared via deprotection of the butyloxycarbonyl (Boc) and tertbutyl groups (tBu) in poly(BocGlu(MA)OtBu) (pBGOMA). The poor compatibility between hydrophilic and hydrophobic polymers often makes it challenging to synthesize amphiphilic diblock copolymers. However, owing to the protecting groups, the precursor polymer pBGOMA is miscible in many organic solvents (tetrahydrofuran, chloroform, toluene, etc.), which makes it possible to synthesize pGlnMA-based amphiphilic diblock copolymers easily. The synthesis of pGlnMA@CP was almost identical to that of PEGx@CPs except that tetra-thiol (TT) and decadiene (DD) were used as crosslinkers. In brief, a micellar solution of pGlnMA-b-p(butylmethacrylate-co-allylmethacrylate (BMA)0.9n-co-(AMA)0.1n)n was mixed with a TT/DD solution to form a nanoemulsion. The thiol–ene reaction between TT and DD produced a crosslinked structure, in which the allyl group in the hydrophobic block formed pGlnMA@CP.

Preparation of pGlnMA-grafted core-crosslinked particles (pGlnMA@CP) using thiol–ene click chemistry and nanoemulsions comprising a tetra-thiol compound (TT) and decadiene (DD) stabilized with pGlnMA-b-p(BMA-co-AMA). Reprinted (adapted) with permission from [75]. Copyright 2022 American Chemical Society
Figure 7a shows the SAXS profile of pGlnMA@CP in a 150 mM aqueous NaCl solution. An apparent amplitude at q = 0.3 nm−1 suggested a relatively narrow particle size distribution. These experimental data were reproduced with a spherical model of Gaussian chains, as shown with a solid red line. This analysis confirmed the core–shell structure of pGlnMA@CP with a 24 nm diameter. The particle structure was confirmed with dry- and cryo-TEM observations, as shown in Fig. 7b.

a SAXS profile of pGlnMA@CP in 150 mM aqueous NaCl. The black circles are experimental data, and the red curve was calculated with a Gaussian chain-attached spherical model with aggregates described using the Guinier function. b Dry- and (c) cryo-TEM images of pGlnMA@CP (5.0 mg mL−1) in 150 mM aqueous NaCl. Reprinted (adapted) with permission from [75]. Copyright 2022 American Chemical Society
Cellular uptake and tumor permeability of pGlnMA@CP
Glutamine-based ZAPs are known to recognize alanine serine cysteine transporter 2 (ASCT2) in cancer cells and induce cellular uptake of those polymers into the cells [72]. Using flow cytometry, we investigated the cellular uptake level of Cy5-labeled pGlnMA@CP in the mouse colon cancer cell line (CT26), which expresses ASCT2 [75, 76]. As shown in Fig. 8a–b, pGlnMA@CP was actively internalized into the CT26 cells, while the cellular uptake levels were lower in the presence of BCH (LAT1 inhibitor) or BzlSer (ASCT2 inhibitor). The BzlSer suppressed cellular uptake more than BCH, suggesting that ASCT2 could be responsible for the cellular uptake of glutamine-based ZAPs, as previously reported [74]. However, it would still be premature to conclude that pGlnMA@CP was internalized into the cells via recognition using ASCT2 since BzlSer is not a specific inhibitor for ASCT2 and can bind to other AATs [74]. Thus, we need a more detailed study of this cellular uptake mechanism in the future.

a Flow cytometry (FCM) histogram of CT26 cells incubated with or without pGlnMA@CPCy5 (25 μg mL−1) in the absence (control) or presence of inhibitors, BCH (blue) or BzlSer (green), for 1 h at 37 °C, and (b) relative fluorescence intensity determined using FCM. c Relative fluorescence intensity of CT26 cells incubated with pGlnMA@CPCy5 in the absence (control) or presence of endocytic inhibitors (GS, CP, Ami, or CCD) for 1 h at 37 °C measured using FCM. The effect of low incubation temperature (4 °C) on the cellular uptake of pGlnMA@CPCy5 was evaluated. Data are represented as the mean ± standard deviation (n = 3). n.s.: not significant. *P < 0.05 and *P < 0.005 (one-way ANOVA with Tukey’s multiple comparison test). d Structured illumination microscopy (SIM) images of CT26 cells after treatment with pGlnMA@CPCy5 for 3 h at 37 °C. Bright-field (top panels) and CLSM images (middle panels) of CT26 spheroids incubated with (e) pGlnMA@CPCy5, (f) pPEGMA@CPCy5, (g) pGlnMA@CPCy5 with BzlSer, and (h) pGlnMA@CPCy5 with EXO1 in PBS for 3 h at 37 °C. Scale bar = 200 μm. The location of pGlnMA@CPCy5 is indicated in red. The bottom panels are the fluorescence intensity profiles for the areas indicated by the white dotted lines in the fluorescence images. Reprinted (adapted) with permission from [75]. Copyright 2022 American Chemical Society
As shown in Fig. 8c, the cellular uptake level was significantly reduced at 4 °C, indicating that the nanoparticles were internalized through an energy-dependent endocytosis pathway. Furthermore, the endocytic inhibitors genistein (GS; a caveolae-medicated endocytosis inhibitor) and chlorpromazine (a clathrin-medicated endocytosis inhibitor) inhibited cellular uptake more than the other inhibitors amiloride (Am; a micropinocytosis inhibitor) and CCP (CCP; a phagocytosis inhibitor). This suggested that both the caveolae- and clathrin-mediated endocytosis pathways could be involved in cellular uptake. Figure 8d displays the structured illumination microscopy (SIM) images of CT26 cells treated with pGlnMA@CP in PBS at 37 °C for 3 h. The Cy5 label (red) indicates the subcellular distribution of pGlnMA@CP, while the nuclei and late endosome–lysosomes were stained blue (4′,6-diamidino-2-phenylindole (DAPI)) and green (lysotracker), respectively. The partial colocalization observed in Fig. 8d for pGlnMA@CP and endosome–lysosomes indicates that internalization of pGlnMA@CP into the cell involved an AAT-mediated endocytosis pathway. We also observed scattered red fluorescence signals in the cytosol, implying that pGlnMA@CP escaped from the endosomes. The escape mechanism is still unclear, but the primary amines and carboxylic acid functional groups in the particle shell might interact with the endosome membranes to enable collapse and escape of the particle into the cytosol.
In a tumor-targeted DDS, the polymer-based nanoparticles that accumulate in the tumor tissue via the EPR effect must penetrate deep into the tumor to deliver the encapsulated drugs to the entire tumor. According to a study of tumor penetration using spheroids as the tumor model, neutral polymers, such as PEG [77] and pHPMA [78], did not penetrate deeply into the spheroids, while zwitterionic polymers based on phosphobetaines, sulfobetaines, and carboxybetaines showed relatively deep penetration [79]. The penetration of pGlnMA@CP into spheroids with CT26 was compared with that of a pPEGMA@CP control sample. As displayed in Fig. 8e–f, pPEGMA@CP was stacked on the spheroid surface, whereas relatively high permeability was observed for pGlnMA@CP, which demonstrated a permeability similar to those of other zwitterionic particles. Furthermore, in the presence of BzlSer (Fig. 8g), pGlnMA@CP still deeply penetrated the spheroid, while the cellular uptake at the outer surface was reduced. The penetration behavior did not change in the presence of the exocytosis inhibitor EXO1 (Fig. 8h), indicating that penetration depends on paracellular pathways between cell–cell tight junctions in the spheroid.
PK and PD of pGlnMA@CP
Finally, the in vivo PK and PD of pGlnMA@CP and pPEGMA@CP were compared after intravenous administration into tumor-bearing BALB/c mice. As shown in Fig. 9a, the PK profiles of both samples were almost identical. Both nanoparticles showed relatively longer blood circulation than conventional polymer micelles [35, 46]. However, approximately half of the nanoparticles were eliminated from the blood within 1 h after administration, whereas only >30% of the PEGx@CPs were eliminated within 1 h after administration. This difference might be related to the difference in their core-crosslink structures. The core-crosslinked network structure of D4H/DD in PEG@CPs was synthesized via a hydrosilylation reaction, while the TT/DD network in pGlnMA@CP and pPEGMA@CP was formed via a thiol–ene reaction. Both chemistries are known as “click chemistry,” but different mechanisms are involved, which may have resulted in a difference in the final network structure. The impact of the core-crosslinked structures in polymeric CPs on their PK profiles is an important issue in CP-based DDSs, which will constitute our future research project.

a In vivo pharmacokinetics of pGlnMA@CPCy5 (red) and pPEGMA@CPCy5 (blue) after intravenous (IV) administration into mice. b Biodistribution of pGlnMA@CPCy5 (red) and pPEGMA@CPCy5 (blue) after 24 h of IV administration into mice. Data are represented as the mean ± standard deviation (n = 5). n.s.: not significant. *P < 0.05 and **P < 0.005 (Student’s t test). Reprinted (adapted) with permission from [75]. Copyright 2022 American Chemical Society
The biocompatibility of pPEGMA is known to be similar to or even better than that of PEG [80]. The biocompatibilities of ZAPs were studied by observing the antifouling behaviors of ZAP-grafted surfaces against proteins [69], which were similar to or better than those of other zwitterionic polymer surfaces and PEG surfaces. The comparable blood circulation behaviors of pGlnMA@CP to pPEGMA@CP might be related to the stealth properties of pGlnMA against serum proteins, even in vivo.
In contrast to the PK profiles, a difference was observed in the biodistribution levels of pGlnMA@CP and pPEGMA@CP. First, the level of pGlnMA@CP accumulation in most healthy organs was significantly lower than that of pPEGMA@CP. Although PEG has been used to avoid accumulation of many DDS nanoparticles in the liver, pGlnMA demonstrated a more remarkable ability to inhibit accumulation in the liver than PEG. However, pGlnMA@CP showed higher accumulation in the spleen compared to pPEGMA@CP. The reasons for this are still unclear, but this indicates that a delivery system targeting the spleen might be feasible with pGlnMA-based nanoparticles. Furthermore, the level of pGlnMA@CP accumulation in tumor tissues was approximately twice as high as that of pPEGMA@CP. This difference in their accumulation levels within tumor tissues was due to differences in the subsequent penetration and internalization behaviors of the nanoparticles; the abilities of both pGlnMA@CP and pPEGMA@CP to enter the tumor tissue based the EPR effect were comparable since both show quite identical PK. Another way to explain the tumor accumulation level is that the permeability of pGlnMA into the blood vessels around the tumor was superior to that of pPEGMA, which enhanced the EPR effect of pGlnMA. Thus, pGlnMA has various advantages over pPEGMA and will be used in designing DDS nanoparticles in the future.
Conclusion
This review has summarized recent research trends for polymeric CPs and was focused on their structural properties and functions. The CPs formed from nanoemulsions composed of amphiphilic block copolymers and crosslinkers in the oil phase are useful for preparing physically stable polymeric CPs for application as DDS carriers. The particle structures, including the polymer chain conformations in the particle shell, were readily defined with various characterization methods, such as light scattering (LS), SAXS, and cryo-TEM measurements. For PEGylated nanoparticles, which have been frequently used as DDS carriers, the relationship between the PEG conformation and their in vivo fate has often been discussed. However, in PEGyalted CPs, the PEG conformation does not determine the in vivo PK or PD of the particles, but it is important in determining the in vivo release kinetics of the encapsulated drugs. Instead of PEG-based polymers, we employed pGlnMA as a biocompatible pilot polymer in a polymeric CP with cancer-targeting ability, and we improved the tumor permeability and the specific interactions with the targeted cancer cells, which resulted in relatively high accumulation levels in tumor tissues. Thus far, we have summarized the potential of using polymeric CPs as drug carriers; however, there are still issues that need to be addressed. One major issue arises from the extremely high stabilities of CPs. First, the combination of high structural stability and biologically inert polymers, such as in PEGx@CP, might cause them to remain in the bloodstream for a long time (days, weeks, or even months). In addition, even if strategies are incorporated to promote cellular uptake by the target cancer cells, such as with pGlnMA@CP, the nanoparticles may not be degraded by the biological enzymes in the cell owing to their high structural stability. As discussed above, one potential solution is to employ disulfide-based network structures in the particle cores. Since GSH is relatively overexpressed in cancer cells compared to normal cells, CPs with disulfide-based crosslinked cores could be destabilized in cancer cells. There are many other options for utilizing the specific features of cancer cells to address this issue, such as the relatively low pHs around tumor tissues and overexpression of some specific proteins and enzymes in cancer cells. To summarize this discussion of the design of polymeric CP-based DDSs, the ideal CP should contain biocompatible and actively targetable pilot polymers in the shell and crosslinked structures that can be decomposed in response to biological stimuli at the target sites or in the target cells. Rather than physical encapsulation, the anticancer drugs should chemically bonded to the crosslinked structure with bonds that are cleavable with biological stimuli.
References
van Rijt SH, Sadler PJ. Current applications and future potential for bioinorganic chemistry in the development of anticancer drugs. Drug Discov Today. 2009;14:1089–97. https://doi.org/10.1016/j.drudis.2009.09.003
Ghosh S. Cisplatin: The first metal based anticancer drug. Bioorg Chem. 2019;88:102925 https://doi.org/10.1016/j.bioorg.2019.102925
Heishima K, Sugito N, Soga T, Nishikawa M, Ito Y, Honda R, et al. Petasin potently inhibits mitochondrial complex I–based metabolism that supports tumor growth and metastasis. J Clin Investig. 2021;131:e139933 https://doi.org/10.1172/JCI139933
Carvalho C, Santos RX, Cardoso S, Correia S, Oliveira PJ, Santos MS, et al. Doxorubicin: the good, the bad and the ugly effect. Curr Med Chem. 2009;16:3267–85. https://doi.org/10.2174/092986709788803312.
Saraf S, Jain A, Tiwari A, Verma A, Panda PK, Jain SK. Advances in liposomal drug delivery to cancer: An overview. J Drug Delivery Sci Technol. 2020;56:101549 https://doi.org/10.1016/j.jddst.2020.101549
Lee JS, Feijen J. Polymersomes for drug delivery: Design, formation and characterization. J Contr Release. 2012;161:473–83. https://doi.org/10.1016/j.jconrel.2011.10.005
Liu Y, Castro Bravo KM, Liu J. Targeted liposomal drug delivery: a nanoscience and biophysical perspective. Nanoscale Horizons. 2021;6:78–94. https://doi.org/10.1039/D0NH00605J
Ghezzi M, Pescina S, Padula C, Santi P, Del Favero E, Cantù L, et al. Polymeric micelles in drug delivery: An insight of the techniques for their characterization and assessment in biorelevant conditions. J Contr Release. 2021;332:312–36. https://doi.org/10.1016/j.jconrel.2021.02.031
Hwang D, Ramsey JD, Kabanov AV. Polymeric micelles for the delivery of poorly soluble drugs: From nanoformulation to clinical approval. Adv Drug Delivery Rev. 2020;156:80–118. https://doi.org/10.1016/j.addr.2020.09.009
Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharmacy Pharmacol. 2012;65:157–70. https://doi.org/10.1111/j.2042-7158.2012.01567.x. (acccessed 1/20/2023)
Kataoka K, Harada A, Nagasaki Y. Block copolymer micelles for drug delivery: Design, characterization and biological significance. Adv Drug Delivery Rev. 2012;64:37–48. https://doi.org/10.1016/j.addr.2012.09.013
Tanner P, Baumann P, Enea R, Onaca O, Palivan C, Meier W. Polymeric Vesicles: From Drug Carriers to Nanoreactors and Artificial Organelles. Acc Chem Res. 2011;44:1039–49. https://doi.org/10.1021/ar200036k
Nishimura T, Akiyoshi K. Biotransporting Biocatalytic Reactors toward Therapeutic Nanofactories. Adv Sci. 2018;5:1800801 https://doi.org/10.1002/advs.201800801
Yue J, Liu S, Xie Z, Xing Y, Jing X. Size-dependent biodistribution and antitumor efficacy of polymer micelle drug delivery systems. J Mater Chem B. 2013;1:4273–80. https://doi.org/10.1039/C3TB20296H
Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387–92.
Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Controlled Release. 2000;65:271–84. https://doi.org/10.1016/S0168-3659(99)00248-5
Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6:815–23. https://doi.org/10.1038/nnano.2011.166
Sindhwani S, Syed AM, Ngai J, Kingston BR, Maiorino L, Rothschild J, et al. The entry of nanoparticles into solid tumours. Nat Mater. 2020;19:566–75. https://doi.org/10.1038/s41563-019-0566-2
Kingston BR, Lin ZP, Ouyang B, MacMillan P, Ngai J, Syed AM, et al. Specific Endothelial Cells Govern Nanoparticle Entry into Solid Tumors. ACS Nano. 2021;15:14080–94. https://doi.org/10.1021/acsnano.1c04510
Matsumura Y. Preclinical and clinical studies of NK012, an SN-38-incorporating polymeric micelles, which is designed based on EPR effect. Adv Drug Delivery Rev. 2011;63:184–92. https://doi.org/10.1016/j.addr.2010.05.008
Maeda H. The 35th Anniversary of the Discovery of EPR Effect: A New Wave of Nanomedicines for Tumor-Targeted Drug Delivery—Personal Remarks and Future Prospects. J Personalized Med. 2021;11:229.
Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33:941–51. https://doi.org/10.1038/nbt.3330
D’souza AA, Shegokar R. Polyethylene glycol (PEG): a versatile polymer for pharmaceutical applications. Exp Opin Drug Delivery. 2016;13:1257–75. https://doi.org/10.1080/17425247.2016.1182485
Matsumura Y. 35 years of discussions with Prof. Maeda on the EPR effect and future directions. J Controlled Release. 2022;348:966–9. https://doi.org/10.1016/j.jconrel.2022.06.035
Shi D, Beasock D, Fessler A, Szebeni J, Ljubimova JY, Afonin KA, et al. To PEGylate or not to PEGylate: Immunological properties of nanomedicine’s most popular component, polyethylene glycol and its alternatives. Adv Drug Delivery Rev. 2022;180:114079 https://doi.org/10.1016/j.addr.2021.114079
Fujii S, Sakuragi M, Sakurai K. Characterizing PEG. Chains Tethered onto Micelles and Liposomes Applied as Drug Delivery Vehicles Using Scattering Techniques. Control of Amphiphile Self-Assembling at the Molecular Level: Supra-Molecular Assemblies with Tuned Physicochemical Properties for Delivery Applications, ACS Symposium Series. 1271. American Chemical Society; 2017. p. 115–29. https://doi.org/10.1021/bk-2017-1271.ch005.
Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4:145–60. https://doi.org/10.1038/nrd1632
Klimek L, Novak N, Cabanillas B, Jutel M, Bousquet J, Akdis CA. Allergenic components of the mRNA-1273 vaccine for COVID-19: Possible involvement of polyethylene glycol and IgG-mediated complement activation. Allergy. 2021;76:3307–13. https://doi.org/10.1111/all.14794
Ouyang B, Poon W, Zhang Y-N, Lin ZP, Kingston BR, Tavares AJ, et al. The dose threshold for nanoparticle tumour delivery. Nat Mater. 2020;19:1362–71. https://doi.org/10.1038/s41563-020-0755-z
Liu J, Zeng F, Allen C. In vivo fate of unimers and micelles of a poly(ethylene glycol)-block-poly(caprolactone) copolymer in mice following intravenous administration. Eur J Pharma Biopharma. 2007;65:309–19. https://doi.org/10.1016/j.ejpb.2006.11.010
Ebrahim Attia AB, Yang C, Tan JPK, Gao S, Williams DF, Hedrick JL, et al. The effect of kinetic stability on biodistribution and anti-tumor efficacy of drug-loaded biodegradable polymeric micelles. Biomaterials. 2013;34:3132–40. https://doi.org/10.1016/j.biomaterials.2013.01.042
Wang Y, Pisapati AV, Zhang XF, Cheng X. Recent Developments in Nanomaterial-Based Shear-Sensitive Drug Delivery Systems. Adv Healthcare Mater. 2021;10:2002196 https://doi.org/10.1002/adhm.202002196
Owen SC, Chan DPY, Shoichet MS. Polymeric micelle stability. Nano Today. 2012;7:53–65. https://doi.org/10.1016/j.nantod.2012.01.002
Feiner-Gracia N, Glinkowska Mares A, Buzhor M, Rodriguez-Trujillo R, Samitier Marti J, Amir RJ, et al. Real-Time Ratiometric Imaging of Micelles Assembly State in a Microfluidic Cancer-on-a-Chip. ACS Appl Bio Mater. 2021;4:669–81. https://doi.org/10.1021/acsabm.0c01209
Sun X, Wang G, Zhang H, Hu S, Liu X, Tang J, et al. The Blood Clearance Kinetics and Pathway of Polymeric Micelles in Cancer Drug Delivery. ACS Nano. 2018;12:6179–92. https://doi.org/10.1021/acsnano.8b02830
O’Reilly RK, Hawker CJ, Wooley KL. Cross-linked block copolymer micelles: functional nanostructures of great potential and versatility. Chem Soc Rev. 2006;35:1068–83. https://doi.org/10.1039/B514858H
Liao C, Chen Y, Yao Y, Zhang S, Gu Z, Yu X. Cross-Linked Small-Molecule Micelle-Based Drug Delivery System: Concept, Synthesis, and Biological Evaluation. Chem Mater. 2016;28:7757–64. https://doi.org/10.1021/acs.chemmater.6b02965
Yoo D, Magsam AW, Kelly AM, Stayton PS, Kievit FM, Convertine AJ. Core-Cross-Linked Nanoparticles Reduce Neuroinflammation and Improve Outcome in a Mouse Model of Traumatic Brain Injury. ACS Nano. 2017;11:8600–11. https://doi.org/10.1021/acsnano.7b03426
Gebrie HT, Addisu KD, Darge HF, Birhan YS, Thankachan D, Tsai H-C, et al. pH/redox-responsive core cross-linked based prodrug micelle for enhancing micellar stability and controlling delivery of chemo drugs: An effective combination drug delivery platform for cancer therapy. Biomater Adv. 2022;139:213015 https://doi.org/10.1016/j.bioadv.2022.213015
Parent LR, Bakalis E, Ramírez-Hernández A, Kammeyer JK, Park C, de Pablo J, et al. Directly Observing Micelle Fusion and Growth in Solution by Liquid-Cell Transmission Electron Microscopy. J Am Chem Soc. 2017;139:17140–51. https://doi.org/10.1021/jacs.7b09060
Tian Q, Fei C, Yin H, Feng Y. Stimuli-responsive polymer wormlike micelles. Prog Polymer Sci. 2019;89:108–32. https://doi.org/10.1016/j.progpolymsci.2018.10.001
Lund R, Willner L, Monkenbusch M, Panine P, Narayanan T, Colmenero J, et al. Structural Observation and Kinetic Pathway in the Formation of Polymeric Micelles. Phys Rev Lett. 2009;102:188301 https://doi.org/10.1103/PhysRevLett.102.188301
Lund R, Brun G, Chevallier E, Narayanan T, Tribet C. Kinetics of Photocontrollable Micelles: Light-Induced Self-Assembly and Disassembly of Azobenzene-Based Surfactants Revealed by TR-SAXS. Langmuir. 2016;32:2539–48. https://doi.org/10.1021/acs.langmuir.5b04711
Iijima M, Nagasaki Y, Okada T, Kato M, Kataoka K. Core-Polymerized Reactive Micelles from Heterotelechelic Amphiphilic Block Copolymers. Macromolecules. 1999;32:1140–6. https://doi.org/10.1021/ma9815962
Bontha S, Kabanov AV, Bronich TK. Polymer micelles with cross-linked ionic cores for delivery of anticancer drugs. J Controlled Release. 2006;114:163–74. https://doi.org/10.1016/j.jconrel.2006.06.015
Rijcken CJ, Snel CJ, Schiffelers RM, van Nostrum CF, Hennink WE. Hydrolysable core-crosslinked thermosensitive polymeric micelles: Synthesis, characterisation and in vivo studies. Biomaterials. 2007;28:5581–93. https://doi.org/10.1016/j.biomaterials.2007.08.047
Tanaka R, Arai K, Matsuno J, Soejima M, Lee JH, Takahashi R, et al. Furry nanoparticles: synthesis and characterization of nanoemulsion-mediated core crosslinked nanoparticles and their robust stability in vivo. Polymer Chem. 2020;11:4408–16. https://doi.org/10.1039/D0PY00610F
Matsuno J, Kanamaru T, Arai K, Tanaka R, Lee JH, Takahashi R, et al. Synthesis and characterization of nanoemulsion-mediated core crosslinked nanoparticles, and in vivo pharmacokinetics depending on the structural characteristics. J Controlled Release. 2020;324:405–12. https://doi.org/10.1016/j.jconrel.2020.05.035
Zheng P, McCarthy TJ. D4H/D4V Silicone: A Replica Material with Several Advantages for Nanoimprint Lithography and Capillary Force Lithography. Langmuir. 2011;27:7976–9. https://doi.org/10.1021/la201141k
Fujii S, Yamada S, Matsumoto S, Kubo G, Yoshida K, Tabata E, et al. Platonic Micelles: Monodisperse Micelles with Discrete Aggregation Numbers Corresponding to Regular Polyhedra. Sci Rep. 2017;7:44494 https://doi.org/10.1038/srep44494
Klein M, Menta M, Dacoba TG, Crecente-Campo J, Alonso MJ, Dupin D, et al. Advanced nanomedicine characterization by DLS and AF4-UV-MALS: Application to a HIV nanovaccine. J Pharma Biomed Analy. 2020;179:113017 https://doi.org/10.1016/j.jpba.2019.113017
Écija-Arenas Á, Román-Pizarro V, Fernández-Romero JM. Separation and characterization of liposomes using asymmetric flow field-flow fractionation with online multi-angle light scattering detection. J Chromatogr A. 2021;1636:461798 https://doi.org/10.1016/j.chroma.2020.461798
Yang Q, Jones SW, Parker CL, Zamboni WC, Bear JE, Lai SK. Evading Immune Cell Uptake and Clearance Requires PEG Grafting at Densities Substantially Exceeding the Minimum for Brush Conformation. Mol Pharma. 2014;11:1250–8. https://doi.org/10.1021/mp400703d
Du X-J, Wang J-L, Liu W-W, Yang J-X, Sun C-Y, Sun R, et al. Regulating the surface poly(ethylene glycol) density of polymeric nanoparticles and evaluating its role in drug delivery in vivo. Biomaterials. 2015;69:1–11. https://doi.org/10.1016/j.biomaterials.2015.07.048
Zhao Z, Ukidve A, Krishnan V, Mitragotri S. Effect of physicochemical and surface properties on in vivo fate of drug nanocarriers. Adv Drug Delivery Rev. 2019;143:3–21. https://doi.org/10.1016/j.addr.2019.01.002
Cao Z-T, Gan L-Q, Jiang W, Wang J-L, Zhang H-B, Zhang Y, et al. Protein Binding Affinity of Polymeric Nanoparticles as a Direct Indicator of Their Pharmacokinetics. ACS Nano. 2020;14:3563–75. https://doi.org/10.1021/acsnano.9b10015
Wang J-L, Du X-J, Yang J-X, Shen S, Li H-J, Luo Y-L, et al. The effect of surface poly(ethylene glycol) length on in vivo drug delivery behaviors of polymeric nanoparticles. Biomaterials. 2018;182:104–13. https://doi.org/10.1016/j.biomaterials.2018.08.022
Perry JL, Reuter KG, Kai MP, Herlihy KP, Jones SW, Luft JC, et al. PEGylated PRINT Nanoparticles: The Impact of PEG Density on Protein Binding, Macrophage Association, Biodistribution, and Pharmacokinetics. Nano Lett. 2012;12:5304–10. https://doi.org/10.1021/nl302638g
Kanamaru T, Sakurai K, Fujii S. Impact of Polyethylene Glycol (PEG) Conformations on the In Vivo Fate and Drug Release Behavior of PEGylated Core-Cross-Linked Polymeric Nanoparticles. Biomacromolecules. 2022;23:3909–18. https://doi.org/10.1021/acs.biomac.2c00730
Nagarajan R, Ruckenstein E. Theory of surfactant self-assembly: a predictive molecular thermodynamic approach. Langmuir. 1991;7:2934–69. https://doi.org/10.1021/la00060a012
Li Y, Xiao K, Luo J, Xiao W, Lee JS, Gonik AM, et al. Well-defined, reversible disulfide cross-linked micelles for on-demand paclitaxel delivery. Biomaterials. 2011;32:6633–45. https://doi.org/10.1016/j.biomaterials.2011.05.050
Zhao J, Yan C, Chen Z, Liu J, Song H, Wang W, et al. Dual-targeting nanoparticles with core-crosslinked and pH/redox-bioresponsive properties for enhanced intracellular drug delivery. J Coll Interfac Sci. 2019;540:66–77. https://doi.org/10.1016/j.jcis.2019.01.021
Talelli M, Barz M, Rijcken CJ, Kiessling F, Hennink WE, Lammers T. Core-Crosslinked Polymeric Micelles: Principles, Preparation, Biomedical Applications and Clinical Translation. Nano Today. 2015;10:93–117. https://doi.org/10.1016/j.nantod.2015.01.005. From NLM
Zalba S, ten Hagen TLM, Burgui C, Garrido MJ. Stealth nanoparticles in oncology: Facing the PEG dilemma. J Controlled Release. 2022;351:22–36. https://doi.org/10.1016/j.jconrel.2022.09.002
Miura Y, Hoshino Y, Seto H. Glycopolymer Nanobiotechnology. Chem Rev. 2016;116:1673–92. https://doi.org/10.1021/acs.chemrev.5b00247
Zhong Y, Meng F, Deng C, Zhong Z. Ligand-Directed Active Tumor-Targeting Polymeric Nanoparticles for Cancer Chemotherapy. Biomacromolecules. 2014;15:1955–69. https://doi.org/10.1021/bm5003009
Zununi Vahed S, Fathi N, Samiei M, Maleki Dizaj S, Sharifi S. Targeted cancer drug delivery with aptamer-functionalized polymeric nanoparticles. J Drug Targeting. 2019;27:292–9. https://doi.org/10.1080/1061186X.2018.1491978
Li W, Liu Q, Liu L. Antifouling Gold Surfaces Grafted with Aspartic Acid and Glutamic Acid Based Zwitterionic Polymer Brushes. Langmuir. 2014;30:12619–26. https://doi.org/10.1021/la502789v
Alswieleh AM, Cheng N, Canton I, Ustbas B, Xue X, Ladmiral V, et al. Zwitterionic Poly(amino acid methacrylate) Brushes. J Am Chem Soc. 2014;136:9404–13. https://doi.org/10.1021/ja503400r
Fuchs BC, Bode BP. Amino acid transporters ASCT2 and LAT1 in cancer: Partners in crime. Semin Cancer Biol. 2005;15:254–66. https://doi.org/10.1016/j.semcancer.2005.04.005
Häfliger P, Charles R-P. The L-Type Amino Acid Transporter LAT1—An Emerging Target in Cancer. Int J Mol Sci. 2019;20:2428.
Yamada N, Honda Y, Takemoto H, Nomoto T, Matsui M, Tomoda K, et al. Engineering Tumour Cell-Binding Synthetic Polymers with Sensing Dense Transporters Associated with Aberrant Glutamine Metabolism. Sc Rep. 2017;7:6077 https://doi.org/10.1038/s41598-017-06438-y
Takano S, Sakurai K, Fujii S. Internalization into cancer cells of zwitterionic amino acid polymers via amino acid transporter recognition. Polym Chem. 2021;12:6083–7. https://doi.org/10.1039/D1PY01010G
Leiske MN, Mazrad ZAI, Zelcak A, Wahi K, Davis TP, McCarroll JA, et al. Zwitterionic Amino Acid-Derived Polyacrylates as Smart Materials Exhibiting Cellular Specificity and Therapeutic Activity. Biomacromolecules. 2022;23:2374–87. https://doi.org/10.1021/acs.biomac.2c00143
Fujii S, Sakurai K. Zwitterionic Amino Acid Polymer-Grafted Core-Crosslinked Particle toward Tumor Delivery. Biomacromolecules. 2022;23:3968–77. https://doi.org/10.1021/acs.biomac.2c00803
Zhao L-P, Chen S-Y, Zheng R-R, Kong R-J, Rao X-N, Chen AL, et al. Self-Delivery Nanomedicine for Glutamine-Starvation Enhanced Photodynamic Tumor Therapy. Adv Healthcare Mater. 2022;11:2102038 https://doi.org/10.1002/adhm.202102038. (acccessed 2023/01/29)
Müllner M, Yang K, Kaur A, New EJ. Aspect-ratio-dependent interaction of molecular polymer brushes and multicellular tumour spheroids. Polym Chem. 2018;9:3461–5. https://doi.org/10.1039/C8PY00703A
Nakamura H, Koziolová E, Chytil P, Etrych T, Haratake M, Maeda H. Superior Penetration and Cytotoxicity of HPMA Copolymer Conjugates of Pirarubicin in Tumor Cell Spheroid. Mol Pharma. 2019;16:3452–9. https://doi.org/10.1021/acs.molpharmaceut.9b00248
Fujii S, Takano S, Nakazawa K, Sakurai K. Impact of Zwitterionic Polymers on the Tumor Permeability of Molecular Bottlebrush-Based Nanoparticles. Biomacromolecules. 2022;23:2846–55. https://doi.org/10.1021/acs.biomac.2c00216
Ozer I, Kelly G, Gu R, Li X, Zakharov N, Sirohi P, et al. Polyethylene Glycol-Like Brush Polymer Conjugate of a Protein Drug Does Not Induce an Antipolymer Immune Response and Has Enhanced Pharmacokinetics than Its Polyethylene Glycol Counterpart. Adv Sci (Weinh). 2022;9:e2103672 https://doi.org/10.1002/advs.202103672.
Acknowledgements
This work was supported by JSPS KAKENHI: Grant-in-Aid for Scientific Research (Grant Number 19K15394) and Grant-in-Aid for Scientific Research B (22H01913).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Fujii, S. Polymeric core-crosslinked particles prepared via a nanoemulsion-mediated process: from particle design and structural characterization to in vivo behavior in chemotherapy. Polym J 55, 921–933 (2023). https://doi.org/10.1038/s41428-023-00793-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41428-023-00793-6
This article is cited by
-
Design of functional soft interfaces with precise control of the polymer architecture
Polymer Journal (2024)
