
Abstract
Biocomposites of poly(lactic acid) (PLA) and pectin, which are low-cost organic materials, were prepared using an internal mixing machine in various pectin contents, i.e., 2, 4, 6 and 8% w/w. When pectin was added as a nucleating agent, the mechanical properties of the biocomposites, such as tensile and impact testing, were considerably improved, particularly following the annealing process. In addition, the PLA–pectin annealed at 4% w/w showed the highest strength and thermal stability. This can be explained by the fact that PLA containing 4% pectin by weight had the best dispersion, as indicated by scanning electron microscopy (SEM) and synchrotron-based 2D chemical mapping FT-IR. Moreover, pectin not only serves as a reinforcing material to improve mechanical characteristics but also aids in the crystallization of PLA, which was confirmed by in situ synchrotron-based wide-angle X-ray scattering (SR-WAXS). The crystallization rate and crystallinity were maximum at 8% w/w pectin addition according to the SR-WAXS results. This shows that pectin dispersion is the most important factor in determining the mechanical and thermal properties of biocomposites.
Similar content being viewed by others


Introduction
Synthetic polymers such as low-density polyethylene, poly(vinyl chloride), polybutylene and copolymers of ethylene with vinyl acetate are now used in the food industry, medication packaging, seed coatings, and controlled-release fertilizer applications. Despite the fact that these materials pose issues such as contamination during harvesting, removal challenges, and pollution, they are nevertheless used. Therefore, biodegradable polymers such as poly(butylene succinate), poly(3-hydroxybutyrate) (PHB), poly(ε-caprolactone), and poly(lactic acid) (PLA) have been widely employed [1,2,3,4].
PLA is a biodegradable polymer made from renewable biomass, such as sugar, corn, beet and cassava, which can be made by allowing microorganisms to be produced. When this biopolymer degrades, it transforms into an inert humus-like material that is less hazardous to the environment and blends nicely with natural soil [5, 6]. However, the problems of PLA include its mechanical brittleness, flexibility, long degradation periods and high production costs.
Several strategies can be used to improve the impact strength and degradation rate. Blending is a typical technique for improving polymer qualities by combining it with other materials. Various substances have been used as the nucleating agent in the blending process to improve the crystallinity of PLA [7,8,9,10], including hydrophilic polymers that create physical hydrogels and cause a wide variation in physical and mechanical properties. Several polysaccharide systems, such as cellulose, can also substantially reduce the total cost while improving the quality of the physical structure and brittleness of their composites [11, 12]. Adding these organic substances not only improves the nucleation of PLA but also creates desirable biocomposites that are environmentally friendly and can completely degrade.
Pectin is a hydrophilic polymer that is abundantly available from an agricultural resource and has been recently considered as an alternative addition to producing biopolymer blending. Pectin is a part of natural complex structural polysaccharides that give the shape of soft, nonwoody sections of plant cell walls in vegetables and fruits. It is also one of the most common dietary fiber sources in human nutrition. On various sugar residues, pectin has sophisticated functional groups, such as carboxylic groups, methyl ester groups and methyl amide groups. The hydrophilicity of pectin can be exploited to speed up the decomposition of some degradable hydrophobic polymers [13]. Microorganisms or enzymes can hydrolyze it into glucose, which is then converted into carbon dioxide and water [14]. Therefore, pectin is an ideal material to modify or mix with PLA to increase the structure and biodegradability of PLA–pectin composites. However, only a few studies have investigated the role of pectin on the mechanical properties and structures of PLA biocomposites, especially the crystallinity in heterogeneous nucleated composites.
In this work, a series of PLA–pectin composites were characterized by extrusion and mold injection techniques for mechanical tests. The morphology and dispersion of the pectin were examined by scanning electron microscopy (SEM) and synchrotron-based 2D chemical mapping FT-IR. Moreover, the crystallization kinetics of the biocomposites were traced during isothermal treatment via in situ SR-WAXS.
Experimental setup
PLA–pectin blending preparation
Injection grade PLA (2021D) was provided by Natural Work (USA), while pectin was provided by Fluka (Switzerland). Various ratio compositions between PLA and pectin (100:0, 98:2, 96:4, 94:6 and 92:8% w/w) were prepared, and then the mixture pellets were dried in an oven at 70 °C for 8 h to remove humidity in materials before mixing. The mixture pellets were blended using an internal mixer (Haake Rheomix 6000p, Germany) at a speed of 50 rpm at 180 °C for 20 min. After that, the blends were cooled by air for 30 min before grinding with a mechanical grinder (Retsch grinder machine, Thailand). The samples for tensile and impact testing were injected into a mold (Gotech model GT-7014-A30, USA) at 180 °C. The blends were preheated for 10 min and then pressed for 10 min. After that, the samples were cooled at room temperature.
Annealing process
To increase the strength of the polymer material, an annealing process was introduced to alter the polymer structure and change the physical properties. After injection into the mold, the biopolymer specimens were kept at room temperature for a day before the annealing process. The biopolymer specimens were heated from room temperature (25 °C) to 100 °C and then hold for 60 min. After that, the specimens were slowly cooled to room temperature for 24 h (Fig. 1) [15].

Thermal step for the annealing process of PLA composite to enhance the crystallinity
Tensile testing
The tensile testing samples of the samples were prepared by pressing the blends in a dogbone-shaped stainless steel mold. Tensile testing was performed according to the ASTM D638 (1998) standard test method. Standard type I specimens with a thickness of 4 mm, a width at a narrow section of 3.18 mm, an overall width of 9.53 mm, a gauge length of 7.62 mm and an overall length of 63.5 mm were prepared. The tensile properties of the samples were determined using a universal testing machine (Instron model 5569, Chicago, USA) with a load cell of 5 kN and a crosshead speed of 1 mm/min at 25 °C. The testing was performed in triplicate. Tensile force and elongation were recorded. The elongation at break, elastic modulus and tensile toughness were determined.
Impact testing
The impact testing specimens were prepared by pressing the blends in a rectangular stainless steel mold. The specimens had a width of 12.7 mm, a length of 64 mm and a depth of 4 mm.
All specimens were stored overnight at room temperature after that. The notched Izod impact test was performed according to the ASTM 256 standard test method using an Atlas testing machine (model BPI, USA). A pendulum energy of 2.7 J was used at 25 °C. The impact toughness (J/m2) was reported by the failure energy divided by the samples across a section area after being notched. This reflects the amount of energy absorption from the beginning mechanical load to the final fracture. Five specimens were tested for each composition.
Scanning electron microscopy (SEM)
The morphology of the impact fracture surfaces of PLA and PLA–pectin blends both with and without the annealing process was observed using scanning electron microscopy (SEM, JEOL model JSM 6400, Japan) at room temperature. The fracture surface samples were coated with gold for 3 min before analysis. An acceleration voltage of 20 kV was used to collect SEM images of the sample.
Thermal properties
The thermal properties of PLA and PLA–pectin at various ratios were determined using thermogravimetric analysis (TGA, TA Instrument; model SDT 2960, USA). Each sample was heated from room temperature to 600 °C at a rate of 20 °C/min. The thermal decomposition temperature of each sample was examined under a nitrogen atmosphere.
Synchrotron radiation wide-angle x-ray scattering (SR-WAXS)
To determine the development of crystallization in the PLA–pectin composite, SR-WAXS at a beamline of 1.3 W of 1.2 A GeV synchrotron light source at Synchrotron Light Research Institute, SLRI, was used. An X-ray at 9 keV (wavelength 1.3776 Å) from a multipole wiggler and a double multilayer monochromator was delivered to track in situ structural evolution [16]. An experimental station equipped with a Rayonix SX165 CCD detector was set up for a flight tube length of 120 mm. From the information of the TGA experiment, the SR-WAXS measurements were performed at 100 °C to investigate the development of PLA crystals under isothermal conditions, and the X-ray scattering pattern was recorded every 1 min with 30 s of exposure time.
Synchrotron-based fourier transform infrared spectroscopy (FT-IR)
FTIR data were collected at the infrared beamline (BL4-1 IR Spectroscopy and Imaging) at SLRI. Spectra were acquired with a Vertex 70 FTIR spectrometer coupled with an IR microscope with a mercury cadmium telluride detector cooled in liquid nitrogen. Data were collected from 10 square grids, and the detector area for each measurement was ~20 × 20 µm2, thus covering a sample area of 200 × 200 µm2 at a resolution of 4 cm−1 with 64 scans co-added. Spectral acquisition and instrument control were performed using OPUS 7.5 software (Bruker Optics Ltd., Ettlingen, Germany).
Results and discussion
Tensile properties
The results of the tensile properties are shown in Fig. 2. When comparing the tensile strength of pure PLA before and after annealing, the pure PLA had a higher tensile strength after annealing. The result of the PLA tensile test before annealing was found at 54.2 MPa. The tensile strength was enhanced to 59.9 MPa following the annealing process. This improvement in the tensile strength is due to a polymer recrystallization process that can create microspherical crystal structures during the annealing process [17, 18]. Moreover, the PLA–pectin composite demonstrated that the highest tensile strength was 2.5 times that of pure PLA with 4% pectin added. The increase in impact strength value partly resulted from the recrystallization process, as previously mentioned. This might be related to subsequent chemical interactions between pectin and PLA, especially when the composite contains 4% pectin. On the other hand, the higher levels of pectin addition at 6 and 8% resulted in PLA–pectin composites with lower tensile strength. Nevertheless, the annealing process increases the tensile strength in PLA–pectin composites with pectin contents of 6 and 8% compared to composites without annealing at the same pectin content. The decline in tensile strength at higher pectin contents of 6 and 8% can be affected by the larger grain particles of pectin in the PLA matrix. Li and Favis [19] showed that excessive amounts of compatibilizer higher than 5% w/w can create interface saturation, which does not improve tensile characteristics. In addition, larger spherulites also generate brittleness with low energy absorption during the debonding process following annealing [20, 21].

Tensile strength of pure PLA, PLA–pectin 2%, PLA–pectin 4%, PLA–pectin 6% and PLA–pectin 8% compared between annealing and nonannealing processes
Impact testing
The results of the impact reinforcement before the annealing process are shown in Fig. 3.

Impact strength of pure PLA, PLA–pectin 2%, PLA–pectin 4%, PLA–pectin 6%, and PLA–pectin 8% compared between annealing and nonannealing processes
The impact strength of the PLA–pectin composites was shown to decrease with increasing pectin concentration. The impact strength was reduced to 4.6 KJ/m2 compared to 7.0 KJ/m2 for pure PLA. This might be the effect from the increased level of heterogeneous nucleation between PLA and the pectin matrix that was provided with the gap in the structure and destroyed the composite structure under impact force [22, 23]. In contrast, the PLA–pectin composite improved its impact strength after the annealing process. The recrystallization process plays an important role in improving the impact strength to 11.2 KJ/m2 when compared to nonannealing of pure PLA at 7.0 KJ/m2. The result of 2% pectin addition provided impact strength up to 12.5 KJ/m2 higher than that of annealed bare PLA. The highest impact strength of the composite was found with 4% pectin addition up to 16.1 KJ/m2. The higher impact strength might be related to the improvement of the crystallinity matrix and the spherulitic morphologies of the composite [24]. However, the impact strength decreased when increasing the pectin content up to 6 and 8%. A large number of pectin particles could be created at the larger grain boundaries of spherulites and provide a more brittle composite structure [25].
Morphology
SEM micrographs of the tensile fracture surface of pure and PLA–pectin composites (nonannealing process) are presented in Fig. 4. The surfaces after tensile tests were smooth, with no stack cracked layer appearing, indicating that the pectin had been completely embedded and provided the high interfacial matrix in PLA. These composites would not have formed a crystalline structure without the annealing process (Fig. 4a–e).

SEM micrographs of tensile fractured surfaces for the nonannealing process. a pure PLA, (b) PLA–pectin 2%, (c) PLA–pectin 4%, (d) PLA–pectin 6%, and (e) PLA–pectin 8%
A comparison between the surfaces of pure PLA before and after the annealing process is shown in Fig. 5. The PLA surface exhibits steady crack and propagation after the annealing process (Fig. 5a, b). However, the stack cracked line surface occurred when pectin was added to the PLA matrix (Fig. 5c–f). The fractured surface of the stack after the tensile test revealed that the direction obstructed the crack from the crystalline stack [26,27,28].

SEM micrographs of tensile fractured surfaces of pure PLA before the annealing process. a Pure PLA after the annealing process (b), PLA–pectin 2% after crystallization (c), PLA–pectin 4% after crystallization (d), PLA–pectin 6% after crystallization (e) and PLA–pectin 8% after crystallization (f). Red arrows show the crystalline stack after the annealing process
Thermal stability
TGA can obtain important information on thermal decomposition from polymeric material structures by investigating the weight loss of annealing polymers in a temperature range between 25 and 600 °C [29]. The TGA curves for pure PLA and PLA–pectin composites as a function of temperature are shown in Fig. 6. The thermal stability of pure PLA demonstrated single-step weight loss from 230 to 325 °C. The reason for this is that PLA is hydrophobic, and no initial weight loss from moisture phenomena occurs. The TGA results of PLA–pectin composites at 2 and 4% were demonstrated to be nearly similar to those of pure PLA; however, the thermal stabilities were shifted toward lower temperatures. This information corresponds to other physical properties. The deterioration of the composite material with 6 and 8% pectin occurred in three main stages. The moisture content was eliminated by the initial temperature during the melting process over 100 °C, which was the first step of degradation. Disintegration of the pectin composite was shown in the second step at 50% weight loss. The polysaccharide linkage and major chain structure might be destroyed by the depolymerization process. Evaporative products such as CO2, CO, CH4, and organic compounds were released in the last step at ~30% weight loss. The organic compound composite residue was found in the form of char residue PLA [30]. These several steps of degradation and the volatile residue of the composite indicated that pectin is a significant factor in the thermal stability of this composite. Pectin is involved in strength formation by causing interfacial bonding between PLA and pectin. The overabundant pectin at 6 and 8% demonstrated the rupture of glycosidic linkages, the major compounds of pectin, at ~30% weight reduction. The presence of char residue in the sample following the TGA experiment indicated incomplete degradation.

TGA of PLA–pectin 2%, PLA–pectin 4%, PLA–pectin 6 and PLA–pectin 8%
SR-WAXS
The structural evolution and crystallization of pure PLA and PLA–pectin composites at 2, 4, 6 and 8% w/w were shown in an isothermal crystallization experiment with in situ SR-WAXS measurements at 100 °C. 2D-scattering patterns were recorded every 1 min with 30s of exposure time. An example of a series of scattering patterns elucidating the structural evolution in PLA–pectin in the 4% composite is shown in Fig. 7. After that, the intensities of the scattering patterns of each condition were analyzed by SAXSIT software developed by SLRI. The 1D profile was extracted by radially averaging the intensity from φ = 0° to φ = 270°. Only the plot of crystallization evolution analyzed from the SR-WAXS scattering patterns under the best conditions of the PLA-pectin composite at 4% is shown in Fig. 8 due to space limitations. Furthermore, the evolution of crystallinity for each pectin-added composite was estimated using only data from the peaks at 14.8°, 16.6°, 19.0°, and 22.3°, which were contributed from the α-phase of PLA. The areas under these peaks were fitted with the pseudo-Voigt function and then divided by the total area under all curves and the background of the halo-amorphous peaks fitted with a Gaussian distribution function to elucidate the crystallinities of the biocomposites, as plotted in Fig. 9. The crystallinity of pectin-added composites clearly exhibited an improvement in both the quantity and time of PLA crystallization. While pure PLA crystallized gradually over time, various composites with pectin added crystallized rapidly and saturated in 6–8 mins. While pure PLA and PLA–pectin 2% have not fully crystallized in the experiment, PLA–pectin 4%, 6 and 8% can reach their saturation with crystallization halftime (t1/2) at 5.83, 5.82 and 5.48 min, respectively. The plot also showed that with the increase in pectin added into PLA, a higher level of crystallinity is observed. In addition, the crystal sizes of the PLA–pectin composites have been determined using the Debye–Scherrer equation [31, 32] as in [Eq. 1];
where τ is the size of the crystallites, K is the Scherrer constant (K = 0.89), λ is the wavelength of the X-ray in SR-WAXS (0.13776 nm), and β is the full width at half maximum of the peaks. The result showed that the sizes of the crystallites at each PLA–pectin composite can be comparably varying in the range of 12.1–13.0 nm.

Example of a series of scattering patterns elucidating the structural evolution in the PLA–pectin 4% w/w composite at 100 °C. The series is presented at 2 min intervals to emphasize the evolution of the composite

Crystallization evolution was analyzed from the SR-WAXS scattering patterns under the best conditions of the PLA–pectin composite at 4%. The plots analyzed from the WAXS recorded every 1 min at a constant temperature of 100 °C

Development of crystallinity of pectin-added PLA composites vs. time. The crystallization was analyzed from the SR-WAXS experiment
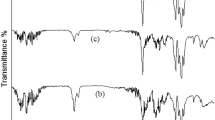
FT-IR
From the synchrotron ATR-FTIR, the average spectra of each group of experiments were determined to represent the FT-IR spectra of pectin, PLA, and PLA–pectin composites at 110 °C (blending temperature) and 180 °C (completely decomposed temperature). The temperature of thermal decomposition was chosen when the composites appeared in dark brown shade. The chemical bonding between PLA and pectin occurred, as seen in the shape, and band peaks from PLA at 1178 and 1127 cm−1 disappeared after blending. The characteristic band of the C=O carbonyl also changed as it broadened (1780–1740 cm−1), indicating that the interaction occurred, which could be from hydrogen bonding with PLA and pectin. The broadening band in PLA–pectin also occurs due to the chemical environments observed by different intermolecular bonding (Fig. 10) [33,34,35]. With the high brightness of SR, we can measure the sample at high spatial resolution with a high signal-to-noise ratio. Therefore, the differences between each sample were clearly elucidated.

Synchrotron-based FT-IR spectrum of 1900–900 cm−1 region of the commercial pectin, pure PLA–pectin 110 and 180 °C, respectively
The peak for the hydroxyl group was exhibited at ~3436 cm−1, which reflected typical water absorption and is shown elsewhere. The pectin region bands situated in the range of 1000–2000 cm−1 were caused by the signal of galacturonic acid. The functional groups of COO− and COOR corresponded to 1735 and 1617 cm−1, respectively (Fig. 10). The characteristic bands of C-O-C vibrations (glycosidic linkages) were observed at 1149 and 1045 cm−1, as mentioned in [36]. Further investigation showed that the heterogeneous distribution of pectin in the mapping showed major components in the composite (Fig. 11). The FT-IR chemical mapping image was analyzed to discover the distribution of pectin by using the characteristic absorption bands of the pectin composite phase. In this research, the absorption bands at 1633 cm−1 were used to characterize the N-H group of pectin in the matrix. The pectin was dispersed over the whole measuring area with a particle size of ~80–100 µm, as seen in Fig. 11. The 4% pectin added to the PLA-pectin composite is consistently and thoroughly distributed in the PLA matrix (Fig. 11b). On the other hand, the excessive amount of pectin caused the agglomeration shown in Fig. 11c and d. Consistent with the morphology results obtained via SEM and the mechanical properties obtained via tensile/impact testing, the influence of pectin dispersion in the PLA matrix plays an important role in the mechanical properties of the PLA–pectin composite. While the pectin was perfectly dispersed in the composite, it induced a perfect crystalline structure. Nevertheless, the excessive amount of pectin composite provided contradicts this result. Oversaturated pectin formed agglomerates and reduced the mechanical characteristics of the composites. This may lead to pectin debonding during mechanical testing or the poor interaction between pectin and PLA in the composites.

2D chemical imaging scanning dispersion of pectin in biopolymer blending, PLA–pectin 2%. a PLA–pectin 4% (b), PLA–pectin 6% (c) and PLA–pectin 8% (d) composites generated from the integrated area under the pectin specific absorption band (N-H group). The color scale is a rainbow scale with blue (low intensity) and pink (high intensity)
Conclusion
The results of biocomposites with various pectin ratios added to PLA polymer were investigated. An internal mixer and twin-screw extrusion were used to successfully produce the composites, which were then injected into the mold. Tensile testing was carried out, and it was discovered that adding up to 4% pectin to the PLA biocomposite can improve its tensile strength. For the impact testing, the impact strength decreases at various pectin contents. After the annealing process, the tensile and impact strengths both increased and reached a maximum at a pectin content of 4% w/w. The morphologies of the composites were elucidated using SEM. The result shows that adding pectin to composites gives them a smooth surface, especially after annealing. SEM also showed the highest amount of crystalline lamellar stacking at the 4% pectin concentration. The TGA experiment showed that the thermal decomposition temperature of the composites decreased while the pectin content in the PLA/pectin composite increased. To investigate the crystallization of the biocomposites, the SR-WAXS experiment was explored. The SR-WAXS results demonstrate that as the pectin content increases, the crystallinities are dramatically increased, and the crystallization is accelerated. To further understand pectin dispersion in PLA composites, researchers used synchrotron-based FT-IR to show that the pectin particles caused a highly heterogeneous nucleation dispersion and enhanced the crystallinity of the composite. However, when the pectin level was oversaturated by up to 6–8% w/w, some agglomerations developed and caused a decrease in mechanical properties. Therefore, pectin addition not only improves the mechanical and thermal properties of PLA composites, especially after annealing, but also widens PLA composite utilization, as it has become completely “green” to use pectin as a bionucleating agent.
References
Arrieta MP, Fortunati E, Dominici F, Rayón E, López J, Kenny JM. Multifunctional PLA-PHB/cellulose nanocrystal films: processing, struc- tural and thermal properties. Carbohydr Polym. 2014;107:16–24. https://doi.org/10.1016/j.carbpol.2014.02.044. PMID 24702913.
Basbagill J, Flager F, Lepech M, Fischer M. Application of life-cycle assessment to early stage building design for reduced embodied environmental impacts. Build Environ. 2013;60:81–92. https://doi.org/10.1016/j.buildenv.2012.11.009
Roes AL, Patel MK. Ex-ante environmental assessments of novel technologies improved caprolactam catalysis and hydrogen storage. J Clean Prod. 2011;19:1659–67. https://doi.org/10.1016/j.jclepro.2011.05.010
Jem KJ, Tan B. The development and challenges of poly (lactic acid) and poly (glycolic acid). Adv Ind Eng Polym Res. 2020;3:60–70. https://doi.org/10.1016/j.aiepr.2020.01.002
Woodruff MA, Hutmacher DW. The return of a forgotten polymer-polycaprolactone in the 21st century. Prog Polym Sci. 2010;35:1217–56. https://doi.org/10.1016/j.progpolymsci.2010.04.002
Fu W, Xu X, Wu H. Mechanical and biodegradable properties of L-lactide-grafted sisal fiber reinforced polylactide composites. J Reinf Plast Compos. 2014;33:2034–45. https://doi.org/10.1177/0731684414552684
Liao R, Yang B, Yu W, Zhou C. Isothermal cold crystallization kinetics of polylac- tide/nucleating agents. J Appl Polym. 2006;104:310–7.
Li H, Huneault MA. Effect of nucleation and plasticization on the crystallization of poly(lactic acid). Polymer 2007;48:6855–66. https://doi.org/10.1016/j.polymer.2007.09.020
Lotz B, Li G, Chen X, Puiggali J. Crystal polymorphism of polylactides and poly (Pro- alt-CO): the metastable beta and gamma phases. Formation of homochiral PLLA phases in the PLLA/PDLA blends. Polymer. 2017;115:204–10. https://doi.org/10.1016/j.polymer.2017.03.018
Tang Z, Zhang C, Liu X, Zhu J. The crystallization behavior and mechanical properties of polylactic acid in the presence of a crystal nucleating Agent. J Appl Polym Sci. 2012;125:1108–15. https://doi.org/10.1002/app.34799
Fortunati E, Armentano I, Zhou Q, Iannoni A, Saino E, Visai L, et al. Multifunctional bionanocomposite films of poly(lactic acid), cellulose nanocrystals and silver nanoparticles. Carbohydr Polym. 2012;87:1596–605. https://doi.org/10.1016/j.carbpol.2011.09.066
Shazleen SS, Yasim-Anuar TAT, Ibrahim NA, Hassan MA, Ariffin H. Functionality of Cellu- lose nanofiber as Bio-Based Nucleating Agent and Nano-Reinforcement Material to Enhance Crystallization and Mechanical Properties of polylactic acid nanocomposite. Polymers 2021;13:389 https://doi.org/10.3390/polym13030389. PMID 33513688
Alves VD, Castelló R, Ferreira AR, Costa N, Fonseca IM, Coelhoso IM. Barrier properties of carrageenan/pectin biodegradable composite films. Procedia Food Sci. 2011;1:240–5. https://doi.org/10.1016/j.profoo.2011.09.038
Einhorn-Stoll U, Kunzek H. The influence of storage conditions heat and humidity on conformation, state transitions and degradation behavior of dried pectins. Food Hydrocoll. 2009;23:856–66. https://doi.org/10.1016/j.foodhyd.2008.05.001
Claire B, Andrew A, Jacek A, Manjusri M, Amar KM.Improving the Impact Strength and Heat Resistance of 3D Printed Models: structure, Property, and processing Correlation- ships during Fused Deposition Modeling (FDM) of poly (Lactic acid). ACS Sustain Chem Eng.2018;3:4400
Rungswang W, Jarumaneeroj C, Chivatanasoontorn V. Application of Small-Angle X-ray Scattering (SAXS) via Synchrotron Radiation for Structural Analysis of Semi-Crystalline Polymer: study Cases from Polypropylene for Industrial Usage. Suranaree J Sci Technol. 2015;22:265–76.
Frone AN, Berlioz S, Chailan JF, Panaitescu DM. Morphology and thermal proper- ties of PLA-cellulose nanofibers composites. Carbon Polym. 2013;91:377–84.
Nagarajan V, Mohanty AK, Misra M. Sustainable green composites: value addition to agricultural residues and perennial grasses. ACS Sustain Chem Eng. 2013;1:325–33. https://doi.org/10.1021/sc300084z
Li J, Favis BD. Strategies to measure and optimize the migration of the interfacial modifier to the interface in immiscible polymer blends. Polymer. 2002;43:4935–45. https://doi.org/10.1016/S0032-3861(02)00277-X
Uday KC, Dabade RRG, Rajesham BM, Tensile S. properties of sun hemp, banana and sisal fiber reinforced with polyester composites. J Reinf Plast Compos. 2007;26:1043–50.
Zhang Z, Cui H. Biodegradability and biocompatibility study of poly(chitosan-glacticacid) scaffolds. Molecules 2012;17:3243–58. https://doi.org/10.3390/molecules17033243. PMID 22418927
Vuillaume PY, Elkoun S, Robert M. Physical and mechanical properties of PLA composites reinforced by TiO2 grafted flax fibers. F, R Mater Des. 2016;106:295–304.
Xian XJ, Choy CL. Impact resistance and damage evaluation of carbon fiber reinforced modified bismaleimide composites. J Reinf Plast Compos. 1994;12:1135–53.
Alix S, Colasse L, Morvan C, Lebrun L, Marais S. Pressure impact of autoclave treatment on water sorption and pectin composition of flax cellulosic-fibres. Carbohydr Polym. 2014;102:21–9. https://doi.org/10.1016/j.carbpol.2013.10.092. PMID 24507251
Nagarajan J, Krishnamurthy NP, Nagasundara Ramanan R, Raghunandan ME, Galanakis CM, Ooi CW. C.W.A facile water-induced complexation of lycopene and pectin from pink guava by-product: extraction, characterization and kinetic studies. Food Chem. 2019;296:47–55. https://doi.org/10.1016/j.foodchem.2019.05.135. PMID 31202305
Prakalathan K, Mohanty S, Nayak SK. Polylactide modified layered silicates nanocom- posites: a critical analysis of morphological, mechanical and thermal properties. J Reinf Plast Compos. 2012;31:1300–10. https://doi.org/10.1177/0731684412457597
Siengchin S, Pohl T, Medina L, Mitschang P. Structure and properties of flax/polylactide/alumina nanocomposites. J Reinf Plast Compos. 2013;32:23–33. https://doi.org/10.1177/0731684412464586
Sung SH, Chang Y, Han J. Development of polylactic acid nanocomposite films re- inforced with cellulose nanocrystals derived from coffee silverskin. Carbohydr Polym. 2017;169:495–503. https://doi.org/10.1016/j.carbpol.2017.04.037. PMID 28504172
Zhu BL, J M,J,W. Thermal dielectric and compressive properties of hollow glass microsphere filled epoxy-matrix composites. Reinf Plast. 2012;31:1311–26.
Bledzki AK, Jaszkiewicz A, Scherzer D. Mechanical properties of PLA composites with man-made cellulose and abaca fibres. Composites. 2009;40:404–12. https://doi.org/10.1016/j.compositesa.2009.01.002
Masoud A, Mohammad K, Seyed MA, Milad J. Poly lactic acid-akermanite composite scaffolds prepared by fused filament fabrication for bone tissue engineering. J Mater Res. 2020;9:14540.
Liu Y, Jiang S, Yan W, He M, Qin J, Qin S, et al. Crystallization morphology regulation on enhancing heat resistance of polylactic acid. Polymers. 2020;12:1563 https://doi.org/10.3390/polym12071563
Jia L, et al. Crystallization and fame-retardant properties of polylactic acid composites with polyhedral octaphenyl silsesquioxane. Polym Adv Technol. 2019;30:648–65.
Niu YQ, Zhu Y, Gao R, Yu W, Li L, Xu K. Synthesis, characterizations and biocompatibility of novel block polyurethanes based on poly(lactic acid) (PLA) and poly(3-hydroxybutyrate-co-4- hydroxybutyrate) (P3/4HB). J Inorg Organomet Polym Mater. 2015;25:81–90. https://doi.org/10.1007/s10904-014-0081-5
Martinez-Felipe A, Brebner F, Zaton D, Concellon A, Ahmadi S, Piñol M, et al. Molecular recognition via hydrogen bonding in supramolecular complexes: a fourier transform infrared spectroscopy study. Molecules 2018;23:2278 https://doi.org/10.3390/molecules23092278. PMID 30200584
Proctor GRA. Determination of pectin degree of esterification by diffuse reflectance Fourier transform infrared spectroscopy. Food Chem. 2000;68:327–32. https://doi.org/10.1016/S0308-8146(99)00191-0
Acknowledgements
The author would like to thank Suranaree University of Technology for supporting this research and Synchrotron Light Research Institute (Public organization) Nakhon Ratchasima for grant support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Satsum, A., Busayaporn, W., Rungswang, W. et al. Structural and mechanical properties of biodegradable poly(lactic acid) and pectin composites: using bionucleating agent to improve crystallization behavior. Polym J 54, 921–930 (2022). https://doi.org/10.1038/s41428-022-00637-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41428-022-00637-9
This article is cited by
-
A review on bio-based polymer polylactic acid potential on sustainable food packaging
Food Science and Biotechnology (2024)
-
Optimising the mechanical properties of additive-manufactured recycled polylactic acid (rPLA) using single and multi-response analyses methods
The International Journal of Advanced Manufacturing Technology (2023)
-
The effects of polylactic acid-whey protein isolated bi-layer film incorporated with ZnO nanoparticles on the quality of common carp Cyprinus carpio
Journal of Food Measurement and Characterization (2023)
-
Evaluation of pectin extractions and their application in the alkaline Maillard reaction
Scientific Reports (2022)
