
Abstract
The precise integration of functional materials, such as organic–inorganic hybrid building blocks or π-conjugated units, is a key issue in the development of bio- and nanoelectronic devices. This focus review highlights the author’s recent study of the nanoscale assembly of polymer materials based on the Langmuir–Blodgett (LB) technique, which allows high-density integration of functional units. (1) The nanoassembly of silsesquioxane-containing polymers can be used to produce uniform monolayers, which are precursors of ultrathin SiO2 films with controlled porosity. The SiO2 films were used as resistive switching devices with low set/reset voltages. (2) The nanoassembly of inorganic nanoparticles and metal-organic framework films on polymer films demonstrates the importance of the spatial alignment of anchoring units in the polymer films depending on adsorption mechanisms. (3) The nanoassembly of π-conjugated units can be used for high-density and low-dimensional assembly, using newly proposed amphiphilic homopolymers with π-conjugated units in the side chains. These examples demonstrate the potential of LB-based fabrication as a candidate strategy for functional device fabrication.
Similar content being viewed by others
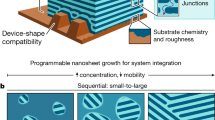

Introduction
The nanoscale assembly of functional materials on solid substrates is a key technology in the nanometer-scale fabrication of optoelectronic and bioelectronic device applications [1, 2]. Studies of the versatile, precise, and single-molecule-level assembly of functional materials constitute a developing field in nanoelectronics applications. For example, single-molecule electronics have been widely investigated using precise techniques such as the break junction method and scanning tunneling spectroscopy [3,4,5,6,7,8]. Although these methods produce well-defined molecular bridges beneficial to detailed evaluations in the nanoelectronics science fields, it is difficult to extend these techniques to the large-scale production of functional devices based on nanoelectronics. In contrast, single-molecule-level fabrication related to organic materials through approaches such as the Langmuir–Blodgett (LB) and self-assembled methods has been conducted for many years [9]. The LB technique can be used to introduce noble amphiphilic materials including amphiphilic polymers [10,11,12,13,14,15,16,17,18]. Studies of self-assembled films were conducted using self-assembled monolayers based on reactive molecules such as thiol or silane derivatives, as well as by the layer-by-layer deposition of polymeric materials based on electrostatic, charge transfer, or other attractive interactions [19,20,21,22,23,24]. In this focus review, nanoassembly based on the LB technique is described and shown to be a versatile and facile approach for producing functional materials (Fig. 1).

Chemical structures of amphiphilic polymers discussed in this focus review
Preparation of oxide nanofilms from amphiphilic polymer nanosheets
The nanoassembly of hybrid polymers is an approach used for the precise assembly of hybrid materials in a direct manner. The nanoassembly of cage-type silsesquioxane (SQ) units, which have a well-defined structure compared to random- and ladder-type SQ units [25,26,27] and are good precursors to SiO2–based materials obtained through oxidation processes [28, 29], based on the LB technique with copolymerization to N-dodecylacrylamide (DDA) was reported to show excellent LB film formation properties [30]. The SQ-containing copolymer p(DDA/SQ) formed a stable monolayer at the air–water interface, and the SQ units were closely packed in the monolayer upon compression, which were confirmed by surface pressure(π)–area (A) isotherm measurements. The monolayer was successively transferred onto hydrophobic substrates by a vertical dipping method, with a high transfer ratio of near unity. Furthermore, atomic force microscopy (AFM) measurements demonstrated that the film thickness increased linearly as the number of layers increased. These highly ordered p(DDA/SQ) nanosheets are a good template for ultrathin SiO2 films [31]. We reported photooxidation by UV-ozone treatment, with the UV fluence as low as several milliwatts per square centimeter through the aid of reactive species such as ozone or atomic oxygen [32]. This method largely reduced the UV fluence through a direct UV irradiation process (~100 mW cm−2) in the previous study. This fabrication technique enables the preparation of ultrathin SiO2 films on polymer substrates such as poly(ethylene terephthalate) (PET) films, with precisely controlled thicknesses. The formation of ultrathin SiO2 films was traced by Fourier-transform infrared (FT-IR) spectroscopy measurements, which revealed that the conversion from silsesquioxane to SiO2 began after the decomposition of organic moieties and that the time constant of this conversion was dependent upon the initial film thickness. This stepwise and thickness-dependent conversion indicates that the conversion from SQ to SiO2 is controlled by the diffusion of oxidizing species such as ozone and atomic oxygen into the film. The thicknesses of the SiO2 films were tuned by 0.4 nm per monolayer by simply changing the deposition number of the p(DDA/SQ) nanosheets. Furthermore, the SiO2 film showed good optical transparency and high electric resistance in a dry state. The resistance on flexible PET substrates also showed a low value (10−11 S cm−1) and was unchanged after a repeated bending test. As described above, we successfully prepared ultrathin SiO2 films on polymer substrates from SQ-containing polymer nanosheets.
On the basis of the technique described above, the structures of ultrathin SiO2 films can be tuned by changing the structure of the precursor polymer nanosheets. The use of a block copolymer is a promising approach. We initially investigated block copolymer synthesis by reversible addition fragmentation chain transfer (RAFT) polymerization [33]. The controlled polymerization of pDDA was achieved by using trithiocarbonate chain transfer agents (CTAs) at low [CTA]/[initiator] ratios, suggesting competition with the termination reaction and the importance of suppressing the noncontrolled free radical polymerization pathway. Based on this work, the SQ-containing block copolymer p(DDA/SQ)-b-pDDA was synthesized by RAFT polymerization using trithiocarbonate CTAs [34]. p(DDA/SQ)-b-pDDA forms a stable monolayer with a high collapse pressure of πc = 45.8 mN m−1. The p(DDA/SQ)-b-pDDA monolayers were deposited onto various hydrophobic substrates with a controlled monolayer film thickness of 2.3 nm, which was similar to using a random copolymer, p(DDA/SQ). Photooxidation by UV−ozone treatment of this block copolymer affords SiO2 nanofilms with a controlled monolayer film thickness of 0.38 nm, which is a similar value to that obtained using a random copolymer, and the nanofilms were prepared successfully by the photooxidation of p(DDA/SQ)-b-pDDA LB films. Notably, the film density of the obtained SiO2 nanofilms was 1.48 g cm−3, which is approximately 2/3 of that of ultrathin SiO2 films formed from p(DDA/SQ) random copolymers (2.24 g cm−3) and bulk SiO2 [35]. Thus, porous SiO2 nanofilms were obtained from block copolymers by using a photooxidation process. For pore characterization, ion permeability tests were conducted based on cyclic voltammetry measurements, using these SiO2 films as a blocking layer over the Au working electrodes and the [Fe(CN)6]3−/4− redox couple as a probe ion (Fig. 2). As a result, a redox reaction was observed for the blocking SiO2 layer from the block copolymer, demonstrating that the pores in the SiO2 nanofilms obtained from the block copolymer had continuous and ion-permeable structures. Very weak redox signals were observed for the blocking SiO2 layer obtained from the random copolymer, indicating that the SiO2 films from the random copolymer were weakly ion-conductive. This research provided a unique method for preparing porous oxide nanofilms with the desired ion permeability by using block copolymer LB films as a template.

Cyclic voltammograms of [Fe(CN)6]3–/[Fe(CN)6]4– for bare Au (black), SiO2 nanofilms obtained from 20-layer p(DDA/SQ26)-b-pDDA (red) and p(DDA/SQ14) (blue) LB films on a Au electrode. The ion source is a K4[Fe(CN)6] aqueous solution (1.25 mM), with KCl (0.1 M) as the supporting electrolyte. The scan rate is 10 mV s–1. (Reprinted with permission from Ishizaki, Y. et al. Langmuir 34, 8007–8014 (2018). Copyright (2018) American Chemical Society)
The application of these SiO2 films to electronic devices was also investigated alongside the preparation and structural control of the SiO2 films described above. We fabricated resistive switching devices using SiO2 films as a resistive switching layer [36]. The SiO2 films from the random copolymer nanosheets satisfy the requirements for the resistive layer, which are being pinhole-free and having thickness controllability and a smooth surface. The resistive switching devices were composed of an Ag (50 nm) | SiO2 | PEDOT:PSS (150 nm) sandwich architecture. The current density (J) – voltage (V) characteristics showed hysteresis in the negative-biased region, although no hysteresis was found in the positive-biased region. The set voltage Vset of the device was dependent on the SiO2 layer thickness. We observed that Vset = −0.3 V for a device with an SiO2 layer thickness of 3.2 nm, which is among the lowest values reported in the literature [37,38,39]. In contrast, no resistive switching was observed in a vacuum, under dry N2, or under dry O2. This suggests that the uptake of water molecules at the PEDOT:PSS/SiO2 interface is necessary for the resistive switching behavior. Thus, water migration coupled with ion migration in the SiO2 layer is crucially important in the resistive switching mechanism. Such ion-based resistive switching is achieved by combining the ion-source (PEDOT:PSS) and weakly ion-transmitting layer (SiO2) with the precise control of the SiO2|PEDOT:PSS interface structure. This example indicates the possibility of using the polymer nanosheets-templated process for optoelectronic applications.
Precise deposition of hybrid materials on polymer films
Another approach for the nanoassembly of hybrid materials is based on adhesion and absorption techniques. We reported polymer nanosheets with catechol as an adhesive functional group [40]. Catechol groups are used as functional groups in adhesive biofunctional materials including in adhesive proteins of marine mussels [41,42,43,44,45,46,47,48,49]. The multimodal adsorption behavior of catechol units was demonstrated by examining the adsorption of different oxide nanoparticles onto polymer nanosheets with catechol groups. Catechol-based polymers p(DDA/DMA) were synthesized, and stable monolayer formation and deposition on solid substrates was confirmed, with nearly unity transfer ratios for both the upstroke and downstroke of nanosheets with a DMA content below 32 mol%. The orientations of DMA units in the nanosheets were investigated by π–A isotherm measurements and IR reflection absorption spectroscopy (IR-RAS), which showed that the DMA units in the p(DDA/DMA) nanosheets had a face-on alignment because of their hydrophilic nature. Thus, the catechol units in the nanosheets were not exposed to the film surface. Furthermore, mid-IR spectroscopy [50, 51] of p(DDA/DMA) nanosheets showed that the nanosheets trapped molecular water, which was tightly bound via hydrogen bonding among neighboring water molecules and the hydroxyl groups of catechol units. Such tightly bound water molecules serve as anchoring points for nanoparticle adsorption through a hydrogen-bonding network, even when the catechol units are buried in the film. The surface potentials of the polymer nanosheets were measured by electrophoresis light scattering (ELS). The p(DDA/DMA32) nanosheets showed drastic changes at approximately pH 6 (from −50 mV at pH 8 to −30 mV at pH 4), although no remarkable change was observed in the pDDA nanosheets. The surface potential transition was ascribed to the chemical change of the catechol form into the o-benzoquinone form, which agrees with previous reports [52, 53]. The adsorption properties of the nanosheets were tested by immersion into an aqueous dispersion of nanoparticles. Scanning electron microscopy (SEM) images of immersed nanosheets showed that SiO2 nanoparticles were adsorbed onto the p(DDA/DMA) nanosheets and that the surface coverage was dependent on the DMA content. These results demonstrate that p(DDA/DMA) nanosheets behave as an adhesive layer for inorganic oxide nanoparticles. The pH dependency of SiO2 nanoparticle adsorption was monitored by a quartz crystal microbalance, demonstrating that SiO2 nanoparticle adsorption occurred only in the region of pH values lower than 6 (Fig. 3). This pH region corresponds to the catechol–quinone transition in p(DDA/DMA) nanosheets, as monitored by ELS measurements. These results indicate that the catechol form in the p(DDA/DMA) nanosheets strongly affected adsorption, although the DMA units were not exposed directly on the surface of the p(DDA/DMA) nanosheets. In the detailed analysis of the adsorption mechanism, Al2O3, which is positively charged at pH values of 2–9, and WO3, which can form coordination bonds to catechol units, were used to examine the electrostatic and coordination mechanisms. Al2O3 nanoparticles were adsorbed onto p(DDA/DMA) nanosheets when the pH value was less than 9, indicating that Al2O3 nanoparticles adsorbed onto the negatively charged p(DDA/DMA) nanosheets through electrostatic interactions. Similar absorption behavior was obtained for pDDA nanosheets without DMA units. Therefore, positively charged Al2O3 nanoparticles were adsorbed by long-range electrostatic interactions with the negatively charged nanosheets, irrespective of the DMA unit chemical structure, rather than through hydrogen bonding to DMA units in the p(DDA/DMA) nanosheets. In contrast, no WO3 nanoparticle adsorption was observed on the p(DDA/DMA) nanosheets when HCl was used to control the pH. Complexation between the DMA units and WO3 nanoparticles did not occur because of the spatial restriction of DMA units, as the catechol units were buried in the DDA side chains. In contrast, the adsorption of WO3 nanoparticles was observed when citric acid was used to control the pH. Citric acid forms coordination bonds to WO3 nanoparticles and hydrogen bonds to surrounding water molecules. In this situation, water molecules trapped in the nanosheets bridge the gap between citric acid on the WO3 nanoparticles and catechol units in the nanosheet through hydrogen bonding. Therefore, we concluded that WO3 nanoparticles can be adsorbed onto p(DDA/DMA) nanosheets when assisted by the hydrogen-bonding interactions of water molecules. This study highlights the importance of the spatial arrangements of functional groups of materials in the monolayer in developing functional materials based on nanoparticle adsorption.

Weights of adsorbed amounts of (top) SiO2 (blue), (middle) Al2O3 (red), and (bottom) WO3 (green) NPs on two-layer p(DDA/DMA32) (circles) and pDDA (triangles) nanosheets detected using the QCM technique. The pH was controlled by HCl (closed symbols) and citric acid (open symbols). (Yamamoto, S. et al. [40]. Reproduced by permission of The Royal Society of Chemistry)
Molecular-level bottom-up processes are another approach for the nanoassembly of hybrid materials by adsorption-based approaches. We reported a bottom-up preparation method using a surface-attached metal-organic framework (SURMOF) by alternating immersion of polymer-coated substrates into solutions of a metal complex and an organic linker molecule for the facile preparation of SURMOF films on various substrates (Fig. 4) [54]. Previous studies of SURMOF fabrication using liquid-phase epitaxy employed substrates modified by self-assembled monolayers with polar groups such as hydroxyl and carboxyl groups as anchoring groups [55,56,57,58,59,60,61]. We used pDDA nanosheets as templates for the SURMOF layer because it is easy to control the surface polarity of Y-type films. Amide groups, which can serve as anchoring groups, were exposed to odd-numbered layers of LB films. The 9-layered LB films were alternately immersed in solutions of copper(II) acetate (Cu(OAc)2) and 1,3,5-benzenetricarboxylic acid (H3btc) to form an MOF consisting of Cu3(btc)2(H2O)3, named HKUST-1. The FT-IR spectra showed a characteristic peak at 1374 cm−1, which increased upon alternate immersion into the solutions, indicating the formation of the HKUST-1 film on the pDDA nanosheets as a two-dimensional scaffold. The deposition process was traced by FT-IR spectroscopy, which revealed a linear increase in absorbance at 1374 cm−1, indicating that HKUST-1 growth proceeded homogeneously during the step-by-step immersion processes. The structure of the HKUST-1 film was confirmed by X-ray diffraction measurements, demonstrating that the growth proceeded in the (111) direction. These crystallographic directions indicate that film growth starts from the [{Cu(OAc)2}2] paddlewheel complex, which is anchored by hydrogen bonding to the amide groups in the pDDA nanosheet. The morphology of the HKUST-1 films was observed by SEM and AFM, which revealed a homogeneous and smooth film structure composed of domains of 30–40 nm in diameter. These results indicate that the HKUST-1 thin film on pDDA was polycrystalline and had a highly uniform morphology free from defects, achieved by using densely packed amide groups on the surface of the pDDA nanosheets. Notably, self-standing MOF nanofilms (pDDA | HKUST-1) were prepared by using the sacrificial layer (cellulose acetate) underneath the pDDA nanosheets. This method provides a facile preparation process for MOF-based nanofilms on various substrates.

Schematic illustration of fabrication of HKUST-1 nanofilms on the amide groups of pDDA polymer nanosheets (blue dots) through alternating immersion into solutions of a metal complex ([{Cu(OAc)2}2], green dots) and an organic linker molecule (H3btc, gray wires)
Nanoassembly of π-conjugated units by polymer nanosheets
In addition to hybrid materials, π-conjugated units are key functional moieties for optoelectronic device applications [62,63,64]. Previous attempts at LB-based nanoassembly of π-conjugated units involved amphiphilic copolymers [65,66,67], conjugated polymers with amphiphilic sidechains [68, 69], and stabilizer-assisted film formation from conjugated polymers [70, 71]. These approaches were used to successfully transfer the monolayer onto the solid substrates; however, it remains challenging to assemble a wide range of π-conjugated units in monolayers to cover the crossover regime from one-dimensional to three-dimensional systems. We propose a versatile and facile low-dimensional integration approach for π-conjugated units using newly designed acrylamide-based polymers and π-conjugated units in side chains. Poly(9-ethyl-3-carbazolyl acrylamide) (pCzAA) was designed based on the structure of pDDA, which has excellent LB film-forming properties. Multistacking, including hetero junction formation at the single-molecule level, is easily achieved by tuning the monolayer sequence for LB film deposition. The monolayer behavior of the amphiphilic polymer pCzAA was investigated by comparison with the hydrophobic polymer poly(N-vinylcarbazole). The π–A isotherms and Brewster angle microscope images indicated that pCzAA can form a monolayer at the air–water interface and that the hydrophilic amide group plays a key role in monolayer formation, in contrast to the random aggregation of poly(N-vinylcarbazole) on the water surface. Although pCzAA forms a stable monolayer, it is difficult to transfer onto substrates without the blending of a stabilizing agent. For the uniform transfer of monolayers, pCzAA:pDDA blend monolayers were formed by spreading each solution separately (co-spreading method), which showed that even a small amount of pDDA stabilizes the pCzAA monolayer (Fig. 5). Namely, the π* value at which the compressibility modulus Cs−1 = –A(dπ/dA) reached a local maximum drastically increased upon the addition of 10 mol% pDDA and reached a value comparable to that of neat pDDA LB films. This behavior depends on the blending method. A 40 mol% addition of pDDA is required to stabilize pCzAA:pDDA monolayers by the mixed solution spreading method. This indicates that the phase-separated structure of the Langmuir film is a key factor in monolayer stabilization by pDDA. Remarkably, the high pCzAA content exceeded that of the percolation threshold in the two-dimensional plane (45 mol%) [72, 73], indicating the involvement of the charge transport pathway in the in-plane direction. The area per monomer unit for pCzAA SCz is independent on the pCzAA content from 50 to 90 mol % pCzAA, at ~0.28 nm2 per unit, which corresponds to the projected area of carbazole groups in edge-on orientation. The pCzAA:pDDA co-spread monolayers were transferred onto hydrophobic substrates for both the upstroke and downstroke, indicating Y-type film deposition. AFM images of the nanosheets showed a phase-separated structure with disk-like domains of 100–500 nm in diameter achieved by the co-spreading method, indicating that the pCzAA:pDDA monolayers undergo phase separation. The structures of the pCzAA:pDDA nanosheets were investigated by FT-IR spectroscopy, which demonstrated that the amide group packing was slightly disturbed by the addition of pCzAA. Nevertheless, the transferred nanosheets formed a highly ordered layered structure in the out-of-plane direction, as confirmed by X-ray reflectivity measurements. The co-spreading of an amphiphilic carbazole homopolymer is effective for hydrogen bond formation with large-scale phase-separated structures to enhance monolayer stability and transferability. This example shows the possibility to control the dimensionality of π-conjugated nanoassemblies by the LB technique using acrylamide-based polymers with π-conjugated units.

(a–e) π–A isotherms of pDDA:pCzAA monolayers measured at 20 °C: co-spreading (red) and mixed solution spreading (blue). The pCzAA contents are (a) 20, (b) 50, (c) 80, (d) 90, and (e) 95 mol %. (f) Plots of π* and (g) SCz as a function of pCzAA contents: co-spreading (red) and mixed solution spreading (blue). Black symbols correspond to single-component spreading. (Reprinted with permission from Yamamoto, S. et al. Langmuir 34, 10491–10497 (2018). Copyright (2018) American Chemical Society)
Closing remarks
This focus review describes the author’s recent study of nanoscale fabrication techniques associated with organic–inorganic hybrid nanomaterials and organic functional nanomaterials based on the LB technique. The LB technique does not require large-scale facilities and is a process that can be carried out under ambient conditions at room temperature. The author’s approaches show potential for applications in electronic devices in wet environments based on both electron- and ion-transporting properties. Such wet electronic devices combined with nanostructure control can be developed for bio-oriented applications in healthcare monitoring or neuromorphic devices. Further studies of nanofabrication using classical LB techniques will provide cutting-edge approaches for developing functional electronic devices.
References
Wolf EL. Nanophysics and Nanotechnology: an introduction to modern concepts in nanoscience. 2nd ed. Weinheim: Wiley-VCH; 2008.
Waser R (ed). Nanoelectronics and information technology: advanced electronic materials and novel devices. 3rd ed. Weinheim: Wiley-VCH; 2012.
Hong W, Li H, Liu S-X, Fu Y, Li J, Kaliginedi V, et al. Trimethylsilyl-terminated oligo(phenylene ethynylene)s: an approach to single-molecule junctions with covalent Au–C σ-bonds. J Am Chem Soc. 2012;134:19425–31.
Uji H, Morita T, Kimura S. Molecular direction dependence of single-molecule conductance of a helical peptide in molecular junction. Phys Chem Chem Phys. 2013;15:757–60.
Perrin ML, Frisenda R, Koole M, Seldenthuis JS, Gil JAC, Valkenier H, et al. Large negative differential conductance in single-molecule break junctions. Nat Nanotechnol. 2014;9:830–4.
Smith CE, Odoh SO, Ghosh S, Gagliardi L, Cramer CJ, Frisbie CD. Length-dependent nanotransport and charge hopping bottlenecks in long thiophene-containing π-conjugated molecular wires. J Am Chem Soc. 2015;137:15732–41.
Perrin ML, Galán E, Eelkema R, Thijssen JM, Grozema F, van der Zant HSJ. A gate-tunable single-molecule diode. Nanoscale. 2016;8:8919–23.
Yuan L, Wang L, Garrigues AR, Jiang L, Annadata HV, Anguera Antonana M, et al. Transition from direct to inverted charge transport Marcus regions in molecular junctions via molecular orbital gating. Nat Nanotechnol. 2018;13:322–9.
Ulman A. An introduction to ultrathin organic films: from langmuir–blodgett to self-assembly. London: Academic Press; 1991.
Pretty MC. Langmuir-Blodgett films: an introduction. Cambridge: Cambridge University Press; 1996.
Miyashita T, Yatsue T, Mizuta Y, Matsuda M. Langmuir-Blodgett films of preformed copolymers of N-alkylacrylamides with vinylcarbazole. Thin Solid Films. 1989;179:439–43.
Miyashita T, Mizuta Y, Matsuda M. Studies on Langmuir–Blodgett multilayer formation from preformed poly(N-alkylacrylamides). Br Polym J. 1990;120:327–31.
Mizuta Y, Matsuda M, Miyashita T. Reactivity ratios of N-dodecylacrylamide in copolymerization with methyl methacrylate and Langmuir-Blodgett multilayer formation of the copolymers. Polym J. 1991;23:1387–91.
Ito S, Kanno K, Ohmori S, Onogi Y, Yamamoto M. Fluorescence polarization method for studying molecular orientation of mono- and multilayered polyimide films prepared by the Langmuir-Blodgett technique. Macromolecules. 1991;24:659–65.
Mabuchi M, Kawano K, Ito S, Yamamoto M. Structural relaxation of Langmuir-Blodgett films of poly(vinyl octanal acetal) with various tacticities. Macromolecules. 1998;31:6077–82.
Talham DR. Conducting and magnetic Langmuir-Blodgett films. Chem Rev. 2004;104:5479–501.
Mccullough DH, Regen SL. Don’t forget Langmuir–Blodgett films. Chem Commun. 2004;21:2787–91.
Mitsuishi M, Matsui J, Miyashita T. Functional organized molecular assemblies based on polymer nano-sheets. Polym J. 2006;38:877–96.
Decher G. Fuzzy nanoassemblies: toward layered polymeric multicomposites. Science. 1997;277:1232–7.
Ariga K, Hill JP, Ji Q. Layer-by-layer assembly as a versatile bottom-up nanofabrication technique for exploratory research and realistic application. Phys Chem Chem Phys. 2007;9:2319–40.
Shimazaki Y, Mitsuishi M, Ito S, Yamamoto M. Preparation of the layer-by-layer deposited ultrathin film based on the charge-transfer interaction. Langmuir. 1997;13:1385–7.
Benten H, Ogawa M, Ohkita H, Ito S. Design of multilayered nanostructures and donor-acceptor interfaces in solution-processed thin-film organic solar cells. Adv Funct Mater. 2008;18:1563–72.
Srivastava S, Kotov NA. Composite layer-by-layer (LBL) assembly with inorganic nanoparticles and nanowires. Acc Chem Res. 2008;41:1831–41.
Richardson JJ, Björnmalm M, Caruso F. Technology-driven layer-by-layer assembly of nanofilms. Science. 2015;348:aaa2491.
Lee LH, Chen WC, Liu WC. Structural control of oligomeric methyl silsesquioxane precursors and their thin-film properties. J Polym Sci Part A Polym Chem. 2002;40:1560–71.
Park E, Ro H, Nguyen C, Jaffe R, Yoon D. Infrared spectroscopy study of microstructures of poly(silsesquioxane)s. Chem Mater. 2008;20:1548–54.
Araki H, Naka K. Syntheses and properties of star- and dumbbell-shaped POSS derivatives containing isobutyl groups. Polym J. 2012;44:340–6.
Hessel CM, Henderson EJ, Veinot JGC. Hydrogen silsesquioxane: a molecular precursor for nanocrystalline Si–SiO2 composites and freestanding hydride-surface-terminated silicon nanoparticles. Chem Mater. 2006;18:6139–46.
Kowarik S, Lim S-H, Caster AG, Leone SR, Nicolet O, Schwartzberg AM. Observing hydrogen silsesquioxane cross-linking with broadband CARS. J Raman Spectrosc. 2009;40:770–4.
Mitsuishi M, Zhao F, Kim Y, Watanabe A, Miyashita T. Preparation of ultrathin silsesquioxane nanofilms via polymer Langmuir−Blodgett films. Chem Mater. 2008;20:4310–6.
Kim Y, Zhao F, Mitsuishi M, Watanabe A, Miyashita T. Photoinduced high-quality ultrathin SiO2 film from hybrid nanosheet at room temperature. J Am Chem Soc. 2008;130:11848–9.
Yamamoto S, Sonobe K, Miyashita T, Mitsuishi M. Flexible SiO2 nanofilms assembled on poly(ethyleneterephthalate) substrates through a room temperature fabrication process for nanoscale. J Mater Chem C. 2015;3:1286–93.
Yamamoto S, Miyashita T, Mitsuishi M. Amphiphilic acrylamide block copolymer: RAFT block copolymerization and monolayer behaviour. RSC Adv. 2017;7:44954–60.
Ishizaki Y, Yamamoto S, Miyashita T, Mitsuishi M. Synthesis and porous SiO2 nanofilm formation of the silsesquioxane-containing amphiphilic block copolymer. Langmuir. 2018;34:8007–14.
Sze SM. Physics of semiconductor devices. 2nd ed. New York: Wiley; 1981.
Yamamoto S, Kitanaka T, Miyashita T, Mitsuishi M. Resistive switching of organic–inorganic hybrid devices of conductive polymer and permeable ultra-thin SiO2 films. Nanotechnology. 2018;29:26LT02.
Bersuker G, Gilmer DC, Veksler D, Kirsch P, Vandelli L, Padovani A, et al. Metal oxide resistive memory switching mechanism based on conductive filament properties. J Appl Phys. 2011;110:124518.
Raeis Hosseini N, Lee J. Resistive switching memory based on bioinspired natural solid polymer electrolytes. ACS Nano. 2015;9:419–26.
Krishnan K, Tsuruoka T, Mannequin C, Aono M. Mechanism for conducting filament growth in self-assembled polymer thin films for redox-based atomic switches. Adv Mater. 2016;28:640–8.
Yamamoto S, Uchiyama S, Miyashita T, Mitsuishi M. Multimodal underwater adsorption of oxide nanoparticles on catechol-based polymer nanosheets. Nanoscale. 2016;8:5912–9.
Waite JH, Tanzer ML. Polyphenolic substance of mytilus edulis: novel adhesive containing l-dopa and hydroxyproline. Science. 1981;212:1038–40.
Waite JH, Qin X. Polyphosphoprotein from the adhesive pads of Mytilus edulis. Biochemistry. 2001;40:2887–93.
Lee H, Lee BP, Messersmith PB. A reversible wet/dry adhesive inspired by mussels and geckos. Nature. 2007;448:338–41.
Yuen AKL, Hutton Ga, Masters AF, Maschmeyer T. The interplay of catechol ligands with nanoparticulate iron oxides. Dalt Trans. 2012;41:2545.
Ji X, Palui G, Avellini T, Na HBin, Yi C, Knappenberger KL, et al. On the pH-dependent quenching of quantum dot photoluminescence by redox active dopamine. J Am Chem Soc. 2012;134:6006–17.
Heo J, Kang T, Jang SG, Hwang DS, Spruell JM, Killops KL, et al. Improved performance of protected catecholic polysiloxanes for bioinspired wet adhesion to surface oxides. J Am Chem Soc. 2012;134:20139–45.
Sedó J, Saiz-Poseu J, Busqué F, Ruiz-Molina D. Catechol-based biomimetic functional materials. Adv Mater. 2013;25:653–701.
Wang R, Li J, Chen W, Xu T, Yun S, Xu Z, et al. A Biomimetic mussel-inspired ε-poly-L-lysine hydrogel with robust tissue-anchor and anti-infection capacity. Adv Funct Mater. 2017;27:1604894.
Han L, Lu X, Liu K, Wang K, Fang L, Weng LT, et al. Mussel-inspired adhesive and tough hydrogel based on nanoclay confined dopamine polymerization. ACS Nano. 2017;11:2561–74.
Fornés V, Chaussidon J. An interpretation of the evolution with temperature of the ν 2+ν 3 combination band in water. J Chem Phys. 1978;68:4667–71.
Malsam J, Aksan A. Hydrogen bonding and kinetic/thermodynamic transitions of aqueous trehalose solutions at cryogenic temperatures. J Phys Chem B. 2009;113:6792–9.
Laviron E. Electrochemical reactions with protonations at equilibrium: Part X. The kinetics of the p-benzoquinone/hydroquinone couple on a platinum electrode. J Electroanal Chem Interfacial Electrochem. 1984;164:213–27.
Deakin MR, Wightman RM. The kinetics of some substituted catechol/o-quinone couples at a carbon paste electrode. J Electroanal Chem. 1986;206:167–77.
Ohara H, Yamamoto S, Onodera T, Kasai H, Oikawa H, Miyashita T, et al. Nanoscale deposition of metal-organic framework films on polymer nanosheets. RSC Adv. 2016;6:74349–53.
Shekhah O, Wang H, Zacher D, Fischer RA, Wöll C. Growth mechanism of metal-organic frameworks: Insights into the nucleation by employing a step-by-step route. Angew Chem - Int Ed. 2009;48:5038–41.
Makiura R, Motoyama S, Umemura Y, Yamanaka H, Sakata O, Kitagawa H. Surface nano-architecture of a metal-organic framework. Nat Mater. 2010;9:565–71.
Nan J, Dong X, Wang W, Jin W, Xu N. Step-by-step seeding procedure for preparing HKUST-1 membrane on porous α-alumina support. Langmuir. 2011;27:4309–12.
Heinke L, Cakici M, Dommaschk M, Grosjean S, Herges R, Bräse S, et al. Photoswitching in two-component surface-mounted metal-organic frameworks: Optically triggered release from a molecular container. ACS Nano. 2014;8:1463–7.
Talin AA, Centrone A, Ford AC, Foster ME, Stavila V, Haney P, et al. Tunable electrical conductivity in metal-organic framework thin-film devices. Science. 2014;343:66–9.
Stavila V, Schneider C, Mowry C, Zeitler TR, Greathouse JA, Robinson AL, et al. Thin film growth of nbo MOFs and their Integration with Electroacoustic Devices. Adv Funct Mater. 2016;26:1699–707.
Müller K, Fink K, Schöttner L, Koenig M, Heinke L, Wöll C. Defects as color centers: the apparent color of metal-organic frameworks containing Cu2+-based paddle-wheel units. ACS Appl Mater Interfaces. 2017;9:37463–7.
Pope M, Swenberg CE. Electronic processes in organic crystals and polymers. 2nd ed. New York: Oxford University Press; 1999.
Malliaras G, Friend R. An organic electronics primer. Phys Today. 2005;58:3.
Someya T, Bao Z, Malliaras GG. The rise of plastic bioelectronics. Nature. 2016;540:379–85.
Miyashita T, Sakai J, Mizuta Y, Matsuda M. Photochemical studies of Langmuir-Blodgett films fabricated from N-alkylacrylamide copolymers with various vinyl aromatic monomers. Thin Solid Films. 1994;244:718–22.
Matsui J, Mitsuishi M, Miyashita T. Characterization of the molecular environment of polymer Langmuir−Blodgett films using a pyrene fluorescent probe. Macromolecules. 1999;32:381–6.
Mitsuishi M, Tanuma T, Matsui J, Chen J. Anthracene chromophores in polymer Langmuir–Blodgett films by an integrated optical waveguide technique. Langmuir. 2001;17:7449–51.
Reitzel N, Greve DR, Kjaer K, Howes PB, Jayaraman M, Savoy S, et al. Self-assembly of conjugated polymers at the air / water interface. structure and properties of langmuir and langmuir–blodgett films of amphiphilic regioregular polythiophenes. J Am Chem Soc. 2000;122:5788–800.
Ito Y, Miyake T, Suginome M, Katakura R, Mitsuishi M, Miyashita T. Langmuir–Blodgett films of helical rigid-rod poly(quinoxaline-2,3-diyl)s. Polym J. 2010;42:406–10.
Matsui J, Yoshida S, Mikayama T, Aoki A, Miyashita T. Fabrication of polymer langmuir–blodgett films containing regioregular poly(3-hexylthiophene) for application to field-effect transistor. Langmuir. 2005;21:5343–8.
Nagano S, Kodama S, Seki T. Ideal spread monolayer and multilayer formation of fully hydrophobic polythiophenes via liquid crystal hybridization on water. Langmuir. 2008;24:10498–504.
Scher H, Zallen R. Critical density in percolation processes. J Chem Phys. 1970;53:3759–61.
Kirkpatrick S. Percolation and conduction. Rev Mod Phys. 1973;45:574–88.
Acknowledgements
The author acknowledges the support from Grants-in-Aid for Young Scientists ((B) 26810111) and for Early-Career Scientists (18K14294) from JSPS, the Sumitomo Foundation, the Murata Science Foundation, and the JGC-S Scholarship Foundation. This work was also supported by the Cooperative Research Program “Network Joint Research Center for Materials and Devices: Dynamic Alliance for Open Innovation Bridging Human, Environment and Materials (MEXT).” The author expresses special thanks to Prof. Masaya Mitsuishi (IMRAM, Tohoku University) for his continuous encouragement and kind support throughout this work. The author is also grateful to all the members in the Polymer Hybrid Nanomaterials Laboratory (IMRAM, Tohoku University) for their contributions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that he has no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yamamoto, S. Development of Interfacial Nanoassembly Techniques in Functional Nanomaterials. Polym J 51, 731–738 (2019). https://doi.org/10.1038/s41428-019-0190-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41428-019-0190-7
