
Abstract
Functionalized acrylic materials derived from methyl methacrylate (MMA), 2-hydroxyethyl methacrylate (HEMA), and diethyl 2-(methacryloyloxy)ethyl phosphate (DMP) were synthesized by successive free-radical polymerizations using benzoyl peroxide (BPO) as the initiator and N,N-dimethyl-p-toluidine (DMpT) as the activator. NMR and GPC data showed that successful random copolymerization of MMA and DMP was carried out using the BPO-DMpT system. Moreover, the resulting products were able to react with HEMA monomer, yielding polymers with P(MMA-co-DMP)-b-PHEMA chain structures. Interestingly, the glass transition of DMP/PHEMA-containing copolymers was reduced with respect to the PMMA homopolymer. In addition, the thermal stability was enhanced with increasing DMP content in P(MMA-co-DMP) copolymers and was further enforced by incorporating PHEMA blocks. The presence of DMP/PHEMA segments should improve the thermal behavior of acrylic materials, which is of great interest in the design of versatile bone cements for total joint arthroplasty and functional coatings in targeted drug delivery.
Similar content being viewed by others

Introduction
Synthetic polymethacrylates derived from methyl methacrylate (MMA) and 2-hydroxyethyl methacrylate (HEMA) are considered valuable in the fields of orthopedic surgery and targeted drug delivery [1,2,3]. PMMA-based bone cement has a pivotal role in fixating implants to bone tissues in total joint arthroplasty [1]. In the pharmaceutical industry, PMMA derivatives are commonly used as film-coating agents for both tablet and capsule dosages [2]. Alternatively, PHEMA has been used in soft contact lenses and artificial cornea; furthermore, it is used as a substrate to prevent cellular adhesion and dispersion [3]. The block copolymerization of MMA and HEMA could be an interesting route to modify the intrinsic properties of PMMA and synthesize novel PMMA-based acrylic materials. Block copolymerization could increase the selection of polymers with variable properties available for the design and fabrication of cements and/or pharmaceuticals to achieve versatile applications.
It is well-known that PMMA bone cement materials are biologically inert and do not show activity in bone augmentation. However, biomimetic modifications via phosphorylation have greatly enhanced the heterogeneous nucleation of apatite and cell adhesion behavior [4]. Moreover, PMMA polymer systems bearing different functional groups can potentially be used in broad-spectrum drug release [5, 6]. Previous reports have shown a remarkably high affinity between the amine groups of PMMA derivatives and the phosphate groups of a model drug dexamethasone, which provided the desired release profile of the drug [6].
According to the literature, copolymers of diethyl 2-(methacryloyloxy)ethyl phosphate (DMP) and several alkyl acrylates were synthesized by radical polymerization in benzene using azobis(isobutyronitrile) (AIBN) as an initiator [7, 8]. Reghunadhan Nair et al. [7] studied the copolymerization of DMP with MMA, methyl acrylate, ethyl acrylate, and butyl acrylate, as initiated by AIBN. In this reaction system, the DMP content in the resulting polymer was very close to that in the feed, revealing that DMP underwent a completely random copolymerization with MMA; however, DMP entered the polymer chain preferentially when reacted with the other acrylates. It is to be expected that the glass transition temperature (Tg) of the copolymers containing DMP may be influenced by the sequence distribution of monomers in the polymer chains. Moreover, the authors found that the Tg decreased with an increase in the weight fraction of DMP in the MMA/DMP copolymers [7]. The effect of two different initiators, AIBN and benzoyl peroxide (BPO), on the graft copolymerization of MMA onto poly(ethylene-co-propylene) was investigated in toluene [9]. The authors found that BPO-initiated copolymerization yields an appreciable degree of grafting compared to the AIBN initiator. Therefore, it would be interesting to study whether the block copolymerization of MMA/HEMA can be achieved using BPO as the initiator, as is typically used in the polymerization of acrylic bone cement in total joint arthroplasty [1]. Furthermore, the effect of DMP/PHEMA segments in the resulting copolymers on the glass transition properties and the thermal decomposition behaviors that are intrinsic to PMMA is still unknown, but this effect is an important parameter that dictates the quality of film coatings and success of total joint replacement surgeries.
In this study, we report the synthesis of acrylic polymers with phosphate functional groups by the successive addition of MMA/HEMA/DMP monomers, using BPO as an initiator and DMpT as an activator for free-radical polymerization (Fig. 1). The resulting copolymers were characterized by gel permeation chromatography (GPC), nuclear magnetic resonance (NMR) spectroscopy, Fourier-transform infrared spectroscopy (FT-IR), differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), and elemental analysis. The results are discussed in comparison to PMMA homopolymers.

Schematic illustration showing the successive free-radical polymerization of MMA and HEMA with phosphate functional groups in THF, as initiated by BPO-DMpT
Materials and methods
Materials
Methyl methacrylate (MMA), 2-hydroxyethyl methacrylate (HEMA), benzoyl peroxide (BPO), N,N-dimethyl-p-toluidine (DMpT), diethyl chlorophosphate, trimethylamine, copper(I) chloride (CuCl), and magnesium sulfate (MgSO4) were purchased from Acros Organics (Geel, Belgium). All solvents were of analytical grade. DMP was synthesized according to the literature [7, 8]. Typically, HEMA (22.5 g, 0.175 mol) was dissolved in 100 mL of anhydrous ethyl ether in a reaction vessel with a magnetic stirrer. A molar equivalent of trimethylamine (1 M solution in THF) was added to the reaction solution, and the system was lowered to 0 °C. Then, 150 mg of CuCl was added to the solution, followed by a dropwise addition of diethyl chlorophosphate (30 g, 0.175 mol), and the mixture was allowed to stir continuously at this temperature for 2 h. The reaction system was returned to room temperature and kept overnight. The filtrate was extracted twice with an aqueous solution of NaOH (2 wt%). The organic phase was then washed twice with distilled water, dried over magnesium sulfate, and evaporated under reduced pressure. A conversion rate of 90% was determined by 1H-NMR. Yield: 30 g (65%). 1H-NMR (DMSO-d6): δ (ppm) = 1.2 (t, 6H, –CH3 on phosphate ester), 1.9 (s, 3H, CH3 on allyl), 3.9–4.4 (m, 8H, –O–CH2−), 5.7 (s, 1H, –CH vinyl trans to carbonyl), 6.0 (s, 1H, –CH vinyl cis to carbonyl).
Synthesis of PMMA
MMA (9.3 g, 0.093 mol), BPO (2 wt%, 0.000826 mol), and DMpT (2.5 wt%, 0.001722 mol) were dissolved in 50 mL of THF in a round-bottomed vessel equipped with a mechanical stirrer, and the solution was allowed to stir at 70 °C under a dry argon atmosphere for 5 h. Polymerization of MMA at different BPO/MMA ratios and reaction times was carried out using the same procedure. The product was collected by re-precipitation with THF as the solvent and diethyl ether as the non-solvent, followed by filtration and vacuum drying. 1H-NMR (DMSO-d6): δ (ppm) = 0.7–1.0 (d, 3H, CH3), 1.6–2.0 (d, 2H, CH2), 3.5 (s, 3H, OCH3).
Synthesis of P(MMA-co-DMP)
To synthesize the copolymer with an MMA/DMP weight ratio of 95/5, MMA (9.5 g, 0.095 mol), DMP (0.5 g, 0.00187 mol), BPO (2 wt%, 0.00083 mol), and DMpT (2 wt%, 0.00148 mol) were dissolved in 50 mL of THF in a round-bottomed vessel equipped with a mechanical stirrer, and the solution was allowed to stir at 70 °C under a dry argon flow for 5 h. Copolymerization of MMA and DMP at different weight ratios was carried out using the same procedure. All polymers were recovered by re-precipitation with THF as the solvent and diethyl ether as the non-solvent, followed by filtration and vacuum drying.
Synthesis of PMMA-b-PHEMA
The block copolymerization of MMA and HEMA follows a two-step process. Typically, in the first step, MMA (2.2 g, 0.022 mol), BPO (2 wt%, 0.000272 mol), and DMpT (2 wt%, 0.000463 mol) were dissolved in 50 mL of THF in a round-bottomed vessel equipped with a mechanical stirrer, and the solution was allowed to stir at 70 °C under a dry argon atmosphere for 3 h. In the second step, HEMA (2.2 g, 0.017 mol) was added dropwise through a dropping funnel, and the reaction solution was maintained at the same temperature under a dry argon environment for another 2 h. The product was recovered by re-precipitation with THF as the solvent and diethyl ether as the non-solvent, followed by filtration and vacuum drying. 1H-NMR (DMSO-d6): δ (ppm) = 0.75–1.05 (d, 6H, CH3), 1.76 (d, 4H, CH2), 3.5 (s, 5H, OCH3; CH2), 3.9 (s, 2H, OCH2), 4.8 (s, 1H, OH).
Synthesis of P(MMA-co-DMP)-b-PHEMA
A terpolymer comprised of P(MMA-co-DMP) and PHEMA blocks was synthesized in the same manner as PMMA-b-PHEMA, with a mixture of MMA/DMP monomers instead of only MMA in the first step. Typically, MMA (1.1 g, 0.011 mol), DMP (1.1 g, 0.041 mol), BPO (2 wt%, 0.000272 mol), and DMpT (2 wt%, 0.000463 mol) were dissolved in 50 mL of THF in a round-bottomed vessel equipped with a mechanical stirrer, and the solution was allowed to stir at 70 °C under a dry argon atmosphere for 3 h. In the second step, HEMA (1.1 g, 0.0085 mol) was added dropwise through a dropping funnel, and the reaction solution was maintained at the same temperature under a dry argon environment for another 2 h. The product was recovered by re-precipitation with THF as the solvent and diethyl ether as the non-solvent, followed by filtration and vacuum drying. 1H-NMR (DMSO-d6): δ (ppm) = 0.75–1.05 (d, 9H, CH3), 1.28 (t, 6H, CH3), 1.76 (d, 6H, CH2), 3.54 (s, 5H, OCH3; CH2), 3.9 (s, 2H, OCH2), 4.03–4.11 (m, 8H, OCH2), 4.8 (s, 1H, OH).
Measurements
GPC was performed at 40 °C using a setup composed of an HPLC pump, a refractive index detector, and two size-exclusion columns connected in series: one guard column (PLgel 5 μm, 7.5 × 50 mm) and one Mixed-D column (PLgel 5 μm, 7.5 × 300 mm) (Agilent Technologies, Inc. UK). The mobile phase was THF, and the flow rate was 0.8 mL min−1. The weight-average molecular weight (Mw) and polydispersity index (Mw/Mn) of the polymer were expressed with respect to PMMA standards (Polymer Standards Service, Inc., Amherst, MA, USA). 1H-NMR and two-dimensional diffusion ordered spectroscopy (2D DOSY) NMR measurements were performed at 300 K on a 600 MHz Varian VNMRS-600 NMR spectrometer (Varian, Palo Alto, CA, USA) with DMSO-d6 as the solvent and tetramethylsilane (TMS) as the reference. 31P NMR spectra were obtained with a Varian UNITYINOVA-500 NMR spectrometer. IR spectra were recorded with a PerkinElmer Spectrum 100 FT-IR spectrometer (PerkinElmer, Santa Clara, CA, USA). DSC measurements were obtained on a LT-Modulate DSC 2920 calorimeter (TA Instruments, New Castle, DE, USA). The samples were first heated from 25 to 105 °C under a nitrogen atmosphere and then kept at 105 °C for 5 min, followed by rapid cooling to room temperature. A second heating was performed from 30 °C to 200 °C at a heating rate of 10 °C min−1. TGA was carried out on a TA Instruments SDT Q600 thermogravimetric analyzer (TA Instruments, New Castle, DE, USA). The samples were heated from 25 to 600 °C at a rate of 10 °C min−1. Elemental analyses for carbon (C) and hydrogen (H) were recorded on a Vario EL cube apparatus (Elementar, Germany), and the amount of oxygen (O) was recorded on a Flash 2000 Instrument (Thermo Fisher Scientific Inc., Italy). From these data, the amount of phosphorus (P) was quantified.
Results and discussion
A series of functionalized acrylic materials derived from MMA, HEMA and DMP were synthesized by successive free-radical polymerization in THF using a BPO-DMpT-initiated system (Fig. 1). When BPO and DMpT are mixed together, DMpT causes the decomposition of BPO in a reduction/oxidation electron-transfer process, resulting in benzoyl and DMpT radicals [10]. In the first step of the polymerization, one free-radical attacks the alkene double bond of a methacrylate monomer, MMA or DMP, propagating the polymerization process until it is terminated by another free radical. In the second step, the unterminated polymeric-free radical serves as a macro-initiator and is allowed to react with HEMA, rendering copolymers with PMMA/PHEMA segments bearing phosphate functional groups. Table 1 shows the composition and molecular characteristics of the various homo/copolymers.
A pilot study concluded that the free-radical polymerization of MMA initiated by the formation of benzoyl/DMpT radicals is an efficient and convenient method to form PMMA (Supplementary Figure S1 and Table S1). The conversion of monomer to polymer was very fast, as the increase in molecular weight occurred dramatically during the first 3 h, and more than 90% of the loaded MMA monomer was converted into PMMA. After this, the molecular weight increased slightly and reached a steady state after 5 h. An increase in the BPO/monomer ratio led to a decrease in the molecular weight of the polymer, from Mw = 53,000 g mol−1 for 0.2 wt% BPO to Mw = 20,000 g mol−1 for 5 wt% BPO. From these preliminary experiments, it was concluded that the free-radical polymerization of MMA initiated by the formation of the benzoyl radical is an efficient and convenient method to form PMMA.
We next investigated the synthesis of MMA/DMP random copolymers by the free-radical polymerization of a mixture containing MMA and DMP, using BPO as an initiator and DMpT as an activator. The reaction was performed for 5 h at a fixed BPO/monomer ratio of 2 wt%. After purification by dissolution/precipitation, the BPO-initiated MMA/DMP copolymers were recovered as a dough-like mass and changed from white to yellow as the DMP content increased. As the solvent evaporated, a hard, cement-like complex was formed. The MMA/DMP weight ratios of the feeds were selected to be 95/5, 90/10 and 50/50.
As shown in Table 1, the molecular weight of the MMA/DMP copolymers did not change significantly as the DMP content increased from 0 to 10% under the same conditions (2 wt% BPO and 2 wt% DMpT). However, when the DMP content in the MMA/DMP copolymer reached 50%, a strong decrease in the molecular weight of the copolymer was observed. This finding may result from the bulky structure of the DMP monomers, which causes difficulties when MMA and DMP were incorporated into the polymer chain. Additionally, the hydrodynamic volume of the MMA/DMP copolymers bearing DMP segments changed compared with the parent PMMA homopolymer.
Figure 2a shows a typical 1H-NMR spectrum and the chemical structure of the resulting copolymer. Typical signals for PMMA were observed as follows: 1.8 ppm (protons a), 0.7–1.0 ppm (protons b), and 3.6 ppm (protons c) [11, 12]. The presence of DMP units was confirmed by the appearance of signals at 4.0–4.2 ppm (protons f + g + h) and 1.3 ppm (protons i) and by the increase in the signals at 1.8 ppm (protons d) and 0.7–1.0 ppm (protons e). The MMA/DMP weight ratio was calculated from the integration of signal c of the MMA units at 3.6 ppm and signals f + g + h of the DMP units at 4.0–4.2 ppm. The results are summarized in Table 1. It appears that the MMA/DMP weight ratios in the copolymers, as obtained by 1H-NMR, are very close to those of the initial feeds, indicating a very good conversion of both MMA and DMP monomers. Figure 2b shows the 31P NMR spectrum of the P(MMA-co-DMP) copolymer (sample P3). It contains three split peaks, which were attributed to different triads of MMA and DMP repeating units. Notably, only a single peak can be observed for the DMP monomer.

Chemical structures and a 1H-NMR and b 31P NMR analyses of a P(MMA-co-DMP) copolymer (sample P3) in DMSO-d6 (*: DMSO; #: H2O)
PMMA-b-PHEMA block copolymer was synthesized by the free-radical polymerization of successively added MMA and HEMA in the presence of BPO-DMpT initiator. The process of block copolymerization involves two steps. In the first step, the polymerization of MMA, initiated by BPO-DMpT, occurred in the first 3 h. Then, in the second step, the obtained PMMA-free radical served as a macro-initiator and was allowed to react with HEMA over the next 2 h. P(MMA-co-DMP)-b-PHEMA terpolymer was synthesized in the same manner using a predetermined amount of a MMA/DMP mixture, instead of only MMA monomer (Fig. 1). The GPC chromatograms of PMMA-b-PHEMA and P(MMA-co-DMP)-b-PHEMA tend towards a broad bimodal molecular weight distribution, suggesting the presence of two species with different molecular weights (Fig. 3a). Figure 4a shows a DOSY NMR spectrum of the P(MMA-co-DMP)-b-PHEMA copolymer. Typical signals for PMMA, PDMP, and PHEMA components were observed. The presence of PHEMA units was confirmed by the appearance of signals at 4.7 ppm (protons n). Signals characteristic of MMA, DMP, and HEMA diads or triads were detected in the 31P NMR spectra (Fig. 4b), indicating that copolymerization occurred. Figure 3b shows the FT-IR spectra of PMMA and the synthesized copolymers. In the spectrum of PMMA, the bands at 1726 and 1150 cm−1 were assigned to C=O and C–O stretching vibrations. The spectrum also shows two characteristic absorptions at 2955 and 1450 cm−1, which are attributed to the stretching vibrations of C–H and C–O, respectively [12,13,14]. MMA/DMP copolymers exhibited bands at 982 and 1027 cm−1, which is in agreement with the presence of DMP units [15, 16]. In the spectrum of P(MMA-co-DMP)-b-PHEMA, another C–O stretching band appeared at 1075 cm−1, and an O–H stretching band appeared at 3400–3500 cm−1, which is in agreement with the presence of PHEMA blocks [15]. In the present study, the MMA/DMP weight ratios in the resulting polymers, as obtained by 1H-NMR (Table 1) and elemental analyses (Table 2), are very close to those of the initial feeds, indicating that a successful random copolymerization was carried out using the BPO-DMpT initiator-activator system. Moreover, the resulting products were able to react with the HEMA monomer, rendering a polymer with a chain structure of P(MMA-co-DMP)-b-PHEMA. We also attempted to synthesize a block copolymer of MMA/HEMA via the successive addition of HEMA and MMA; however, no product could be recovered after dissolution/precipitation. This result was attributed to the fact that the products from the polymerization of HEMA led to cross-linked aggregates in the solvent (THF), rather than dissolution. This result revealed that block copolymerization of MMA/HEMA cannot be achieved using this procedure.

a GPC traces, b FT-IR spectra of PMMA and its copolymers. PMMA (sample M4), P(MMA-co-DMP) (samples P2 or P3), PMMA-b-PHEMA (sample E1), and P(MMA-co-DMP)-b-PHEMA (sample E2)

Chemical structure and a DOSY and b 31P NMR analyses of a P(MMA-co-DMP)-b-PHEMA copolymer (sample E2) in DMSO-d6 (*: DMSO; #: H2O)
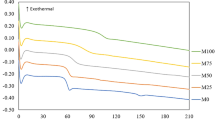
The thermal properties of the various copolymers were investigated using DSC and TGA and were compared with the corresponding PMMA homopolymer. For DSC measurements, samples were first heated from 25 °C to 105 °C and then rapidly cooled to room temperature to erase the thermal history. For Tg measurements, a second heating step from 30 °C to 200 °C was carried out at a heating rate of 10 °C min−1. The DSC thermograms are shown in Fig. 5a. The Tg of the PMMA homopolymer (sample M4) was detected at 118 °C, indicating that PMMA is an organic glass at room temperature. Notably, the Tg values agree with those predicted by the Flory–Fox equation, i.e., the Tg increased with increasing molecular weight (Supplementary Table S1). Introducing 5% DMP into the PMMA chain (sample P1) slightly decreased the Tg to 116 °C. However, when another 5% DMP was incorporated into the PMMA (i.e., sample P2), the Tg decreased to 109 °C. Interestingly, sample P3 showed a Tg value very close to that of sample P2, indicating that no significant effect on the Tg occurred by increasing the DMP content from 10 to 50%. For the PHEMA-containing copolymers, similar thermograms were found with slightly lower Tg values. These findings showed that PHEMA blocks decreased the chain mobility of the PMMA or P(MMA-co-DMP) blocks. It is worth noting that the Tg of a random copolymer is a monotonic function of the composition, while block copolymers can present two or more transitions [17].

Thermal behavior of PMMA and its copolymers. a DSC and b TGA thermograms: PMMA (sample M4), P(MMA-co-DMP) (sample P3), PMMA-b-PHEMA (sample E1), and P(MMA-co-DMP)-b-PHEMA (sample E2)
TGA thermograms of the samples were recorded from 25 to 600 °C, as shown in Fig. 5b. PMMA exhibited uniform thermal degradation behavior. For the copolymers, a two-step degradation profile was detected. It is assumed that the first degradation step corresponds to the MMA moiety. The value of the decomposition temperature (Td) was reported at 5% weight loss of the sample. A Td value of 208 °C was obtained for PMMA, and values of 223, 237 and 250 °C were obtained for the P(MMA-co-DMP) copolymer samples P1, P2, and P3, respectively (Table 1). The TGA curve of the MMA/DMP copolymer shifted to a higher temperature compared to PMMA, i.e., the overall stability increased due to the presence of DMP segments.
In pharmaceutical formulations, Tg is a critical parameter in terms of sustained release dosage forms. It is generally believed that the coating process must be carried out at a temperature (called the minimum film-forming temperature) above the Tg of the film-coating materials, which facilities uniform distribution of the coating [2]. In total joint replacement, radical polymerization is an exothermic reaction that consequently increases the temperature of the cement mixture. This factor can cause serious complications in vivo, such as aseptic loosening by necrosis of the surrounding tissues [1]. Therefore, next-generation bone cements need to incorporate monomers with low heats of reaction in order to reduce thermal necrosis and enhance biocompatibility. Sabino et al. reported the curing properties of bone cements with MMA as the base monomer and either methacrylic acid (MAA) or diethyl amino ethyl methacrylate (DEAEMA) as co-monomers [18]. The addition of MAA to MMA produced bone cements with a maximum temperature during polymerization (Tmax) of 73 °C, while the addition of DEAEMA resulted in a Tmax of 38 °C. In addition, they also found that low Tmax values corresponded to a low Tg for the bone cement. Therefore, investigating the transition temperature of homo/copolymers is important for predicting the curing properties of new bone cement formulations. In this work, DSC analysis demonstrated that the Tg of the MMA/DMP copolymers was lower than that of the PMMA homopolymer and was further decreased by the incorporation of PHEMA blocks (Table 1). For this reason, using DMP or PHEMA to modify PMMA improves the glass transition properties for bone cement applications. Regarding treatment by thermal processes, Yang et al. studied the thermal degradation behavior of hybrid copolymers of MMA and ε-caprolactone (CL) under a nitrogen atmosphere [19]. All MMA/CL copolymers exhibited improved thermostability in comparison with the two homopolymers, and the Td increased with the MMA molar fraction over the range of 30 mol% to 70 mol%. In the present study, we also found that the Td increased with increasing DMP content in P(MMA-co-DMP) copolymers, and was further increased by incorporating PHEMA blocks. From a processing viewpoint, block copolymers of PMMA/PHEMA with phosphate functional groups provide a wide temperature range for thermal treatment.
Conclusion
Herein, we reported the synthesis and characterization of functionalized acrylic materials prepared by the successive addition of MMA/DMP/HEMA monomers, using BPO-DMpT-initiated polymerization. At low DMP/MMA ratios, DMP underwent a random copolymerization with MMA; however, the bulky structure of DMP restricted chain propagation at high DMP contents. In this reaction system, the resulting products were able to react with the HEMA monomer, rendering polymers with a chain structure of P(MMA-co-DMP)-b-PHEMA. Analysis of the thermal properties found that the incorporation of DMP or PHEMA in PMMA rendered a copolymer with a low Tg and high Td compared to the PMMA homopolymer, thus increasing the overall stability of the main chain polymer. These features are of great interest for designing novel biomedical materials and particularly show potential for applications in bone cements and coating system additives.
References
Webb JC, Spencer RF. The role of polymethyl methacrylate bone cement in modern orthopaedic surgery. J Bone Jt Surg Br. 2007;89:851–7.
Patra CN, Priya R, Swain S, Jena GK, Panigrahi KC, Ghose D. Pharmaceutical significance of Eudragit: a review. Future J Pharma Sci. 2017;3:33–45.
Casadio YS, Brown DH, Chirila TV, Kraatz H, Baker MV. Biodegradation of poly(2-hydroxyethyl methacrylate) (PHEMA) and poly{(2-hydroxyethyl methacrylate)-co-[poly(ethylene glycol) methyl ether methacrylate]} hydrogels containing peptide-based cross-linking agents. Biomacromolecules. 2010;11:2949–59.
Sailaja GS, Kumary TV, Varma HK. Biomimetically modified poly(2-hydroxy ethyl methacrylate-co-methylmethacrylate) microspheres for bone augmentation. Trends Biomater Artif Organs. 2006;20:3–6.
Monge S, Canniccioni B, Graillot A, Robin JJ. Phosphorus-containing polymers: a great opportunity for the biomedical field. Biomacromolecules. 2011;12:1973–82.
Guzman ML, Manzo RH, Olivera ME. Eudragit E100 as a drug carrier: the remarkable affinity of phosphate ester for dimethylamine. Mol Pharm. 2012;9:2424–33.
Reghunadhan Nair CP, Clouet G, Brossas J. Copolymerization of diethl 2-(methacryloyloxy) ethyl phosphate with alkyl acrylates: reactivity ratios and glass transition temperatures. J Polym Sci A Polym Chem. 1988;26:1791–807.
Ni SC, Kuo PL. Chemical and physical adsorption of polymers containing thiophosphate, amino, or polysiloxane groups at the oil/metal interface under extreme pressure. J Polym Sci A Polym Chem. 2003;41:106–15.
Tomasek L, Jukic A, Janovic Z. Free radical grafting of methyl methacrylate onto ethylene-propylene amorphous copolymer. Croat Chem Acta. 2009;82:825–32.
Milner R. The development of theoretical relationships between some handling parameters (setting time and setting temperature), composition (relative amounts of initiator and activator) and ambient temperature for acrylic bone cement. J Biomed Mater Res Part B Appl Biomater. 2004;68B:180–5.
Wang TL, Liu YZ, Jeng BC, Cai YC. The effect of initiators and reaction conditions on the polymer syntheses by atom transfer radical polymerization. J Polym Res. 2005;12:67–75.
Islam MR, Bach LG, Park JM, Hong SS, Lim KT. Synthesis and characterization of poly(HEMA-co-MMA)-g-POSS nanocoposites by combination of reversible addition fragmentation chain transfer polymerization and click chemistry. J Appl Polym Sci. 2013;127:1569–77.
Ahmad S, Ahmad S, Agnihotry SA. Synthesis and characterization of in situ prepared poly(methy methacrylate) nanocomposites. Bull Mater Sci. 2007;30:31–35.
Zhao X, Cheng J, Chen S, Zhang J, Wang X. Hydrophilic modification of poly(vinylidene fluoride) (PVDF) by in situ polymerization of methyl methacrylate (MMA) monomer. Colloid Polym Sci. 2010;288:1327–32.
Sailaja GS, Kumari TV, Yokogawa Y, Varma HK. In vitro mineralization and cell adhesion on surface modified poly (2-hydroxy ethyl methacrylate-co-methyl methacrylate). Key Eng Mater. 2006;309-311:493–6.
Jang J, Jeong YK. Synthesis and flame-retardancy of UV-curable methacryloyloxy ethyl phosphates. Fiber Polym. 2008;9:667–73.
Kim J, Mok MM, Sandoval RW, Woo DJ, Torkelson JM. Uniquely broad glass transition temperatures of gradient copolymers relative to random and block copolymers containing repulsive comonomers. Macromolecules. 2006;39:6152–60.
Sabino MA, Ajami D, Salih V, Nazhat SN, Vargas-Coronado R,Cauich-Rodriguez JV, Ginebra MP. Physicochemical, mechanical, and biological properties of bone cements prepared with functionalized methacrylates. J Biomater Appl. 2004;19:147–61.
Yang H, Xu J, Pispas S, Zhang G. Hybrid copolymerization of ε-caprolactone and methyl methacrylate. Macromolecules. 2012;45:3312–7.
Acknowledgements
This study was supported by the National Health Research Institutes of Taiwan (Grant Number: 107A1-IVPP22-014). Yun-Fen Peng carried out her thesis research under the auspices of the Ph.D. Program in Tissue Engineering and Regenerative Medicine, National Chung Hsing University and National Health Research Institutes. We are grateful to the NHRI for awarding a summer research scholarship to Ashley Tsai, an undergraduate student in the Departments of Bioengineering and Materials Science & Engineering, University of California, Berkeley. Additionally, we are grateful to Mr. Chun-Wei Chang, Institute of Biotechnology and Pharmaceutical Research of NHRI, for his help with NMR analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Peng, YF., Tsai, A. & Huang, MH. Synthesis and thermal investigation of phosphate-functionalized acrylic materials. Polym J 50, 967–974 (2018). https://doi.org/10.1038/s41428-018-0086-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41428-018-0086-y
