
Abstract
Molecular simulations are powerful tools for revealing the properties of polymers at the molecular level. In particular, coarse-grained molecular dynamics simulations are useful for elucidating the deformation and fracture processes of polymers. However, in the case of crystalline polymers, it is difficult to reproduce experimentally observed structures and mechanical properties using these models. This review describes our recent investigations into the deformation and fracture processes of crystalline polymers using coarse-grained molecular dynamics simulations. We were able to successfully reproduce the lamellar structure of polyethylene, which is a fundamental structural feature of this polymer, and obtain a stress–strain curve that exhibited good consistency with that observed experimentally. The molecular dynamics simulations revealed that void generation in the amorphous layers was caused by the movement of the chain ends, which is difficult to observe through experiments. The conditions required to reproduce the experimentally observed structure and mechanical properties using molecular simulations are also discussed.
Similar content being viewed by others
Introduction
The mechanical properties of polymers and their composites play a crucial role in the applications of these materials. Because crystalline polymers such as polyethylene, polypropylene, polyvinyl alcohol, and polyoxymethylene have hierarchical structures, their deformation and fracture processes are complex but of great importance. Understanding these processes is vital for improving the mechanical properties of the polymers, and thus, they have been actively studied [1,2,3,4,5,6]. Alongside experimental approaches, computational simulations represent a powerful tool for elucidating the deformation and fracture processes of polymers at the molecular level.
In terms of computational simulations, coarse-grained molecular dynamics simulations have been extensively used to reveal the fracture processes of polymers because polymer dynamics occur over extended timescales and lengths. The fracture processes of polymer glasses have been studied in bulk materials [7,8,9,10] and thin films [11], as reviewed previously [12]. It should be noted that a full experimental system cannot be modeled by molecular simulations, even by using coarse-grained models; it is therefore difficult to directly compare the results of experiments and simulations. Molecular simulations typically rely on modeling a small region of experimental samples. For example, coarse-grained molecular dynamics simulations have been used to simulate the fracture process around a crack and monitor crack propagation in polymer glasses at the molecular level [7, 8].
Although the fracture processes of polymer glasses have been successfully revealed, those of crystalline polymers remain unclear at the molecular level. To our knowledge, there have been few studies of the fracture processes of crystalline polymers using molecular simulations; these are summarized briefly. Rutledge et al. first simulated the fracture processes of crystalline polymers. They modeled the lamellar structure of polyethylene, in which amorphous layers are sandwiched by crystalline layers, and stretched it using a coarse-grained molecular dynamics simulation [13,14,15,16]. Yamamoto also used a coarse-grained molecular dynamics approach to simulate the crystallization of polyethylene and study its fracture process [17]. These studies revealed the generation of voids in the amorphous layer. However, the simulation sizes used in these studies were still too small to unravel the entire fracture process of the lamellar structure. A large-scale simulation consisting of 4.3 × 106 beads was performed by Jabbari-Faouji et al. [18, 19] using a coarse-grained model of polyvinyl alcohol. The authors adopted a polycrystalline structure and revealed the orientation of polymer chains along the stretching direction. The results demonstrated the effectiveness of the simulation approach for understanding fracture processes at the molecular level, although the structure used in the simulation was not in agreement with the experimental one. All-atomic models have also been investigated. Monasse et al. studied the fracture process of the lamellar structure of polyethylene and revealed the stretching process of the tie chains [20]. O’Connor et al. modeled fiber structures of polyethylene based on completely straight polymer chains and determined their yield stress [21]. However, problems with these methods persist. In particular, the lamellar structure generally observed experimentally has not been constructed in large-scale simulations; therefore, the obtained stress–strain curves are not in agreement with the experimental ones.
To elucidate the deformation and fracture processes of crystalline polymers by molecular dynamics simulations, both atomic information and a large simulation size are essential. The atomic information is required for the crystallization of the polymer chains, while a large-scale simulation is crucial for constructing the hierarchical structure and modeling the high-molecular mass of crystalline polymers. There are two feasible approaches for meeting these requirements. One is to use an all-atomic model and increase the simulation size. The other is to use a coarse-grained model and gradually incorporate atomic information. In the latter case, the problem is determining how to consider the atomic-scale information and how much to enlarge the simulation size. Recently, we successfully constructed the lamellar structure of polyethylene and demonstrated that the stress–strain curve agrees with that obtained experimentally [22]. This review discusses our recent work in this area [22,23,24]. A simple coarse-grained model is used, and atomic-scale information is added. Then, the factors required to reproduce the mechanical properties of crystalline polymers using molecular simulations are discussed.
Coarse-grained model
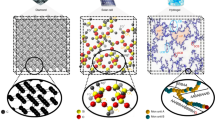
In this section, coarse-grained models of polyethylene are discussed, which are depicted in Fig. 1. In an all-atomic model (Fig. 1a), each of the atoms is represented by one sphere. In the coarse-grained model shown in Fig. 1b, which is referred to as the united atom model, each CH2 group is represented by one sphere. Coarse graining therefore decreases the total number of spheres, which reduces the computational cost. Consequently, the size and timescale of the simulation can be increased. In the coarse-grained model depicted in Fig. 1c, which is referred to as the bead-spring model, several CH2 groups are represented by one sphere. In this model, atomic information is absent, and the polymer behaves as a chain. The degree of coarse-graining was changed. Initially, the most coarse-grained model discussed in this review is adopted. Then, to reflect the properties of polyethylene, the bending potential required to realize the gauche conformation is used. Finally, the attractive interaction is increased by decreasing the average distance between bonding monomers. In this procedure, the model gradually approaches the actual structure of polyethylene. In other words, the model transitions from Fig. 1b, c. This method can be used to reveal important information about the properties of crystalline polymers.

a All-atomic model of polyethylene. Each carbon or hydrogen atom is represented by one sphere. b United atom model of polyethylene. Each CH2 group is represented by one sphere. c Bead-spring model. Several CH2 groups are represented by one sphere. The size and timescale of the simulation are increased, but atomic information is lost
In all of these models, the potential energy of the system consists of bonding, bending, and attractive and repulsive terms. The details of the potentials and coefficients have been previously described [22, 23]. For the attractive and repulsive terms, the Lennard–Jones potential is used. The bonding and bending terms are briefly explained here. These potentials are important for the properties of polyethylene and affect the polymer’s crystal structure. For the bonding term, the following equation is typically used:
where l is the bond length between neighboring monomers, l0 is the equilibrium bond length, and \(k_{{\mathrm{bond}}}^{\mathrm{a}}\) is the force constant for the spring. To allow for bond breaking during the stretching process, the following equation is used:
where R0 is the length at which the bond breaks, R1 is the average bond length, and U0 is the energy barrier to bond breaking. The coefficient \(k_{{\mathrm{bond}}}^{\mathrm{b}}\) is determined by R0, R1, and U0. This type of potential has also been used to study the fracture of glassy polymers upon stretching [8]. In two previous reports [23, 24], the coefficients were set as follows: \(k_{{\mathrm{bond}}}^{\mathrm{b}} = 3600{\it{\epsilon }}/a^2\), R0 = 1.5a, \(R_1 = \frac{{2.5}}{{3.0}}a\), and U0 = 75.0ε, where the length and energy units are normalized by a and ε, respectively. The energy barrier to bond breaking was 75.0ε, the average bond length was approximately 1.0a, and the length at which the bond breaks was 1.5a. In another study [22], the coefficients were set as follows: \(k_{{\mathrm{bond}}}{\mathrm{ = }}1.1 \times 10^5\epsilon /{\mathrm{a}}^2\), R0 = 0.75a, R1 = 0.27a, and U0 = 643.7ε. The average bond length was ~0.39a, and the length at which the bond breaks was 0.75a.
For the bending term, the following equation is used in the simplest case:
where θa is the angle between adjacent bond vectors. The coefficient kθ determines the rigidity of the polymer chains. The linear shape is the most stable, and the energy gradually increases with bending. Schematic depictions of the polymer chains are presented in Fig. 2. When the bending potential described by Eq. 3 is applied, the polymer chain bends into a circular shape (Fig. 2a). In a previous study [23], the coefficient was set to kθ = 20ε. For the bending elasticity, the persistence length was \({l}_{\mathrm{p}} \simeq 4.5a\). This polymer chain is slightly rigid and can therefore be considered a semiflexible chain. According to this model, the single-polymer chain exhibits a toroidal crystal structure [25, 26].

Schematic images of polymer chains modeled using various potentials. a The polymer chain bends in a circular shape upon applying Eq 3. b, c The polymer chain bends orthogonally upon applying Eq 4. The smaller bond length in the case of c leads to a cylindrical shape with the monomers closer together
To adequately reproduce the properties of polyethylene, the gauche conformation should be incorporated. In this case, the following equation is used for the bending term:
where θb is the C–C–C bond angle. The parameters are selected to realize the following characteristics: the linear state is the most stable, the perpendicular state is a less stable local minimum, and an energy barrier exists between the two states. When Eq. 4 is applied, the polymer chain bends orthogonally as shown in Fig. 2b. In previous studies [22, 24], the coefficients were set as follows: \(k_{{\mathrm{bend}}}^{\mathrm{a}} = 12.428\epsilon\), \({\mathrm{k}}_{{\mathrm{bend}}}^{\mathrm{b}} = 38.370\epsilon\), \({\mathrm{k}}_{{\mathrm{bend}}}^{\mathrm{d}} = 123.38\epsilon\), and θ0 = 108.78 (deg). To increase the density of attractive interactions, the average bond length was decreased, leading to the cylindrical shape shown in Fig. 2c. In the united atom model (Fig. 1b), the monomers are positioned in a zigzag manner, and the polymer chain is shaped like a plate.
The length of each polymer chain remains the same throughout the simulations. In previously developed coarse-grained molecular dynamics simulations, the chain lengths were N = 1078 [17], N = 513 [15], and N = 300 [18, 19]. In our simulations, the chain lengths were set to N = 400 (Fig. 2a) [23], N = 2000 (Fig. 2b) [24], and N = 1000 (Fig. 2c) [22], which are long compared with those in previously reported simulations. In the united atom model of polyethylene [15, 17], a monomer (bead) corresponds to a CH2 group. In the model shown in Fig. 2c, a monomer also corresponds to a CH2 group. In more coarse-grained models, a monomer is composed of several CH2 groups.
Crystal structure in the previous studies
The conformation of a polymer chain and the corresponding crystal structure depend on the potentials. The details of the crystal growth process in molecular simulations were previously reviewed [27]. To elucidate the mechanical properties of crystalline polymers, it is essential to construct the polymers’ crystal structure. However, it has been reported that molecular dynamics simulations realize spinodal-assisted crystallization in poly(vinylidene fluoride) and polyethylene models, owing to the simulation time limitation (rapid cooling rate) [28]. Therefore, modeling methods for constructing the crystalline structure of polymers are required. This focus review discusses the relationship between the coarse-grained model and crystal structure.
In previous studies focusing on the mechanical properties of crystalline polymers, various methods were used to construct the crystal structure, which are summarized as follows. To more accurately reproduce the crystallization process, Yamamoto [17] modeled the crystallization process in fluids and successfully constructed a lamellar structure using a coarse-grained molecular dynamics simulation. The Monte Carlo method has also been used to construct lamellar structures [13,14,15]. These studies adopted the united atom model of polyethylene. To prepare a large-scale structure consisting of 4.3 × 106 beads, a small crystal grain was obtained and then copied [18]; the authors of this study used a coarse-grained model of polyvinyl alcohol similar to the model presented in Fig. 2c.
Polycrystal structure
Efforts have been made to construct crystal structures, and our findings are summarized here. The most coarse-grained model (Fig. 2a) was initially used. To enhance crystallization, the method proposed by Koyama et al. [29] was adopted, in which the cell is elongated in one direction with the orientation of the polymer chains. Figure 3a, b show a constructed crystal structure and a typical polymer chain in the crystal structure, respectively. Although the polymer chains are able to fold, the crystal domains are distributed randomly and the crystallinity is low. Because the polymer chains bend circularly, the amorphous regions become large.

Fracture process of a crystalline polymer upon stretching. a Cross-sectional and b typical single-chain snapshots, where the blue and green regions represent the crystalline and amorphous domains, respectively. The stretching speed was set to 0.06a/τ
Figure 3a shows the fracture process of a crystalline polymer containing randomly distributed crystalline domains. Upon stretching, a void is generated and then grows parallel to the stretching direction. Figure 3b shows a typical polymer chain during the fracture process. The figure shows that some bonds dissociated during stretching. The melt state was also stretched; however, in this case bond dissociation was not observed. Therefore, in the crystal structure, the polymer chains do not easily relax in response to stretching, and the bonds become elongated. In the crystalline structure, the stress strongly depends on the stretching velocity, whereas this dependence is weak in the melt state. This observation also confirms that the polymer chains in the folded state do not easily relax in response to stretching. The obtained structure is not consistent with the typical experimental result; however, this simple model demonstrates that the relaxation time of the folded structure is long compared with that of a random coil [23].
We examined the influence of the stretching velocity on the mechanical properties. The velocity was set to 0.06a/τ, 0.01a/τ, 0.006a/τ, 0.002a/τ, and 0.001a/τ, where a and τ are the units of length and time. The stress critically decreased with a decrease in the stretching velocity, whereas the amorphous polymers were not sensitive to the stretching velocity. This finding also shows that the relaxation of crystalline polymers is much slower than that of amorphous polymers.
Lamellar structure with low crystallinity
To solve the problem of circular bending related to the polycrystal structure of polymers, a model based on orthogonal bending, as shown in Fig. 2b, was used. Furthermore, a cyclic force was applied to construct the crystal structure because it is difficult to obtain the lamellar structure without excessive computational cost. Figure 4a and b show the constructed crystal structures consisting of 4.0 × 104 and 1.0 × 106 monomers, respectively. Whereas a blurred lamellar structure was observed in the smaller model (Fig. 4a), the lamellar structure was successfully constructed in the larger model (Fig. 4b). Figure 4c shows a typical snapshot of a folded polymer chain. Therefore, when the polymer chain bends orthogonally, it is possible to construct the lamellar structure using cyclic force. However, the crystalline regions are still narrow.

Snapshots of the lamellar structures consisting of a 4 × 104 and b 1 × 106 monomers. c A typical polymer chain. Reproduced with permission from Higuchi et al. [24], ©(2016) by the Information Processing Society of Japan
The lamellar structure was stretched along the oriented direction, and the fracture process is shown in Fig. 5. Fragmentation of the lamellar layers was observed [24] in agreement with the experimental observations made using electron microscopy [30]. However, the stress–strain curve was not consistent with that obtained experimentally [31].

Fracture process of the lamellar structure upon stretching at (a) t= 0τ, (b) t = 150τ, (c) t = 225τ, (d) t= 300τ, (e) t = 375τ, (f) t= 450τ, and (g) t = 675τ. The stretching speed was set to 0.06a/τ. Reproduced with permission from Higuchi et al. [24], © (2016) by the Information Processing Society of Japan
Lamellar structure with high crystallinity
To increase the length of the crystalline domain, we increased the attractive interaction by adopting the model shown in Fig. 2c. Upon applying a cyclic force, a lamellar structure was not obtained from the melt state. The reason is that entanglements are conserved during the crystallization process [1]. To decrease these entanglements prior to the crystallization, we positioned all of the polymer chains linearly and then performed the relaxation simulation using the NPT ensemble method. Subsequently, we applied a cyclic force to construct the lamellar structure and copied the cell to increase the simulation size. Figure 6 shows the constructed lamellar structure consisting of 3.0 × 106 beads. The size of the crystalline regions increased compared with that shown in Fig. 4b. Therefore, the lamellar structure was successfully constructed. The lamellar thickness was set to 15 nm, which is a typical experimental value [32, 33].

Deformation and fracture processes of the lamellar structure upon stretching a parallel and b perpendicular to the oriented direction. The stretching speed was set to 0.03a/τ. Reproduced with permission from Higuchi et al. [22], ©(2017) by the American Chemical Society
Structures at the molecular level, such as loops, entanglements and tie chains, were compared with those in the lamellar structure constructed by Rutledge et al. [13,14,15]. The number densities of the loops and entanglements in the structure shown in Fig. 6 were similar to those reported in the previous studies by Rutledge et al., whereas the number density of tie chains was higher. These differences were ascribed to the fact that the crystallinity of our constructed lamellar structure was higher than that obtained in previous studies and the amorphous thickness obtained in our work was lower. Therefore, our constructed lamellar structure is reasonable for elucidating the mechanical properties of this polymer.
Figure 6a, b show the fracture processes of the lamellar structure upon stretching parallel and perpendicular to the oriented direction, respectively. Upon stretching parallel to the oriented direction, the polymers became thinner, and the amorphous layers then deformed further. A void was generated in the amorphous layers and subsequently grew larger. The difference between the crystal and amorphous parts is related to the energy dissipation and heat via stretching. In the lamellar model, heat-up in the amorphous region was confirmed. This finding indicates that the crystalline layer is solid against stretching. Upon stretching perpendicular to the oriented direction, the polymers also became thinner, although voids were not generated and the amorphous and crystalline layers were conserved. Figure 7a, b shows the stress–strain curves and the changes in the crystallinity during stretching. Upon stretching parallel to the oriented direction, the stress increased for strains ranging from 0.0 to 0.6. The crystallinity also increased during stretching, which indicates that the polymers crystallized upon stretching. In contrast, upon stretching perpendicular to the oriented direction, the stress increased more gradually. The crystallinity also increased in this case; however, the increase was smaller than that observed upon stretching parallel to the oriented direction. These trends observed for the stress–strain curves and the crystallinity changes are consistent with previous experiments [31]. Therefore, the lamellar structure and its fracture process obtained in our studies were successfully validated.

a Stress–strain curves for the lamellar structure upon stretching parallel and perpendicular to the oriented direction, which are indicated by z and x, respectively. b Crystallinity versus strain curves upon stretching parallel and perpendicular to the oriented direction. Reproduced with permission from Higuchi et al. [22], © (2017) by the American Chemical Society
Discussion
Based on the results described above, rigid polymer chains can form crystal structures, but it is difficult to realize lamellar structures. Orthogonal bending is required to construct lamellar structures. However, the following two conditions must be noted: (i) natural crystallization to lamellar structures is impossible, and therefore, cyclic forces are essential to enhancing crystallization; (ii) entanglements prior to crystallization severely hinder the construction of lamellar structures and thus must be reduced. To increase the crystallinity, the average bond length should be reduced, and the attractive interaction should be increased. Furthermore, it is conceivable that a cylindrical polymer chain also increases the crystallinity. In contrast to those in a polymer chain with a cylindrical shape, the spaces between adjacent monomers in the other models allow for easy movement of the polymer chains and lead to fluctuations in the crystal region. In the united atom model, the polymer chain is shaped like a plate; therefore, the orientation of the polymer chains increases due to the suppressed movement. It is desirable to adopt a reasonable model to limit computational costs. The reason is that long simulation times are essential for elucidating the dynamics of polymer chains, and atomic information is also crucial to studying crystalline polymers. Therefore, our approach, in which a coarse-grained model is used initially and then atomic information is gradually added, contributes to understanding the conditions required to reproduce the lamellar structure of crystalline polymers.
Several treatments can be applied to the axes perpendicular to the stretching direction in stretching simulations: (i) both axes are fixed, (ii) both axes are relaxed using the NPT ensemble method, (iii) both axes are relaxed using a constant strain rate, (iv) vacuum spaces are located in one direction and the other axis is fixed, (v) vacuum spaces are located in one direction and the other axis is relaxed using the NPT ensemble method, and (vi) vacuum spaces are located in both directions. These treatments influence the mechanical properties. In case (i), the condition is very severe because deformation perpendicular to the stretching direction is not permitted, which leads to fracture of the polymers. This model conceivably corresponds to the enlargement of the small area around a crack in experiments. Rottler et al. demonstrated the validity of a coarse-grained molecular dynamics model by successfully estimating the fracture energy of polymer glasses [8]. We also modeled the fracture processes of semicrystalline polymers [23]. The fragmentation of the lamellar structure to the block structure was also observed [24], in agreement with experimental observations made using electron microscopy [30]. This method is useful for revealing the fracture processes of polymers; however, the deformation process is difficult to determine. Therefore, the stress–strain curve is not consistent with that obtained experimentally. In case (ii), polymers can deform naturally. However, both soft and hard parts are forcibly compressed. This compression is not a problem for elastic strain. After yielding, the polymers partially deform, or voids are generated. The non-deformed parts are also compressed. Therefore, the stress–strain curve is not consistent with that obtained experimentally after yielding. Makke et al. [34, 35] observed buckling of a lamellar structure consisting of amorphous and glassy layers, which are soft and hard, respectively. This interesting behavior is a periodic phenomenon, and therefore, boundary conditions are needed in the corresponding molecular simulations. Rutledge et al. also used this method to investigate the deformation and fracture processes of polyethylene [15, 16]. The authors prepared many initial conditions to determine the effects of molecular structures such as tie chains and entanglements. Therefore, this method is conceivably useful for elucidating deformation and fracture processes with limited computational cost. Case (iii) is similar to case (ii). The choice of the constant strain is difficult. Neither the option of using a fixed volume nor that of selecting the value using Poisson’s ratio leads to good consistency with the experimental values. Discrepancies exist between simulations and experiments in terms of size, structure, etc. Yamamoto adopted this method to determine void generation in the amorphous layer [17]. Compared with the method used by Rutledge et al., [13,14,15,16] the processes are similar. In case (iv), the polymers can deform naturally in one direction but not in the other direction. Therefore, the deformation and fracture processes are unnatural. In case (v), the polymers can deform naturally. However, the thickness in the direction with the vacuum space should be sufficient. In Fig. 6, the vacuum space is set in one direction. This treatment is considered suitable for revealing mechanical properties, as long as the simulation size is sufficiently large. Case (vi) is optimum if the simulation size is sufficiently large, because the sample can deform naturally throughout the entire process. However, it is difficult to construct such a large model, owing to the excessive computational cost.
Herein, void generation and rupture are discussed. As shown in Fig. 6a, voids were generated and then grew in the lamellar structure consisting of 3.0 × 106 monomers. In contrast, void generation was not observed in a lamellar structure consisting of 5.0 × 105 monomers. This discrepancy was observed because the thickness was too narrow in the latter case and there was not sufficient space for void formation. Therefore, a large simulation size is required to adopt case (v). After voids grow, rupture should be observed. However, rupture is difficult to observe in simulations due to the limited simulation size. When one part of the polymer breaks, the polymers do not rupture suddenly but rather break gradually. The broken part is large compared with the total simulation size. Therefore, the ruptures observed in molecular simulations cannot be directly compared with those observed in experiments.
Finally, this review discusses the main priorities considered when performing molecular simulations. Many factors affect the mechanical properties of crystalline polymers. In particular, the structure at the molecular level is important. It has been suggested that crystalline layers are connected through tie chains and entanglements in the amorphous layers [3, 4, 6, 36]. Therefore, Humbert et al. [37, 38] referred to these points of contact as stress transmitters. To determine the influence of these factors on mechanical properties, molecular simulations are useful. Rutledge et al. used coarse-grained molecular dynamics simulations to analyze molecular structures such as tie chains and entanglements to determine their influence on mechanical properties [13,14,15,16]. In addition, Monasse et al. [20] used an all-atomic model and performed molecular dynamics simulations, which indicated that stress increases with an increasing number of tie chains. We have also analyzed molecular structures and determined that the movement of chain ends from amorphous layers to crystal layers causes void generation [22]. This finding indicates that the chain ends act as defects. Indeed, we have also shown that amorphous layers with a high concentration of chain ends fracture, whereas those with low concentrations of chain ends do not. These findings are difficult to observe experimentally.
Conclusion
This review has summarized our recent work [22,23,24] concerning the deformation and fracture processes of crystalline polymers using coarse-grained molecular dynamics simulations. First, we constructed a polycrystalline model and revealed the long relaxation time of folded chains against stretching. Then, to construct a lamellar structure, we modified the model from circularly bending chains to orthogonally bending chains and applied cyclic forces to enhance crystallization. The constructed lamellar structure fractured into block structures, as observed experimentally using electron microscopy. Finally, we reduced the number of entanglements and increased the attractive interaction density between the polymer chains. These modifications allowed for the construction of a lamellar structure with high crystallinity. By using this model and locating vacuum spaces perpendicular to the stretching direction, we were able to obtain a stress–strain curve that agreed well with that obtained experimentally.
The conditions required to reproduce the crystalline structure and mechanical properties observed experimentally have also been discussed, which are summarized as follows. To achieve the crystallization of lamellar structures, (i) the gauche conformation should be included in the model; (ii) the attractive interaction density should be high, which means that the average bond length between monomers should be low; (iii) cyclic force is required to enhance crystallization and construct lamellar structures; and (iv) entanglements should be reduced prior to crystallization. To reproduce the stress–strain curves, (i) the vacuum spaces should be located in one direction, and the cell in the other direction should be relaxed using NPT ensembles; and (ii) the simulation size should be sufficiently large to at least allow for the observation of void generation and growth.
It has also been revealed that voids are generated by the movement of chain ends from amorphous to crystalline layers, which is difficult to observe experimentally. Molecular simulations are powerful tools for elucidating the mechanisms involved in the deformation and fracture processes of crystalline polymers at the molecular level. It is hoped that our recent work and this review will contribute to the development of this field of research.
References
Strobl G. The physics of polymers: concepts for understanding their structures and behavior. Heidelberg, Springer; 2007.
Peterlin A. Molecular model of drawing polyethylene and polypropylene. J Mater Sci. 1971;6:490–508.
Séguéla R. Critical review of the molecular topology of semicrystalline polymers: the origin and assessment of intercrystalline tie molecules and chain entanglements. J Polym Sci B. 2005;43:1729–48.
Séguéla R. On the natural draw ratio of semi-crystalline polymers: review of the mechanical, physical and molecular aspects. Macromol Mater Eng. 2007;292:235–44.
Galeski A. Strength and toughness of crystalline polymer systems. Prog Polym Sci. 2003;28:1643–99.
Patlazhan S, Remond Y. Structural mechanics of semicrystalline polymers prior to the yield point: a review. J Mater Sci. 2012;47:6749–67.
Rottler J, Barsky S, Robbins MO. Cracks and crazes: on calculating the macroscopic fracture energy of glassy polymers from molecular simulations. Phys Rev Lett. 2002;89:148304.
Rottler J, Robbins MO. Growth, microstructure, and failure of crazes in glassy polymers. Phys Rev E. 2003;68:011801.
Papakonstantopoulos GJ, Riggleman RA, Barrat J-L, de Pablo JJ. Molecular plasticity of polymeric glasses in the elastic regime. Phys Rev E. 2008;77:041502.
Makke A, Perez M, Lame O, Barrat J-L. Mechanical testing of glassy and rubbery polymers in numerical simulations: role of boundary conditions in tensile stress experiments. J Chem Phys. 2009;131:014904.
Yoshimoto K, Jain TS, Nealey PF, de Pablo JJ. Local dynamic mechanical properties in model free-standing polymer thin ˝lms. J Chem Phys. 2005;122:144712.
Rottler J. Fracture in glassy polymers: a molecular modeling perspective. J Phys Condens Matter. 2009;21:463101.
Lee S, Rutledge GC. Plastic deformation of semicrystalline polyethylene by molecular simulation. Macromolecules. 2011;44:3096–108.
Kim JM, Locker R, Rutledge GC. Plastic deformation of semicrystalline polyethylene under extension, compression, and shear using molecular dynamics simulation. Macromolecules. 2014;47:2515–28.
Yeh I-C, Andzelm JW, Rutledge GC. Mechanical and structural characterization of semicrystalline polyethylene under tensile deformation by molecular dynamics simulations. Macromolecules. 2015;48:4228–39.
Yeh I-C, Lenhart JL, Rutledge GC, Andzelm JW. Molecular dynamics simulation of the effects of layer thickness and chain tilt on tensile deformation mechanisms of semicrystalline polyethylene. Macromolecules. 2017;50:1700–12.
Yamamoto T. Molecular dynamics in fiber formation of polyethylene and large deformation of the fiber. Polymer. 2013;54:3086–97.
Jabbari-Farouji S, Rottler J, Lame O, Makke A, Perez M, Barrat J-L. Plastic deformation mechanisms of semicrystalline and amorphous polymers. ACS Macro Lett. 2015;4:147–50.
Jabbari-Farouji S, Rottler J, Lame O, Makke A, Perez M, Barrat J-L. Correlation of structure and mechanical response in solid-like polymers. J Phys: Condens Matter. 2015;27:194131.
Monasse B, Queyroy S, Lhost O. Molecular Dynamics prediction of elastic and plastic deformation of semi-crystalline polyethylene. Int. J. Mater Form. 2008; 1:1111–4.
O’Connor TC, Robbins MO. Chain ends and the ultimate strength of polyethylene fibers. ACS Macro Lett. 2016;5:263–7.
Higuchi Y, Kubo M. Deformation and fracture processes of a lamellar structure in polyethylene at the molecular level by a coarse-grained molecular dynamics simulation. Macromolecules. 2017;50:3690–702.
Higuchi Y, Kubo M. Coarse-grained molecular dynamics simulation of the void growth process in the block structure of semicrystalline polymers. Modell Simul Mater Sci Eng. 2016;24:055006.
Higuchi Y, Kubo M. Large-scale coarse-grained molecular dynamics simulation based on MPI parallel computing: mechanical property of polymers in molecular scale. HPCS. 2016;2016:9–14.
Higuchi Y, Sakaue T, Yoshikawa K. Chain length dependence of folding transition in a semiflexible homo-polymer chain: appearance of a core-shell structure. Chem Phys Lett. 2008;461:42–46.
Higuchi Y, Yoshikawa K, Iwaki T. Confinement causes opposite effects on the folding transition of a single polymer chain depending on its stiffness. Phys Rev E. 2011;84:021924.
Yamamoto T. Computer modeling of polymer crystallization - toward computer-assisted materials’ design. Polymer. 2009;50:1975–85.
Gee RH, Lacevic N, Fried LE. Atomistic simulations of spinodal phase separation preceding polymer crystallization. Nat Mater. 2009;8:159–159.
Koyama A, Yamamoto T, Fukao K, Miyamoto Y. Molecular dynamics simulation of polymer crystallization from an oriented amorphous state. Phys Rev E. 2002;65:050801.
Adams W, Yang D, Thomas E. Direct visualization of microstructural deformation processes in polyethylene. J Mater Sci. 1986;21:2239–53.
Zhou H, Wilkes GL. Orientation-dependent mechanical properties and deformation morphologies for uniaxially melt-extruded high-density polyethylene films having an initial stacked lamellar texture. J Mater Sci. 1998;33:287–303.
Mandelkern L. The relation between structure and properties of crystalline polymers. Polym J. 1985;17:337–50.
Kennedy MA, Peacock AJ, Mandelkern L. Tensile properties of crystalline polymers: linear polyethylene. Macromolecules. 1994;27:5297–310.
Makke A, Perez M, Lame O, Barrat J-L. Nanoscale buckling deformation in layered copolymer materials. Proc Natl Acad Sci USA. 2012;109:680–5.
Makke A, Lame O, Perez M, Barrat J-L. Nanoscale buckling in lamellar block copolymers: a molecular dynamics simulation approach. Macromolecules. 2013;46:7853–64.
Fukuoka M, Aya T, Saito H, Ichihara S, Sano H. Role of amorphous region on the deformation behavior of crystalline polymers. Polym J. 2006;38:542547.
Humbert S, Lame O, Chenal JM, Rochas C, Vigier G. New insight on initiation of cavitation in semicrystalline polymers: in-situ SAXS measurements. Macromolecules. 2010;43:7212–21.
Humbert S, Lame O, Vigier G. Polyethylene yielding behaviour: what is behind the correlation between yield stress and crystallinity? Polymer. 2009;50:3755–61.
Acknowledgements
This research was supported by JSPS KAKENHI (Grant No. JP17K14534) and MEXT as “Exploratory Challenge on Post-K Computer” (Challenge of Basic Science-Exploring Extremes through Multi-Physics and Multi-Scale Simulations). The author acknowledges computational resources of the K computer provided by the RIKEN Advanced Institute for Computational Science through the HPCI System Research project (Project ID: hp160271 and hp170245) and SR16000 supercomputing resources from the Center for Computational Materials Science of the Institute for Materials Research, Tohoku University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Higuchi, Y. Fracture processes of crystalline polymers using coarse-grained molecular dynamics simulations. Polym J 50, 579–588 (2018). https://doi.org/10.1038/s41428-018-0067-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41428-018-0067-1
This article is cited by
-
Charge-transfer electronic states in organic solar cells
Nature Reviews Materials (2019)
-
Visco- and plastoelastic fracture of nanoporous polymer sheets
Polymer Journal (2019)
