
Abstract
Inorganic–organic hybrid materials have promising properties for bone repair because of the covalent bonding between their inorganic and organic phases. This desirable interaction allows the limitations of composite materials, such as inhomogeneous biodegradation rates and nonbiointeractive surfaces, to be overcome. In this study, a polycaprolactone (PCL)-based polyurethane (PU) with an organosilane functional group was synthesized for the first time. Thereafter, a biodegradable PU-silica hybrid was produced through the sol-gel process. The PU-silica hybrid was not only flexible and fully biodegradable but also possessed shape memory ability. In addition, allophanate bonding enabled the silane coupling agent to induce increased crosslinking between the polymer and silica network, as well as between polymer and polymer. Accordingly, the sol-to-gel gelation time required to produce the hybrids was very short, which allowed the production of 3D porous hybrid scaffolds through a simple salt-leaching process. A hybrid scaffold with a 30 wt. % silica composition was the most ideal bone regenerative scaffold since it was able to withstand thermal deformation with promising mechanical properties. Moreover, the hybrid scaffold induced osteogenic differentiation and angiogenesis to accelerate bone regeneration.
Similar content being viewed by others

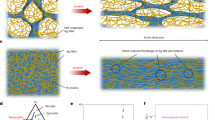

Introduction
Aging and trauma, such as that caused by falling and car crashes, are the main causes of bone defects. The acceleration of global aging and industrialization has rapidly increased the demand for bone grafts and bone substitutes. Allografts and autografts are common in bone transplantation because they provide three-dimensional templates and biological support for bone regeneration. However, limitations with regard to harvest sites and graft quantity have led to the development of synthetic bone grafts (bioceramics, polymers, and composites). Inorganic materials have bioactivity but are too brittle to withstand cyclic loads. Polymers are biodegradable and tough, but they are bioinert. Therefore, numerous studies have focused on formulating ideal bone-regenerative composite materials. Nevertheless, composites are subjected to macroscale phase separation owing to the weak bonding between the organic and inorganic phases. This makes the cracks in the composite vulnerable to external forces and may lead to a loss of mechanical properties. In addition, the polymer matrix tends to mask the inorganic content, which reduces bioactivity1. To overcome these shortcomings, various inorganic–organic hybrids, or polymers interconnected to silica networks via covalent bonding, have been explored by numerous researchers1,2,3,4,5. The organic polymers were copolymerized with silane coupling agents, such as (3-glycidyloxypropyl)trimethoxysilane (GPTMS)2,4 and 3-(trimethoxysilyl)propyl methacrylate (TMSPMA)3, to form conetworks with the silica matrix via the sol-gel process. However, most biodegradable hybrids require a long gelation time, making them difficult to manufacture into bone scaffolds with porous structures. They were usually produced into cylindrical shapes, since the gelling and drying stages during the sol-gel process took several days6,7.
In this study, a biodegradable polyurethane (PU)-silica hybrid was designed to fabricate a porous bone regenerative scaffold that had the advantages of the inorganic‒organic hybrid properties mentioned above. PUs are versatile polymers capable of being incorporated with various combinations of different types and ratios of polyols, isocyanates, and crosslinkers8, which allows for the functionalization of the polymers. The developed PU was thermoplastic and biodegradable as a result of polymerization with polycaprolactone (PCL)-diol, hexamethylene diisocyanate (HDI), and (3-aminopropyl)triethoxysilane (APTES). PCL-diol and aliphatic HDI have been used to synthesize thermoplastic, biodegradable, and biocompatible PUs9,10. A thermoplastic polymer was required for the sol-gel process since polymers must exist in a sol state before forming covalent bonds with the silica network. The introduction of APTES during polymerization allowed for the functionalization of PU with alkoxysilane groups, which enabled covalent bonding to the silica network. A homogeneous hybrid was produced because the PU was dissolvable in solvents that did not phase-separate from the silica network precursor (hydrolyzed tetraethyl orthosilicate, TEOS). In addition, the formation of allophanate branches due to the high temperatures of the prepolymer synthesis reaction was able to increase the number of exposed functional groups. Increasing the number of silane groups, which participate in the reaction of branched PU with hydrolyzed TEOS, in the polymer chains can significantly shorten the gelation time compared to previously reported studies2,4,11. This allowed the production of biomimetic hybrid scaffolds with a 3D porous structure using the facile salt-leaching method. Interestingly, the hybrid scaffold possessed shape memory ability since the crystalline PCL backbone in the PU was chemically cross-linked in the hybrid system. This enabled the hybrid scaffold to switch its crystalline structure when an appropriate temperature was applied. The shape memory property of the hybrid scaffold allowed to fill up irregular bone defects, which enhanced bone repair by increasing the biointeractive surface area between the scaffold and defect.
Finally, the effects of using different silica ratios in the hybrids on the bone regenerative ability were examined using a rat distal condyle defect model. Histological evaluation was performed to identify differences in the cell types of the new bone, and mineralization of the newly formed bone was revealed using micro-computed tomography (µCT). Furthermore, immunostaining for expression markers of inflammatory and bone healing events, such as angiogenesis and osteogenic differentiation, were evaluated, which are of crucial importance in bone remodeling.
Results and discussion
Polyurethane and hybrid confirmations
Similar to previous studies9,10, PCL with two terminated hydroxyl groups at opposite ends, namely, PCL-diol, was used as the polyol to synthesize biodegradable PU. The diol groups were reacted with HDI, a diisocyanate, to produce urethane bonds between the PCL-diols and polyurethane12,13. For the formation of covalent bonds with the silica matrix, APTES, an organoalkoxysilane, was introduced during the polymer synthesis to react with the HDI-terminated PCL-diol (Fig. 1A). The polymer structure was confirmed using proton nuclear magnetic resonance spectroscopy (1H-NMR). As shown in Fig. 1B, PCL (4.08 and 2.34 ppm), HDI (3.18 ppm), and APTES (0.66 ppm) peaks were present in the NMR spectrum. Biodegradable polyurethane was synthesized with three different molar ratios of APTES to confirm the effect of organoalkoxysilane on polymer synthesis since covalent bonds between the polymers and silica network can affect the physiological and chemical properties of hybrids14. Despite introducing different molar ratios of APTES, the molar ratio (in percentile) of each component and the number average molecular weight (Mn) of the polymers did not change significantly (Table 1). The molecular weight of the polymers suggested that polymerization did not occur, as schematically represented in Fig. 1B; PCL-diol reacted at opposite ends with HDI followed by APTES (expected molecular weight of 2.8 kDa). As shown in Fig. S1, the signal of C = O stretching from the allophanate linkages suggested that the polymers underwent a secondary reaction. Due to this allophanate formation, the molecular weight of P(CL-co-APTES) was higher than expected, and the alkoxysilane group content increased in the polymer chain. The allophanate formation was possibly due to the high polymerization temperature (90 °C) used to synthesize PU15. The polymer was brittle, similar to PCL. However, after the hydrolysis and condensation reactions, which formed Si-O-Si bonds between the polymer chains, it became tough. The increase in the molecular weight, which led to more entanglements between the polymer chains, affected the physical and mechanical properties.

A Schematic illustration of P(CL-co-APTES) synthesis. B 1H NMR spectra of P(CL-co-APTES). Inorganic–organic hybrid characterizations. C Rheology measurements of BPH0, BPH10, BPH20, and BPH30 during the sol-to-gel transition. The crossover point of the lines, G' and G'', represents gelation time. D FTIR spectra and (E) TGA of the polymer and hybrids.
After confirming the polymer synthesis, alkoxysilane-functionalized biodegradable polyurethane was made into inorganic–organic hybrids via the sol-gel process. TEOS, a silica network precursor, was hydrolyzed to silicic acid (Si(OH)4), and the polymer was introduced before the condensation reaction. APTES from the polymer and Si(OH)4 mixture formed siloxane bonds, producing covalently bonded hybrids. Hybrids with four silica ratios (0, 10, 20, and 30 wt. %) were produced. The hybrid samples were referred to as BPH0, BPH10, BPH20, and BPH30 (BPH corresponds to ‘biodegradable polyurethane hybrid,’ and the number corresponds to the inorganic content in wt. %). As the average molar ratio of APTES in the polymer was 4.3%, gelation occurred within 10 min of the sol-gel process (Fig. 1C). This showed that APTES functionalization contributed to the rapid gelation of the inorganic–organic hybrid material. Additionally, since the PU synthesis at a high temperature increased the number of APTES molecules (crosslinker) per chain, the gelation time of the hybrid was shortened. The storage modulus (G’) and loss modulus (G'') are indicative of the liquid-to-solid phase transition. The crossover moduli of BPH0, BPH10, BPH20, and BPH30 occurred 520, 80, 240, and 330 s after introducing the polymer to the hydrolyzed TEOS, respectively. For BPH0, the sol-gel transition time was the longest compared to all the other groups because the crosslinking points were fewer compared to those of the hybrids with silica content. The amount of TEOS added affected the gelation time of the hybrids; the higher the silica content was, the higher the gelation time. This was because Si(OH)4 has a slower condensation rate under acidic conditions, which requires more time for the silica matrix to form16. The transition of the PU-silica hybrids from the sol-to-gel state was faster than that of other biodegradable hybrids from previous studies, which took 1–2 weeks to gel2,4. This rigorous process was necessary because a rapid drying method can induce capillary stress, which leads to crack formation. Consistent with this, the rheological properties of a PCL-silica composite (PCL-diol with no organoalkoxysilane functional group) were measured to compare their gelation time with that of the hybrids. As shown in Fig. S2, the composite with 30 wt. % silica content remained as a sol after 1 h of mixing. Since BPH does not require a lengthy gelation time, it can be fabricated to a porous 3D scaffold through various approaches, such as the salt-leaching method and 3D printing technique17.
The chemical structures of the hybrids were confirmed using Fourier transform infrared spectroscopy (FTIR) (Fig. 1D). Representative polymer and silica spectra were obtained from the hybrid samples. The signals of C = O and C-O stretchings of the aliphatic hydrocarbons of the polymer were found at 1163–1210 cm−1 and 1085–1150 cm−1, respectively. The bands between 798 cm−1 and 1094 cm−1 indicated the silica matrix or Si-O-Si bonding of the hybrids. As the inorganic content was increased from 0 to 30 wt. %, the absorption intensity of hydrocarbons decreased, while the silica bands increased. The actual inorganic content of the hybrids was confirmed using thermogravimetric analysis (TGA) (Fig. 1E). The residual mass of each sample was close to the target inorganic ratios of 5.2, 8.1, 21.7, and 30 wt. % for BPH0, BPH10, BPH20, and BPH30, respectively. The thermal decomposition of BPH0, or PU alone, occurred between 285 and 410 °C, whereas the inorganic–organic hybrids decomposed between 370 and 500 °C. This was due to siloxane bonding within the hybrids, which enhanced the thermal stability because silica can act as an insulator and mass transport barrier18. FTIR and TGA analyses confirmed that the polymer and silica were both present in the hybrids. The endothermic peaks, which represent the melting temperature, of the silica-containing hybrids were lower than those of the polymer-only hybrids, as seen from the differential scanning calorimetry (DSC) curves of both BPH and BPHS (BPHS corresponds to ‘biodegradable polyurethane hybrid scaffold,’ and the number corresponds to the inorganic content in wt. %) conformations (Fig. S3 and Fig. 2Ai). This was possibly due to the silica network impeding the semicrystalline structure of the polymer within the hybrid system and the sol-gel silica network having an amorphous structure19. The crystallinity of the samples was determined using the enthalpy of fusion. BPH and BPHS with 0 and 10 wt. % silica content had no significant differences in their crystallinity: the crystallinity values were 28.84 and 29.63% for BPH0 and BPH10, and 21.83 and 23.66% for BPHS0 and BPHS10, respectively. However, BPHS20 and BPHS30 exhibited significantly lower crystallinity than the BPH samples; the values decreased from 10.65 and 4.74% to 0.99 and 1.61%, respectively. Previous studies have shown that porous scaffolds (produced by electrospinning and salt-leaching methods) can have a lower crystallinity than bulk samples, since a higher surface-area-to-volume ratio with thin struts and a low polymer content can reduce crystalline formation20.
Annealing effects on hybrid scaffolds
3D porous hybrid scaffolds (BPHS) were synthesized with a silica content below 40 wt. % using the salt-leaching method, since the scaffold with 40 wt. % silica was brittle (Fig. S4). The hybrid scaffolds were produced with four different silica ratios, and the scaffold samples were referred to as BPHS0, BPHS10, BPHS20, and BPHS30 (number corresponds to the inorganic content in wt. %). PCL is a semicrystalline polymer; therefore, heat treatment or annealing can modify the crystallinity, mechanical properties, and physical structure of BPHS. The melting point temperature of the hybrids was 30–50 °C, which was close to the melting point temperature of PCL (the main polymer backbone of the biodegradable polyurethane). As the hybrids exhibited a melting point near body temperature, which could lead to difficulties in maintaining their physical and mechanical properties in vivo, DSC curves, pore size, porosity, and mechanical properties of BPHS before and after annealing were compared. The annealing temperature was set to 60 °C, which is above the melting point of BPH.
The total volume, pore size, and mechanical properties of BPHS were affected by the annealing process. Following annealing, the DSC curves (Fig. 2Ai and ii) showed that the double melting peaks of BPHS transformed into a single peak. The annealing process was able rearrange the semicrystalline structure of PCL-based polyurethane. The enthalpy of fusion of BPHS10 decreased significantly after annealing, and the crystallinity decreased from 23.66 to 1.78%. The heat treatment was able to produce a more organized crystalline structure for BPHS0, while the recrystallization of BPHS10 was hindered due to the silica matrix. Conversely, both BPHS20 and BPHS30 maintained low crystallinity after undergoing the heat treatment: the crystallinity values were 1.13 and 2.01% for BPHS20 and BPHS30, respectively.
As shown in Fig. 2Bi, there was no change in the tubular shape of BPHS after the heat treatment because the silica network acted as a robust structural support or a net point21. Higher shrinkage in diameter was observed as the silica content decreased; the diameters of BPHS0, BPHS10, and BPHS20 shrank by ~32.3, 6.4 and 1.8%, respectively. On the other hand, the diameter of BPHS30 expanded by 3.5% compared to the original diameter. The expansion phenomenon of BPHS30 was due to the formation of a disordered structure in the amorphous region. The high concentration of silica network possibly suppressed the movement of polymer chains during the relaxation process22. The pore sizes were also influenced by annealing. The scanning electron microscopy (SEM) images in Fig. 2Bii show that BPHS0 had a significantly reduced pore size after annealing. As shown in Fig. 2Biii, the pore sizes before annealing were 344.6 ± 22.7, 430.1 ± 35.1, 340.8 ± 79.0, and 387.2 ± 47.4 μm for BPHS0, BPHS10, BPHS20, and BPHS30, respectively, which were in good agreement with the NaCl particle sizes (300–500 μm) that were used for the salt-leaching method. The pore diameters of all the BPHS samples decreased after annealing (226.4 ± 12.3, 266.7 ± 40.3, 248.5 ± 19.9, and 327.6 ± 58.8 μm for BPHS0, BPHS10, BPHS20, and BPHS30, respectively). The average shrinkage ratios were 34.3, 38.0, 27.1, and 15.4% for BPHS0, BPHS10, BPHS20, and BPHS30, respectively. This was due to the rearrangement of the crystalline region during the annealing process23,24,25. BPHS0, which had a high crystallinity, displayed high shrinkage ratio after the annealing process, similar to the highly cross-linked PCL scaffolds from a previous study26. In contrast, scaffolds with a higher silica content (≥20 wt. %) with low crystallinity (Fig. 2Aii) experienced lower shrinkage. A minimal pore size of 100–200 μm is known to facilitate vascularization and cartilage maintenance27,28. All BPHS pore sizes were over 100–200 µm, meeting the hypothesized minimum requirements for vascularization. Specifically, the silica-containing scaffolds had pore sizes of >250 μm. Scaffolds with a pore size of >250 μm were able to enhance the osteogenesis and differentiation of mesenchymal stem cells (MSCs) in a previous study28. Although the annealing process affected the pore size, the porosity remained consistent (Fig. 2Biii). This confirmed that the porous structure formed by the salt-leaching method was well interconnected. In addition, strong covalent bonding between the polymer and silica network prevented the scaffolds from collapsing.
The mechanical properties of the hybrid scaffolds were evaluated using compression tests. As shown in Fig. 2C and supplementary video 1 (a recording of a representative compression test of BPHS30), the stress‒strain properties of all the BPHS samples indicated ductility and toughness. The mechanical properties, however, were significantly affected by the crystallinity and silica content. A more organized crystalline structure and an increase in crystallinity generally led to improvements in the compression strength and modulus29. Moreover, the silica network enhanced the mechanical properties of the hybrids.

A DSC curves of BPHS (i) before annealing and (ii) after annealing. B Effect of annealing on BPHS. (i) Representative BPHS images after annealing (scale bar: 4 mm), (ii) cross-sectional scanning electron microscope images of BPHS (scale bar: 390 μm), and (iii) quantification of the average pore diameter (n = 23) and interstitial porosity (n = 3) of BPHS before and after annealing (**p < 0.01). C Mechanical properties of BPHS. Representative stress/strain curves of BPHS before annealing (left) and after annealing at 60 °C (right).
The Young’s modulus and ultimate strength of BPHS0, BPHS10, and BPHS20 were not significantly different prior to the annealing process (Table 2). The mechanical properties of BPHS30 were >3.4-fold higher than those of the other compositions. After annealing, the compression modulus and ultimate strength of the hybrids were changed as a result of polymer chain rearrangement, such as that which occurs during shrinkage, changes in the degree of crystallinity, and crystalline orientation. All the hybrid scaffolds retained their toughness after the heat treatment. A significant increase in the Young’s modulus and ultimate strength of BPHS0 was observed. This was expected because shrinkage and a more ordered crystalline structure enhance the mechanical properties. In contrast, the Young’s modulus and ultimate strength of BPHS10 decreased by 10.61- and 9.79-fold, respectively. This was due to the decrease in crystallinity by 13.29-fold after annealing. It is presumed that the homogeneously distributed silica network in the hybrids interfered with the crystalline rearrangement, which resulted sharp decrease in the crystallinity after annealing. In contrast, BPHS20 and BPHS30 were less affected by the heat treatment. There were no significant differences in the mechanical properties for BPHS20 and BPHS30 after annealing. Although the mechanical properties of BPHS0 improved after annealing, it was not considered as a bone regenerative scaffold because of the drastic shrinkage and lack of a silica matrix. BPHS30 exhibited the strongest mechanical properties among the hybrid scaffolds while it was not affected by the heat treatment. Therefore, the authors believed that it was the most suitable hybrid composition for the bone scaffold.
BPHS30 was also confirmed to have shape memory properties since the hybrid scaffold possessed a chemically cross-linked matrix with a crystalline structure. Schematic representations of the shape memory evaluation test and mechanisms are illustrated in Fig. S5A, B. As shown in Fig. S5Biv, the endothermic peaks of BPHS30 were observed before heating and after cooling at temperatures ranging from 20 to 60 °C. Heating above the melting point disordered the crystalline structure of BPHS30, while cooling to room temperature reordered the crystalline structure. As shown in supplementary videos 2 and 3, the original shapes of the BPH30 and BPHS30 were recovered upon reheating. The shape memory ability of BPHS30 allows its volume to shrink during an implantation and then expand to fill up the irregular bone defect after the implantation (Fig. S5C). This reduces the burden of surgeons to cut or shape bone scaffolds to exactly match the shape of the irregular defect. In addition, shape memory ability can enhance bone regeneration by allowing close interactions with the surrounding bone tissue30.
In vitro biocompatibility and bioactivity evaluations of the hybrid scaffolds
After confirming the physical and mechanical properties of the BPHSs, in vitro cytotoxicity tests were conducted to evaluate their biocompatibility in accordance with ISO 10993-5 and ISO 10993-12. As shown in Fig. 3A, the CCK-8 assay confirmed that all the scaffolds passed the ISO standards for cytotoxicity; the viability of the MC3T3-E1 preosteoblast cells was above 70% of the viability of the control media when exposed to the scaffold extract. This proved that all the organic solvents and excess chemicals (i.e., unreacted monomers) were removed by drying and precipitation. As shown in Fig. 3B, the MC3T3-E1 cells were stained with phalloidin dye conjugates; expression of the microfilament protein, F-actin (red), and DAPI staining (blue) of the nuclei was observed. The MC3T3-E1 cells adhered to all the BPHS samples, regardless of the silica content. The cell clusters of BPHS20 and BPHS30 were denser than those of BPHS0 and BPHS10. Consistently, DNA quantification measurements confirmed that more cells adhered to the BPHS with a higher silica content after 1 d of culture (Fig. S6): these values were 0.509 ± 0.103, 0.751 ± 0.023, 1.167 ± 0.108, and 1.333 ± 0.082 mg for BPHS0, BPHS10, BPHS20, and BPHS30, respectively. Cell adherence to the scaffolds stabilized after 7 days of culture. The DNA quantification measurements for all the scaffolds were similar.

A CCK-8 assay performed in accordance with ISO 10993. MC3T3-E1 cells were cultured with the dissolution products of BPHS. Polyethylene (PE) media conditioned with a nontoxic substance was used as the negative control, and polyurethane (PU) was used as the positive control. B Confocal microscopy images of F-actin and nuclei stained MC3T3-E1 (F-actin labeling in red, and DAPI nuclear counterstain in blue) for (i) BPHS0, (ii) BPHS10, (iii) BPHS20, and (iv) BPHS30 (Scale bar: 200 μm). C Accelerated degradation test conducted on BPHS. Change in the mass of BPHS after the accelerated degradation test (n = 4) (*p < 0.05 and **p < 0.01). BPHS was immersed in 1 M NaOH at 25 °C for 0, 8, 24, 48, 72, and 168 h. D Gene expression of osteogenic differentiation markers (n = 3): (i) RUNX2, (ii) ALPL, and (iii) COL1A1 (*p < 0.05 and **p < 0.01). E Silica dissolution profile of BPHS30 upon immersion in PBS solution over 1, 2, and 4 weeks (n = 3).
Degradation tests were performed to confirm the biodegradability of BPHS and to compare the biodegradation rates of BPHS samples with different inorganic–organic ratios. The degradation mechanism of PCL, the main chain of PU, involves the cleavage of the ester bond by hydrolysis. Because PCL is known to have a slow degradation rate, which is ~1–2 years depending on the implant environment31, the biodegradability of BPHS was evaluated under accelerated degradation conditions32. BPHS was immersed in 1 M NaOH at 25 °C. After the degradation test, the pore and pore interconnection sizes of BPHS gradually increased as the strut size decreased, and all the BPHS samples were fully degraded (Fig. S7). As shown in Fig. 3C, the biodegradability of porous scaffolds under accelerated degradation was measured for the samples before annealing. BPHS0 and BPHS10 decomposed rapidly and disappeared after 168 h. BPHS20 and BPHS30 were fully degraded after 72 h and 48 h of immersion, respectively. Scaffolds with a higher silica content exhibited more rapid degradation rates. This was possibly due to the low polymer crystallinity and high hydrophilic silica content33. BPHS with a low silica content (BPHS0 and BPHS10) had a lower degradation rate than BPHS with a higher silica composition (BPHS20 and BPHS30), as the hybrid scaffolds with a low silica content had higher crystallinity than BPHS20 and BPHS30 (Fig. 2A). A crystalline structure decomposes more slowly than an amorphous structure because it absorbs less solution than an amorphous structure. In addition, the larger pore size of BPHS30 results in a higher surface area to volume ratio, which induces faster degradation (Fig. 2Biii)34. As a slow degradation rate impedes cell migration and bone regeneration, it is advantageous for bone scaffolds to have a degradation rate that matches the bone regeneration rate35. The degradation test showed that the degradation rate of BPHS was tailorable by its silica content, which is beneficial for designing bone scaffolds.
Since the mechanical properties and initial cell attachment of BPHS30 were superior to those of the other compositions, the in vitro osteogenic differentiation of BPHS0 and BPHS30 was further evaluated. The expression of osteogenic marker genes differed between the hybrid scaffolds. The osteogenic markers (RUNX2, ALPL, and COL1A1) in preosteoblast cells were analyzed to evaluate the osteogenesis of MC3T3-E1 and the early osteogenic differentiation levels induced by BPHS. MC3T3-E1 cells, which adhered to BPHS, were harvested after 1, 7, and 28 days of culture to assess gene expression. The cells were cultured in basal medium for the first 7 days and thereafter in differentiation medium for up to 28 days. The gene expression levels were normalized to 18 S ribosomal RNA and BPHS0. As shown in Fig. 3D, there were no differences in the gene expression levels of BPHS0 and BPHS30 before the induction of osteogenic differentiation. However, BPHS30 had significantly higher expression levels of RUNX2, ALPL, and COL1A1 than BPHS0 after 28 d of culture. As shown in Fig. 3Di, RUNX2 expression increased as the silica content increased on Day 28: these values were 3.98 ± 0.27 and 42.4 ± 11.9 for BPHS0 and BPHS30, respectively. RUNX2 is essential for osteoblast proliferation as well as the preosteoblast cell cycle, which induces the differentiation of MSCs and preosteoblast cells36. Several studies have demonstrated that silicate-based bioceramics37 and silicate ions38 activate the bone morphogenetic protein (BMP) signaling cascade and the BMP downstream cascade, which includes RUNX2. ALPL encodes tissue-nonspecific alkaline phosphatase, which enhances the deposition of calcium and phosphorous in the bones and teeth. The gene expression of ALPL, as shown in Fig. 3Dii, was higher in BPHS30 (766.80 ± 225.78) after 28 days of culture. COL1A1 encodes the α1 chain of type I collagen, which is one of the main constituents of the bone extracellular matrix (ECM). The gene expression levels of COL1A1 in BPHS0 were 7.49 ± 0.25, while BPHS30 showed the highest expression level of 20.80 ± 6.67 after 28 days of culture (Fig. 3Diii). Previous studies have reported that COL1A1, COL1A2, RUNX2, ALPL, and OCN expression levels increase when osteoblasts are exposed to silica ions. Soluble silica has been hypothesized to enhance osteogenic differentiation39,40. To confirm the release of silica ions from BPHS, BPHS30 was immersed in 1X PBS (pH 7.4) at 37 °C. As shown in Fig. 3E, the concentrations of silicate ions released from BPHS30 were 5.66 ± 0.02, 8.18 ± 0.79, and 19.94 ± 1.27 mg/L after 1, 2, and 4 weeks of immersion, respectively. Silica degradation has also been shown to affect the mechanical properties of hybrid scaffolds. The mechanical properties of BPHS30 significantly decreased after 4 weeks of immersion in the PBS solution (Fig. S8 and Table S1). These results demonstrate that silica ion release from the hybrid scaffold can contribute to upregulating the expression of osteogenic markers.
In vivo assessment of hybrid scaffolds for bone regeneration
The in vivo osteogenic properties of the hybrid scaffold were evaluated using a distal condyle defect model with a defect size of 3 mm in diameter and 4 mm in depth. This defect size is known as a critical size defect, as complete bone regeneration does not occur 4 weeks after injury41,42. Three experimental groups, namely, empty defects (control), BPHS0, and BPHS30, were compared to evaluate the bioactivity of the hybrid scaffolds.
To quantify the mineralization of the newly formed bones, the bone volume fraction in the total volume (BV/TV) was calculated from the reconstructed 3D µCT volume of interest (Fig. 4A and Table 3). After 2 weeks, there was no difference in the BV/TV values between the groups. However, 4 weeks after implantation, the BV/TV value of the BPHS30 group was significantly higher than that of the BPHS0 group. Although there was no significant difference in terms of mineralization between the same groups at two time points, the amount of mineralized tissue increased by 3.04-, 1.63-, and 3.16-fold in the control, BPHS0, and BPHS30 groups, respectively. The authors would like to point out that no increase in mineralization was observed in the central defects with the BPHS0 implant. This suggests that the PU scaffold with no silica release exhibited slower bone formation than BPHS30 due to its bioinertness.

A Representative top- and side-view images of the defect site. The red dotted lines in the top-view images represent the sliced region in the side-view images. The yellow dotted lines in the side-view images represent the defect sites. Bone volume fraction (BV/TV, %) of the newly formed bones at 2 and 4 weeks postsurgery (n = 3) (*p < 0.05). B Hematoxylin and eosin (H&E) staining of the distal condyle bone defects with enlarged images within the defect sites (scale bar: 500 μm) and morphology of the defects stained with Masson’s trichrome (MT) staining (scale bar: 500 μm).
After decalcifying the bone tissues, hematoxylin and eosin (H&E) staining and Masson’s trichrome (MT) staining were used to further evaluate bone regeneration at the defect site (Fig. 4B). As shown in the H&E images, most of the newly formed tissues regenerated from the periphery of the defect site 2 weeks after surgery. Defects without implants were mainly composed of fibrous tissue. The BPHS0 group showed fibrous tissue in the peripheral regions of the defect and only blood cells in the region of the defect. In contrast, osteoblasts were formed around the connective tissue throughout the defects in the BPHS30 group, and blood vessels were observed. At 4 weeks after implantation, the tissue composition between groups was histologically compared. In the control group, new tissues gradually infiltrated toward the center of the defect from the peripheral region, but the central region was not filled with tissues. Additionally, the density of the newly formed tissues was not uniform. In contrast, the scaffolds with porous structures were able to promote guided tissue regeneration; newly formed tissues were intruded into the pores, and the entire porous region was filled. The transformation of tissue from mesenchyme to trabecular was observed in the BPHS0 group, whereas a greater number of trabeculae of woven tissues were observed in the BPHS30 group. Less differentiated bone tissues were observed for BPHS0 than for BPHS30. In addition, osteocytes, which regulate mineral homeostasis and bone formation, were found in the trabeculae observed for BPHS30. After MT staining, collagen-rich areas (stained blue) and normal cells (stained red) were observed in the defects. Fibrous tissue and collagen (callus) are known to form in the early bone healing stage, after which the callus is replaced by trabecular bone43. However, the overexpression of fibrous tissue and collagen in the bone defect site is known to be the result of a dysregulated wound healing process44 and was observed in the defects without a scaffold. 2 weeks postsurgery, the control and BPHS0 groups had low collagen deposition, while fibrous tissue and collagen were observed (violet color: overlapped blue and red colors) in the BPHS30 group. 4 weeks after implantation, irregular collagen formation was observed in the control group. Densely formed collagen was observed for the BPHS30 group, whereas low collagen deposition was observed in the BPHS0-implanted defect. Based on the µCT values and histological staining results, fibrosis occurred in the defects without scaffolds, and BPHS30 implantation accelerated bone tissue regeneration and collagen synthesis compared to BPHS0 implantation.
To confirm that the osteogenic differentiation and bone formation were a result of hybrid scaffold implantation, immunostaining of the osteogenic markers, collagen type 1 (COL I) and osteocalcin (OCN), was performed (Fig. 5A). COL I is known to provide strength by forming an oriented layer, while OCN is a linker between the inorganic and organic components within the bone matrix. As shown in Fig. 5B, the mean values of COL I at 4 weeks postsurgery for the control group were 5.4-fold higher than those at the 2-week time point. BPHS30 implantation resulted in an increase in COL I expression at 4 weeks postsurgery, which was 6.1-fold higher than that observed at the 2 week time point. However, there were no apparent differences observed in the BPHS0 group. Similar trends of OCN expression were detected in the control and BPHS30 groups (Fig. 5C); a 5.3- and 2.9-fold increase in expression at 4 weeks postsurgery compared to the 2 week time point was observed, respectively. OCN expression was the highest in the BPHS30 group at 4 weeks postsurgery. Interestingly, differences in protein expression were observed between the defects with and without the hybrid scaffolds. The protein expression in the defects with hybrid scaffolds was mainly observed within the scaffold pores. In contrast, protein expression was concentrated in the central region of the unhealed defects in the control group. This was possibly due to fibrosis surrounding the unhealed defect area, as the accumulation of COL I is known as a characteristic of fibrosis. The silica content has also been shown to affect protein expression. The quantified proteins in the BPHS30-implanted defects were higher than those in the BPHS0 group. Silica ions have been shown to be able to stimulate osteogenic differentiation, including COL I and OCN markers38,45. Specifically, silicate-based composites46,47 and silicate ions48 have been shown to activate the Wnt/β-catenin pathway, which acts as an important factor in MSC activation and new bone formation48. Downstream genes of the Wnt/β-catenin pathway, including COL1A1 and OCN, possibly enabled the hybrid scaffolds with a higher silica content to enhance bone regeneration.

A Representative immunofluorescence staining of type I collagen (COL I) and osteocalcin (OCN) at 2 and 4 weeks postsurgery. COL I (red) and OCN (green) coimmunolabeling was used to assess bone regeneration (scale bar: 200 μm). B COL I-positive staining areal density and C OCN-positive staining areal density measured by ImageJ (*p < 0.05 and **p < 0.01).
The bone healing process involves new blood vessel formation from preexisting vessels and the conversion of neovessels to mature vessels in the defect area. To evaluate vessel maturation and angiogenesis, α-SMCs and endothelial cells were subjected to immunofluorescence staining (Fig. 6A) using α-SMA (red) and the von Willebrand factor (vWF, green). As shown in Fig. 6B, C, α-SMA- and vWF-positive densities were higher in the BPHS30 samples, while the other groups displayed lower densities with small improvements over the implantation time. Based on the double immunofluorescence staining results, the maturation index was determined (Fig. 6D). Similar to the angiogenic activity, the maturation index value of the BPHS30 group was the highest at 4 weeks postsurgery, and this group was the only group whose values increased over the implantation period. This indicated that the blood vessels formed gradually in the BPHS30 group by recruiting SMCs, possibly due to the release of silicate ions from the hybrid scaffold. Silicate ions are also known to upregulate vascular endothelial growth factor signaling pathways, which stimulate angiogenesis, such as neoblood vessel and tubule network formation45,49. The angiogenesis and mature vessel formation results were in accordance with those of a previous study on polymethacrylate-silica hybrid scaffolds for bone repair45. Enhancing angiogenesis could stimulate damaged bones by supplying nutrients and oxygen50.

A Representative immunofluorescence staining for angiogenic evaluation at 2 and 4 weeks postsurgery. α-SMA (red) and vWF (green) coimmunolabeling was used to assess the maturation of the vessels (scale bar: 200 μm). Quantitative analysis of the (B) α-SMA expression (*p < 0.05), C vWF expression, and D maturation index was performed using ImageJ. The maturation index was based on a comparison of the α-SMA-positive vessel area to the total vessel area (†p < 0.05).
In the early bone healing phase, inflammatory cells are activated for ~1 week51,52. To recruit inflammatory cells, the presence of macrophages and their polarization are important in the bone healing process. Additionally, they are critical for implantable biomedical devices. Macrophages are known to polarize into two distinct functional phenotypes, M1 and M2, in response to microenvironmental signals. M1 macrophages are proinflammatory agents, whereas M2 macrophages are regenerative and anti-inflammatory agents. Proinflammatory cells phagocytose foreign bodies during the early stages of inflammation, while anti-inflammatory cells contribute to tissue regeneration. Immunolabeling of CD68 (pan macrophage, red) and CD206 (M2 marker, green) was performed (Fig. 7A) to quantify and assess the phenotypic profiles of the macrophages within the defect sites. The population of CD68, which represents both M1 and M2 macrophages, was similarly lower in all the groups except for the control group at 4 weeks postsurgery (Fig. 7B). CD68 expression in the control group was significantly higher at 4 weeks. The ratio of CD68 to CD206 was measured (Fig. 7C) to quantify the M2 macrophages. Anti-inflammatory cells (M2 macrophages) are found in the late inflammatory stages compared to proinflammatory cells (M1 macrophages), which contribute to osteogenic effects, such as mineralization and vessel maturation53,54. The ratio values of the control group at the 2 and 4 week time points were 58.8 ± 5.8 and 46.2 ± 6.5%, respectively. Half of the macrophages in the control groups were polarized toward the M1 phenotype, regardless of the time point. In contrast, over 96.3% of M2 macrophage expression was observed in the defects with hybrid scaffolds. In the bone healing process, the M1 macrophages were typically shown to disappear within a few days54, but the M1 macrophages found in the control group were half of the total macrophage density at 2 and 4 weeks postsurgery. In addition, a greater number of macrophages was observed in the control group at 4 weeks postsurgery. In the severe bone defect model, a persistent proinflammatory reaction, which leads to fibrosis and delayed bone healing, was observed for defects without any implants51. On the other hand, macrophages were polarized to the M2 phenotype, and chronic inflammation was not observed within the defects in the BPHS0 and BPHS30 groups. The biocompatible and 3D porous scaffold structure induced macrophage polarization toward the M2 phenotype; these results were in good agreement with those of previous studies evaluating macrophage phenotypes45,55,56. It was speculated that the interconnected porous structure with macropores and high porosity can promote M2 macrophage polarization56,57.

A Representative immunofluorescence staining of macrophages at 2 and 4 weeks postsurgery. CD68 (red) and CD206 (green) coimmunolabeling was used to evaluate the phenotypic profiles of the macrophages (scale bar: 200 μm). B CD68-positive staining (*p < 0.05 and **p < 0.01), and C the CD206/CD68 ratio measured using ImageJ (**p < 0.01).
The in vivo evaluations showed that BPHS30 was capable of enhancing osteogenic differentiation and angiogenesis without inflammatory responses at an early bone formation phase (within 4 weeks). Although a long-term in vivo implantation test should be conducted to evaluate the impact of hybrid scaffolds on a complete bone repair, numerous studies have shown that angiogenesis and inflammatory responses are activated in the early bone healing process, while bone formation and repair occur within 20 days to several months43,58,59,60. In addition, when bone repair is aided by bone scaffolds, bone regenerates by infiltrating into the porous structure of scaffolds at early time points. This augments the stability and rate of bone formation in later stages of the bone healing process61,62. Therefore, it is important to confirm the bioactivity and inflammatory responses of bone scaffolds at an early stage. Based on these results, the authors believe that evaluating bone repair with hybrid scaffolds using a long-term implantation of >12 weeks in medium-sized animal models will be a prominent future study.
Conclusions
In this study, a tough, biodegradable, and porous PU-silica hybrid scaffold with shape memory ability was successfully fabricated for bone repair. Class II hybrid synthesis was achieved by polymerizing PU through a polycondensation reaction and introducing APTES during the polymer synthesis. The sol-gel transition time was shortened by increasing the crosslinking density via allophanate bonding. This allowed the production of hybrid scaffolds with interconnected porous structures through the facile salt-leaching technique. BPHS30 exhibited the toughest mechanical properties among all the hybrid scaffolds and had shape memory properties to fill irregular bone defects. In vivo evaluation confirmed that BPHS30 enhanced bone mineralization, angiogenesis, and vascular maturation in the early bone formation phase without fibrosis and chronic inflammation. The authors believe that the biodegradable PU-silica hybrid is a promising biomaterial for bone repair.
Materials and methods
Synthesis of biodegradable polyurethane
The polymer was synthesized via a two-step polymerization process. PCL-diol (number average molecular weight of 2000, Sigma) in toluene (anhydrous, 99.8%, Sigma) was added to a flask under an argon atmosphere; thereafter, hexamethylene diisocyanate (HDI, ≥ 99.0%, Sigma) and tin(II) 2-ethyl hexanoate (SnOct2, 97%, Sigma) were added to the flask. After 2 h of stirring at 90 °C, (3-aminopropyl)triethoxysilane (APTES, ≥ 98%, Sigma‒Aldrich) was added. The molar feed ratio of PCL-diol/HDI/APTES was 1:2:2. After stirring for 1 h at 90 °C, the flask was maintained at room temperature (RT) overnight. The product was purified via precipitation in methanol (99.8%) and vacuum-dried overnight. The molecular weights (Mn and Mw) of the polymers were measured using gel permeation chromatography (GPC). The dn/dc of the polymer in CHCl3 was 0.0759. A Bruker AscendTM 400 proton nuclear magnetic resonance spectrometer (1H NMR) at 400 MHz was used to determine the structure of the polymer.
Synthesis of inorganic–organic hybrids and sample preparation
Biodegradable polyurethane was dissolved in tetrahydrofuran (THF, ≥ 99.9%, Sigma), and TEOS (98%, Sigma) was hydrolyzed in the following molar ratio: TEOS:water:HCL = 1:4:0.6. The polymer was mixed with the hydrolyzed TEOS at a weight ratio of 70–100 wt. % (with 10 wt. % increments). The mixture was stirred vigorously, and 1.9 g of sodium chloride (NaCl, ≥99.5%, Biosesang) was gradually added to the 1 ml mixture before gelation. The NaCl particles were collected using a 300–500 µm sieve. The mixture/salt paste was placed in a mold and stored overnight at 40 °C. After air drying at RT for 1 h, the NaCl was leached by soaking in deionized water (DW) for 5 days. For all the tests, except the compression test of the 0–40 wt. % silica hybrids, the samples were cut into cylindrical shapes that were 8.66 mm in diameter and 1 to 2 mm in thickness.
Hybrid characterization
TGA (TA Instruments Q50) was used to measure the weight percentages of the silica and polymer in the hybrids; the samples were heated at a rate of 20 °C/min from 0 to 150 °C in N2. The transmittance spectra of the hybrids were measured using a Fourier transform infrared (FTIR) spectrometer (Nicolet iS20 FTIR spectrometer). For the polymer, ester stretching and aliphatic ether peaks appeared at 1210–1163 cm−1 and 1150–1085 cm−1, respectively. For the hydrolyzed TEOS, the Si-O-Si bond peaks appeared at 1094 cm−1 and 798 cm−1. The gelation time of the hybrids was determined by determining the cross-points of the storage modulus (G’) and loss modulus (G’) using a rheometer (MSC 102, Anton Paar, DE). All the samples were measured immediately after mixing the hydrolyzed TEOS with the polymer solution. The measurements were conducted at an oscillating frequency of 1 Hz and 2% of the oscillating strain at 25 °C.
Experimental analysis of the annealing effect
Crystalline structure characterization
Differential scanning calorimetry (DSC) measurements were performed to measure the enthalpy change (ΔΗ) of the hybrids using a Q10 differential scanning calorimeter. The hybrids were heated at a rate of 10 °C/min from 0 to 150 °C in N2. The enthalpy change (∆Hm) was measured to determine the silica-polymer ratio and the scaffold morphology. The enthalpy of fusion of the 100% crystalline PCL (\(\Delta H_m^0\)) was 139.5 J/g. The percent crystallinity of the samples was calculated as follows:
Accelerated degradation test
Accelerated degradation was assessed at 5 time points (8, 24, 48, 72, and 168 h). The hybrids were immersed in 1 M sodium hydroxide (NaOH, Junsei Chemical Co., Ltd.) at 25 °C, rinsed with DW three times, and dried overnight under vacuum. The percentage of mass loss was then measured.
Porous structure evaluation
Cross-sectional images of the hybrids before and after annealing were obtained using SEM after the hybrids were cross-sectioned and sputtered with gold. The pore sizes of the hybrids were measured from the SEM images using ImageJ. The interstitial porosity was measured using an AutoPore IV 9500 Series.
Mechanical properties
The compression tests were performed using a 5966 universal testing machine (5966 UTM). The compression speed was 1 mm/min, and the samples were compressed to 60% of the original sample height before and after annealing.
BPHS30 was immersed in 1X PBS and incubated at 37 °C in an orbital shaker at a rpm of 50 for 2 or 4 weeks. After immersion, the samples were washed three times with DW to remove the salt from the PBS solution and dried overnight in a vacuum oven. Compression tests were performed as described above.
Shape memory behavior
BPH30 with a rectangular shape (35 mm × 14 mm × 3 mm) and BPHS30 with a cylindrical shape (11 mm diameter × 6.25 mm thickness) were soaked in a water bath at 60 °C (above melting temperature) for 5 min, bent and pressed to deform the shape and cooled at room temperature for 5 min to fix the temporary shapes. Shape recovery was observed when the samples were reheated above their melting points.
Silica ions released from hybrid scaffolds
The silica ions released from BPHS30 were measured using inductively coupled plasma-optical emission spectroscopy (ICP‒OES, iCAP 6500 Duo). A standard curve of the silica ions was prepared at 0.5, 1, 5, and 10 ppm. The hybrid scaffolds (30 mg) were dissolved in 20 ml of PBS (1X, pH 7) and incubated in a shaking incubator for 1, 2, and 4 weeks (at 37 °C and 50 rpm). The solution was mixed with 2 M nitric acid in a 1:4 volume ratio. The silica content was measured at a wavelength of 212.412 nm.
In vitro test
Cell culture and cell seeding
Mouse preosteogenic precursor cells (MC3T3-E1, ATCC) were harvested in α-MEM (Gibco) supplemented with 10% v/v FBS (HyClone) and 5 ng·mL−1 penicillin/streptomycin. The hybrids were sterilized in 70% ethanol for 5 min and washed with α-MEM for 30 min before cell seeding. The MC3T3-E1 cells were seeded at a density of 250,000 cells on the hybrids and cultured in basal medium for 7 days and thereafter in osteogenic differentiation medium (α-MEM supplemented with 10 v/v % FBS, 5 ng·mL−1 penicillin/streptomycin, 5 µg·mL−1 L-ascorbic acid (Sigma), and 2 mM β-glycerophosphate disodium salt (Sigma)). Ascorbic acid and β-glycerophosphate disodium salt induced the osteogenic differentiation of MC3T3-E1 cells.
Cytotoxicity test
MC3T3-E1 cells were used to perform in vitro cytotoxicity tests in accordance with ISO 10993-5 and ISO 10993-12 protocols. The hybrids were dissolved in α-MEM at 37 °C for 72 h and thereafter sterilized using a 0.2 µm filter. The negative control for the toxicity test was a high-density polyethylene (PE) film (Hatano Research Institute, FDSC), and the positive control was polyurethane (PU) containing 0.1% zinc diethyldithiocarbamate (ZDEC; Hatano Research Institute, FDSC), which is cytotoxic. The MC3T3-E1 cells were cultured in 96-well plates in α-MEM for 24 h. The medium was changed with dissolution products and incubated for 24 h. The cell viability was measured using a CCK-8 (DOjinDO) cell viability assay kit; 10% CCK-8 of the total volume was added to each well, and the absorbance was measured at 450 nm using Spectramax 384.
Cell attachment test
An inorganic–organic mixture (sol–state hybrid) was coated onto a glass substrate and dried in an oven at 40 °C to perform the cell attachment test. MC3T3-E1 cells were seeded on hybrid-coated glass substrates at a density of 106 cells/ml and incubated for 3 days in basal α-MEM. The samples were washed in PBS, and thereafter, 4% paraformaldehyde (PFA) was added to fix the cells at 4 °C overnight before immunohistochemical (IHC) staining. The cells were then treated with 0.5% Triton X-100 in PBS for 20 min to permeabilize the cell membranes. After permeation, they were blocked with 10 mg/ml BSA in PBS at 4 °C for 30 min. All the samples were stained with Alexa Fluor™ 594 Phalloidin (1:200 dilution, 10 mg/ml BSA in PBS) and DAPI nuclei staining (0.1 μg/mL in PBS). The labeled F-actin and cell nuclei were obtained using a Zeiss LSM 700 confocal microscope (Carl Zeiss).
DNA quantification
The MC3T3-E1 cells were harvested after being cultured on the hybrid scaffolds for 1, 7, and 28 days to measure the DNA concentration, as described in the “Cell culture and cell seeding” section above. The samples were washed in 1X PBS and thereafter left in 0.1% v/v Triton-X; freeze‒thaw cycles were repeated for cell lysis. DNA from the cell lysate was quantified using Hoechst staining 33258 (≥98%, Sigma), and calf thymus DNA was used as the DNA standard.
qRT‒PCR for mRNA quantification
The hybrid scaffolds were analyzed for the expression of two osteogenic markers, RUNX2 and COL1A1, in MC3T3-E1 preosteoblast cells using qPCR. All the expression levels were normalized to the housekeeping gene 18 S. RNA from the cells on the hybrid scaffold was extracted using an RNeasy Mini Kit (Qiagen) and converted into cDNA using Maxime RT Premix (iNtRON). The primer sequences are listed in Table 4. Real-time PCT was performed using SYBR Premix Ex Taq (Applied Biosystems 7500 real-time cyclers).
In vivo testing
Animal surgical procedure
All animals were treated according to the protocols of the Institutional Animal Care and Use Committee at the Biomedical Research Institute of the Korea Institute of Science and Technology. The distal femoral defect model was adapted from Tielinen et al.63. Sprague‒Dawley rats (12 weeks old, male, 350–400 g; DBL) were used in three different experimental groups (n = 5 animals/group): control (defect without scaffold), BPHS0, and BPHS30. Inflammation and bone regeneration were assessed at two time points (2 and 4 weeks postsurgery). The animals were anesthetized using isoflurane. The lateral epicondyles of the distal femur were shaved and disinfected using povidone-iodine. After incision of the skin and muscles, cylindrical defects were created with a 3 mm diameter and 4 mm depth in the lateral condyles. The defects were washed with 1X PBS and filled with the samples or left empty. (BPHS0 and BPHS30 were preannealed at 60 °C for 2 h to prevent the sample from shrinking). The incision was closed using a 3–0 black silk suture. At 2 and 4 weeks postsurgery, the animals were sacrificed, and the distal femurs were explanted. The femurs were fixed in 10% (v/v) formalin for 3 days.
Microcomputed tomography for the evaluation of bone mineralization
The explanted specimens were analyzed using µCT (Skyscan 1172). The X-ray source was set at 56 kV, 179 μA, and 10 W to transmit through the samples at 30%. An aluminum filter (0.5 μm) was used to scan the femurs (high-density tissues). The newly formed bone volume (BV) and total bone volume (TV) of the samples were quantified using CTAn software (Skyscan Ltd). Thereafter, 3D images were generated from the scanned images using CTvox software.
Histological and immunofluorescence analyses
The specimens were decalcified using a decalcification solution (Sigma, USA) for 36 h, followed by rinsing with tap water. Thereafter, the samples were dehydrated with increasing concentrations of ethanol (70, 80, 90, 95, and 100%) for 1 h each. The samples were embedded in paraffin and sectioned into 7 μm-thick slices for histological and fluorescence staining. The slides for histological analysis were stained with H&E and MT after deparaffinization and rehydration. The collagen density was defined as the ratio of blue intensity to the total area using ImageJ software (n = 3 in each group). To evaluate bone regeneration in the defects, immunofluorescence staining was performed to study osteogenesis, phagocytic activity, vasculogenesis, and angiogenesis. For osteogenesis evaluation, the cross-sectioned slices were stained with antibodies against type I collagen (ab34710, 1:200; Abcam) and osteocalcin (sc-390877 FITC, Santa Cruz Biotechnology, USA, 1:50). Alexa Fluor 594 (anti-rabbit IgG) was used as the secondary antibody against type I collagen. The nuclei were counterstained with DAPI (Microcular Probes, USA). The positive areas of the stained sections in three random fields were quantified using ImageJ software (n = 3 in each group). To evaluate phagocytic activity, the samples were stained with antibodies against CD68 (ab955, Abcam, UK, 1:60) and CD206 (sc-34577, Santa Cruz Biotechnology, 1:100). Alexa Fluor 594 (anti-mouse IgG) and Alexa Fluor 488 (anti-goat IgG) were used as the secondary antibodies. The CD68- and CD206-positive areas in the three fields were quantified using ImageJ software (n = 3 in each group). The CD206+/CD68+ ratio indicates the proportion of M2 macrophages. To verify vasculogenesis, the primary antibodies used were the monoclonal mouse anti-human α-smooth muscle actin antibody (α-SMA, sc-53142 AF594, Santa Cruz Biotechnology, 1:50) for staining the α-smooth muscle cells (SMCs) and the polyclonal rabbit anti-human von Willebrand factor antibody (vWF, sc-365712 FITC, Santa Cruz Biotechnology, 1:50) for staining the endothelial cells. To evaluate vascular cell recruitment in the defect areas, the α-SMA- and vWF-positive areas in the three fields were quantified using ImageJ software (n = 3 in each group). The vessel maturation index (%) was calculated using the α-SMA-positive/vWF-positive area ratio.
Statistical analysis
The statistical analysis was performed using one-way ANOVA and Tukey’s test for multiple comparisons. The results are shown as the means ± standard errors of the means (SEMs) and were considered statistically significant at *p < 0.05 and **p < 0.01.
References
Jones, J. R. Review of bioactive glass: from Hench to hybrids. Acta Biomater. 9, 4457–4486 (2013).
Mahony, O. et al. Silica-gelatin hybrids with tailorable degradation and mechanical properties for tissue regeneration. Adv. Funct. Mater. 20, 3835–3845 (2010).
Chung, J. J. et al. Effect of comonomers on physical properties and cell attachment to silica-methacrylate/acrylate hybrids for bone substitution. Macromol. Rapid Commun. https://doi.org/10.1002/marc.201700168 (2017).
Sang, T. et al. Hybrids of Silica/Poly(caprolactone coglycidoxypropyl trimethoxysilane) as Biomaterials. Chem. Mater. 30, 3743–3751 (2018).
Tallia, F. et al. Bouncing and 3D printable hybrids with self-healing properties. Mater. Horiz. 5, 849–860 (2018).
Rhee, S.-H. Bone-like apatite-forming ability and mechanical properties of poly (ε-caprolactone)/silica hybrid as a function of poly (ε-caprolactone) content. Biomaterials 25, 1167–1175 (2004).
Chung, J. J., Li, S., Stevens, M. M., Georgiou, T. K. & Jones, J. R. Tailoring mechanical properties of sol–gel hybrids for bone regeneration through polymer structure. Chem. Mater. 28, 6127–6135 (2016).
Akindoyo, J. O. et al. Polyurethane types, synthesis and applications—a review. RSC Adv. 6, 114453–114482 (2016).
Hong, S. M. et al. Hyperelastic, shape‐memorable and ultra‐cell‐adhesive degradable polycaprolactone‐polyurethane copolymer for tissue regeneration. Bioeng. Transl. Med. 7, e10332 (2022).
Rubio Hernández-Sampelayo, A. et al. Biodegradable and biocompatible thermoplastic poly (ester-urethane) s based on poly (ε-caprolactone) and novel 1, 3-propanediol bis (4-isocyanatobenzoate) diisocyanate: synthesis and characterization. Polymers 14, 1288 (2022).
Poologasundarampillai, G. et al. Poly (γ‐glutamic acid)/silica hybrids calcium incorporated silica Netw. use a calcium alkoxide precursor. Chemistry 20, 8149–8160 (2014).
Lee, S. Y. et al. Synthesis and characterization of polycaprolactone-based polyurethanes for the fabrication of elastic guided bone regeneration membrane. Biomed. Res. Int. 2018, 3240571 (2018).
Xu, C. C. et al. Development of dopant-free conductive bioelastomers. Sci. Rep. 6, 34451 (2016).
Ravarian, R. et al. Molecular interactions in coupled PMMA-bioglass hybrid networks. J. Mater. Chem. B 1, 1835–1845 (2013).
Guney, A. & Hasirci, N. Properties and phase segregation of crosslinked PCL-based polyurethanes. J. Appl. Polym. Sci. https://doi.org/10.1002/app.39758 (2014).
Novak, B. M. Hybrid nanocomposite materials—between inorganic glasses and organic polymers. Adv. Mater. 5, 422–433 (1993).
Parak, A. et al. Functionalizing bioinks 3D bioprinting applications. Drug Discov. Today 24, 198–205 (2019).
Yu, L. Y., Xu, Z. L., Shen, H. M. & Yang, H. Preparation and characterization of PVDF-SiO2 composite hollow fiber UF membrane by sol-gel method. J. Membr. Sci. 337, 257–265 (2009).
Jiang, X. F., Tang, X. N., Tang, L. H., Zhang, B. & Mao, H. M. Synthesis and formation mechanism of amorphous silica particles via sol-gel process with tetraethylorthosilicate. Ceram. Int. 45, 7673–7680 (2019).
Huang, R., Zhu, X. M., Tu, H. Y. & Wan, A. The crystallization behavior of porous poly(lactic acid) prepared by modified solvent casting/particulate leaching technique for potential use of tissue engineering scaffold. Mater. Lett. 136, 126–129 (2014).
Panahi-Sarmad, M. et al. Deep focusing on the role of microstructures in shape memory properties of polymer composites: a critical review. Eur. Polym. J. 117, 280–303 (2019).
Karatrantos, A., Clarke, N., Composto, R. J. & Winey, K. I. J. S. M. Polymer conformations in polymer nanocomposites containing spherical nanoparticles. Soft Matter 11, 382–388 (2015).
Chen, J., Deng, C. J., Hong, R., Fu, Q. & Zhang, J. Effect of thermal annealing on crystal structure and properties of PLLA/PCL blend. J. Polym. Res. https://doi.org/10.1007/s10965-020-02206-1 (2020).
Saffar, A., Ajji, A., Carreau, P. J. & Kamal, M. R. The impact of new crystalline lamellae formation during annealing on the properties of polypropylene based films and membranes. Polymer 55, 3156–3167 (2014).
Benedetti, L., Brule, B., Decreamer, N., Evans, K. E. & Ghita, O. Shrinkage behaviour of semi-crystalline polymers in laser sintering: PEKK and PA12. Mater Design. https://doi.org/10.1016/j.matdes.2019.107906 (2019).
Pfau, M. R., McKinzey, K. G., Roth, A. & Grunlan, M. A. PCL-based shape memory polymer semi-IPNs: the role of miscibility in tuning the degradation rate. Biomacromolecules 21, 2493–2501 (2020).
Duan, P. G. et al. The effects of pore size in bilayered poly(lactide-co-glycolide) scaffolds on restoring osteochondral defects in rabbits. J. Biomed. Mater. Res. A 102, 180–192 (2014).
Swanson, W. B. et al. Macropore design of tissue engineering scaffolds regulates mesenchymal stem cell differentiation fate. Biomaterials https://doi.org/10.1016/j.biomaterials.2021.120769 (2021).
Zarebski, K., Putyra, P. & Mierzwinski, D. Effect of annealing on the impact resistance and fracture mechanism of PNC-60 sinters after cold plastic deformation. J. Mater. Eng. Perform. 28, 2439–2450 (2019).
Zhang, D. et al. A bioactive “self-fitting” shape memory polymer scaffold with potential to treat cranio-maxillo facial bone defects. Acta Biomater. https://doi.org/10.1016/j.actbio.2014.07.020 (2014).
Woodruff, M. A. & Hutmacher, D. W. The return of a forgotten polymer-polycaprolactone in the 21st century. Prog. Polym. Sci. 35, 1217–1256 (2010).
Ang, K. C., Leong, K. F., Chua, C. K. & Chandrasekaran, M. Compressive properties and degradability of poly(epsilon-caprolatone)/hydroxyapatite composites under accelerated hydrolytic degradation. J. Biomed. Mater. Res A 80a, 655–660 (2007).
Limsukon, W., Auras, R. & Smith, T. Effects of the three-phase crystallization behavior on the hydrolysis of amorphous and semicrystalline poly(lactic acid)s. ACS Appl Polym. Mater. 3, 5920–5931 (2021).
Jones, J. R., Ehrenfried, L. M., Saravanapavan, P. & Hench, L. L. Controlling ion release from bioactive glass foam scaffolds with antibacterial properties. J. Mater. Sci. Mater. Med. 17, 989–996 (2006).
Woodard, L. N., Kmetz, K. T., Roth, A. A., Page, V. M. & Grunlan, M. A. J. B. Porous poly (ε-caprolactone)–poly (L-lactic acid) semi-interpenetrating networks as superior, defect-specific scaffolds with potential for cranial bone defect repair. Biomacromolecules 18, 4075–4083 (2017).
Miyazono, K., Maeda, S. & Imamura, T. Coordinate regulation of cell growth and differentiation by TGF-beta superfamily and Runx proteins. Oncogene 23, 4232–4237 (2004).
Zhai, D., Xu, M. C., Liu, L. Q., Chang, J. & Wu, C. T. Silicate-based bioceramics regulating osteoblast differentiation through a BMP2 signalling pathway. J. Mater. Chem. B 5, 7297–7306 (2017).
Uribe, P. et al. Soluble silica stimulates osteogenic differentiation and gap junction communication in human dental follicle cells. Sci. Rep. https://doi.org/10.1038/s41598-020-66939-1 (2020).
Varanasi, V. G. et al. Si and Ca individually and combinatorially target enhanced MC3T3-E1 subclone 4 early osteogenic marker expression. J. Oral. Implantol. 38, 325–336 (2012).
Costa-Rodrigues, J., Reis, S., Castro, A. & Fernandes, M. H. Bone anabolic effects of soluble Si: in vitro studies with human mesenchymal stem cells and CD14+ osteoclast precursors. Stem Cells Int. 2016, 5653275 (2016).
Jia, B. et al. Biodegradable Zn-Sr alloy for bone regeneration in rat femoral condyle defect model: In vitro and in vivo studies. Bioact. Mater. 6, 1588–1604 (2021).
Ishack, S., Mediero, A., Wilder, T., Ricci, J. L. & Cronstein, B. N. Bone regeneration in critical bone defects using three -dimensionally printed beta-tricalcium phosphate/hydroxyapatite scaffolds is enhanced by coating scaffolds with either dipyridamole or BMP-2. J. Biomed. Mater. Res. B 105, 366–375 (2017).
Bahney, C. S. et al. Cellular biology of fracture healing. J. Orthop. Res. 37, 35–50 (2019).
Wynn, T. A. & Ramalingam, T. R. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat. Med 18, 1028–1040 (2012).
Chung, J. J. et al. 3D Printed porous methacrylate/silica hybrid scaffold for bone substitution. Adv. Healthc Mater. https://doi.org/10.1002/adhm.202100117 (2021).
Jia, Y. C. et al. Regeneration of large bone defects using mesoporous silica coated magnetic nanoparticles during distraction osteogenesis. Nanomed. Nanotechnol. https://doi.org/10.1016/j.nano.2019.102040 (2019).
Sun, T. et al. Biomimetic composite scaffold containing small intestinal submucosa and mesoporous bioactive glass exhibits high osteogenic and angiogenic capacity. Tissue Eng. Part A 24, 1044–1056 (2018).
Han, P. P., Wu, C. T. & Xiao, Y. The effect of silicate ions on proliferation, osteogenic differentiation and cell signalling pathways (WNT and SHH) of bone marrow stromal cells. Biomater. Sci. 1, 379–392 (2013).
Dashnyam, K. et al. Promoting angiogenesis with mesoporous microcarriers through a synergistic action of delivered silicon ion and VEGF. Biomaterials 116, 145–157 (2017).
Dashnyam, K. et al. Angiogenesis-promoted bone repair with silicate-shelled hydrogel fiber scaffolds. Biomater. Sci. 7, 5221–5231 (2019).
Hesketh, M., Sahin, K. B., West, Z. E. & Murray, R. Z. Macrophage phenotypes regulate scar formation and chronic wound healing. Int. J. Mol. Sci. https://doi.org/10.3390/ijms18071545 (2017).
Jin, S. S. et al. A biomimetic hierarchical nanointerface orchestrates macrophage polarization and mesenchymal stem cell recruitment to promote endogenous bone regeneration. ACS Nano 13, 6581–6595 (2019).
Qiao, W. et al. Sequential activation of heterogeneous macrophage phenotypes is essential for biomaterials-induced bone regeneration. Biomaterials https://doi.org/10.1016/j.biomaterials.2021.121038 (2021).
Maruyama, M. et al. Modulation of the inflammatory response and bone healing. Front. Endocrinol. https://doi.org/10.3389/fendo.2020.00386 (2020).
Won, J. E. et al. Hierarchical microchanneled scaffolds modulate multiple tissue-regenerative processes of immune-responses, angiogenesis, and stem cell homing. Biomaterials. https://doi.org/10.1016/j.biomaterials.2019.119548 (2020).
Tylek, T. et al. Precisely defined fiber scaffolds with 40 mu mporosity induce elongation driven M2-like polarization of human macrophages. Biofabrication. https://doi.org/10.1088/1758-5090/ab5f4e (2020).
Liu, Z.-Z., Xu, N.-Y., Wang, M.-L., Tang, R.-Z. & Liu, X.-Q. Physical confinement in alginate cryogels determines macrophage polarization to a M2 phenotype by regulating a STAT-related mRNA transcription pathway. Nanoscale 10, 2315–2327 (2022).
Wang, W. & Yeung, K. W. K. Bone grafts and biomaterials substitutes for bone defect repair: a review. Bioact Mater. 2, 224–247 (2017).
Schell, H. et al. The haematoma and its role in bone healing. J. Exp. Orthop. 4, 5 (2017).
Maruyama, M. et al. Modulation of the inflammatory response and bone healing. Front. Endocrinol. (Lausanne) 11, 386 (2020).
Hing, K. A., Revell, P. A., Smith, N. & Buckland, T. Effect of silicon level on rate, quality and progression of bone healing within silicate-substituted porous hydroxyapatite scaffolds. Biomaterials 27, 5014–5026 (2006).
Wang, Y. et al. Bone remodeling-inspired dual delivery electrospun nanofibers for promoting bone regeneration. Nanoscale 11, 60–71 (2019).
Tielinen, L. et al. Inability of transforming growth factor-beta 1, combined with a bioabsorbable polymer paste, to promote healing of bone defects in the rat distal femur. Arch. Orthop. Trauma Surg. 121, 191–196 (2001).
Acknowledgements
The authors would like to acknowledge the National Research Foundation of Korea (NRF) grant (NRF-2020R1C1C1012881) funded by the Ministry of Science and ICT (MSIT) and KIST institutional program (2E32351).
Author information
Authors and Affiliations
Contributions
Conceptualization: S.J.P., S.H.K., J.J.C. Methodology: S.J. P., S.H.I., D.K., D.P., Y.J., H.s.H. Investigation: S.J.P., S.H.I., D.K., D.P. Visualization: S.J.P. Funding acquisition: Y.J., S.H.K., J.J.C. Project administration: S.H.K., J.J.C. Supervision: S.H.K., J.J.C. Writing—original draft: S.J.P., J.J.C. Writing—review and editing: S.J.P., Y.J., H. Han, S.H. Kim, J.J. Chung.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Park, S.J., Im, S.H., Kim, D. et al. Tough and biodegradable polyurethane-silica hybrids with a rapid sol-gel transition for bone repair. NPG Asia Mater 15, 28 (2023). https://doi.org/10.1038/s41427-023-00475-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41427-023-00475-y
