
Abstract
The contradictory nature of increasing the crystallization speed while extending the amorphous stability for phase-change materials (PCMs) has long been the bottleneck in pursuing ultrafast yet persistent phase-change random-access memory. Scandium antimony telluride alloy (ScxSb2Te3) represents a feasible route to resolve this issue, as it allows a subnanosecond SET speed but years of reliable retention of the RESET state. To achieve the best device performances, the optimal composition and its underlying working mechanism need to be unraveled. Here, by tuning the doping dose of Sc, we demonstrate that Sc0.3Sb2Te3 has the fastest crystallization speed and fairly improved data nonvolatility. The simultaneous improvement in such ‘conflicting’ features stems from reconciling two dynamics factors. First, promoting heterogeneous nucleation at elevated temperatures requires a higher Sc dose to stabilize more precursors, which also helps suppress atomic diffusion near ambient temperatures to ensure a rather stable amorphous phase. Second, however, enlarging the kinetic contrast through a fragile-to-strong crossover in the supercooled liquid regime should require a moderate Sc content; otherwise, the atomic mobility for crystal growth at elevated temperatures will be considerably suppressed. Our work thus reveals the recipe by tailoring the crystallization kinetics to design superior PCMs for the development of high-performance phase-change working memory technology.
Similar content being viewed by others
Introduction
Commercialized phase-change random-access memory (PCRAM) stores digital information in the amorphous and crystalline phases of chalcogenide phase-change materials (PCMs), such as Ge2Sb2Te51,2,3. Reversible switching between these two phases of Ge2Sb2Te5 in a PCRAM device is typically induced by fast electrical pulse (Joule) heating for tens of nanoseconds4. The encoded data, in the power-off state, can be reliably stored for tens of years at ambient temperatures5. Such swift and nonvolatile features, as well as the merits4,6 of high scalability, low power consumption, and long cycling endurance, make PCRAM the best candidate for realizing an ideal “universal memory” to renovate the current computing system based on the classic von Neumann architecture7. Since 2015 until very recently, Intel’s Optane DC8 and Micron’s X100 NVMe (https://www.micron.com/products/advanced-solutions/3d-xpoint-technology/x100) chips, both employing 3D Xpoint PCRAM technology, served as storage-class memory to mitigate the widening performance gap (memory wall) between volatile dynamic random-access memory (DRAM) and nonvolatile solid-state drive flash memory. Nevertheless, substitution of traditional working memories, i.e., static random-access memory and DRAM, has long been recognized as impossible7 owing to the stringent requirements of even faster (sub ~1–10 ns) operating speeds and years of data nonvolatility for PCRAM devices9,10,11.
Such a speed bottleneck of the PCRAM devices originates from the relatively sluggish crystallization process in the SET operation, not from the amorphization procedure for the RESET operation2,12. Generally, the whole crystallization process consists of two stages: the nucleation of small crystallites and their subsequent growth. Note that the crystal growth velocity in the supercooled liquid of Ge2Sb2Te5 can be rather fast, reaching a maximum of ~0.5–3.0 m s−1 at ~600–700 K13,14,15. In a relatively miniaturized PCRAM device of ~7.5 × 17.0 nm2 in cross section, even when using Sb-rich Ge2Sb2Te5 to further strengthen the growth momentum, the shortest SET operation still takes ~30 ns16. Such a ‘limited’ speed performance suggests that in addition to the strong (inward) growth momentum from peripheral crystalline interfaces, it would be beneficial to also have copious small crystallites inside the glassy matrix that can quickly grow simultaneously; the two processes would work together to facilitate an even faster crystallization9,10,17. However, the nucleation of Ge2Sb2Te5 faces an unavoidable ‘time’ issue, namely, it is stochastic in nature because the time for incubating stable nuclei over the critical size is distributed over a broad scale, from several hundreds of picoseconds to many nanoseconds9,18. The stochasticity is associated with the short lifetime of subcritical nuclei that consist of cubic motifs constructed by Ge(Sb)–Te bonds11,12,19. These soft chemical bonds constantly form and rupture at high crystallization temperatures, e.g., ~500–700 K; therefore, these nucleation embryos fluctuate severely, surviving for only several picoseconds11. One may thus resort to a common solution by introducing stable heterogeneities into a glassy phase to elevate the nucleation rate. However, a difficulty arises here from the contradictory nature of Ge2Sb2Te5-like PCMs that (most of the) doping20,21,22 always extends the stability of the amorphous phase for long-term data retention but inadvertently slows down the crystallization. The fast and persistent features of the phase-change working memory are seemingly irreconcilable, and a trade-off seems inevitable.
To solve this problem, we designed a Sc0.2Sb2Te3 alloy and validated its superior functions in a conventional PCRAM device, which enabled a 0.7-ns SET speed and a comparable data retention ability to the Ge2Sb2Te5 device of the same geometry11. The high-strength Sc–Te bonds effectively enhance the stability of nucleation embryos, prolonging the lifetime against thermal fluctuations. Moreover, the embedded Sc–Te embryos are geometrically conformable with their crystalline counterpart, i.e., rocksalt Sb2Te3, lowering the surface free energy. These two traits together prompt a dramatic decrease in the energy barrier for nucleation, reducing the stochasticity and thus shortening the wait time for incubation11. We have also elucidated that Sc addition can create an enlarged kinetic contrast inside the supercooled liquid state over a fairly narrow temperature range23. This guarantees high atomic mobility at elevated temperatures for rapid crystal growth and suppressed atomic diffusion near room temperature for good data retention.
At this point, one may ask another question that is worth exploring: is there a better composition than Sc0.2Sb2Te3 to achieve an even faster SET speed and better data nonvolatility of the PCRAM device at the same time? In this work, we provide direct evidence to answer this question by systematically characterizing the structural, electrical, and thermal properties of a series of ScxSb2Te3 compounds. We reveal that as the Sc content increases, the amorphous stability of ScxSb2Te3 films can be enhanced monotonically, whereas balancing the nucleation rate with the growth velocity leads to the optimal Sc dose of x ~0.3, which enables the fastest crystallization speed and improved data retention ability. Extra Sc doping restrains the kinetics substantially throughout the whole temperature range, reducing the crystallization speed. Our work may serve as a useful example demonstrating how to optimize the composition of PCMs through tailoring of their crystallization kinetics, aiming at the development of high-performance phase-change working- and storage-class memories.
Materials and methods
Film preparation and characterization
Approximately 300-nm-thick ScxSb2Te3 (x = 0.1, 0.2, 0.3, and 0.36) films were deposited on SiO2/Si substrates at room temperature by cosputtering pure Sc and Sb2Te3 targets in ultrahigh vacuum with a base pressure of <~2 × 10−7 mTorr. The deposition rate was controlled to ~5 nm min−1. Ge2Sb2Te5 films of the same thickness were fabricated by sputtering the pure alloy target. The compositions of all the films were determined by using X-ray fluorescence spectroscopy (Rigaku RIX 2100). The sheet resistance of all the films was studied using a Linkam HFS600E-PB4 hot stage with a temperature accuracy of ~0.1 K. The morphology and crystallinity of ~220 °C-annealed ScxSb2Te3 films (~20 nm in thickness) were confirmed by high-resolution transmission electron microscopy (HRTEM, JEOL JEM-2100F) at a high tension of 200 kV. An ~10-nm-thick SiO2 capping layer was in situ grown on top of each film inside the vacuum chamber to avoid oxidation for HRTEM and thermal measurement.
Device fabrication and electrical measurement
Conventional T-shaped PCRAM devices with a tungsten bottom electrode contact (BEC) of ~80–190 nm in diameter (φ) were fabricated using the 0.13 μm node complementary metal-oxide semiconductor technology11. The thickness of the PCM films in all devices was controlled to ~150 nm. Approximately 15-nm-thick TiN and ~300-nm-thick Al films were used as the top electrode in all devices. All electrical measurements were performed by using a Keithley 2400C source meter (measuring the cell resistance), a Tektronix AWG5002B pulse generator (generating a voltage pulse with a minimum width of ~6 ns), a homemade constant current driver (generating a current pulse with a maximum magnitude of ~10 mA) and a Tektronix 7054 digital phosphor oscilloscope (measuring the transient voltage drop across the cell when the current pulse was applied).
Flash differential scanning calorimetry (FDSC) measurements
Power-compensation differential scanning calorimetry (DSC) was performed using a Mettler-Toledo FDSC 1 instrument with sensor chips (USF-1), each containing working and reference areas. ScxSb2Te3 small flakes were scraped off from the substrate and then transferred onto the working area of the chip sensor. The heating rate (Φ) was varied from 10 to 40,000 K s−1. At each Φ, measurements were repeated at least three times for low Φ and 5–10 times for high Φ, as the values of the crystallization temperature become more scattered at high Φ. The thermal lag and temperature calibration of the FDSC are evaluated in detail in the Supplementary information. The detailed methodology of Kissinger fitting can be seen in our previous work24. The viscosity model used in this work is illustrated in the Supplementary information. Since the ScxSb2Te3 flakes were only one-side capped, it is appropriate to compare the FDSC results with those of uncapped Ge2Sb2Te5 films13,23.
Density functional theory simulations
Density functional theory (DFT) simulations were carried out with the Vienna ab initio Simulations Package (VASP)25. The Perdew–Burke–Ernzerhof functionals26 and the projector augmented wave pseudopotentials27 were used for VASP. Rocksalt Sc0.3Sb1.7Te3 supercells with 180 (11 Sc, 61 Sb, and 108 Te) atoms were simulated with periodic boundary conditions by NVT DFT-based molecular dynamics (DFMD). The model was heated from 300 to 1150 K at a heating rate of 15 K ps−1. The energy cutoff was 180 eV, and the time step was 3 fs.
Results and discussion
Structural transformation and stability of the amorphous phase
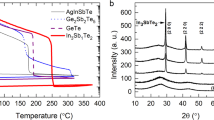
Temperature-dependent electrical resistance measurements were first performed for as-deposited and ~300-nm-thick Ge2Sb2Te5 and ScxSb2Te3 (x = 0.1, 0.2, and 0.3) films upon in situ annealing at a heating rate of 10 °C min−1 (Fig. 1a) since this technique is very sensitive to structural changes in PCMs28. All three as-deposited ScxSb2Te3 films are in amorphous states, and the initial magnitude of the sheet resistance becomes larger as the Sc content (x) increases from 0.1 to 0.3, corresponding to the widened energy band gap of the amorphous semiconductor28. When heated beyond the crystallization temperature (Tc) to ~350 °C, the amorphous semiconductor finally transforms into a narrow-gap crystalline semiconductor of a stable crystalline (hexagonal) phase, with a much (~3–4 orders of magnitude) smaller sheet resistance.

a Temperature dependence of the sheet resistance of ~300-nm-thick Ge2Sb2Te5 and ScxSb2Te3 (x = 0.1, 0.2, and 0.3) films at the same heating rate of 10 °C min−1. The crystallization temperature (Tc) is indicated by the arrow: ~138, ~170, ~199, and ~163 °C for the Sc0.1Sb2Te3, Sc0.2Sb2Te3, Sc0.3Sb2Te3, and Ge2Sb2Te5 films, respectively. b–d BF-TEM images of ~20-nm-thick ScxSb2Te3 films annealed at ~220 °C. The SAED pattern of each sample is shown in the corresponding inset. e Raw radially integrated diffraction curves of the electron diffraction intensity extracted from the respective SAED patterns shown in (b–d). f Ten-year data retention abilities for Ge2Sb2Te5 and ScxSb2Te3 devices. The data were fitted using the Arrhenius equation t = Aexp(Ea/kBT), where t is the time to failure when the cell resistance in the RESET state, at a certain isothermal heating temperature, falls to half of its initial magnitude, A is a proportionality constant, Ea is the activation energy, and kB is the Boltzmann constant.
We selected three annealed (at ~220 °C) samples to assess the microstructural details by HRTEM. The bright-field (BF) HRTEM image and the corresponding selected-area electron diffraction (SAED) pattern of each ScxSb2Te3 film show a homogeneous polycrystalline morphology with many nanosized crystal grains, indicating a nucleation-dominant crystallization behavior (Fig. 1b–d). Figure 1e shows the raw radially integrated diffraction curves of the electron diffraction intensity extracted from the inset SAED patterns in Fig. 1b–d. All the diffraction peaks located in the range from ~1 to ~4 Å well match the featured positions of pure rocksalt Sb2Te329, proving that all the annealed ScxSb2Te3 samples are of the rocksalt phase. As the Sc content increases, the diffraction intensity decreases, accompanied by peak broadening, correlating to smaller crystal grain sizes and poorer crystallinity. It is observable that some tiny crystallites are embedded in the amorphous networks of Sc0.2Sb2Te3 (Fig. 1c) and Sc0.3Sb2Te3 (Fig. 1d). Note that each ScxSb2Te3 film has a comparably wide temperature window (from Tc to ~Tc + 100 °C) to that of Ge2Sb2Te5 for retaining the metastable rocksalt phase11,29. This is beneficial for guaranteeing the amorphous-to-rocksalt transition for the SET operation of ScxSb2Te3 devices through the design of a suitable heating profile. High-speed PCRAM application should avoid the involvement of a stable hexagonal phase because its formation requires a longer SET time, and its melting requires extra RESET energy29.
Since the Sc0.2Sb2Te3 and Ge2Sb2Te5 films have close Tc values (Fig. 1a), the 10-year data retention ability of the RESET state for the Sc0.2Sb2Te3 device (~87 °C) is slightly better than that of the Ge2Sb2Te5 device (~82 °C), as shown in Fig. 1f. It is obvious that increasing (decreasing) the Sc content results in a higher (lower) Tc of the Sc0.3Sb2Te3 (Sc0.1Sb2Te3) film; correspondingly, the 10-year data retention ability of the RESET state becomes superior (~107 °C) and inferior (~68 °C) for the Sc0.3Sb2Te3 and Sc0.1Sb2Te3 devices, respectively. The improved data retention ability should also be attributed to the increased crystallization activation energy (Ea) of the Sc0.2Sb2Te3 and Sc0.3Sb2Te3 devices, i.e., ~2.38 and ~2.50 eV, respectively, compared to that (~2.21 eV) of the Ge2Sb2Te5 device (Fig. 1f). These data demonstrate that once the Sc content is enriched, the crystallization is drastically suppressed in amorphous ScxSb2Te3 near room temperature. Does this also implicate remarkably weakened driving forces for crystallization at elevated temperatures, causing a prolonged SET time? To clarify this issue, we performed electrical measurements on Ge2Sb2Te5 and ScxSb2Te3 devices to compare the SET speed.
RESET energy and SET speed of PCRAM devices
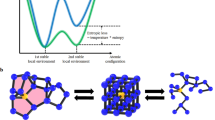
Inside the conventional T-shaped PCRAM device (inset of Fig. 2a), a mushroom-shaped amorphous region can be formed through the RESET operation, i.e., melt quenching of the rocksalt PCM film (~150 nm in thickness) immediately above the tungsten BEC. The amorphous mushroom regions of different sizes with various RESET resistances will significantly affect the crystallization process in the SET operation6. To accurately measure the speed of each SET operation, we preprogrammed all the devices to a fully RESET state by using a very wide constant electric current pulse of 1000 ns. Decreasing the BEC diameter (φ) from 190 (130) to 80 nm markedly reduces the RESET energy of the ScxSb2Te3 and Ge2Sb2Te5 devices (Fig. 2a and Figure S1 in the Supplementary information). In particular, the ScxSb2Te3 devices possess ~85–90% lower RESET energy (0.64–0.48 nJ) than the Ge2Sb2Te5 device (4.20 nJ) with a φ of 80 nm. Such a dramatic reduction in power consumption should be ascribed to the easier melting of the rocksalt lattice of ScxSb2Te3 that contains a higher concentration of cationic vacancies than Ge2Sb2Te511,29. Upon melting of the rocksalt ScxSb2Te3, as revealed by DFMD simulations (Fig. 2b), the central Sc–Te-based cubic motifs remain ordered throughout the heating procedure, whereas the surrounding Sb–Te parts can already be fully liquefied. The Sc–Te motif with a strong bond strength promotes disordering of the adjacent Sb–Te lattices30, through which increasing the Sc content helps lower the RESET energy, although the change is not strikingly large. This also suggests that a slight doping will not significantly change the melting temperature (Tm) of rocksalt ScxSb2Te3. However, it can still essentially alter the crystallization (including both nucleation and growth) behavior23. The DFMD simulation here qualitatively illustrates that numerous subcritical Sc–Te nucleation embryos can survive the melt-quenching process, along with the quenched-in nuclei of larger size, acting as intrinsic (robust) seeds11,30 to speed up the nucleation-dominant crystallization (inset of Fig. 2c) in amorphous ScxSb2Te3.

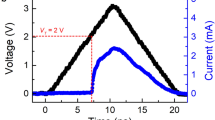
a RESET energy (E) as a function of the bottom electrode contact (BEC) diameter (φ) for Ge2Sb2Te5 and ScxSb2Te3 devices. The inset shows a schematic of conventional T-shaped PCRAM devices with the following geometry: ~150 nm-thick PCM layer, ~20-nm-thick TiN top electrode, and ~80–190 nm in diameter W bottom electrode. The rocksalt PCM (yellow) immediately above the BEC is undergoing a melt-quenching process, where the red area represents the supercooled liquid phase. A fully RESET state refers to a large amorphous phase mushroom, as marked by the black contour. The transient RESET voltage (VRst) across the device is recorded once the RESET state is reached. The input RESET energy is calculated as E = IRst × VRst × t, where IRst is the RESET current, VRst is the RESET voltage, and t is fixed at 1000 ns. b DFMD simulations of the melting process of rocksalt ScxSb2Te3 showing that the initial structure is still maintained at 900 K and that the peripheral Sb2Te3 lattice begins disordering at 1100 K. The surrounding Sb2Te3 part becomes rather disordered at 1150 K after 15 ps, while the central Sc–Te precursor (Sc: red, Te blue), throughout the whole annealing process, remains in the cubic configuration. c SET operation speed for Ge2Sb2Te5 and ScxSb2Te3 devices with the same geometry. (Inset) Inside the mushroom of the supercooled liquid phase (red), small nuclei/crystallites (yellow) are generated before subsequent crystal growth. Upon the SET operation, all the PCMs show nucleation-dominated crystallization behavior.
Voltage pulses of a fixed magnitude but increasing width were imposed on Ge2Sb2Te5 and ScxSb2Te3 devices to assess the SET time (Figure S2 in the Supplementary information). As the magnitude of the applied pulses increases, the SET time can be accordingly shortened (Fig. 2c). We need to note that for the convenience of device fabrication and electrical measurements, we did not extend the testing range to the subnanosecond level; however, the current comparison already suffices to distinguish the differences in the SET speed among the devices. In the Ge2Sb2Te5 device, before the largest resistance drop to reach the SET state, the RESET state takes ~20–50 ns to descend sluggishly to an intermediate (partial) state; in contrast, the resistance drop in all the ScxSb2Te3 devices is relatively steep (Fig. S2 in the Supplementary information), denoting a faster crystallization process. In Fig. 2c, at a low bias of ~1.5–1.6 V, the Sc0.1Sb2Te3 and Ge2Sb2Te5 devices have an approximate SET speed of ~70–80 ns; however, at ~1.9 V, the former needs only ~6 ns to reach SET, whereas the latter still requires ~55 ns. The Sc0.1Sb2Te3 device is ~9 times faster than the Ge2Sb2Te5 device. Similarly, at ~1.5 V, the Sc0.2Sb2Te3 device (~6 ns) is over one order of magnitude faster than the Sc0.1Sb2Te3 device (~62 ns). Regarding the Sc0.3Sb2Te3 device (~6 ns) at ~1.1 V, it is almost four times faster than the Sc0.2Sb2Te3 device (~23 ns). Clearly, enriching the Sc content to x = 0.3 does not seem to hinder the speed of the SET operation. Note that the SET times of all the ScxSb2Te3 devices only reach 6 ns because the minimum pulse width employed here is 6 ns. A further reduction in the SET time for the Sc0.3Sb2Te3 device can be expected if picosecond-pulse programming is applied. Although the kinetics near room temperature are largely arrested in Sc0.3Sb2Te3, the driving forces for crystallization at elevated temperatures must not have been essentially attenuated; otherwise, the SET speed of the device would be slower than those of the Sc-deficient devices. To lend credence to this hypothesis, we performed FDSC measurements on ScxSb2Te3 thin films to assess the crystallization kinetics as a function of temperature.
Enlarged kinetic contrast through a fragile-to-strong crossover
The chip sensor of the Mettler-Toledo Flash DSC 1 shown in the right inset of Fig. 3a was employed in the present study. It has a square active area with a length of ~0.3 mm and allows controlled heating and cooling of samples with a typical mass of ~100 ng up to tens of thousands Kelvin per second13. The as-deposited amorphous ScxSb2Te3 films of ~300 nm thickness were crystallized by varying the heating rate (Φ) from 10 to the maximum of 40,000 K s−1. Several representative FDSC traces of the Sc0.1Sb2Te3 sample after background subtraction are shown in Fig. 3a, where the crystallization peak temperature (Tp, circled) shifts from ~148 to ~206 °C upon increasing Φ from 50 to 20,000 K s−1. The shift of Tp allows for the investigation of the crystallization kinetics at elevated temperatures. Note that the lowest 2 to 3 Tp values at each Φ were regarded as the most credible data because they correspond to the best thermal contact between the stripped-off ScxSb2Te3 films and chip sensors14,23. The crystallization data of ScxSb2Te3 obtained via FDSC measurements are portrayed in a Kissinger plot (Fig. 3b). Previous Ge2Sb2Te5 data obtained by FDSC13 and conventional DSC31 are also included for a direct comparison. All the ScxSb2Te3 films obey a linear relation across a wide Φ range in the Kissinger plot, complying with the Arrhenius behavior up to Φ ~1000–10,000 K s−1, as illustrated by the yellow dashed lines, above which the Arrhenius behavior breaks down. Within the Arrhenius range, the fitted Kissinger plot gives a slope of Ea ~2.08, ~2.35, and ~2.66 eV for Sc0.1Sb2Te3, Sc0.2Sb2Te3, and Sc0.3Sb2Te3, respectively. The values of Ea are reasonably comparable to those obtained in Fig. 1f. In contrast, Ge2Sb2Te5 departs from the Arrhenius behavior (with Ea ~2.19 eV) at a rather low heating rate (Φ ~100 K s−1). Such a broader Arrhenius behavior of ScxSb2Te3 is analogous to those observed in AgIn-doped Sb2Te films17 and Ge2Sb2Te5 nanoparticles14. We thereby adopted the same treatments as those in the literature, i.e., the generalized MYEGA model for viscosity combined with Johnson–Mehl–Avrami–Kolmogorov theory to numerically simulate and fit the FDSC data of ScxSb2Te3 and Ge2Sb2Te5 supercooled liquids (see Supplementary information for details).

a Representative FDSC traces of Sc0.1Sb2Te3 thin films with Φ ranging from 50 to 20,000 K s−1. The left inset shows a zoom-in view of the crystallization peak temperature (Tp) at low Φ. The right inset shows the FDSC chip sensor employed in this study. b Kissinger plots showing the FDSC data of ScxSb2Te3 and Ge2Sb2Te5 (obtained by Orava et al.)13 thin films, as well as several data points derived from conventional DSC for Ge2Sb2Te5 (obtained by Park et al.)27 samples. The crystallization activation energy Ea for ScxSb2Te3 and Ge2Sb2Te5 is derived via linear fitting of the respective data in the lower Φ range, as marked by the yellow dashed line. c Temperature-dependent viscosity η of ScxSb2Te3 and Ge2Sb2Te5 films. The Tg of each film is set as the temperature where η is 1012 Pa s. The η of Ge2Sb2Te5 at Tm is also plotted in the figure, with the cross (×) indicating the viscosity data measured by an oscillating-cup viscometer32 and the open diamond (◊) the simulated viscosity data19, showing good agreement with our results. d The viscosity activation energy Eη for ScxSb2Te3 reaches a plateau when quenched toward Tg, while that of Ge2Sb2Te5 increases monotonically. The inflection temperature, i.e., ~424, ~445, and ~510 K, is determined as the FTS crossover point of Sc0.1Sb2Te3, Sc0.2Sb2Te3, and Sc0.3Sb2Te3, respectively.
Figure 3c depicts the temperature dependency of the viscosity (η) determined from the generalized MYEGA model (see Equation S3, Table S1 and discussions in the Supplementary information). The modeled viscosity shows an excellent match with simulated and experimental data at Tm reported previously19,32. Note that in the generalized MYEGA model, the glass transition temperature (Tg) is not directly provided but is set as the temperature where η reaches 1012 Pa s. The fitted Tg of Sc0.1Sb2Te3, Sc0.2Sb2Te3, and Sc0.3Sb2Te3 is 342, 371, and 438 K, respectively, and our Tg of Ge2Sb2Te5, i.e., 378 K, is comparable to the previously reported value of ~383 K13. In between Tg and Tm, strong liquids such as silica follow the Arrhenius behavior with a nearly constant activation energy for viscous flow, whereas fragile liquids such as o-terphenyl33 exhibit a rather high activation energy near Tg but become markedly less viscous as the temperature increases. The fragility m, defined as the slope of the η at Tg, i.e., m = d[log10η(T)]/d(Tg/T)|T=Tg, is derived as ~56, ~64, and ~64 for Sc0.1Sb2Te3, Sc0.2Sb2Te3, and Sc0.3Sb2Te3, respectively. Ge2Sb2Te5 (m ~102) is apparently more fragile than ScxSb2Te3 when approaching Tg. If a single-fragility model is employed to describe the viscosity behavior of ScxSb2Te3, then it can only be applicable to the high-temperature regime above ~400–500 K, generating rather large fragilities of over ~170. Instead, the generalized MYEGA model can nicely fit not only the curved (non-Arrhenius behavior) but also the linear (Arrhenius behavior) parts in the Kissinger plot14,23. The high-to-low change in the fragility strongly indicates that an enlarged contrast in the kinetics occurs below and above these inflection temperatures at ~400–500 K, manifesting as an apparent fragile-to-strong (FTS) crossover in the deeply supercooled ScxSb2Te3 liquid (Fig. 3c, d).
The FTS crossover can be assessed by monitoring Eη = kBdln(η/η0)/d(1/T), where kB is the Boltzmann constant and Eη is the viscosity activation energy. ScxSb2Te3 approximately obeys the Arrhenius behavior along a clear plateau in Eη from Tg to each inflection point, i.e., ~424, ~445, and ~510 K, beyond which Eη sharply drops, approaching zero at Tg/T ≈ 0.65, thus presenting the super-Arrhenius behavior of increased fluidity (Fig. 3d). These inflection points can be taken as the FTS temperatures for ScxSb2Te3. In contrast, our fitting did not identify any clear FTS crossover in Ge2Sb2Te5 above its Tg, despite a potentially existing crossover at even lower temperatures17. The FTS crossover may originate from the increase in the Peierls distortion in the short range and the formation of an energetically favorable network at or beyond the medium range during quenching of the supercooled liquid34,35,36. As the Peierls distortion increases, electrons become more localized between atoms, exerting constraints on atomic migration and consequently increasing the activation barrier for viscous flow. The enhanced kinetic contrast due to the FTS crossover enables high atomic mobility at elevated temperatures for rapid crystal growth in a less-viscous matrix while suppressing atomic diffusion near room temperature for good data retention in a more-viscous state. If the FTS crossover occurs at either below Tg (e.g., in Ge2Sb2Te5)17 or above Tm (e.g., in Ge15Te85)37, it would cause relatively less- or more-viscous flow with inferior data retention or slower crystal growth, respectively. Regarding this, a desirable FTS crossover for an ultrafast and persistent PCM should occur immediately below the typical programming temperatures for crystallization (<~600–700 K) and considerably above its Tg (>~400 K), as exemplified by the cases of ScxSb2Te3 (x = ~0.1–0.3). Note that an increase in the Sc dose produces a more intensive change in the kinetics (with a larger contrast in Eη) upon a relatively small temperature range from the close Tm to the higher FTS temperature (Fig. 3d). This will undoubtedly essentially alter not only the amorphous stability when approaching room temperature but also, most importantly, the crystallization speed at elevated temperatures.
Crystal growth velocity and heterogeneous nucleation rate
Generally, in the crystallization of a glass through continuous heating, the peak in the nucleation rate usually precedes the peak in the growth velocity. Accordingly, in analyzing the FDSC data of PCMs to derive the crystal growth velocity (U), a reasonably simplified method was usually applied by assuming that the nucleation has already finished before the peak growth of a fixed population of small crystallites occurs13,14. It is known that this simplification does not significantly alter the kinetics results derived compared to the method adopting a more intricate nucleation model14. The temperature-dependent U (Fig. 4a) can then be obtained from Kissinger analysis of the shift of the peak position in Fig. 3b (see Equation S1 and discussions in the Supplementary Information). The maximum growth velocity (Umax) is ~3.14, ~2.07, and ~1.45 m s−1 for Sc0.1Sb2Te3 (at ~0.70 Tm), Sc0.2Sb2Te3 (at ~0.73 Tm), and Sc0.3Sb2Te3 (at ~0.80 Tm), respectively, where Tm (~890 K) is assumed to be roughly equal to that of Sb2Te3. The Umax (~1.92 m s−1) of Ge2Sb2Te5 appears at ~0.70 Tm, with Tm being ~900 K. Such a Umax for Ge2Sb2Te5 well matches previous results measured by FDSC and on real PCRAM devices, ranging from ~0.5 to 3 m s−1,13,14,15.

a Crystal growth velocities U of ScxSb2Te3 and Ge2Sb2Te5 supercooled liquids between Tg and Tm. The maximum velocity Umax value of each material is also shown near the corresponding peak. As the Sc content increases, the peak becomes shorter and shifts to higher temperatures. b Steady-state nucleation rate Iss of Ge2Sb2Te5 (homogeneous, abbreviated as Homo.) and ScxSb2Te3 (heterogeneous, abbreviated as Hetero.) with a contact angle θ of 20° taking the size effect into account.
According to the Stokes–Einstein relation in the Arrhenius case, the atomic diffusivity D is inversely proportional to the viscosity η, i.e., U∝D∝η1. When this relation breaks down, there will be decoupling between U and η, resulting in U∝η−ξ with ξ < 1, i.e., even faster crystal growth. Via fitting, the ξ for Sc0.1Sb2Te3, Sc0.2Sb2Te3, and Sc0.3Sb2Te3 is determined to be ~0.71, ~0.65, and ~0.63, respectively, and that for Ge2Sb2Te5 is ~0.67. In Fig. 3c, from its Tg (~438 K) to ~700 K (i.e., 1,000/T = ~1.43), Sc0.3Sb2Te3 has a very large η many orders of magnitude larger than those of the other materials; unsurprisingly, its U remains the smallest (Fig. 4a). Sc0.1Sb2Te3 is the opposite, behaving as the least-viscous liquid from its Tg (~342 K) to Tm, which ensures the largest U (up to ~1.6 times that of Ge2Sb2Te5). For Sc0.2Sb2Te3, its η is also considerably larger (< ~100-fold) than that of Ge2Sb2Te5 between ~400 and 570 K (i.e., 1,000/T = ~1.75–2.50, in Fig. 3c), corresponding to a markedly slower U (< ~120-fold) than that of the latter in the T range from ~0.44 to ~0.64 Tm (Fig. 4a). However, beyond ~570 K, due to the almost equal η and a slightly smaller ξ, the U of Sc0.2Sb2Te3 becomes comparable to (i.e., ~1.1 times larger than) that of Ge2Sb2Te5. Obviously, it would be irrational to ascribe the significant differences (over 10 times) in the SET speed only to the crystal growth momentum, especially when the weakest material (Sc0.3Sb2Te3) becomes the fastest. Nucleation must play a decisive role in accelerating the whole crystallization process.
Previously, we proved that homogeneous nucleation is invalid in interpreting the much faster crystallization nature of ScxSb2Te323. Regarding heterogeneous nucleation, if we choose a fairly small contact angle θ (say less than 30°, resembling the situation in supercooled water)38, Sc0.3Sb2Te3, strangely, has an extremely lower heterogeneous steady-state nucleation rate (\(I_{{\mathrm{het}}}^{{\mathrm{ss}}}\)) than the other two materials below ~630 K, which just slightly surpasses their rates at higher temperatures (Fig. S3 in the Supplementary information). Note that below ~630 K, Sc0.3Sb2Te3 possesses approximately twofold to to approximately seven orders of magnitude lower U than the other two materials (Fig. 4a). Combining these two characteristics, a contradictory scenario against Fig. 2c is depicted in that Sc0.3Sb2Te3 should crystallize much slower in the device than the other materials. We thus modified the common description of heterogeneous nucleation by taking the size effect of the nucleation precursor38 into consideration (see Equations S14–S19, Table S2 and discussions in the Supplementary information).
Figure 4b shows the comparison among the homogeneous steady-state nucleation rate (\(I_{\mathrm{hom}}^{{\mathrm{ss}}}\)) of Ge2Sb2Te5 and the modified \(I_{{\mathrm{het}}}^{{\mathrm{ss}}}\) of ScxSb2Te3. Although very subtle, it can be determined that the \(I_{{\mathrm{het}}}^{{\mathrm{ss}}}\) of Sc0.1Sb2Te3 is actually slightly larger than the \(I_{\mathrm{hom}}^{{\mathrm{ss}}}\) of Ge2Sb2Te5, e.g., ~2–6-fold at ~500–700 K, and in this range, the former also has an up to ~1.5 times higher U than the latter (Fig. 4a). In this way, the Sc0.1Sb2Te3 device can be ~9 times faster than the Ge2Sb2Te5 device in the SET operation. In contrast, it is easier to distinguish that the \(I_{{\mathrm{het}}}^{{\mathrm{ss}}}\) of Sc0.2Sb2Te3 is constantly larger, i.e., by ~2–5 orders of magnitude, than that of Sc0.1Sb2Te3 in the regime of ~500–700 K. Despite the relatively low growth velocity, Sc0.2Sb2Te3 easily outperforms Sc0.1Sb2Te3 in SET speed owing to the plenty of robust Sc–Te nuclei compared to the latter. The most interesting message here is from Sc0.3Sb2Te3: only beyond ~550 K can its \(I_{{\mathrm{het}}}^{{\mathrm{ss}}}\) exceed that of Sc0.2Sb2Te3, but the latter decays noticeably faster than the former, where the difference enlarges steadily from approximately tenfold at ~600 K to ~3–5 orders of magnitude at ~700–750 K. In light of the merely < ~4 times slower U at ~600–700 K (i.e., T/Tm = ~0.67–0.79 in Fig. 4a), Sc0.3Sb2Te3 undoubtedly can crystallize much faster than Sc0.2Sb2Te3 in the SET operation.
In a very-viscous supercooled liquid near above Tg, nucleation becomes rather difficult17,39. This is the case here for Sc0.3Sb2Te3, whose viscosity below the FTS temperature of ~510 K is very large with respect to the others (Fig. 3c). However, on the other hand, enriching the Sc content is helpful for increasing the number or size of the Sc–Te-based nucleation precursors, by which the heterogeneous nuclei can be more robust (of reduced stochasticity), withstanding higher-temperature fluctuations. However, one cannot arbitrarily use heavy Sc doping to pursue a very high nucleation rate40 because the atomic mobility throughout a wide supercooled liquid regime would be considerably suppressed, especially within the typical SET programming window of ~600–700 K. Thereby, the growth velocity would be severely reduced, and both the nucleation rate and growth velocity peaks would be shifted to an even higher-temperature zone. Since the driving forces for crystallization weaken as the temperature approaches Tm1,2, the nucleation and subsequent growth would strongly decay to rather low levels, leaving a quite narrow programming window for achieving a fast SET speed. To support this point, we further checked the FDSC data of Sc0.36Sb2Te3 (Fig. S4 in the Supplementary information). It is certainly a more viscous liquid than Sc0.3Sb2Te3 in the deeply supercooled regime, due to which its crystal growth is dramatically restrained with a peak value of only ~0.75 m s−1 postponed to ~735 K (at ~0.83 Tm), while preceding major growth, its nucleation rate is approximately the same as or even significantly lower than that of Sc0.3Sb2Te3. As the temperature further increases to ~800 K (at ~0.90 Tm), both the nucleation tendency and growth momentum are sharply weakened. Clearly, Sc0.36Sb2Te3 has little chance to outperform Sc0.3Sb2Te3 in crystallization speed.
Conclusions
Faster crystallization speed and better amorphous stability are the two ‘conflicting’ properties in pursuing ultrafast and persistent phase-change working memory. ScxSb2Te3 is the most promising PCM system for reconciling the contradiction. We proved that as the Sc content increases from x = 0.1–0.3 in the ScxSb2Te3 device, the data retention ability of the RESET state can be monotonically enhanced, whereas the SET operation also becomes faster accordingly. We thus performed FDSC experiments to unravel the seemingly strange temperature-dependent crystallization dynamics in the supercooled liquid regime. We found that enriching Sc increases the viscosity of the supercooled liquid and shapes broader Arrhenius-like kinetics from Tg up to an apparent FTS crossover. The rather suppressed atomic diffusion in the ‘strong’ liquid greatly restrains the crystallization near room temperature, strengthening the amorphous phase to provide long-term nonvolatility, while above the inflection point in the kinetics, the ‘strong’ liquid crossovers into a ‘fragile’ liquid of less-viscous flow, allowing high atomic mobility for fast crystal growth at elevated temperatures. The maximum crystal growth velocity of ScxSb2Te3 (x = 0.1–0.3) is ~3.14–1.45 m s−1, occurring at ~623–712 K, which is more or less comparable to that of Ge2Sb2Te5 (~1.92 m s−1 at ~630 K). Nonetheless, in the typical programming temperature range for crystallization, i.e., ~600–700 K, the heterogeneous nucleation rate of ScxSb2Te3 (x = 0.1–0.3) overwhelms the homogeneous nucleation rate in Ge2Sb2Te5 by several fold to many orders of magnitude; therefore, one can witness a hierarchy in the SET speed from the slowest Ge2Sb2Te5 to the fastest Sc0.3Sb2Te3 device. Note, however, that doping extra Sc over Sc0.3Sb2Te3 may have a negative impact on the crystallization speed because the driving forces would be substantially suppressed below ~735 K. Overall, our work illustrates a route to design superior PCMs for the development of phase-change working memory; that is, while promoting the nucleation rate at elevated temperatures, the kinetic contrast between elevated and ambient temperatures should be enlarged in a cautious manner, preventing the high-temperature crystal growth velocity from seriously decreasing.
References
Wuttig, M. & Yamada, N. Phase-change materials for rewriteable data storage. Nat. Mater. 6, 824–832 (2007).
Raoux, S., Welnic, W. & Ielmini, D. Phase change materials and their application to nonvolatile memories. Chem. Rev. 110, 240–267 (2010).
Kolobov et al. Understanding the phase-change mechanism of rewritable optical media. Nat. Mater. 3, 703–708 (2004).
Atwood, G. Phase-change materials for electronic memories. Science 321, 210–211 (2008).
Ielmini, D. & Lacaita, A. L. Phase change materials in non-volatile storage. Mater. Today 14, 600–607 (2011).
Ding, K. Y. et al. Phase-change heterostructure enables ultralow noise and drift for memory operation. Science 366, 210–215 (2019).
Wong, H. S. & Salahuddin, S. Memory leads the way to better computing. Nat. Nanotechnol. 10, 191–194 (2015).
Choe, J. Tech insights: Intel 3D XPoint memory die removed from Intel Optane PCM (phase change memory), https://www.techinsights.com/blog/intel-3d-xpoint-memory-die-removed-inteloptanetm-pcm-phase-change-memory.
Loke, D. et al. Breaking the speed limits of phase-change memory. Science 336, 1566–1569 (2012).
Rao, F. et al. Direct observation of titanium-centered octahedra in titanium–antimony–tellurium phase-change material. Nat. Commun. 6, 10040 (2015).
Rao, F. et al. Reducing the stochasticity of crystal nucleation to enable subnanosecond memory writing. Science 358, 1423–1427 (2017).
Hegedüs, J. & Elliott, S. R. Microscopic origin of the fast crystallization ability of Ge-Sb-Te phase-change memory materials. Nat. Mater. 7, 399–405 (2008).
Orava, J., Greer, A. L., Gholipour, B., Hewak, D. W. & Smith, C. E. Characterization of supercooled liquid Ge2Sb2Te5 and its crystallization by ultrafast-heating calorimetry. Nat. Mater. 11, 279–283 (2012).
Chen, B., Brink, G. H., Palasantzas, G. & Kooi, B. J. Crystallization kinetics of GeSbTe phase-change nanoparticles resolved by ultrafast calorimetry. J. Phys. Chem. C. 121, 8569–8578 (2017).
Sebastian, A., Le Gallo, M. & Krebs, D. Crystal growth within a phase change memory cell. Nat. Commun. 5, 4314 (2014).
Kim, I. S. et al. High performance PRAM cell scalable to sub-20 nm technology with below 4F2 cell size, extendable to DRAM applications. Symposium on VLSI Technology, 203–204 (2010).
Orava, J., Hewak, D. W. & Greer, A. L. Fragile-to-strong crossover in supercooled liquid Ag-In-Sb-Te studied by ultrafast calorimetry. Adv. Funct. Mater. 25, 4851–4858 (2015).
Kalikka, J., Akola, J. & Jones, R. O. Crystallization processes in the phase change material Ge2Sb2Te5: Unbiased density functional/molecular dynamics simulations. Phys. Rev. B 94, 134105 (2016).
Akola, J. & Jones, R. O. Structural phase transitions on the nanoscale: The crucial pattern in the phase-change materials Ge2Sb2Te5 and GeTe. Phys. Rev. B 76, 23501 (2007).
Navarro, G. et al. Trade-off between SET and data retention performance thanks to innovative materials for phase-change memory. IEEE International Electron Devices Meeting (IEDM), 570–573 (2013).
Cheng, H. Y. et al. A high performance phase change memory with fast switching speed and high temperature retention by engineering the GexSbyTez phase change material. IEEE International Electron Devices Meeting (IEDM), 51–54 (2011).
Zhou, X. L. et al. Understanding phase-change behaviors of carbon-doped Ge2Sb2Te5 for phase-change memory application. ACS Appl. Mater. Interfaces 6, 14207–14214 (2014).
Chen, B. et al. Kinetics features conducive to cache-type nonvolatile phase-change memory. Chem. Mater. 31, 8794–8800 (2019).
Chen, B., de Wal, D., ten Brink, G. H., Palasantzas, G. & Kooi, B. J. Resolving crystallization kinetics of GeTe phase-change nanoparticles by ultrafast calorimetry. Cryst. Growth Des. 18, 1041–1046 (2018).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Rao, F. et al. Investigation of changes in band gap and density of localized states on phase transition for Ge2Sb2Te5 and Si3.5Sb2Te3 materials. Acta Mater. 60, 323–328 (2012).
Zheng, Y. et al. Direct observation of metastable face-centered cubic Sb2Te3 crystal. Nano Res 9, 3453–3462 (2016).
Wang, X. P. et al. Time-dependent density-functional theory molecular-dynamics study on amorphization of Sc-Sb-Te alloy under optical excitation. NPJ Comput. Mater. 6, 31 (2020).
Park, J., Kim, M. R., Choi, W. S., Seo, H. & Yeon, C. Characterization of amorphous phases of Ge2Sb2Te5 phase-change optical recording material on their crystallization behavior. Jpn. J. Appl. Phys. 38, 4775–4779 (1999).
Schumacher, M. et al. Structural, electronic and kinetic properties of the phase-change material Ge2Sb2Te5 in the liquid state. Sci. Rep. 6, 27434 (2016).
Angell, C. A. Formation of glasses from liquids and biopolymers. Science 267, 1924–1935 (1995).
Rao, F., Zhang, W. & Ma, E. Catching structural transition in liquids. Science 364, 1032–1033 (2019).
Zalden, P. et al. Femtosecond X-ray diffraction reveals a liquid-liquid phase transition in phase-change materials. Science 364, 1062–1067 (2019).
Sun, Z. M., Zhou, J., Mao, H. K. & Ahuja, R. Peierls distortion mediated reversible phase transition in GeTe under pressure. Proc Natl Acad. Sci. USA 109, 5948–5952 (2012).
Weber, H., Orava, J., Kaban, I., Pries, J. & Greer, A. L. Correlating ultrafast calorimetry, viscosity, and structural measurements in liquid GeTe and Ge15Te85. Phys. Rev. Mater. 2, 093405 (2018).
Fletcher, N. H. Size effect in heterogeneous nucleation. J. Chem. Phys. 29, 572–576 (1958).
Orava, J. & Greer, A. L. Classical-nucleation-theory analysis of priming in chalcogenide phase-change memory. Acta Mater. 139, 226–235 (2017).
Zewdie, G. M. et al. Chemical design principles for cache-type Sc-Sb-Te phase-change memory materials. Chem. Mater. 31, 4008–4015 (2019).
Acknowledgements
F.R. gratefully thanks the Major Provincial Basic Research Program of Guangdong (2017KZDXM070) and the Science and Technology Foundation of Shenzhen (JCYJ20180507182248605) for the support. K.D. thanks the Science and Technology Foundation of Shenzhen (JCYJ20190808150605474) for the support. Y.C. thanks the National Natural Science Foundation of China (61904091) for the support. J.W. thanks the National Natural Science Foundation of China (51771216) for the support. X.S. thanks the National Natural Science Foundation of China (61775111) for the support. The supercomputer time was provided by the National Supercomputer Center in Tianjin, and the calculations were performed on TianHe-1 (A).
Author information
Authors and Affiliations
Contributions
K.D. and B.C. fabricated the PCM film samples. K.D. carried out sheet resistance characterizations and TEM experiments. KD. and F.R. prepared the device samples and carried out electrical measurements. Y.C. performed FDSC measurements. B.C. performed simulation and fitting of FDSC data. F.R., B.C., and K.D. carried out theoretical analysis and together wrote this paper with help from Y.C., J.W., and X.S. The project was initiated and conceptualized by F.R.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ding, K., Chen, B., Chen, Y. et al. Recipe for ultrafast and persistent phase-change memory materials. NPG Asia Mater 12, 63 (2020). https://doi.org/10.1038/s41427-020-00246-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41427-020-00246-z
This article is cited by
-
Top-down patterning of topological surface and edge states using a focused ion beam
Nature Communications (2023)
-
Some novel perspectives of iso-conversional analysis in the study of Meyer–Neldel energy for thermally governed crystallization by using Johnson–Mehl–Avrami (JMA) theory
Journal of Thermal Analysis and Calorimetry (2023)
-
Rules of hierarchical melt and coordinate bond to design crystallization in doped phase change materials
Nature Communications (2021)
-
Ab initio molecular dynamics and materials design for embedded phase-change memory
npj Computational Materials (2021)
