
Abstract
The emergence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in 2019 prompted scientific, medical, and biotech communities to investigate infection- and vaccine-induced immune responses in the context of this pathogen. B-cell and antibody responses are at the center of these investigations, as neutralizing antibodies (nAbs) are an important correlate of protection (COP) from infection and the primary target of SARS-CoV-2 vaccine modalities. In addition to absolute levels, nAb longevity, neutralization breadth, immunoglobulin isotype and subtype composition, and presence at mucosal sites have become important topics for scientists and health policy makers. The recent pandemic was and still is a unique setting in which to study de novo and memory B-cell (MBC) and antibody responses in the dynamic interplay of infection- and vaccine-induced immunity. It also provided an opportunity to explore new vaccine platforms, such as mRNA or adenoviral vector vaccines, in unprecedented cohort sizes. Combined with the technological advances of recent years, this situation has provided detailed mechanistic insights into the development of B-cell and antibody responses but also revealed some unexpected findings. In this review, we summarize the key findings of the last 2.5 years regarding infection- and vaccine-induced B-cell immunity, which we believe are of significant value not only in the context of SARS-CoV-2 but also for future vaccination approaches in endemic and pandemic settings.
Similar content being viewed by others


Introduction
In late 2019, a cluster of pneumonia cases of unknown etiology was reported from Wuhan, China [1, 2]. It soon became clear that a novel coronavirus was the causative agent, rapidly leading to an increasing number of infections and deaths. The virus is thought to have originated from zoonotic spillover from bats via an intermediate host associated with Huanan Seafood Wholesale Market [3, 4]. In ensuing months, SARS-CoV-2 encountered an immunologically naïve human population, resulting in the most severe pandemic outbreak since the 1918 Spanish Flu.
To date, more than 767 million confirmed cases of coronavirus disease 2019 (COVID-19) and 6.9 million associated deaths have been reported worldwide [5]. As large parts of the human population have developed immunity to the virus through infection and/or vaccination, the pandemic phase is waning. However, SARS-CoV-2 may become a recurrent, seasonal pathogen, requiring induction of durable immunity or periodic booster vaccinations to protect those at risk. Moreover, after SARS-CoV-1 in 2002 and the Middle East respiratory syndrome (MERS) coronavirus in 2012, SARS-CoV-2 is the third zoonotic betacoronavirus infecting the human population in the last two decades, underscoring the fact that we may face newly emerging CoVs in the future. An in-depth understanding of vaccine- and infection-induced immune responses against SARS-CoV-2 is therefore highly relevant for postpandemic mitigation as well as pandemic preparedness.
The majority of symptomatic SARS-CoV-2 infections result in mild to moderate disease with prototypical symptoms of a respiratory infection, including fever, fatigue, and dry cough [6]. However, a significant proportion of infections progress to more severe and critical disease involving dyspnea, acute respiratory distress syndrome (ARDS) or multiorgan failure. The fatality rate has been estimated to be 0.23% but varies considerably across locations, probably reflecting different population characteristics [7]. Older age and comorbidities are important factors contributing to disease severity, but a range of other variables, including sex, race, and socioeconomic status, have also been discussed [8,9,10,11]. In addition to acute illness, SARS-CoV-2 infection can lead to persistent health problems affecting multiple organs, which is referred to as long COVID. This inconsistent, multifaceted disease manifestation is estimated to occur in at least 10% of symptomatic infections, but vaccination prior to infection significantly reduces the risk [12,13,14].
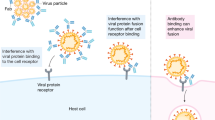
SARS-CoV-2 is a betacoronavirus that encodes four structural proteins from its large positive-sense RNA genome: spike (S), nucleocapsid (N), matrix (M), and envelope (E). Cell entry is mediated by the trimeric S glycoprotein, which binds to its entry receptor angiotensin-converting enzyme 2 (ACE2) [15]. To allow for fusion of the viral and host cell membranes, the S protein must be primed by furin-mediated cleavage at the S1/S2 site, generating S1 and S2 fragments. A second cleavage at the S2’ site by transmembrane protease serine subtype 2 (TMPRSS2) or cathepsin B/L after attachment or endocytosis, respectively, is required to liberate the fusion peptide [15]. Each S protein monomer contains a receptor-binding domain (RBD) that can be buried within the N-terminal domain (NTD; RBD “down”) or exposed (RBD “up”), with only the RBD in the up position being able to interact with ACE2 [15, 16]. Similar to other viral glycoproteins, S has an energetically unfavorable and unstable prefusion conformation that can spontaneously refold into a postfusion state [16, 17]. The postfusion conformation lacks or hides important epitopes for neutralizing antibodies [17]. Insertion of two proline residues into the C-terminal S2 fusion domain (S-2P) stabilizes the S protein in its prefusion conformation to stably expose the RBD [16]. This strategy is widely used in licensed SARS-CoV-2 vaccines.
The S protein is the exclusive target of SARS-CoV-2-specific nAbs and therefore the most relevant antigen for vaccines. Ninety percent of neutralizing activity in convalescent sera is mediated by RBD-specific nAbs [18,19,20,21,22,23]. However, nAbs targeting other epitopes in the S protein, such as the S1 NTD or regions important for protease cleavage or conformational changes, have been described [24,25,26,27,28]. S2-specific antibodies are mostly nonneutralizing, with exceptions such as those targeting the S2 stem helix or the fusion peptide [26, 29,30,31,32]. Nevertheless, the RBD remains the most prominent target for nAbs, and the resulting immune pressure is reflected by an increasing number of escape mutations in the RBD, which is particularly the case for the omicron variant [33, 34].
In general, vaccines were instrumental in controlling the recent pandemic. In Europe and the US, mRNA-, adenoviral vector-, and protein-based vaccines were licensed and demonstrated efficacies between 74% and 95% against symptomatic disease in clinical trials [35,36,37,38]. However, protection against infection decreases over time due to waning immunity and the emergence of variants escaping antibody responses. In particular, nAbs induced by the initially licensed vaccines are less potent against newly emerging VOCs, which eventually led to adaptation of the mRNA vaccines [39, 40]. There is clear evidence of vaccine-mediated protection, at least against severe COVID-19, when ancestral or adapted vaccine boosters are administered at appropriate intervals [41, 42]. These and other insights generated in the course of the recent pandemic will help to optimize vaccine strategies to provide long-term immunity against SARS-CoV-2 but may also support pandemic preparedness against emerging CoVs or other viruses.
Humoral response to infections
Adaptive immunity consists of antigen-specific antibodies and T cells, which fulfill distinct and complementary roles in viral infections. Although antibody responses can prevent infection by neutralizing the viral inoculum, cytotoxic T cells are critically involved in eliminating virus-infected cells. A key feature of the humoral immune response is generation of highly specific and affine immunoglobulins (Ig) to almost any foreign antigen encountered. B cells, particularly B-cell receptors (BCRs), are the source of this staggering adaptability. V(D)J recombination and clonal selection followed by an iterative affinity maturation process results in a broad and flexible B-cell receptor repertoire, eventually giving rise to plasma cells (PCs) that produce the corresponding antibody response [43].
In the bone marrow, precursor B cells undergo somatic recombination by joining individual V, D, and J gene segments that are present in multiple copies [44]. This process generates diversity at the complementary determining regions of the heavy and light chains, creating germline-encoded BCRs. Following positive and negative selection mechanisms that ensure proper functionality and limited self-reactivity, mature B cells migrate to the spleen to form the naïve B-cell pool [45].
B-cell responses in the context of infection or vaccination can be divided into canonical germinal center (GC) and noncanonical extrafollicular (EF) responses. In GC responses, specific B-cell clones expand within the dark zone of the GC. During this expansion, activation-induced cytidine deaminase (AID) activity leads to mutagenesis of the variable region of the BCR, a mechanism termed somatic hypermutation (SHM). De novo-generated B-cell clones then enter the GC light zone and undergo a Darwinian selection process. The most affine BCRs outcompete other clonotypes in capturing antigens from follicular dendritic cells (FDCs) and receive help from CD4+ T follicular helper cells (TFHs) via the CD40 ligand and IL-21 to survive and expand [43, 46,47,48]. The progressive increase in the affinity of BCRs for a given antigen through iterative mutation and selection in dark and light zones is termed affinity maturation. AID not only promotes mutagenesis of the variable region but also induces class-switch recombination toward IgG subclasses (IgG1, IgG2, IgG3, IgG4), IgE, and IgA [49, 50]. To produce these class-switched, affinity-matured immunoglobulins in serum or mucosal fluids, GC B cells must leave the GC as antibody-secreting, short-lived plasmablasts (PBs) or long-lived PCs [51]. However, GC B cells can also differentiate into MBCs that circulate until antigen re-exposure leads to their re-entry into the GC, followed by new rounds of affinity maturation [52, 53]. Compared to PBs or PCs, MBCs appear to leave GCs earlier with a lower level of SHM, possibly creating greater flexibility in the B-cell memory pool for recognition of related or even unrelated antigens [54,55,56].
In noncanonical EF responses, B cells expand massively upon contact with antigen-presenting FDCs outside of GCs but without involvement of follicular CD4+ T cells. These EF B cells rapidly differentiate into short-lived PBs that cause a prompt but short-lived antibody response. Despite a common misconception that EF B cells do not undergo class-switch recombination or SHM, there is considerable evidence for this, as discussed elsewhere [57]. Nevertheless, the extent of SHM and BCR diversification is not comparable to GC responses, resulting in near germline-encoded BCRs and shared, public antibody responses [58].
The result of these qualitatively different B-cell responses is a division of labor. (i) Extrafollicular B-cell responses generate an early antibody response within a few days without time-consuming SHM and without inducing immunological memory in terms of long-term antibody production or MBCs. (ii) GC B cells differentiate into long-lived PCs that produce mature, high-affinity nAbs to provide a first (and often sufficient) layer of immune memory against reinfection. (iii) MBCs produce a new generation of affinity-matured nAbs in the event of breakthrough infections. The affinity of such GC-mediated antibody responses increases with time after a single antigen exposure but also progressively with each new cognate antigen contact. Many infections and vaccines induce persistent, sometimes lifelong, immunity (though this immunity may not be provided exclusively by B-cell or antibody responses). Typical examples are protective immune responses against measles, mumps, and smallpox; in contrast, immunity to respiratory viruses is much less durable and protective [59]. Immunization with inactivated influenza vaccines induces bone marrow PCs that decline significantly within one year of vaccination [60], whereas infection with respiratory syncytial virus does not induce any protective B-cell memory [61]. Thus, humoral immune responses can effectively protect against infection, but respiratory pathogens may have evolved strategies to undermine these mechanisms.
Antibody responses to primary SARS-CoV-2 infection
Early in the pandemic, the repertoire of commercially available and “in-house” serological methods began to grow, including enzyme-linked immunosorbent assays, chemiluminescent assays, flow cytometry-based assays, point-of-care lateral flow tests, and pseudotype/live virus neutralization assays [15, 62,63,64,65]. These methods have become important tools for diagnosing late/postinfection stages, tracing COVID-19 contacts, assessing epidemiologic aspects and evaluating immunity after infection or vaccination.
Systemic antibody responses
Regardless of disease severity, almost all SARS-CoV-2-infected individuals experience seroconversion within two to four weeks postinfection, with a median of 12 days postsymptom onset (PSO). In some asymptomatic infected individuals, seroconversion might be absent or transient, but cellular immune responses appear to be comparable to those of symptomatic patients [66, 67]. In the case of seroconversion, IgM, IgA, and IgG appear rather contemporaneously [19, 68,69,70,71,72]. This is in contrast to some other viral infections, in which IgM precedes IgG responses by several days [73], but is congruent with findings for SARS-CoV-1 infections [74]. The viral S and N proteins are major targets of this serological response [19, 72, 75]. Serum IgG, IgA, and IgM levels reach a plateau between two and four weeks PSO [68, 71, 72, 76], before IgM and IgA decline to preinfection levels in the majority of patients within three months [68, 77,78,79,80]. Iyer et al. reported median times to seroreversion for RBD-specific IgA and IgM of 71 days and 49 days, respectively [68], but there are also reports indicating a low-level persistence of anti-RBD and anti-S IgA for four [81] and eight months [75]. Early reports also suggested rapid decay among IgG responses, with a short half-life of only 36 to 49 days [19, 82, 83], raising concerns about the durability of infection-induced humoral responses. However, this underestimation of IgG persistence may be due to biphasic decay kinetics. Several studies with a longer observation period described an initial rapid decline within the first four months, followed by a more gradual decline in ensuing months [68, 80, 84]. This late and stable plateau phase has been observed for more than eight [80] and eleven [84] months, and most convalescent patients do not experience seroreversion within 10 months postinfection [79]. nAb titers plateau at three to six weeks postinfection and correlate significantly with RBD-specific IgG levels [68, 70, 71, 76].
Stratification of patient cohorts has revealed a strong positive correlation between disease severity and the height of antibody peak levels as well as response duration [75, 76, 79, 83, 85, 86]. Patients with more severe disease have higher levels of S2-, S1-, and RBD-binding antibodies and concomitantly the strongest serum nAb titers at two to four weeks after infection [85]. Hospitalized COVID-19 patients showed higher RBD-specific IgG levels than nonhospitalized convalescents, even after more than 120 days of PSO [75]. This correlation may be the result of higher viral loads and prolonged viral replication in more severely ill patients, resulting in stronger immune responses, though clear evidence for this hypothesis is lacking [87]. In addition, very high serum IgG and IgA titers occur more frequently in critically ill patients and those with SARS-CoV-2-mediated ARDS [88], possibly reflecting EF B-cell and PB responses (see below). This hypothesis is further supported by Woodruff et al., who described pronounced EF B-cell responses at early time points in critically ill patients, and these EF responses produced relatively high levels of nAbs [58]. Of note, although at low titers, nAbs are readily detectable in many patients, suggesting that extensive SHM is not crucial for development of nAbs against SARS-CoV-2 [70, 86, 89, 90]. Consistent with this, early IgA production is an important contributor to the early neutralizing response [72].
Cross-reactive antibodies induced by previous infections with endemic human coronaviruses (hCoVs) were much debated early in the pandemic as potential mediators of cross-protection or induction of antibody-dependent enhancement (ADE) of infection, though in vivo evidence for ADE in coronavirus infections is very limited [91]. Indeed, SARS-CoV-2 reactive antibodies were found to be present in a small proportion of SARS-CoV-2 naïve and unvaccinated individuals, particularly in children and adolescents, but they did not correlate with disease severity in either direction after SARS-CoV-2 infection [29, 92, 93]. These responses mostly involve binding to the more conserved S2 domain of the S protein, and the majority of these antibodies lack neutralizing activity [92, 94,95,96,97].
Mucosal antibody responses
Serum nAb responses induced by vaccination or previous infection correlate with protection against SARS-CoV-2 infection [98,99,100,101]. Although such systemic immune responses are easily assessed in blood samples, they do not allow for conclusions to be drawn about local immune responses. Nonetheless, mucosal antibodies and tissue-resident memory T cells play a critical role in the defense against prototypical respiratory viruses such as influenza A virus or respiratory syncytial virus, as shown in preclinical [102,103,104] and clinical [61, 105, 106] studies.
At the humoral level, both IgG and secretory dimeric IgA (sIg) may be present in the airway mucosa. Mucosal IgG levels are maintained by neonatal fragment crystallizable (Fc) receptor-mediated transport of systemic IgG across polarized epithelial barriers into luminal secretions [107]. In contrast, secretory IgA is secreted into the luminal space via the polymeric Ig receptor [108] and is thought to depend on tissue-resident MBCs and lung-homing PCs after local antigen exposure via respiratory infection or mucosal vaccination [109]. Consistent with this, SARS-CoV-2 convalescent patients exhibit stronger S- and RBD-specific IgA responses in bronchoalveolar lavage (BAL) and saliva than infection-naïve vaccinees (Fig. 1) [110,111,112]. Similar to the systemic response, salivary antibodies are readily detectable a few days PSO and peak at approximately two weeks [71, 72]. The early mucosal antibody response consists of IgM, IgA, and IgG, all of which correlate with corresponding serum levels [71], but only mucosal fluids contain dimeric sIgA [72]. In addition, IgA appears to constitute the majority of the mucosal nAb response, at least at early time points PSO [72, 113], but its levels decline more rapidly compared to IgG in mucosal samples. In saliva, anti-S IgA responses return to baseline levels within six months in most patients [111, 114]; other studies have found persistent IgA responses in nasal fluid for more than six and nine months [115, 116]. Use of different sampling and detection methods may be one reason why these studies yielded different results [90].

Systemic SARS-CoV-2 vaccination results in mucosal antibody responses dominated by IgG, whereas IgM and secretory dimeric IgA (sIgA) responses are low and very transient (left). After breakthrough infection, vaccinees display elevated IgG levels and a robust induction of sIgA in the respiratory mucosa (center). SARS-CoV-2 convalescent patients display moderate levels of IgG, IgM, and sIgA responses, and vaccination reinvigorates levels of sIgA and IgG in the respiratory tract (right). Antibody kinetics and relative levels are simplified for ease of interpretation. Created with BioRender.com
Wang et al. have shown that monomeric IgA is twofold less potent than IgG in neutralizing SARS-CoV-2 but that dimeric sIgA is 15-fold more potent than IgG [117]. The most likely and straightforward interpretation of these results is increased binding avidity due to the dimeric form. In addition to a higher valency, IgA has other properties that distinguish it from IgG, including the absence of complement activation or ability to engage Fcα receptors [118]. Along these lines, one study showed that nasal IgG is the main correlate of virus phagocytosis and that nasal IgA correlates with virus neutralization [113], supporting the view of some division of labor between different immunoglobulin classes in the airway mucosa. The most intriguing support for the nonredundant functions of IgA is from two studies reporting a positive correlation between mucosal IgA titers and protection against SARS-CoV-2 infection [112, 119]. Havervall et al. examined the risk of breakthrough infection in relation to mucosal IgA and IgG levels in triple-vaccinated health care workers. Individuals with high mucosal IgA levels (>75th percentile) showed a relative risk reduction of 65% compared to 27% for high mucosal IgG levels [112]. For previously infected individuals with high mucosal IgA, the risk reduction was even higher at 79%. Whether IgA acts as a direct COP remains to be investigated. Mucosal IgA may also correlate with the presence of tissue-resident T-cell responses, which may have direct antiviral effects. Such resident T cells are induced at high frequencies in the nasal cavity after SARS-CoV-2 infection and might be directed against several viral antigens [120]. Therefore, it will be important to investigate the contribution of mucosal T-cell and antibody responses to protection by using appropriate sample techniques and study designs, such as nasal immune cell sampling [120].
B-cell responses to primary infection
Serologic assays remain important for tracking infection- and vaccine-induced immune responses, yet analyses of actual B-cell responses are essential for understanding how certain serologic features develop. Use of state-of-the-art methods to analyze B cells has been a hallmark of the response by the scientific community to the recent pandemic. The knowledge gained will undoubtedly help to optimize vaccine strategies to elicit broadly reactive, long-lasting and protective immune responses.
An important observation was the early appearance of potent nAb clones upon SARS-CoV-2 infection, though this does not necessarily coincide with high serum neutralization titers [70, 86, 89, 90, 121, 122]. One would expect that de novo B-cell responses require time to produce potent nAbs through some degree of affinity maturation, and hCoV-specific MBC responses have been considered a source of such rapidly emerging nAbs. However, despite early recruitment of highly mutated B-cell clones reflecting cross-reactive hCoV memory, these responses are mostly nonneutralizing [92, 94,95,96,97] and decline between three and six months after infection, whereas de novo-generated B-cell responses expand and largely contribute to the nAb response [94]. Furthermore, these cross-reactive responses do not appear to confer protection against SARS-CoV-2 [92, 95, 123].
Several studies support the idea that the early nAb response arises de novo from class-switched EF B-cell responses or from GC B cells with limited SHM. B-cell analyses in patients have shown that the infection-induced antibody response is highly polyclonal but that potent neutralizers are close to the germline [23, 28, 124,125,126,127] and show enrichment for specific immunoglobulin heavy-chain variable region (IGHV) genes such as 3–53, 1–2, 3–9, and 3–30 [128]. Near germline nAbs may result from the recent zoonotic origin of SARS-CoV-2 and lack of adaptation to the human B-cell repertoire. Ongoing virus evolution in the form of VOCs gradually allows for evasion of these primary immune responses, with the latest omicron variants being able to evade most of the initially described nAb clones [129]. Regardless, it is currently unknown whether omicron variants induce near germline responses with neutralizing activities in SARS-CoV-2 naïve individuals. Such analysis is certainly worthwhile, though finding respective cohorts to study may be difficult.
Another important finding was that severe SARS-CoV-2 infection coincides with exceptionally strong humoral responses, including high nAb titers [58, 88, 130]. At the B-cell level, these serologic findings are paralleled by a significantly decreased frequency of circulating B cells as part of general lymphopenia but a substantial expansion of PBs, accounting for up to 30% of B cells upon acute infection [131,132,133,134]. These alterations in the immune system normalize at three to six months after infection [133]. One explanation for the marked shift from B cells to PBs upon acute SARS-CoV-2 infection may be loss of GCs in affected patients. Three studies reported hypoplasia or a lack of GCs in lung-draining lymph nodes, including decreased numbers of FDCs, TFHs, and B cells [135,136,137]. Kaneko et al. investigated postmortem thoracic lymph nodes and found a dramatic reduction in GC B cells and TFH cells, whereas IgG-producing, class-switched PBs were increased [135]. The authors attributed the lack of TFH cells to increased levels of TNF-α. In the absence of a TFH response, GCs are not induced, and the B-cell response shifts to T-cell-independent EF responses, resulting in PB expansion and exceptionally high antibody responses (Fig. 2).

Mild COVID-19 leads to extrafollicular (EF) and GC responses (left), whereas severe cases result in defective GC reactions and a shift toward pronounced EF responses (center). In contrast, mRNA vaccines elicit only minimal EF responses but persistent and pronounced GC reactions. Continuous B-cell maturation may cause consecutive class-switch recombination (CSR), which may explain the increased IgG4 levels after repeated mRNA vaccinations. Created with BioRender.com
Despite these EF-like responses early after infection and the absence of GC reactions in critically ill patients, convalescent patients are generally able to mount an MBC evolution over time. Several groups have followed B-cell responses longitudinally and observed an increase in MBCs from early infection to three to eight months PSO, as well as a relatively stable MBC population between six and 12 months after infection [75, 81, 97, 138, 139]. This was true for IgG+ but not IgA+ and IgM+ B cells, which showed a decline from approximately 150 days and 70 days, respectively [75]. S- and especially RBD-specific MBCs exhibited increasing SHM over time, and the corresponding nAbs had higher affinity, potency, resistance to RBD mutations, and neutralization breadth, which explains why neutralization titers remain stable even as RBD IgG levels decline [19, 81, 84, 139,140,141]. In addition, PCs that produce SARS-CoV-2-specific antibodies remain detectable in the bone marrow for seven to eight months [84]. Therefore, it is tempting to speculate that the biphasic decline in serum IgG reflects an initial, rapid decline in short-lived PBs, followed by a second, slower decline in long-lived PCs. Taken together, these studies suggest an ongoing GC reaction over several months in SARS-CoV-2 convalescent patients, at least after noncritical illness (Fig. 2).
Antibody responses to SARS-CoV-2 vaccination
Several SARS-CoV-2 vaccines became available in Europe and in the United States between late 2020 and early 2021, including mRNA vaccines and adenoviral vector vaccines. Clinical trials have demonstrated vaccine efficacies against symptomatic disease ranging from 74% to 95%, but the two mRNA vaccines, mRNA-1273 from Moderna (Spikevax) and Bnt162b2 from Pfizer/Biontech (Comirnaty), outperformed the viral vector vaccines in terms of immunogenicity and vaccine efficacy [35,36,37,38, 142, 143]. There have also been reports of rare cases of thrombocytopenia induced by adenoviral vector vaccines, mainly AstraZeneca’s ChAdOx-1 vaccine [144,145,146], leading to restricted use in certain age groups in some countries. As a result, mRNA vaccines dominated vaccination campaigns in the later stages of the pandemic and booster campaigns in Europe and the United States. For these reasons, most of the studies discussed below focus on mRNA vaccine-induced immunity. However, it is important to note that inactivated SARS-CoV-2 vaccines such as CoronaVac (Sinovac Biotech) have been widely used in Asia, South America and other parts of the world. Although the immunogenicity and vaccine effectiveness of CoronaVac against symptomatic infection with ancestral SARS-CoV-2 strains was lower than that of mRNA vaccines, its effectiveness against severe and fatal omicron infection was noninferior [147, 148]. Broader T-cell responses [149] and trained immunity [150] have been proposed as possible mechanisms but are not discussed in this review.
Basic immunization scheme
Most licensed vaccines were initially licensed for a two-dose schedule. In fact, it quickly became apparent that a single dose of mRNA vaccine did not induce durable antibody responses. nAb responses in vaccinees were reported to peak at approximately 14 days postimmunization, but only 50% actually had detectable nAb titers [142, 151,152,153]. The same studies demonstrated that a second vaccination resulted in seroconversion and nAb detection in 100% of vaccinees. Accordingly, vaccine effectiveness (VE) against illness was shown to be higher after two doses compared to a single dose, though a single dose still provided almost complete protection against hospitalization for up to 16 weeks [154]. S-specific antibody levels and nAb titers peaked at 14 days after the second vaccination but waned substantially within six months, though nAb titers declined less steeply, probably due to higher antibody maturation at later time points [76, 155,156,157,158]. Nevertheless, approximately 16% of twice-vaccinated individuals became seronegative within six months [157].
Due to the varying availability of vaccine doses at the beginning and updated vaccine recommendations, heterologous prime-boost regimens were employed for some vaccinees. Interestingly, heterologous prime-boost vaccination with an adenoviral vector prime followed by mRNA vaccination was superior to homologous vaccine schemes in inducing humoral and cellular immune responses [159,160,161,162,163,164,165,166,167]. The superior immunogenicity was evident not only in S-specific IgG levels but also in nAb and MBC levels. Similar results were observed for mRNA-1273 and Bnt162b2 as well as for ChAdOx1 and Ad26.COV2-S as primary and secondary vaccine modalities, respectively. The generally lower immunogenicity of homologous ChAdOx1-ChAdOx1 schemes might be explained by interference of anti-vector immunity and the lower immunogenicity of the delivered wild-type S as an antigen. Boosting with 2P-S-encoding mRNA vaccines has been shown to focus the humoral response on prefusion epitopes, resulting in higher and broader nAb responses [163]. Similarly, Ad26.COV2-S encodes a prefusion-stabilized S, but with an additional stabilization at the furin cleavage site [168], resulting in more S1-focused immune responses. Therefore, the Ad26.COV2-S-mRNA scheme consists of two different vaccine antigens, likely resulting in differences in the quantity and quality of humoral responses. In addition, some studies have reported different intervals between homologous and heterologous boosting, which may influence immunogenicity [158, 169].
Convalescent individuals demonstrate a much more efficient response to a primary vaccine dose than those who had not previously been infected. Individuals with previous SARS-CoV-2 exposure exhibited immune responses to a single mRNA vaccine dose comparable to those of uninfected individuals receiving two doses [139, 151, 170]. Notably, convalescent patients showed approximately 1000-fold higher nAb titers, broader neutralization, and resistance to escape mutations than twice-vaccinated noninfected individuals [152]. Nonetheless, infection-induced immunity exhibits significant variation, and a subset of those who have recovered fail to develop humoral immunity. Such individuals respond to an initial mRNA vaccine dose with a kinetic comparable to that of vaccination-naïve individuals [171,172,173]. A second vaccine dose given within the recommended three- to four-week interval did not further increase humoral or cellular responses in previously infected individuals [158, 174,175,176,177,178]. However, delaying the second vaccination further into later memory or waning immunity led to an efficient boost of antibody responses [158, 169].
Escape from vaccine-induced humoral immunity
Although the SARS-CoV-2 replication machinery has 3’-5’ exoribonuclease proofreading ability, the virus still exhibits a significant mutation rate of approximately 10-6 mutations per nucleotide per replication cycle, which is similar to that of other betacoronaviruses but ten to 100-fold lower than that of other RNA viruses, such as hepatitis C virus or human immunodeficiency virus [179, 180]. This mutation rate allows SARS-CoV-2 to adapt to different selection pressures. On the one hand, the virus adapts to the human host, leading to increased viral fitness and infectivity, as seen with the D614G mutation or the H69/V70 deletion, neither of which provide significant immune escape [181,182,183]. On the other hand, immune escape mutations became much more prevalent and relevant after large portions of the community had acquired infection- or vaccine-induced immunity against SARS-CoV-2. E484K and N501Y were the first prominent RBD escape mutations that reduced vaccine-induced neutralization of the alpha variant [184, 185]. Immune escape was further observed by the subsequent beta, gamma, and delta variants before the omicron variant with an extensive mutational load in the S gene appeared, leading to exceptional immune escape [180, 186, 187]. The omicron variant and its sublineages BA, BQ, and XBB carry more than 30 amino acid substitutions, insertions, and deletions compared to the ancestral S variant [188, 189]. In light of these developments, vaccine recommendations and booster modalities were repeatedly updated during the pandemic to maintain or increase protective immunity against VOCs.
For pre-omicron variants, a moderate escape from vaccine-induced immunity has been reported [185, 190,191,192,193], but two mRNA vaccinations or prior infection plus one vaccination induces robust nAb responses against viral variants [151, 194] and efficient protection from infection [195]. Vaccination with mRNA, viral vector, or inactivated SARS-CoV-2 vaccines induces a broader IgG response compared to infection [196], covering a higher epitope diversity in the RBD [197]. The situation changed with the emergence of omicron variants. These strains efficiently escape from infection- and vaccination-induced immunity as well as from most monoclonal antibodies in clinical use [129, 198,199,200,201,202,203,204]. As a result, VE against symptomatic omicron infections and hospitalization also declined after the basic immunization cycle, though the latter remained at a relatively high level [201, 205].
Booster immunization
A third vaccination with mRNA vaccines encoding the ancestral S variant, the so-called booster vaccination, has been recommended in most European countries and the United States in response to the emergence of omicron variants and waning immune responses in the population. Booster immunization leads to efficient reinvigoration of nAb levels within one week, even exceeding peak levels after the second vaccination [156, 196]. In addition, an mRNA booster dose increases omicron-specific nAb titers [158, 189, 202, 203, 206, 207] and protection against omicron infection to robust levels [201, 205, 208]. Timing of this booster is relevant to vaccination effectiveness in two regards. First, omicron-specific nAb levels [158] and protection [201] decline within six to seven months after the second vaccination, though they are already low at the peak. A third vaccination restores absolute S1-binding antibody quantities similar to the peak response after the second vaccination. Second, booster vaccination promotes further maturation of antibody quality, resulting in higher IgG avidity and improved nAb titers [158]. Interestingly, omicron nAb levels appear to benefit substantially from timely spaced antigen exposures, suggesting diversification of the B-cell response rather than pure selection based on BCR affinity [158]. Boosting such mature B cells in late memory phases may then lead to specific selection of broadly neutralizing clonotypes. However, the resulting cross-reactive antibody responses are transient, waning within six months after both first and even second booster vaccinations [209, 210].
In light of the substantial antigenic distance between the omicron sublineages and the Wuhan-Hu-1-based vaccines, adapted booster vaccines have recently been employed to provide a better vaccine match and to reinvigorate immunity against contemporary variants. Both Moderna and Biontech/Pfizer have released adapted bivalent mRNA vaccines to deliver original+BA.1 or original+BA.4/5 S proteins, which are currently recommended for booster vaccination in European countries and the United States. Davis-Gardner et al. report that a bivalent original+BA.4/5 booster vaccination induces superior nAb levels against the omicron lineages BA, BQ, and XBB compared to one or even two booster vaccinations with the original, monovalent vaccine [39]. Similarly, BA.1–adapted BNT162b2 induced a more efficient boost of BA.1-specific humoral responses than a booster with the original vaccine [40]. It is important to note that the original vaccines also boosted omicron-specific nAb responses, but to a two- to threefold lesser extent compared to bivalent formulations. Two other studies reported a preferential boost in ancestral S-specific humoral immunity and a comparable increase in BA.5-specific immunity by original monovalent and bivalent adapted booster vaccines [211, 212], supporting the idea of antigenic imprinting. Taken together, these immunogenicity studies do not provide conclusive evidence for the absolute superiority of adapted mRNA vaccines, as both vaccines induce omicron nAbs at similar or only moderately different levels. The most intriguing argument in favor of adapted vaccines is from real-world estimates of booster effectiveness. A retrospective matched cohort study showed that the bivalent BA.4/5 mRNA vaccine provided better protection against severe BA.5 infection than a booster with the original mRNA vaccine [42]. Specifically, the VE of the bivalent original+BA.4/5 was estimated at 51.0%, and the VE of the original+BA.1 and monovalent ancestral vaccines was estimated at 49% and 27%, respectively. Another study concluded that VE against hospitalization was 25.2% for the monovalent booster and 58.7% for the bivalent booster, with 24.9% and 61.8% against death after a monovalent or bivalent booster, respectively [41]. Whether this protection is due to increased induction of omicron-specific nAbs remains to be investigated, as there is also evidence that T-cell responses contribute to viral clearance in breakthrough infections [213, 214].
Mucosal immune responses
SARS-CoV-2 vaccines are primarily evaluated by induction of systemic nAb responses, but mucosal immune responses may also contribute to protection against infection. S-specific IgG is induced in saliva samples after vaccination but at 100-fold lower levels compared to serum levels [196, 215]. In contrast, salivary IgA is not detectable or only at low levels after two or three vaccinations (Fig. 1) [215, 216]. Importantly, the beneficial effect of a third vaccination to induce omicron neutralization in serum is not reflected in saliva samples. Indeed, only 4% of vaccinees show a detectable nAb response after the third mRNA vaccination [216]. As mentioned above, induction of mucosal sIgA is particularly dependent on local antigenic stimuli, and therefore, breakthrough infection after two or three doses of mRNA vaccine results in substantially higher salivary IgA levels than in vacinees without breakthrough infection [112, 119]. In addition, Park et al. reported that omicron breakthrough infection, but not vaccination alone, induced nAb responses in nasal secretions capable of neutralizing omicron and the ancestral SARS-CoV-2 strain [193]. These hybrid nAb responses peak at approximately two to three weeks postinfection and remain detectable for more than 180 days. Importantly, infection-primed mucosal antibody responses can be reinvigorated by parental SARS-CoV-2 vaccination (Fig. 1) [217]. These findings suggest that systemic mRNA vaccination may not be very efficient in establishing mucosal B-cell responses de novo but may be an option to boost anamnestic responses. One explanation may be that tissue-resident B-cell responses induced by previous SARS-CoV-2 infection [218] are reactivated by circulating antigen produced after systemic mRNA vaccination [219] but that the circulating antigen levels are insufficient to initiate mucosal MBCs.
B-cell responses to SARS-CoV-2 vaccination
B-cell response dynamics
S protein-specific PB responses peak at approximately two weeks after primary mRNA vaccination and at approximately one week after a second mRNA dose before declining significantly to background levels within three to four weeks in most individuals [220, 221]. Fine-needle aspiration of lymph nodes has allowed for analysis of vaccine-induced GC B-cell dynamics. Turner et al. reported that levels of S-specific GC B cells from draining axillary lymph nodes were stably maintained for at least 12 weeks after a second vaccination [220]. Such long-lasting GC reactions were not observed after influenza vaccination in humans [222] or after alum-adjuvanted protein immunization in mice [223]. Congruent findings were also reported for TFH responses, which persisted at robust levels for at least six months after mRNA vaccination and showed a significant correlation with GC B-cell responses [224]. Consistent with these observations, mRNA vaccination, but not MF59-adjuvanted protein vaccination, induces potent GC reactions in mice that correlate with an increased TFH response, nAb production, and differentiation of long-lived PCs and MBCs [225]. A consequence of ongoing GC responses is a progressive increase in SHM in MBCs and emergence of nAbs with increased affinity and neutralization of derived nAbs [226]. Thus, mRNA vaccines promote strong and durable GC reactions consisting of persistent GC B and TFH populations in draining lymph nodes, eventually resulting in nAbs with progressively increasing affinity (Fig. 2). The exact mechanisms of the GC-stimulating effects of mRNA vaccines remain to be investigated but might be related to intrinsic adjuvant effects of the lipid nanoparticles or their systemic trafficking [227, 228]. Furthermore, a logical prerequisite for such ongoing GC reactions is persistence of the respective antigen, allowing for selection of affinity-increased B cells. Röltgen et al. confirmed this hypothesis by detecting vaccine mRNA and S protein in the lymph nodes of vaccinated but not previously infected individuals for up to 60 days [196]. Vaccine antigen was detected around GC foci, indicating ongoing antigen presentation by follicular DCs.
B-cell responses in hybrid immunity
By the time the vaccination campaigns began, some individuals had already been infected with SARS-CoV-2, and others experienced breakthrough infections after vaccination. These and even more complicated histories of antigen exposure create “hybrid immunity”. Prior SARS-CoV-2 infection enhances the immunogenicity of primary SARS-CoV-2 vaccination, resulting in a humoral response amplitude comparable to that of two doses of mRNA vaccine in infection-naïve individuals [139, 151, 170]. The level of infection-derived B-cell memory is predictive of the postvaccination response in immunized convalescent patients [229]. Such hybrid immunity also provides broader cross-variant neutralization by targeting more conserved parts or by broadly inducing nAbs through extensive SHM [139, 230, 231]. As described above, omicron evades vaccination- or previous variant infection-induced immunity [232,233,234]. As a consequence of this antigen mismatch, primary infection with an omicron variant mostly induces omicron sublineage-specific immunity in immunologically naïve individuals, which in turn neutralizes earlier variants poorly. Interestingly, preimmune individuals in whom non-omicron immunity is mounted and who experience omicron breakthrough, particularly BA.4 or BA.5 infection, show broad neutralization across all tested variants. Such breakthrough infection primarily activates the MBCs that recognize conserved epitopes between ancestral and omicron lineages rather than promoting de novo B-cell responses [193, 235]. Although such immune imprinting appears to be beneficial for induction of broadly reactive antibodies against known variants, it hinders induction of neutralizing responses against novel epitopes. This may be detrimental if conserved sites escape from immunity in newly emerging variants.
Several more mechanisms are discussed below as explanations for the beneficial features of hybrid immunity. First, vaccine- and infection-primed B-cell responses use different variable region genes, i.e., select different B-cell clones, which might broadly favor nAbs in convalescent patients [236, 237]. Second, SARS-CoV-2 infection induces more MBCs and more pronounced TH1 responses than vaccination [238]. An altered CD4+ T-cell response, especially TFHs, may also contribute to altered GC reactions and B-cell responses, as has been shown in critically ill patients [135]. Third, antibodies generated by the primary response directly modulate secondary responses against SARS-CoV-2 by blocking or enhancing recruitment of naïve B cells [239]. Infection- and vaccination-induced antibody responses may therefore differentially modulate secondary B-cell responses. Fourth, regardless of the actual immunization modality, the timing of repeated antigen contacts directly influences the characteristics of the immune responses provoked. Several studies suggest that timely spaced antigen contacts promote more extensive GC reactions compared to shorter vaccination intervals, resulting in high affinity and cross-reactive nAbs [158, 169, 240]. This effect was also observed with extended intervals between mRNA doses, independent of an infection event [241]. Therefore, the strict three-week interval between the first two mRNA doses may induce a less efficient immune response compared to a single vaccination followed by SARS-CoV-2 infection with longer intervals between antigen exposures. Ultimately, hybrid immunity is more durable and provides the most efficient protection against infection. Goldberg and colleagues showed that at six to eight months after the last antigen exposure, the risk of infection is approximately four to five times lower with hybrid immunity compared with immunity after two or three doses of mRNA vaccine [242]. Further studies are needed to define protective correlates in hybrid immunity, as it consists of more immune parameters than nAbs. Nonneutralizing Abs against viral proteins other than S, T-cell responses, and mucosal immunity may all contribute to the beneficial effects observed with hybrid immunity.
Unusual class switching toward IgG4 after mRNA vaccination
nAb responses are an important COP against SARS-CoV-2 [98]. However, there is accumulating evidence from other viral infections that the neutralizing capacity is not the only function that is important for the antiviral activity of antibodies. For example, Fc functions critically contribute to antibody-mediated protection from Ebola infection [243], and broadly neutralizing hemagglutinin stalk-specific antibodies require Fcγ receptor interactions for protection against influenza [244].
Vaccination with one or two doses of mRNA vaccine induces a humoral response dominated by IgG1 and IgG3 subclass antibodies [245]. These two IgG subclasses mediate prototypical Fc effector functions such as antibody-dependent cytotoxicity, antibody-dependent phagocytosis, or antibody-dependent complement activation, even against the omicron variant that escapes their neutralizing activity. However, in a longitudinal study, we observed that the proportion of S-specific IgG4 increased significantly at approximately six months after the second and even more after the third mRNA vaccine dose [246]. S-specific IgG4 was undetectable in most vaccinees after the first and early after the second vaccination but reached almost 20% of the IgG response late after the third vaccination. Up to 37% of the S-specific MBC response consisted of IgG4+ MBCs. IgG4 is a noninflammatory IgG subclass with poor Fc effector functions [247]. Concomitantly, sera containing high levels of IgG4 provided few antibody-mediated effector functions, such as antibody-dependent phagocytosis or complement deposition, despite increased antibody affinity and neutralizing capacity in our study. Importantly, total IgG4 levels in the sera of vaccinees did not increase after mRNA vaccination (our own unpublished observation).
Buhre et al. confirmed a shift toward IgG4 late after the second mRNA boost with the mRNA-1273 and BNT162b2 vaccines but not after two vaccinations with viral vector vaccines [248]. Interestingly, they also reported a stronger IgG4 induction by mRNA-1273 compared to BNT162b2, in line with the slightly lower immunogenicity of the latter [245, 249]. Therefore, both studies link the profound induction of S-specific IgG4 with repeated vaccination with mRNA lipid nanoparticle vaccines. Similar to our own findings, elevated anti-S IgG4 antibodies were not found when vector-based vaccines were used for primary vaccination [250], and elevated IgG4 anti-S levels were also not common in subjects infected with SARS-CoV-2 prior to vaccination [250]. It appears that processes following mRNA vaccination in the priming phase of the immune response drive the class switch to distal isotypes.
High IgG4 levels have mostly been observed in the context of repeated antigen contacts, such as successful allergen-specific immunotherapy, and in beekeepers after several beekeeping seasons [251,252,253]. These studies describe a slow rise in IgG4 levels after multiple antigen exposures; other IgG subclasses were readily measured at early time points. It is tempting to speculate that IgG4 class-switched B cells emerge after consecutive class-switch recombination events. Of the four γ heavy chain genes (γ3-γ1-γ2-γ4), γ4 is the most downstream. Therefore, IgG4 class-switched MBCs may require multiple antigen contacts and an extensive GC reaction to develop [254]. This idea fits well with mRNA vaccines, which induce persistent GC and TFH responses (Fig. 3) [220, 224]. Another possibility is that vaccine-induced, freely circulating S antigen stimulates MBCs in the absence of inflammatory costimulants [196]. Such a situation would be similar to allergen immunotherapy, whereby repeated low-dose antigen administration in the absence of inflammation results in antigen-blocking IgG4. However, the exact mechanisms of mRNA vaccine-induced IgG4 need to be investigated with high priority. Due to the different organization of the IgH gene locus and the different regulation of class switch recombination, studies in mice may not be adequate. Interestingly, nonhuman primates show similar or even more pronounced IgG4 responses after an immunization regime with the mRNA-1273 vaccine predominantly used in humans during the pandemic, i.e., immunizations at weeks 0 and 4 and boosters months later [255]. Therefore, injection of the second highly potent antigen dose shortly after the first, presumably during the ongoing GC response, may be responsible for the unusual class switch toward downstream γ heavy-chain genes.

S-specific humoral responses induced by the basic immunization cycle with mRNA vaccines are dominated by IgG1 and IgG3, which possess proinflammatory Fc functionalities. Late after the second vaccination, and especially after the booster dose, IgG1 is still dominant, but the fraction of noninflammatory IgG3 and IgG4 is substantially increased. Antibody kinetics and relative levels are simplified for ease of interpretation. Created with BioRender.com
Importantly, despite reduced engagement of Fc-mediated effector functions, the increase in IgG4, as expected, does not negatively impact in vitro neutralizing capacity. This is consistent with reports indicating that mRNA boosters increase protection against infection [41, 42]. However, due to the high selective pressure on neutralizing epitopes in the RBD, VOCs often show immune escape at these sites. As up to 95% of S-specific antibodies are nonneutralizing [256,257,258], there may be a shift in the predominant antiviral function from neutralization to Fc-mediated functions against future immune escape variants. In such a scenario, a substantial fraction of IgG4 would provide less antiviral functions. Along these lines, omicron variants have been shown to escape the RBD-specific nAb response [259, 260], whereas nonneutralizing responses recognizing other parts of S retain antigen- and Fcγ receptor-binding activity [256]. At least in preclinical studies, Fcγ receptor-dependent antibody effector functions are essential for mRNA vaccine-mediated protection [261]. In the context of human SARS-CoV-2 infection, IgG4 has not been well explored, but two studies do describe circumstantial observations. In one, elevated total IgG4 in sera predicted higher lethality in COVID-19 patients [262]. This may be related to the premorbidity of these patients, as high IgG4 levels are a feature of IgG4-related disease, an inflammatory disease of unknown origin [263]. Additionally, elevated S-specific IgG4 antibodies were observed early in infected patients who were more likely to die from COVID-19 [264]. IgG4 was still a minor contributor to all anti-S antibodies in that study, suggesting confounding effects in this cohort. It is important not to draw false or premature conclusions from these studies. mRNA vaccination, especially booster vaccination leading to high IgG4 levels, clearly demonstrates robust VE. Nevertheless, the mechanism behind and the consequences of repeated mRNA vaccination-induced IgG4 remain unclear and require further investigation.
Concluding remarks and perspectives
The recent SARS-CoV-2 pandemic has had a dramatic impact on several aspects of life, including social interaction, the economy, and health care. At the same time, the research community initiated an unprecedented research effort that ultimately led to key insights in immunology, virology, and vaccinology. B-cell and antibody responses were at the center of this research because of their strong association with protective immunity. Similar to the important insights generated by HIV research over more than three decades, SARS-CoV-2-related research will shape human immunology and vaccine design for decades.
Novel vaccine platforms, including mRNA, viral vector, and recombinant protein vaccines, were developed at “light speed” and employed in exceptionally large cohorts. Moreover, use of state-of-the-art technologies allowed for in-depth analyses of vaccine-induced immunity. In particular, mRNA vaccine technology demonstrated impressive immunogenicity but also revealed some surprising features, such as persistent GC reactions and unusual induction of IgG4 antibodies. These phenomena should be further explored in future research, especially as mRNA vaccines are being developed for other infectious diseases.
Although vaccines are critical and effective in controlling the pandemic phase, emergence of escape variants, including the omicron complex, limits VE and demands vaccine adaptations. Now that we are likely to approach an endemic situation with newly emerging virus variants each season, similar to influenza A drift variants, it is desirable to maintain stable protective immunity over several seasons. This applies on the one hand to the amplitude and persistence of vaccine-induced immunity and on the other hand to the specificity of the immune response, which should target conserved sites of the S protein. Optimally, vaccine strategies should establish such durable and broadly reactive immune responses to protect persons at risk over several seasons. The minimal goal is to regularly vaccinate these individuals with adapted vaccines, as is currently the case for adapted mRNA boosters. Regardless, the difficulty in predicting the dominant virus variants in the upcoming season and the inherent risk of vaccine mismatches is evident in seasonal influenza vaccinations.
An additional feature of mRNA and viral vector vaccines is efficient induction of T-cell immunity and not only humoral responses. nAbs correlate with protection from infection, but future studies should also investigate the contribution of other adaptive immune responses to protection, such as nonneutralizing antibodies and T cells. Both immune parameters recognize epitopes that appear to be more conserved among SARS-CoV-2 variants and might become important COPs in the case of strong immune escape of nAb targets.
Mucosal immune responses have also gained attention as potential COPs. sIgA and tissue-resident memory T cells are not efficiently established by current SARS-CoV-2 vaccines but instead require mucosal antigen delivery. Therefore, harnessing mucosal immunity through vaccination will require intensive research on mucosal vaccine platforms. It will be crucial to develop strategies that induce strong but also durable immune responses in the respiratory tract. In addition, there is a lack of non- or minimally invasive routine methods to sample these immune responses in different parts of the respiratory tract, especially for T-cell analyses. These methods and appropriate study designs will be essential to define mucosal COPs and evaluate mucosal vaccines in clinical trials.
References
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506.
Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N Engl J Med. 2020;382:727–33.
Worobey M, Levy JI, Malpica Serrano L, Crits-Christoph A, Pekar JE, Goldstein SA, et al. The Huanan Seafood Wholesale Market in Wuhan was the early epicenter of the COVID-19 pandemic. Science (80-). 2022;377:951–9.
Gostin LO, Gronvall GK. The Origins of Covid-19 — Why It Matters (and Why It Doesn’t). N Engl J Med. 2023;388:2305–8.
WHO WHO. COVID-19 Weekly Epidemiological Update, Edition 150. 2023. https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---6-july-2023 (accessed 13 Jul 2023).
Hu B, Guo H, Zhou P, Shi Z-L. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol. 2021;19:141–54.
Ioannidis JPA. Infection fatality rate of COVID-19 inferred from seroprevalence data. Bull World Health Organ. 2021;99:19–33F.
Bunders MJ, Altfeld M. Implications of sex differences in immunity for SARS-CoV-2 pathogenesis and design of therapeutic interventions. Immunity. 2020;53:487–95.
Yehia BR, Winegar A, Fogel R, Fakih M, Ottenbacher A, Jesser C, et al. Association of race with mortality among patients hospitalized with Coronavirus disease 2019 (COVID-19) at 92 US Hospitals. JAMA Netw Open. 2020;3:e2018039.
Maroko AR, Nash D, Pavilonis BT. COVID-19 and inequity: a comparative spatial analysis of New York City and Chicago Hot Spots. J Urban Heal. 2020;97:461–70.
Rydyznski Moderbacher C, Ramirez SI, Dan JM, Grifoni A, Hastie KM, Weiskopf D, et al. Antigen-specific adaptive immunity to SARS-CoV-2 in acute COVID-19 and associations with age and disease severity. Cell. 2020;183:996–1012.e19.
Brannock MD, Chew RF, Preiss AJ, Hadley EC, Redfield S, McMurry JA, et al. Long COVID risk and pre-COVID vaccination in an EHR-based cohort study from the RECOVER program. Nat Commun. 2023;14:2914.
Davis HE, McCorkell L, Vogel JM, Topol EJ. Long COVID: major findings, mechanisms and recommendations. Nat Rev Microbiol. 2023;21:133–46.
Notarte KI, Catahay JA, Velasco JV, Pastrana A, Ver AT, Pangilinan FC, et al. Impact of COVID-19 vaccination on the risk of developing long-COVID and on existing long-COVID symptoms: a systematic review. eClinicalMedicine. 2022;53:101624.
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–80.e8.
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, Abiona O, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science (80-). 2020;367:1260–3.
Cai Y, Zhang J, Xiao T, Peng H, Sterling SM, Walsh RM, et al. Distinct conformational states of SARS-CoV-2 spike protein. Science (80-). 2020;369:1586–92.
Greaney AJ, Loes AN, Crawford KHD, Starr TN, Malone KD, Chu HY, et al. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe. 2021;29:463–76.e6.
Piccoli L, Park Y-J, Tortorici MA, Czudnochowski N, Walls AC, Beltramello M, et al. Mapping neutralizing and immunodominant sites on the SARS-CoV-2 spike receptor-binding domain by structure-guided high-resolution serology. Cell. 2020;183:1024–42.e21.
Rogers TF, Zhao F, Huang D, Beutler N, Burns A, He W, et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science (80-). 2020;369:956–63.
Zost SJ, Gilchuk P, Chen RE, Case JB, Reidy JX, Trivette A, et al. Rapid isolation and profiling of a diverse panel of human monoclonal antibodies targeting the SARS-CoV-2 spike protein. Nat Med. 2020;26:1422–7.
Liu H, Wilson IA. Protective neutralizing epitopes in SARS‐CoV‐2. Immunol Rev. 2022;310:76–92.
Dejnirattisai W, Zhou D, Ginn HM, Duyvesteyn HME, Supasa P, Case JB, et al. The antigenic anatomy of SARS-CoV-2 receptor binding domain. Cell. 2021;184:2183–2200.e22.
Chi X, Yan R, Zhang J, Zhang G, Zhang Y, Hao M, et al. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science (80-). 2020;369:650–5.
McCallum M, De Marco A, Lempp FA, Tortorici MA, Pinto D, Walls AC, et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell. 2021;184:2332–47.e16.
Poh CM, Carissimo G, Wang B, Amrun SN, Lee CY-P, Chee RS-L, et al. Two linear epitopes on the SARS-CoV-2 spike protein that elicit neutralising antibodies in COVID-19 patients. Nat Commun. 2020;11:2806.
Peter AS, Roth E, Schulz SR, Fraedrich K, Steinmetz T, Damm D, et al. A pair of noncompeting neutralizing human monoclonal antibodies protecting from disease in a SARS‐CoV‐2 infection model. Eur J Immunol. 2022;52:770–83.
Brouwer PJM, Caniels TG, van der Straten K, Snitselaar JL, Aldon Y, Bangaru S, et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science (80-). 2020;369:643–50.
Song G, He W, Callaghan S, Anzanello F, Huang D, Ricketts J, et al. Cross-reactive serum and memory B-cell responses to spike protein in SARS-CoV-2 and endemic coronavirus infection. Nat Commun. 2021;12:2938.
Pinto D, Sauer MM, Czudnochowski N, Low JS, Tortorici MA, Housley MP, et al. Broad betacoronavirus neutralization by a stem helix–specific human antibody. Science (80-). 2021;373:1109–16.
Zhou P, Yuan M, Song G, Beutler N, Shaabani N, Huang D et al. A human antibody reveals a conserved site on beta-coronavirus spike proteins and confers protection against SARS-CoV-2 infection. Sci Transl Med. 2022; 14. https://doi.org/10.1126/scitranslmed.abi9215.
Dacon C, Tucker C, Peng L, Lee C-CD, Lin T-H, Yuan M, et al. Broadly neutralizing antibodies target the coronavirus fusion peptide. Science (80-). 2022;377:728–35.
McCallum M, Czudnochowski N, Rosen LE, Zepeda SK, Bowen JE, Walls AC, et al. Structural basis of SARS-CoV-2 Omicron immune evasion and receptor engagement. Science (80-). 2022;375:864–8.
Viana R, Moyo S, Amoako DG, Tegally H, Scheepers C, Althaus CL, et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature. 2022;603:679–86.
Baden LR, El Sahly HM, Essink B, Kotloff K, Frey S, Novak R, et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N Engl J Med. 2021;384:403–16.
Sadoff J, Gray G, Vandebosch A, Cárdenas V, Shukarev G, Grinsztejn B, et al. Safety and efficacy of single-dose Ad26.COV2.S vaccine against Covid-19. N Engl J Med. 2021;384:2187–201.
Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N Engl J Med. 2020;383:2603–15.
Voysey M, Clemens SAC, Madhi SA, Weckx LY, Folegatti PM, Aley PK, et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet. 2021;397:99–111.
Davis-Gardner ME, Lai L, Wali B, Samaha H, Solis D, Lee M, et al. Neutralization against BA.2.75.2, BQ.1.1, and XBB from mRNA Bivalent Booster. N Engl J Med. 2023;388:183–5.
Winokur P, Gayed J, Fitz-Patrick D, Thomas SJ, Diya O, Lockhart S, et al. Bivalent Omicron BA.1–Adapted BNT162b2 booster in adults older than 55 years. N Engl J Med. 2023;388:214–27.
Lin D-Y, Xu Y, Gu Y, Zeng D, Wheeler B, Young H, et al. Effectiveness of bivalent boosters against severe Omicron Infection. N Engl J Med. 2023;388:764–6.
Mateo-Urdiales A, Sacco C, Fotakis EA, Del Manso M, Bella A, Riccardo F et al. Relative effectiveness of monovalent and bivalent mRNA boosters in preventing severe COVID-19 due to omicron BA.5 infection up to 4 months post-administration in people aged 60 years or older in Italy: a retrospective matched cohort study. Lancet Infect Dis. 2023. https://doi.org/10.1016/S1473-3099(23)00374-2.
Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. 2022;40:413–42.
Tonegawa S. Somatic generation of antibody diversity. Nature. 1983;302:575–81.
Nemazee D. Mechanisms of central tolerance for B cells. Nat Rev Immunol. 2017;17:281–94.
Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, et al. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell. 2010;143:592–605.
Gitlin AD, Mayer CT, Oliveira TY, Shulman Z, Jones MJK, Koren A, et al. T cell help controls the speed of the cell cycle in germinal center B cells. Science (80-). 2015;349:643–6.
Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem. 2007;76:1–22.
Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–63.
Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell. 2000;102:565–75.
Kräutler NJ, Suan D, Butt D, Bourne K, Hermes JR, Chan TD, et al. Differentiation of germinal center B cells into plasma cells is initiated by high-affinity antigen and completed by Tfh cells. J Exp Med. 2017;214:1259–67.
Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, et al. CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat Immunol. 2014;15:631–7.
Shinnakasu R, Inoue T, Kometani K, Moriyama S, Adachi Y, Nakayama M, et al. Regulated selection of germinal-center cells into the memory B cell compartment. Nat Immunol. 2016;17:861–9.
Weisel FJ, Zuccarino-Catania GV, Chikina M, Shlomchik MJ. A temporal switch in the germinal center determines differential output of memory B and plasma cells. Immunity. 2016;44:116–30.
McCarthy KR, Watanabe A, Kuraoka M, Do KT, McGee CE, Sempowski GD, et al. Memory B cells that cross-react with group 1 and group 2 Influenza A viruses are abundant in adult human repertoires. Immunity. 2018;48:174–84.e9.
Williams LD, Ofek G, Schätzle S, McDaniel JR, Lu X, Nicely NI et al. Potent and broad HIV-neutralizing antibodies in memory B cells and plasma. Sci Immunol. 2017; 2. https://doi.org/10.1126/sciimmunol.aal2200.
Elsner RA, Shlomchik MJ. Germinal center and extrafollicular B cell responses in vaccination, immunity, and autoimmunity. Immunity. 2020;53:1136–50.
Woodruff MC, Ramonell RP, Nguyen DC, Cashman KS, Saini AS, Haddad NS, et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID-19. Nat Immunol. 2020;21:1506–16.
Morens DM, Taubenberger JK, Fauci AS. Rethinking next-generation vaccines for coronaviruses, influenzaviruses, and other respiratory viruses. Cell Host Microbe. 2023;31:146–57.
Davis CW, Jackson KJL, McCausland MM, Darce J, Chang C, Linderman SL, et al. Influenza vaccine–induced human bone marrow plasma cells decline within a year after vaccination. Science (80-). 2020;370:237–41.
Habibi MS, Jozwik A, Makris S, Dunning J, Paras A, DeVincenzo JP, et al. Impaired antibody-mediated protection and defective IgA B-cell memory in experimental infection of adults with respiratory syncytial virus. Am J Respir Crit Care Med. 2015;191:1040–9.
Lapuente D, Maier C, Irrgang P, Hübner J, Peter ASAS, Hoffmann M, et al. Rapid response flow cytometric assay for the detection of antibody responses to SARS-CoV-2. Eur J Clin Microbiol Infect Dis. 2020. https://doi.org/10.1007/s10096-020-04072-7.
Brochot E, Demey B, Handala L, François C, Duverlie G, Castelain S. Comparison of different serological assays for SARS-CoV-2 in real life. J Clin Virol. 2020;130:104569.
Amanat F, Stadlbauer D, Strohmeier S, Nguyen THO, Chromikova V, McMahon M, et al. A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat Med. 2020;26:1033–6.
Bewley KR, Coombes NS, Gagnon L, McInroy L, Baker N, Shaik I, et al. Quantification of SARS-CoV-2 neutralizing antibody by wild-type plaque reduction neutralization, microneutralization and pseudotyped virus neutralization assays. Nat Protoc. 2021;16:3114–40.
Le Bert N, Clapham HE, Tan AT, Chia WN, Tham CYL, Lim JM et al. Highly functional virus-specific cellular immune response in asymptomatic SARS-CoV-2 infection. J Exp Med. 2021; 218. https://doi.org/10.1084/jem.20202617.
Sekine T, Perez-Potti A, Rivera-Ballesteros O, Strålin K, Gorin J-B, Olsson A, et al. Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell. 2020;183:158–68.e14.
Iyer AS, Jones FK, Nodoushani A, Kelly M, Becker M, Slater D et al. Persistence and decay of human antibody responses to the receptor binding domain of SARS-CoV-2 spike protein in COVID-19 patients. Sci Immunol. 2020; 5. https://doi.org/10.1126/sciimmunol.abe0367.
Long Q-X, Liu B-Z, Deng H-J, Wu G-C, Deng K, Chen Y-K, et al. Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat Med. 2020;26:845–8.
Wajnberg A, Amanat F, Firpo A, Altman DR, Bailey MJ, Mansour M, et al. Robust neutralizing antibodies to SARS-CoV-2 infection persist for months. Science (80-). 2020;370:1227–30.
Isho B, Abe KT, Zuo M, Jamal AJ, Rathod B, Wang JH et al. Persistence of serum and saliva antibody responses to SARS-CoV-2 spike antigens in COVID-19 patients. Sci Immunol. 2020; 5. https://doi.org/10.1126/sciimmunol.abe5511.
Sterlin D, Mathian A, Miyara M, Mohr A, Anna F, Claër L et al. IgA dominates the early neutralizing antibody response to SARS-CoV-2. Sci Transl Med. 2021; 13. https://doi.org/10.1126/scitranslmed.abd2223.
Landry ML. Immunoglobulin M for acute infection: true or false? Clin Vaccin Immunol. 2016;23:540–5.
Hsueh P-R, Huang L-M, Chen P-J, Kao C-L, Yang P-C. Chronological evolution of IgM, IgA, IgG and neutralisation antibodies after infection with SARS-associated coronavirus. Clin Microbiol Infect. 2004;10:1062–6.
Dan JM, Mateus J, Kato Y, Hastie KM, Yu ED, Faliti CE et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science (80-). 2021; 371. https://doi.org/10.1126/science.abf4063.
Röltgen K, Powell AE, Wirz OF, Stevens BA, Hogan CA, Najeeb J et al. Defining the features and duration of antibody responses to SARS-CoV-2 infection associated with disease severity and outcome. Sci Immunol. 2020; 5. https://doi.org/10.1126/sciimmunol.abe0240.
Wu J, Liang B, Chen C, Wang H, Fang Y, Shen S, et al. SARS-CoV-2 infection induces sustained humoral immune responses in convalescent patients following symptomatic COVID-19. Nat Commun. 2021;12:1813.
Duysburgh E, Mortgat L, Barbezange C, Dierick K, Fischer N, Heyndrickx L, et al. Persistence of IgG response to SARS-CoV-2. Lancet Infect Dis. 2021;21:163–4.
Peghin M, De Martino M, Fabris M, Palese A, Visintini E, Graziano E et al. The fall in antibody response to SARS-CoV-2: a longitudinal study of asymptomatic to critically Ill patients up to 10 months after recovery. J Clin Microbiol. 2021; 59. https://doi.org/10.1128/JCM.01138-21.
Hartley GE, Edwards ESJ, Aui PM, Varese N, Stojanovic S, McMahon J et al. Rapid generation of durable B cell memory to SARS-CoV-2 spike and nucleocapsid proteins in COVID-19 and convalescence. Sci Immunol. 2020; 5. https://doi.org/10.1126/sciimmunol.abf8891.
Gaebler C, Wang Z, Lorenzi JCC, Muecksch F, Finkin S, Tokuyama M, et al. Evolution of antibody immunity to SARS-CoV-2. Nature. 2021;591:639–44.
Ibarrondo FJ, Fulcher JA, Goodman-Meza D, Elliott J, Hofmann C, Hausner MA, et al. Rapid decay of anti–SARS-CoV-2 antibodies in persons with mild Covid-19. N Engl J Med. 2020;383:1085–7.
Long Q-X, Tang X-J, Shi Q-L, Li Q, Deng H-J, Yuan J, et al. Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nat Med. 2020;26:1200–4.
Turner JS, Kim W, Kalaidina E, Goss CW, Rauseo AM, Schmitz AJ, et al. SARS-CoV-2 infection induces long-lived bone marrow plasma cells in humans. Nature. 2021;595:421–5.
Chen X, Pan Z, Yue S, Yu F, Zhang J, Yang Y, et al. Disease severity dictates SARS-CoV-2-specific neutralizing antibody responses in COVID-19. Signal Transduct Target Ther. 2020;5:180.
Chen Y, Zuiani A, Fischinger S, Mullur J, Atyeo C, Travers M, et al. Quick COVID-19 healers sustain Anti-SARS-CoV-2 antibody production. Cell. 2020;183:1496–507.e16.
Dadras O, Afsahi AM, Pashaei Z, Mojdeganlou H, Karimi A, Habibi P et al. The relationship between COVID‐19 viral load and disease severity: a systematic review. Immunity Inflamm Dis. 2022; 10. https://doi.org/10.1002/iid3.580.
Cervia C, Nilsson J, Zurbuchen Y, Valaperti A, Schreiner J, Wolfensberger A, et al. Systemic and mucosal antibody responses specific to SARS-CoV-2 during mild versus severe COVID-19. J Allergy Clin Immunol. 2021;147:545–57.e9.
Robbiani DF, Gaebler C, Muecksch F, Lorenzi JCC, Wang Z, Cho A, et al. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature. 2020;584:437–42.
Muecksch F, Wise H, Batchelor B, Squires M, Semple E, Richardson C, et al. Longitudinal serological analysis and neutralizing antibody levels in Coronavirus Disease 2019 convalescent patients. J Infect Dis. 2021;223:389–98.
Hohdatsu T, Yamada M, Tominaga R, Makino K, Kida K, Koyama H. Antibody-dependent enhancement of feline infectious peritonitis virus infection in feline alveolar macrophages and human monocyte cell line U937 by serum of cats experimentally or naturally infected with feline Coronavirus. J Vet Med Sci. 1998;60:49–55.
Anderson EM, Goodwin EC, Verma A, Arevalo CP, Bolton MJ, Weirick ME, et al. Seasonal human coronavirus antibodies are boosted upon SARS-CoV-2 infection but not associated with protection. Cell. 2021;184:1858–64.e10.
Ng KW, Faulkner N, Cornish GH, Rosa A, Harvey R, Hussain S, et al. Preexisting and de novo humoral immunity to SARS-CoV-2 in humans. Science (80-). 2020;370:1339–43.
Crowley AR, Natarajan H, Hederman AP, Bobak CA, Weiner JA, Wieland-Alter W et al. Boosting of cross-reactive antibodies to endemic coronaviruses by SARS-CoV-2 infection but not vaccination with stabilized spike. Elife. 2022; 11. https://doi.org/10.7554/eLife.75228.
Dugan HL, Stamper CT, Li L, Changrob S, Asby NW, Halfmann PJ, et al. Profiling B cell immunodominance after SARS-CoV-2 infection reveals antibody evolution to non-neutralizing viral targets. Immunity. 2021;54:1290–303.e7.
Brewer RC, Ramadoss NS, Lahey LJ, Jahanbani S, Robinson WH, Lanz TV. BNT162b2 vaccine induces divergent B cell responses to SARS-CoV-2 S1 and S2. Nat Immunol. 2022;23:33–9.
Sakharkar M, Rappazzo CG, Wieland-Alter WF, Hsieh C-L, Wrapp D, Esterman ES et al. Prolonged evolution of the human B cell response to SARS-CoV-2 infection. Sci Immunol. 2021; 6. https://doi.org/10.1126/sciimmunol.abg6916.
Khoury DS, Cromer D, Reynaldi A, Schlub TE, Wheatley AK, Juno JA, et al. Neutralizing antibody levels are highly predictive of immune protection from symptomatic SARS-CoV-2 infection. Nat Med. 2021;27:1205–11.
Addetia A, Crawford KHD, Dingens A, Zhu H, Roychoudhury P, Huang M-L et al. Neutralizing antibodies correlate with protection from SARS-CoV-2 in humans during a fishery vessel outbreak with a high attack rate. J Clin Microbiol. 2020; 58. https://doi.org/10.1128/JCM.02107-20.
Gilbert PB, Montefiori DC, McDermott AB, Fong Y, Benkeser D, Deng W, et al. Immune correlates analysis of the mRNA-1273 COVID-19 vaccine efficacy clinical trial. Science (80-). 2022;375:43–50.
Cromer D, Steain M, Reynaldi A, Schlub TE, Wheatley AK, Juno JA, et al. Neutralising antibody titres as predictors of protection against SARS-CoV-2 variants and the impact of boosting: a meta-analysis. Lancet Microbe. 2022;3:e52–61.
Maier C, Fuchs J, Irrgang P, Wißing MH, Beyerlein J, Tenbusch M et al. Mucosal immunization with an adenoviral vector vaccine confers superior protection against RSV compared to natural immunity. Front Immunol. 2022; 13. https://doi.org/10.3389/fimmu.2022.920256.
Wu T, Hu Y, Lee Y-T, Bouchard KR, Benechet A, Khanna K, et al. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J Leukoc Biol. 2014;95:215–24.
Zohar T, Hsiao JC, Mehta N, Das J, Devadhasan A, Karpinski W, et al. Upper and lower respiratory tract correlates of protection against respiratory syncytial virus following vaccination of nonhuman primates. Cell Host Microbe. 2022;30:41–52.e5.
Jozwik A, Habibi MS, Paras A, Zhu J, Guvenel A, Dhariwal J, et al. RSV-specific airway resident memory CD8+ T cells and differential disease severity after experimental human infection. Nat Commun. 2015;6:10224.
Guvenel A, Jozwik A, Ascough S, Ung SK, Paterson S, Kalyan M, et al. Epitope-specific airway-resident CD4+ T cell dynamics during experimental human RSV infection. J Clin Invest. 2019;130:523–38.
Spiekermann GM, Finn PW, Ward ES, Dumont J, Dickinson BL, Blumberg RS, et al. Receptor-mediated immunoglobulin G transport across mucosal barriers in adult life. J Exp Med. 2002;196:303–10.
Johansen F-E, Pekna M, Norderhaug IN, Haneberg B, Hietala MA, Krajci P, et al. Absence of epithelial immunoglobulin a transport, with increased mucosal leakiness, in polymeric immunoglobulin receptor/secretory component–deficient mice. J Exp Med. 1999;190:915–22.
Bertrand Y, Sánchez-Montalvo A, Hox V, Froidure A, Pilette C IgA-producing B cells in lung homeostasis and disease. Front Immunol. 2023; 14. https://doi.org/10.3389/fimmu.2023.1117749.
Tang J, Zeng C, Cox TM, Li C, Son YM, Cheon IS et al. Respiratory mucosal immunity against SARS-CoV-2 after mRNA vaccination. Sci Immunol. 2022; 7. https://doi.org/10.1126/sciimmunol.add4853.
Denis J, Garnier A, Cheutin L, Ferrier A, Timera H, Jarjaval F et al. Long-term systemic and mucosal SARS-CoV-2 IgA response and its association with persistent smell and taste disorders. Front Immunol. 2023; 14. https://doi.org/10.3389/fimmu.2023.1140714.
Havervall S, Marking U, Svensson J, Greilert-Norin N, Bacchus P, Nilsson P, et al. Anti-spike mucosal IgA protection against SARS-CoV-2 Omicron infection. N Engl J Med. 2022;387:1333–6.
Butler SE, Crowley AR, Natarajan H, Xu S, Weiner JA, Bobak CA et al. Distinct features and functions of systemic and mucosal humoral immunity among SARS-CoV-2 convalescent individuals. Front Immunol. 2021; 11. https://doi.org/10.3389/fimmu.2020.618685.
Alkharaan H, Bayati S, Hellström C, Aleman S, Olsson A, Lindahl K, et al. Persisting salivary IgG against SARS-CoV-2 at 9 months after mild COVID-19: A complementary approach to population surveys. J Infect Dis. 2021;224:407–14.
Fröberg J, Gillard J, Philipsen R, Lanke K, Rust J, van Tuijl D, et al. SARS-CoV-2 mucosal antibody development and persistence and their relation to viral load and COVID-19 symptoms. Nat Commun. 2021;12:5621.
Wright PF, Prevost-Reilly AC, Natarajan H, Brickley EB, Connor RI, Wieland-Alter WF, et al. Longitudinal systemic and mucosal immune responses to SARS-CoV-2 infection. J Infect Dis. 2022;226:1204–14.
Wang Z, Lorenzi JCC, Muecksch F, Finkin S, Viant C, Gaebler C et al. Enhanced SARS-CoV-2 neutralization by dimeric IgA. Sci Transl Med. 2021; 13. https://doi.org/10.1126/scitranslmed.abf1555.
Bakema JE, van Egmond M. The human immunoglobulin A Fc receptor FcαRI: a multifaceted regulator of mucosal immunity. Mucosal Immunol. 2011;4:612–24.
Zuo F, Marcotte H, Hammarström L, Pan-Hammarström Q. Mucosal IgA against SARS-CoV-2 Omicron infection. N Engl J Med. 2022;387:e55.
Lim JME, Tan AT, Le Bert N, Hang SK, Low JGH, Bertoletti A SARS-CoV-2 breakthrough infection in vaccinees induces virus-specific nasal-resident CD8+ and CD4+ T cells of broad specificity. J Exp Med. 2022; 219. https://doi.org/10.1084/jem.20220780.
Dispinseri S, Secchi M, Pirillo MF, Tolazzi M, Borghi M, Brigatti C, et al. Neutralizing antibody responses to SARS-CoV-2 in symptomatic COVID-19 is persistent and critical for survival. Nat Commun. 2021;12:2670.
Lucas C, Klein J, Sundaram ME, Liu F, Wong P, Silva J, et al. Delayed production of neutralizing antibodies correlates with fatal COVID-19. Nat Med. 2021;27:1178–86.
Lin C-Y, Wolf J, Brice DC, Sun Y, Locke M, Cherry S, et al. Pre-existing humoral immunity to human common cold coronaviruses negatively impacts the protective SARS-CoV-2 antibody response. Cell Host Microbe. 2022;30:83–96.e4.
Kreer C, Zehner M, Weber T, Ercanoglu MS, Gieselmann L, Rohde C, et al. Longitudinal isolation of potent near-germline SARS-CoV-2-neutralizing antibodies from COVID-19 patients. Cell. 2020;182:843–54.e12.
Schultheiß C, Paschold L, Simnica D, Mohme M, Willscher E, von Wenserski L, et al. Next-generation sequencing of T and B cell receptor repertoires from COVID-19 patients showed signatures associated with severity of disease. Immunity. 2020;53:442–55.e4.
Nielsen SCA, Yang F, Jackson KJL, Hoh RA, Röltgen K, Jean GH, et al. Human B cell clonal expansion and convergent antibody responses to SARS-CoV-2. Cell Host Microbe. 2020;28:516–25.e5.
Andreano E, Nicastri E, Paciello I, Pileri P, Manganaro N, Piccini G, et al. Extremely potent human monoclonal antibodies from COVID-19 convalescent patients. Cell. 2021;184:1821–35.e16.
Yuan M, Liu H, Wu NC, Lee C-CD, Zhu X, Zhao F, et al. Structural basis of a shared antibody response to SARS-CoV-2. Science (80-). 2020;369:1119–23.
Cao Y, Wang J, Jian F, Xiao T, Song W, Yisimayi A, et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature. 2022;602:657–63.
Legros V, Denolly S, Vogrig M, Boson B, Siret E, Rigaill J, et al. A longitudinal study of SARS-CoV-2-infected patients reveals a high correlation between neutralizing antibodies and COVID-19 severity. Cell Mol Immunol. 2021;18:318–27.
Mathew D, Giles JR, Baxter AE, Oldridge DA, Greenplate AR, Wu JE et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science (80-). 2020; 369. https://doi.org/10.1126/science.abc8511.
Mann ER, Menon M, Knight SB, Konkel JE, Jagger C, Shaw TN et al. Longitudinal immune profiling reveals key myeloid signatures associated with COVID-19. Sci Immunol. 2020; 5. https://doi.org/10.1126/sciimmunol.abd6197.
Shuwa HA, Shaw TN, Knight SB, Wemyss K, McClure FA, Pearmain L, et al. Alterations in T and B cell function persist in convalescent COVID-19 patients. Med. 2021;2:720–35.e4.
Kuri-Cervantes L, Pampena MB, Meng W, Rosenfeld AM, Ittner CAG, Weisman AR et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci Immunol. 2020; 5. https://doi.org/10.1126/sciimmunol.abd7114.
Kaneko N, Kuo H-H, Boucau J, Farmer JR, Allard-Chamard H, Mahajan VS, et al. Loss of Bcl-6-expressing T follicular helper cells and germinal centers in COVID-19. Cell. 2020;183:143–57.e13.
Duan Y, Xia M, Ren L, Zhang Y, Ao Q, Xu S, et al. Deficiency of Tfh cells and germinal center in deceased COVID-19 patients. Curr Med Sci. 2020;40:618–24.
Haslbauer JD, Matter MS, Stalder AK, Tzankov A. Histomorphological patterns of regional lymph nodes in COVID-19 lungs. Pathologe. 2021;42:89–97.
Rodda LB, Netland J, Shehata L, Pruner KB, Morawski PA, Thouvenel CD, et al. Functional SARS-CoV-2-specific immune memory persists after mild COVID-19. Cell. 2021;184:169–83.e17.
Wang Z, Muecksch F, Schaefer-Babajew D, Finkin S, Viant C, Gaebler C, et al. Naturally enhanced neutralizing breadth against SARS-CoV-2 one year after infection. Nature. 2021;595:426–31.
Sokal A, Chappert P, Barba-Spaeth G, Roeser A, Fourati S, Azzaoui I, et al. Maturation and persistence of the anti-SARS-CoV-2 memory B cell response. Cell. 2021;184:1201–13.e14.
Muecksch F, Weisblum Y, Barnes CO, Schmidt F, Schaefer-Babajew D, Wang Z, et al. Affinity maturation of SARS-CoV-2 neutralizing antibodies confers potency, breadth, and resilience to viral escape mutations. Immunity. 2021;54:1853–68.e7.
Walsh EE, Frenck RW, Falsey AR, Kitchin N, Absalon J, Gurtman A, et al. Safety and immunogenicity of two RNA-based Covid-19 vaccine candidates. N Engl J Med. 2020;383:2439–50.
Anderson EJ, Rouphael NG, Widge AT, Jackson LA, Roberts PC, Makhene M, et al. Safety and immunogenicity of SARS-CoV-2 mRNA-1273 vaccine in older adults. N. Engl J Med. 2020;383:2427–38.
Greinacher A, Thiele T, Warkentin TE, Weisser K, Kyrle PA, Eichinger S. Thrombotic thrombocytopenia after ChAdOx1 nCov-19 vaccination. N Engl J Med. 2021;384:2092–101.
Schultz NH, Sørvoll IH, Michelsen AE, Munthe LA, Lund-Johansen F, Ahlen MT, et al. Thrombosis and thrombocytopenia after ChAdOx1 nCoV-19 vaccination. N Engl J Med. 2021;384:2124–30.
Baker AT, Boyd RJ, Sarkar D, Teijeira-Crespo A, Chan CK, Bates E et al. ChAdOx1 interacts with CAR and PF4 with implications for thrombosis with thrombocytopenia syndrome. Sci Adv. 2021; 7. https://doi.org/10.1126/sciadv.abl8213.
McMenamin ME, Nealon J, Lin Y, Wong JY, Cheung JK, Lau EHY, et al. Vaccine effectiveness of one, two, and three doses of BNT162b2 and CoronaVac against COVID-19 in Hong Kong: a population-based observational study. Lancet Infect Dis. 2022;22:1435–43.
Jara A, Undurraga EA, González C, Paredes F, Fontecilla T, Jara G, et al. Effectiveness of an inactivated SARS-CoV-2 vaccine in Chile. N Engl J Med. 2021;385:875–84.
Lim JME, Hang SK, Hariharaputran S, Chia A, Tan N, Lee ES, et al. A comparative characterization of SARS-CoV-2-specific T cells induced by mRNA or inactive virus COVID-19 vaccines. Cell Rep. Med. 2022;3:100793.
Yu S, Lin Y, Li Y, Chen S, Zhou L, Song H et al. Systemic immune profiling of Omicron-infected subjects inoculated with different doses of inactivated virus vaccine. Cell. 2023. https://doi.org/10.1016/j.cell.2023.08.033.
Goel RR, Apostolidis SA, Painter MM, Mathew D, Pattekar A, Kuthuru O et al. Distinct antibody and memory B cell responses in SARS-CoV-2 naïve and recovered individuals after mRNA vaccination. Sci Immunol. 2021; 6. https://doi.org/10.1126/sciimmunol.abi6950.
Wang Z, Schmidt F, Weisblum Y, Muecksch F, Barnes CO, Finkin S, et al. mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. Nature. 2021;592:616–22.
Widge AT, Rouphael NG, Jackson LA, Anderson EJ, Roberts PC, Makhene M, et al. Durability of responses after SARS-CoV-2 mRNA-1273 vaccination. N Engl J Med. 2021;384:80–2.
Carazo S, Talbot D, Boulianne N, Brisson M, Gilca R, Deceuninck G, et al. Single-dose messenger RNA vaccine effectiveness against severe acute respiratory syndrome Coronavirus 2 in healthcare workers extending 16 weeks postvaccination: a test-negative design from Québec, Canada. Clin Infect Dis. 2022;75:e805–13.
Levin EG, Lustig Y, Cohen C, Fluss R, Indenbaum V, Amit S, et al. Waning immune humoral response to BNT162b2 Covid-19 vaccine over 6 months. N Engl J Med. 2021;385:e84.
Falsey AR, Frenck RW, Walsh EE, Kitchin N, Absalon J, Gurtman A, et al. SARS-CoV-2 neutralization with BNT162b2 vaccine dose 3. N Engl J Med. 2021;385:1627–9.
Israel A, Shenhar Y, Green I, Merzon E, Golan-Cohen A, Schäffer AA, et al. Large-scale study of antibody titer decay following BNT162b2 mRNA vaccine or SARS-CoV-2 infection. Vaccines. 2021;10:64.
Wratil PR, Stern M, Priller A, Willmann A, Almanzar G, Vogel E, et al. Three exposures to the spike protein of SARS-CoV-2 by either infection or vaccination elicit superior neutralizing immunity to all variants of concern. Nat Med. 2022;28:496–503.
Tenbusch M, Schumacher S, Vogel E, Priller A, Held J, Steininger P, et al. Heterologous prime–boost vaccination with ChAdOx1 nCoV-19 and BNT162b2. Lancet Infect Dis. 2021;21:1212–3.
Vogel E, Kocher K, Priller A, Cheng C-C, Steininger P, Liao B-H, et al. Dynamics of humoral and cellular immune responses after homologous and heterologous SARS-CoV-2 vaccination with ChAdOx1 nCoV-19 and BNT162b2. eBioMedicine. 2022;85:104294.
Schmidt T, Klemis V, Schub D, Mihm J, Hielscher F, Marx S, et al. Immunogenicity and reactogenicity of heterologous ChAdOx1 nCoV-19/mRNA vaccination. Nat Med. 2021;27:1530–5.
Normark J, Vikström L, Gwon Y-D, Persson I-L, Edin A, Björsell T, et al. Heterologous ChAdOx1 nCoV-19 and mRNA-1273 Vaccination. N Engl J Med. 2021;385:1049–51.
Kaku CI, Champney ER, Normark J, Garcia M, Johnson CE, Ahlm C, et al. Broad anti–SARS-CoV-2 antibody immunity induced by heterologous ChAdOx1/mRNA-1273 vaccination. Science (80-). 2022;375:1041–7.
Klemis V, Schmidt T, Schub D, Mihm J, Marx S, Abu-Omar A, et al. Comparative immunogenicity and reactogenicity of heterologous ChAdOx1-nCoV-19-priming and BNT162b2 or mRNA-1273-boosting with homologous COVID-19 vaccine regimens. Nat Commun. 2022;13:4710.
Barros-Martins J, Hammerschmidt SI, Cossmann A, Odak I, Stankov MV, Morillas Ramos G et al. Immune responses against SARS-CoV-2 variants after heterologous and homologous ChAdOx1 nCoV-19/BNT162b2 vaccination. Nat Med. 2021. https://doi.org/10.1038/s41591-021-01449-9.
Khoo NKH, Lim JME, Gill US, de Alwis R, Tan N, Toh JZN, et al. Differential immunogenicity of homologous versus heterologous boost in Ad26.COV2.S vaccine recipients. Medicine. 2022;3:104–18.e4.
Behrens GM, Cossmann A, Stankov MV, Nehlmeier I, Kempf A, Hoffmann M, et al. SARS-CoV-2 delta variant neutralisation after heterologous ChAdOx1-S/BNT162b2 vaccination. Lancet. 2021;398:1041–2.
Bos R, Rutten L, van der Lubbe JEM, Bakkers MJG, Hardenberg G, Wegmann F, et al. Ad26 vector-based COVID-19 vaccine encoding a prefusion-stabilized SARS-CoV-2 Spike immunogen induces potent humoral and cellular immune responses. npj Vaccines. 2020;5:91.
Bates TA, Leier HC, McBride SK, Schoen D, Lyski ZL, Lee DX et al. An extended interval between vaccination and infection enhances hybrid immunity against SARS-CoV-2 variants. JCI Insight 2023; 8. https://doi.org/10.1172/jci.insight.165265.
Krammer F, Srivastava K, Alshammary H, Amoako AA, Awawda MH, Beach KF, et al. Antibody responses in seropositive persons after a single dose of SARS-CoV-2 mRNA vaccine. N Engl J Med. 2021;384:1372–4.
Koerber N, Priller A, Yazici S, Bauer T, Cheng C-C, Mijočević H, et al. Dynamics of spike-and nucleocapsid specific immunity during long-term follow-up and vaccination of SARS-CoV-2 convalescents. Nat Commun. 2022;13:153.
Gallais F, Velay A, Nazon C, Wendling M-J, Partisani M, Sibilia J, et al. Intrafamilial exposure to SARS-CoV-2 associated with cellular immune response without seroconversion, France. Emerg Infect Dis. 2021;27:113–21.
Le Bert N, Tan AT, Kunasegaran K, Tham CYL, Hafezi M, Chia A, et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature. 2020;584:457–62.
Lozano-Ojalvo D, Camara C, Lopez-Granados E, Nozal P, del Pino-Molina L, Bravo-Gallego LY, et al. Differential effects of the second SARS-CoV-2 mRNA vaccine dose on T cell immunity in naive and COVID-19 recovered individuals. Cell Rep. 2021;36:109570.
Mazzoni A, Di Lauria N, Maggi L, Salvati L, Vanni A, Capone M et al. First-dose mRNA vaccination is sufficient to reactivate immunological memory to SARS-CoV-2 in subjects who have recovered from COVID-19. J Clin Invest. 2021; 131. https://doi.org/10.1172/JCI149150.
Ebinger JE, Fert-Bober J, Printsev I, Wu M, Sun N, Prostko JC, et al. Antibody responses to the BNT162b2 mRNA vaccine in individuals previously infected with SARS-CoV-2. Nat Med. 2021;27:981–4.
Levi R, Azzolini E, Pozzi C, Ubaldi L, Lagioia M, Mantovani A et al. One dose of SARS-CoV-2 vaccine exponentially increases antibodies in individuals who have recovered from symptomatic COVID-19. J Clin Invest. 2021; 131. https://doi.org/10.1172/JCI149154.
Stamatatos L, Czartoski J, Wan Y-H, Homad LJ, Rubin V, Glantz H, et al. mRNA vaccination boosts cross-variant neutralizing antibodies elicited by SARS-CoV-2 infection. Science (80-). 2021;372:1413–8.
Moeller NH, Shi K, Demir Ö, Belica C, Banerjee S, Yin L et al. Structure and dynamics of SARS-CoV-2 proofreading exoribonuclease ExoN. Proc Natl Acad Sci. 2022; 119. https://doi.org/10.1073/pnas.2106379119.
Markov PV, Ghafari M, Beer M, Lythgoe K, Simmonds P, Stilianakis NI, et al. The evolution of SARS-CoV-2. Nat Rev Microbiol. 2023;21:361–79.
Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182:812–27.e19.
Hou YJ, Chiba S, Halfmann P, Ehre C, Kuroda M, Dinnon KH, et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science (80-). 2020;370:1464–8.
Meng B, Kemp SA, Papa G, Datir R, Ferreira IATM, Marelli S, et al. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell Rep. 2021;35:109292.
Garcia-Beltran WF, Lam EC, St. Denis K, Nitido AD, Garcia ZH, Hauser BM, et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell. 2021;184:2372–83.e9.
Cele S, Gazy I, Jackson L, Hwa S-H, Tegally H, Lustig G, et al. Escape of SARS-CoV-2 501Y.V2 from neutralization by convalescent plasma. Nature. 2021;593:142–6.
Carabelli AM, Peacock TP, Thorne LG, Harvey WT, Hughes J, de Silva TI et al. SARS-CoV-2 variant biology: immune escape, transmission and fitness. Nat Rev Microbiol. 2023. https://doi.org/10.1038/s41579-022-00841-7.
Greaney AJ, Starr TN, Gilchuk P, Zost SJ, Binshtein E, Loes AN, et al. Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. Cell Host Microbe. 2021;29:44–57.e9.
Karim SSA, Karim QA. Omicron SARS-CoV-2 variant: a new chapter in the COVID-19 pandemic. Lancet. 2021;398:2126–8.
Garcia-Beltran WF, St. Denis KJ, Hoelzemer A, Lam EC, Nitido AD, Sheehan ML, et al. mRNA-based COVID-19 vaccine boosters induce neutralizing immunity against SARS-CoV-2 Omicron variant. Cell. 2022;185:457–66.e4.
Weisblum Y, Schmidt F, Zhang F, DaSilva J, Poston D, Lorenzi JC et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. Elife. 2020; 9. https://doi.org/10.7554/eLife.61312.
Collier DA, De Marco A, Ferreira IATM, Meng B, Datir RP, Walls AC, et al. Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA vaccine-elicited antibodies. Nature. 2021;593:136–41.
Wang P, Nair MS, Liu L, Iketani S, Luo Y, Guo Y, et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature. 2021;593:130–5.
Park Y-J, Pinto D, Walls AC, Liu Z, De Marco A, Benigni F, et al. Imprinted antibody responses against SARS-CoV-2 Omicron sublineages. Science (80-). 2022;378:619–27.
Planas D, Veyer D, Baidaliuk A, Staropoli I, Guivel-Benhassine F, Rajah MM, et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature. 2021;596:276–80.
Zeng B, Gao L, Zhou Q, Yu K, Sun F. Effectiveness of COVID-19 vaccines against SARS-CoV-2 variants of concern: a systematic review and meta-analysis. BMC Med. 2022;20:200.
Röltgen K, Nielsen SCA, Silva O, Younes SF, Zaslavsky M, Costales C, et al. Immune imprinting, breadth of variant recognition, and germinal center response in human SARS-CoV-2 infection and vaccination. Cell. 2022;185:1025–40.e14.
Greaney AJ, Loes AN, Gentles LE, Crawford KHD, Starr TN, Malone KD et al. Antibodies elicited by mRNA-1273 vaccination bind more broadly to the receptor binding domain than do those from SARS-CoV-2 infection. Sci Transl Med. 2021; 13. https://doi.org/10.1126/scitranslmed.abi9915.
Liu L, Iketani S, Guo Y, Chan JF-W, Wang M, Liu L, et al. Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature. 2022;602:676–81.
Cele S, Jackson L, Khoury DS, Khan K, Moyo-Gwete T, Tegally H, et al. Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature. 2022;602:654–6.
Hoffmann M, Krüger N, Schulz S, Cossmann A, Rocha C, Kempf A, et al. The Omicron variant is highly resistant against antibody-mediated neutralization: Implications for control of the COVID-19 pandemic. Cell. 2022;185:447–56.e11.
Andrews N, Stowe J, Kirsebom F, Toffa S, Rickeard T, Gallagher E, et al. Covid-19 vaccine effectiveness against the Omicron (B.1.1.529) variant. N Engl J Med. 2022;386:1532–46.
Gruell H, Vanshylla K, Tober-Lau P, Hillus D, Schommers P, Lehmann C, et al. mRNA booster immunization elicits potent neutralizing serum activity against the SARS-CoV-2 Omicron variant. Nat Med. 2022;28:477–80.
Dejnirattisai W, Huo J, Zhou D, Zahradník J, Supasa P, Liu C, et al. SARS-CoV-2 Omicron-B.1.1.529 leads to widespread escape from neutralizing antibody responses. Cell. 2022;185:467–84.e15.
Yang J, Hong W, Lei H, He C, Lei W, Zhou Y, et al. Low levels of neutralizing antibodies against XBB Omicron subvariants after BA.5 infection. Signal Transduct Target Ther. 2023;8:252.
Tseng HF, Ackerson BK, Luo Y, Sy LS, Talarico CA, Tian Y, et al. Effectiveness of mRNA-1273 against SARS-CoV-2 Omicron and Delta variants. Nat Med. 2022;28:1063–71.
Nemet I, Kliker L, Lustig Y, Zuckerman N, Erster O, Cohen C, et al. Third BNT162b2 vaccination neutralization of SARS-CoV-2 Omicron infection. N Engl J Med. 2022;386:492–4.
Carreño JM, Alshammary H, Tcheou J, Singh G, Raskin AJ, Kawabata H, et al. Activity of convalescent and vaccine serum against SARS-CoV-2 Omicron. Nature. 2022;602:682–8.
Accorsi EK, Britton A, Fleming-Dutra KE, Smith ZR, Shang N, Derado G, et al. Association between 3 doses of mRNA COVID-19 vaccine and symptomatic infection caused by the SARS-CoV-2 Omicron and delta variants. JAMA. 2022;327:639.
Hein S, Sabino C, Benz NI, Görgülü E, Maier TJ, Oberle D, et al. The fourth vaccination with a non-SARS-CoV-2 variant adapted vaccine fails to increase the breadth of the humoral immune response. Sci Rep. 2023;13:10820.
Bar-On YM, Goldberg Y, Mandel M, Bodenheimer O, Amir O, Freedman L, et al. Protection by a fourth dose of BNT162b2 against Omicron in Israel. N Engl J Med. 2022;386:1712–20.
Collier AY, Miller J, Hachmann NP, McMahan K, Liu J, Bondzie EA, et al. Immunogenicity of BA.5 Bivalent mRNA Vaccine Boosters. N Engl J Med. 2023;388:565–7.
Wang Q, Bowen A, Valdez R, Gherasim C, Gordon A, Liu L, et al. Antibody response to Omicron BA.4–BA.5 bivalent booster. N Engl J Med. 2023;388:567–9.
Koutsakos M, Reynaldi A, Lee WS, Nguyen J, Amarasena T, Taiaroa G, et al. SARS-CoV-2 breakthrough infection induces rapid memory and de novo T cell responses. Immunity. 2023;56:879–92.e4.
Painter MM, Johnston TS, Lundgreen KA, Santos JJS, Qin JS, Goel RR, et al. Prior vaccination promotes early activation of memory T cells and enhances immune responses during SARS-CoV-2 breakthrough infection. Nat Immunol. 2023;24:1711–24.
Azzi L, Dalla Gasperina D, Veronesi G, Shallak M, Ietto G, Iovino D, et al. Mucosal immune response in BNT162b2 COVID-19 vaccine recipients. eBioMedicine. 2022;75:103788.
Azzi L, Dalla Gasperina D, Veronesi G, Shallak M, Maurino V, Baj A, et al. Mucosal immune response after the booster dose of the BNT162b2 COVID-19 vaccine. eBioMedicine. 2023;88:104435.
Sano K, Bhavsar D, Singh G, Floda D, Srivastava K, Gleason C, et al. SARS-CoV-2 vaccination induces mucosal antibody responses in previously infected individuals. Nat Commun. 2022;13:5135.
Poon MML, Rybkina K, Kato Y, Kubota M, Matsumoto R, Bloom NI et al. SARS-CoV-2 infection generates tissue-localized immunological memory in humans. Sci Immunol. 2021; 6. https://doi.org/10.1126/sciimmunol.abl9105.
Ogata AF, Cheng C-A, Desjardins M, Senussi Y, Sherman AC, Powell M, et al. Circulating severe acute respiratory syndrome Coronavirus 2 (SARS-CoV-2) vaccine antigen detected in the plasma of mRNA-1273 vaccine recipients. Clin Infect Dis. 2022;74:715–8.
Turner JS, O’Halloran JA, Kalaidina E, Kim W, Schmitz AJ, Zhou JQ, et al. SARS-CoV-2 mRNA vaccines induce persistent human germinal centre responses. Nature. 2021;596:109–13.
Pape KA, Dileepan T, Kabage AJ, Kozysa D, Batres R, Evert C, et al. High-affinity memory B cells induced by SARS-CoV-2 infection produce more plasmablasts and atypical memory B cells than those primed by mRNA vaccines. Cell Rep. 2021;37:109823.
Turner JS, Zhou JQ, Han J, Schmitz AJ, Rizk AA, Alsoussi WB, et al. Human germinal centres engage memory and naive B cells after influenza vaccination. Nature. 2020;586:127–32.
Kaji T, Ishige A, Hikida M, Taka J, Hijikata A, Kubo M, et al. Distinct cellular pathways select germline-encoded and somatically mutated antibodies into immunological memory. J Exp Med. 2012;209:2079–97.
Mudd PA, Minervina AA, Pogorelyy MV, Turner JS, Kim W, Kalaidina E, et al. SARS-CoV-2 mRNA vaccination elicits a robust and persistent T follicular helper cell response in humans. Cell. 2022;185:603–13.e15.
Lederer K, Castaño D, Gómez Atria D, Oguin TH, Wang S, Manzoni TB, et al. SARS-CoV-2 mRNA vaccines foster potent antigen-specific germinal center responses associated with neutralizing antibody generation. Immunity. 2020;53:1281–95.e5.
Kim W, Zhou JQ, Horvath SC, Schmitz AJ, Sturtz AJ, Lei T, et al. Germinal centre-driven maturation of B cell response to mRNA vaccination. Nature. 2022;604:141–5.
Pardi N, Tuyishime S, Muramatsu H, Kariko K, Mui BL, Tam YK, et al. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J Control Release. 2015;217:345–51.
Alameh M-G, Tombácz I, Bettini E, Lederer K, Ndeupen S, Sittplangkoon C, et al. Lipid nanoparticles enhance the efficacy of mRNA and protein subunit vaccines by inducing robust T follicular helper cell and humoral responses. Immunity. 2021;54:2877–92.e7.
Reynolds CJ, Pade C, Gibbons JM, Butler DK, Otter AD, Menacho K, et al. Prior SARS-CoV-2 infection rescues B and T cell responses to variants after first vaccine dose. Science (80-). 2021;372:1418–23.
Bates TA, McBride SK, Leier HC, Guzman G, Lyski ZL, Schoen D et al. Vaccination before or after SARS-CoV-2 infection leads to robust humoral response and antibodies that effectively neutralize variants. Sci Immunol. 2022; 7. https://doi.org/10.1126/sciimmunol.abn8014.
Bekliz M, Adea K, Vetter P, Eberhardt CS, Hosszu-Fellous K, Vu D-L, et al. Neutralization capacity of antibodies elicited through homologous or heterologous infection or vaccination against SARS-CoV-2 VOCs. Nat Commun. 2022;13:3840.
Medits I, Springer DN, Graninger M, Camp JV, Höltl E, Aberle SW et al. Different neutralization profiles after primary SARS-CoV-2 Omicron BA.1 and BA.2 infections. Front Immunol. 2022; 13. https://doi.org/10.3389/fimmu.2022.946318.
Jeong HW, Kim S-M, Jung MK, Noh JY, Yoo J-S, Kim E-H, et al. Enhanced antibody responses in fully vaccinated individuals against pan-SARS-CoV-2 variants following Omicron breakthrough infection. Cell Rep. Med. 2022;3:100764.
Zhang Y, Li R, Li Y, Yang H, Zhou L, Yuan J, et al. Antibody response and cross-neutralization after Omicron BA.2 infection. Signal Transduct Target Ther. 2023;8:25.
Quandt J, Muik A, Salisch N, Lui BG, Lutz S, Krüger K et al. Omicron BA.1 breakthrough infection drives cross-variant neutralization and memory B cell formation against conserved epitopes. Sci Immunol. 2022; 7. https://doi.org/10.1126/sciimmunol.abq2427.
Andreano E, Paciello I, Pierleoni G, Maccari G, Antonelli G, Abbiento V, et al. mRNA vaccines and hybrid immunity use different B cell germlines against Omicron BA.4 and BA.5. Nat Commun. 2023;14:1734.
Andreano E, Paciello I, Piccini G, Manganaro N, Pileri P, Hyseni I, et al. Hybrid immunity improves B cells and antibodies against SARS-CoV-2 variants. Nature. 2021;600:530–5.
Rodda LB, Morawski PA, Pruner KB, Fahning ML, Howard CA, Franko N, et al. Imprinted SARS-CoV-2-specific memory lymphocytes define hybrid immunity. Cell. 2022;185:1588–601.e14.
Tas JMJ, Koo J-H, Lin Y-C, Xie Z, Steichen JM, Jackson AM, et al. Antibodies from primary humoral responses modulate the recruitment of naive B cells during secondary responses. Immunity. 2022;55:1856–71.e6.
Miyamoto S, Arashiro T, Adachi Y, Moriyama S, Kinoshita H, Kanno T, et al. Vaccination-infection interval determines cross-neutralization potency to SARS-CoV-2 Omicron after breakthrough infection by other variants. Medicine. 2022;3:249–61.e4.
Payne RP, Longet S, Austin JA, Skelly DT, Dejnirattisai W, Adele S, et al. Immunogenicity of standard and extended dosing intervals of BNT162b2 mRNA vaccine. Cell. 2021;184:5699–714.e11.
Goldberg Y, Mandel M, Bar-On YM, Bodenheimer O, Freedman LS, Ash N, et al. Protection and Waning of Natural and Hybrid Immunity to SARS-CoV-2. N Engl J Med. 2022;386:2201–12.
Gunn BM, Yu W-H, Karim MM, Brannan JM, Herbert AS, Wec AZ, et al. A role for Fc function in therapeutic monoclonal antibody-mediated protection against Ebola Virus. Cell Host Microbe. 2018;24:221–33.e5.
DiLillo DJ, Tan GS, Palese P, Ravetch JV. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcγR interactions for protection against influenza virus in vivo. Nat Med. 2014;20:143–51.
Kaplonek P, Cizmeci D, Fischinger S, Collier A, Suscovich T, Linde C et al. mRNA-1273 and BNT162b2 COVID-19 vaccines elicit antibodies with differences in Fc-mediated effector functions. Sci Transl Med. 2022; 14. https://doi.org/10.1126/scitranslmed.abm2311.
Irrgang P, Gerling J, Kocher K, Lapuente D, Steininger P, Habenicht K et al. Class switch toward noninflammatory, spike-specific IgG4 antibodies after repeated SARS-CoV-2 mRNA vaccination. Sci Immunol. 2023; 8. https://doi.org/10.1126/sciimmunol.ade2798.
Rispens T, Huijbers MG The unique properties of IgG4 and its roles in health and disease. Nat Rev Immunol. 2023. https://doi.org/10.1038/s41577-023-00871-z.
Buhre JS, Pongracz T, Künsting I, Lixenfeld AS, Wang W, Nouta J et al. mRNA vaccines against SARS-CoV-2 induce comparably low long-term IgG Fc galactosylation and sialylation levels but increasing long-term IgG4 responses compared to an adenovirus-based vaccine. Front Immunol. 2023; 13. https://doi.org/10.3389/fimmu.2022.1020844.
Naranbhai V, Garcia-Beltran WF, Chang CC, Berrios Mairena C, Thierauf JC, Kirkpatrick G, et al. Comparative Immunogenicity and Effectiveness of mRNA-1273, BNT162b2, and Ad26.COV2.S COVID-19 Vaccines. J Infect Dis. 2022;225:1141–50.
Kiszel P, Sík P, Miklós J, Kajdácsi E, Sinkovits G, Cervenak L, et al. Class switch towards spike protein-specific IgG4 antibodies after SARS-CoV-2 mRNA vaccination depends on prior infection history. Sci Rep. 2023;13:13166.
Boonpiyathad T, Meyer N, Moniuszko M, Sokolowska M, Eljaszewicz A, Wirz OF, et al. High-dose bee venom exposure induces similar tolerogenic B-cell responses in allergic patients and healthy beekeepers. Allergy. 2017;72:407–15.
Aalberse RC, van der Gaag R, van Leeuwen J. Serologic aspects of IgG4 antibodies. I. Prolonged immunization results in an IgG4-restricted response. J Immunol. 1983;130:722–6.
Akdis CA, Blaser K. Mechanisms of allergen-specific immunotherapy. Allergy. 2000;55:522–30.
Jong BG, IJspeert H, Marques L, Burg M, Dongen JJ, Loos BG, et al. Human IgG2‐ and IgG4‐expressing memory B cells display enhanced molecular and phenotypic signs of maturity and accumulate with age. Immunol Cell Biol. 2017;95:744–52.
Routhu NK, Stampfer SD, Lai L, Akhtar A, Tong X, Yuan D et al. Efficacy of mRNA-1273 and Novavax ancestral or BA.1 spike booster vaccines against SARS-CoV-2 BA.5 infection in non-human primates. Sci Immunol. 2023; eadg7015.
Bartsch YC, Tong X, Kang J, Avendaño MJ, Serrano EF, García-Salum T et al. Omicron variant Spike-specific antibody binding and Fc activity are preserved in recipients of mRNA or inactivated COVID-19 vaccines. Sci Transl Med. 2022; 14. https://doi.org/10.1126/scitranslmed.abn9243.
Amanat F, Thapa M, Lei T, Ahmed SMS, Adelsberg DC, Carreño JM, et al. SARS-CoV-2 mRNA vaccination induces functionally diverse antibodies to NTD, RBD, and S2. Cell. 2021;184:3936–48.e10.
Kaplonek P, Fischinger S, Cizmeci D, Bartsch YC, Kang J, Burke JS, et al. mRNA-1273 vaccine-induced antibodies maintain Fc effector functions across SARS-CoV-2 variants of concern. Immunity. 2022;55:355–65.e4.
Planas D, Saunders N, Maes P, Guivel-Benhassine F, Planchais C, Buchrieser J, et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature. 2022;602:671–5.
VanBlargan LA, Errico JM, Halfmann PJ, Zost SJ, Crowe JE, Purcell LA, et al. An infectious SARS-CoV-2 B.1.1.529 Omicron virus escapes neutralization by therapeutic monoclonal antibodies. Nat Med. 2022;28:490–5.
Mackin SR, Desai P, Whitener BM, Karl CE, Liu M, Baric RS, et al. Fc-γR-dependent antibody effector functions are required for vaccine-mediated protection against antigen-shifted variants of SARS-CoV-2. Nat Microbiol. 2023;8:569–80.
Della-Torre E, Lanzillotta M, Strollo M, Ramirez GA, Dagna L, Tresoldi M. Serum IgG4 level predicts COVID-19 related mortality. Eur J Intern Med. 2021;93:107–9.
Kamisawa T, Zen Y, Pillai S, Stone JH. IgG4-related disease. Lancet (Lond, Engl). 2015;385:1460–71.
Moura AD, da Costa HHM, Correa VA, de S Lima AK, Lindoso JAL, De Gaspari E, et al. Assessment of avidity related to IgG subclasses in SARS-CoV-2 Brazilian infected patients. Sci Rep. 2021;11:17642.
Funding
Deutsche Forschungsgemeinschaft (German Research Foundation) - Projektnummer 215346292 [Winkler] Bavarian consortium for research on the pandemic disease COVID-19 (FOR-COVID) [Tenbusch]. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lapuente, D., Winkler, T.H. & Tenbusch, M. B-cell and antibody responses to SARS-CoV-2: infection, vaccination, and hybrid immunity. Cell Mol Immunol 21, 144–158 (2024). https://doi.org/10.1038/s41423-023-01095-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41423-023-01095-w
Keywords
This article is cited by
-
SARS-CoV-2 immunity
Cellular & Molecular Immunology (2024)
