
Abstract
The global coronavirus disease 2019 (COVID-19) pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has caused severe morbidity and mortality in humans. It is urgent to understand the function of viral genes. However, the function of open reading frame 10 (ORF10), which is uniquely expressed by SARS-CoV-2, remains unclear. In this study, we showed that overexpression of ORF10 markedly suppressed the expression of type I interferon (IFN-I) genes and IFN-stimulated genes. Then, mitochondrial antiviral signaling protein (MAVS) was identified as the target via which ORF10 suppresses the IFN-I signaling pathway, and MAVS was found to be degraded through the ORF10-induced autophagy pathway. Furthermore, overexpression of ORF10 promoted the accumulation of LC3 in mitochondria and induced mitophagy. Mechanistically, ORF10 was translocated to mitochondria by interacting with the mitophagy receptor Nip3-like protein X (NIX) and induced mitophagy through its interaction with both NIX and LC3B. Moreover, knockdown of NIX expression blocked mitophagy activation, MAVS degradation, and IFN-I signaling pathway inhibition by ORF10. Consistent with our observations, in the context of SARS-CoV-2 infection, ORF10 inhibited MAVS expression and facilitated viral replication. In brief, our results reveal a novel mechanism by which SARS-CoV-2 inhibits the innate immune response; that is, ORF10 induces mitophagy-mediated MAVS degradation by binding to NIX.
Similar content being viewed by others


Introduction
Coronaviruses (CoVs) infect a variety of host species, including humans, pigs, cattle, horses, rats, and bats. These viruses predominantly cause respiratory and intestinal tract infections and result in a wide range of clinical manifestations [1, 2]. CoVs were formerly considered relatively harmless respiratory pathogens in humans. However, zoonotic CoVs have crossed species barriers, leading to novel CoVs that can spread from animals to humans. For example, SARS-CoV resulted in outbreaks of SARS in 2002 and 2003 [3], Middle East respiratory syndrome CoV (MERS-CoV) emerged in 2012 and continues to circulate in camels [4], and the novel CoV SARS-CoV-2, the causative agent of coronavirus disease 2019 (COVID-19), emerged in December 2019 and is responsible for the current severe outbreak of COVID-19 worldwide, which has led to substantial morbidity and mortality. Therefore, the need to better understand the molecular mechanism underlying viral infection and spread in the body is urgent.
The SARS-CoV-2 genome is a positive-sense, nonsegmented, single-stranded RNA with a length of 29.9 kb. The genome of SARS-CoV-2 contains 14 open reading frames (ORFs) flanked by 5′ and 3′ untranslated regions [5]. ORF1a and ORF1b, which account for approximately two-thirds of the viral RNA genome, encode polyproteins that are further cleaved to form 16 nonstructural proteins (nsps) that form the viral replicase-transcriptase complex [6, 7]. The ORFs in the third of the genome near the 3′ terminus encode four main structural proteins, the spike (S), envelope (E), membrane (M), and nucleocapsid (N) proteins, which are components of the virion [8]. In addition, the SARS-CoV-2 genome encodes eleven accessory proteins: ORF3a, ORF3b, ORF3c, ORF3d, ORF6, ORF7a, ORF7b, ORF8, ORF9b, ORF9c and ORF10 [9]. Although the genome of SARS-CoV-2 (NCBI reference sequence: NC_045512.2) [10] is similar to that of SARS-CoV (NCBI reference sequence: NC_004718.3) [11], SARS-CoV-2 exhibits lower pathogenicity and higher infectivity than SARS-CoV [12]. Compared with the SARS-CoV genome, the SARS-CoV-2 genome uniquely encodes the ORF10 protein, which contains 38 amino acids [13, 14]. ORF10 was reported to bind to an E3 ubiquitin ligase complex containing Cullin-2, Rbx1, Elongin B, Elongin C, and ZYG11B, which ubiquitinates host proteins and degrades them via the 26 S proteasome; ZYG11B is dispensable for SARS-CoV-2 infection, and the interaction between ORF10 and ZYG11B is not relevant for SARS-CoV-2 infection [15]. However, how ORF10 functions in the interaction between the virus and host cells is unclear.
SARS-CoV-2 disrupts the host innate immune response via both its structural proteins and Nsps and induces overt but delayed type I interferon (IFN-I) responses [16]. Upon SARS-CoV-2 infection, IFN-I production is decreased, and the release of proinflammatory cytokines is enhanced, disrupting the balance of the immune response, which is responsible for the great severity of SARS-CoV-2 infection [17]. IFN-I production is rapidly triggered by the recognition of pathogen-associated molecular patterns, such as viral nucleic acids, by host sensors [18]. CoV RNA can be recognized by RIG-I-like receptors (RLRs), including retinoic acid-inducible gene I (RIG-I) and/or melanoma differentiation gene 5 (MDA5), in the cytoplasm [19]. After activation, RIG-I and MDA5 can interact with the adapter mitochondrial antiviral signaling protein (MAVS, also termed IPS-1, VISA, and Cardif) [16, 20]. Subsequently, MAVS recruits two IKK-related kinases, TANK-binding kinase 1 (TBK1) and inducible IκB kinase (IKKi), leading to activation of the transcription factors interferon regulatory factor 3/7 (IRF3/7) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [21]. Then, these proteins trigger signaling cascades to initiate the production of IFNs as well as the downstream expression of multiple IFN-stimulated genes (ISGs) and inflammatory cytokines [22]. The IFN-I response is critical for the response to SARS-CoV-2 infection. The use of IFN combined with ribavirin for the treatment of SARS-CoV infection has shown some beneficial effects [23,24,25]. Furthermore, IFN treatment was found to inhibit SARS-CoV-2 infection in a dose-dependent manner in human Calu-3 cells and simian Vero E6 cells, but the potential of IFN as a therapeutic agent for COVID-19 awaits validation [17]. Recent studies have shown that MDA5, LGP2, and NOD1 are required for the recognition of SARS-CoV-2 by lung epithelial cells and that IRF3, IRF5 and NF-κB/p65 are key transcription factors that regulate the IFN response during SARS-CoV-2 infection [26]. SARS-CoV-2 has developed multiple strategies to antagonize the innate immune response by attacking these key molecules in the IFN-I signaling pathway. For example, SARS-CoV-2 M targets MAVS and impairs its accumulation and recruitment of downstream signaling components [21]. SARS-CoV-2 ORF6 antagonizes the host innate immune response by blocking IRF3 activation and STAT1 nuclear translocation [16]. SARS-CoV-2 Nsp14 targets the type I IFN receptor IFNAR1 for lysosomal degradation, thus blocking activation of the crucial transcription factors STAT1 and STAT2 [27]. However, whether SARS-CoV-2 ORF10 plays a role in antagonizing antiviral immunity is unknown.
In the immune system, proper mitochondrial function is a prerequisite for inflammatory responses and host defense [28]. Viral infection can affect the host mitochondrial network, including mitophagy induction [29]. Some viruses trigger mitophagy through different strategies and control the process of mitophagy [29]. This ability enables viruses to promote persistent infection and attenuate innate immune responses [29]. The study indicates that the matrix protein of HPIV3 induces Parkin-PINK1-independent complete mitophagy, inhibiting host antiviral IFN responses [30]. In addition, HHV-8-encoded viral interferon regulatory factor 1 (vIRF-1) promotes mitochondrial clearance by binding to a mitophagy receptor, NIX, on mitochondria and activating NIX-mediated mitophagy to support viral replication [31]. NIX is a typical autophagy receptor that is able to transport mitochondria to autophagosomes through direct interaction with Atg8 family proteins [32]. NIX-mediated mitophagy is responsible for the elimination of spontaneously aggregated MAVS [33]. Therefore, successful viral infection and replication are largely achieved by the ability of viruses to attenuate the innate antiviral responses mediated by mitochondria [31]. However, there are no relevant studies on the induction of mitophagy by SARS-CoV-2 to escape the host’s innate immune surveillance.
In this study, we demonstrate that SARS-CoV-2 ORF10 inhibits innate immunity and promotes viral replication by inducing mitophagy to degrade MAVS. ORF10 interacts with Nip3-like protein X (NIX) and LC3B and translocates to mitochondria, where it induces mitophagy, leading to MAVS degradation. Further study showed that NIX is involved in the ORF10-mediated degradation of MAVS and blockade of IFN responses.
Materials and methods
Cells and viruses
HEK293T (ATCC CRL-11268) and HeLa (ATCC CCL-2) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; VivaCell) supplemented with 10% fetal bovine serum (FBS; HyClone) and the antibiotics penicillin and streptomycin (both 100 U/ml; New Cell & Molecular Biotech Co., Ltd.). The cells were maintained at 37 °C in a 5% CO2 environment. The human/CHN/Beijing-01/2020 SARS-CoV-2 strain was used in this experiment. Infection experiments were performed in a ABSL-3 laboratory. For SARS-CoV-2 infection, HeLa cells with stable expression of ACE2 were infected with SARS-CoV-2 at an MOI of 0.1. After 1 h, the cells were washed with PBS (Spark Jade) and cultured for an additional 24 h.
HeLa-ACE2 stable cell lines
pLVX-ACE2-Flag-IRES-Puro was transfected into HeLa cells. After 48 h, puromycin was added to the cells at a concentration of 3 μg/mL, and the cells were selected for 4 days until drug-resistant cell clones were obtained. Drug-resistant cell clones were harvested, and the cultures were expanded. Western blot analysis was performed with an anti-ACE2 antibody to detect the expression of ACE2.
Reagents
The proteasome inhibitors MG132, bafilomycin A1 (Baf A1) and Mdivi-1 were purchased from MedChemExpress; chloroquine diphosphate salt (CQ) was purchased from Sigma–Aldrich and used at a final concentration of 40 μM; and poly(I:C) (LMW) was purchased from InvivoGen.
Plasmid constructs
All enzymes used for cloning procedures were purchased from Vazyme (Nanjing, China). The HA-MDA5, Flag-RIG-IN, Flag-MAVS, IRF3 (5D)-HA, TBK1-HA, and IFN-β-Luc plasmids and the pRL-TK internal control luciferase reporter plasmid used in the study were described previously [34, 35]. SARS-CoV-2 ORF10 was cloned into pEGFP-N1 (Clontech Laboratories, Mountain View, CA), GFP-LC3 was cloned into pcDNA3.1 (Clontech Laboratories, Mountain View, CA), ORF10 was cloned into pCMV-HA (Clontech Laboratories, Mountain View, CA), Flag-NIX was cloned into pLVX-IRES-Puro (Clontech Laboratories, Mountain View, CA), and pcDNA3.1-mCherry-GFP-LC3 (mCherry-GFP-LC3) was purchased from Miaoling. pDsRed2-Mito (Mito-DsRed) was purchased from Clontech. All constructs were subjected to restriction digestion profiling and sequencing analysis for confirmation.
qRT–PCR
Total RNA was extracted, and mRNA was quantified as described previously [36]. All expression levels determined by real-time PCR were normalized to β-actin expression levels to avoid systematic and random errors during sample processing; the primer sequences used are listed in Table 1.
Western blotting and antibodies
Cells were lysed with RIPA lysis buffer (containing ProtLytic Protease and Phosphatase Inhibitor Cocktail (NCM Biotech)) on ice for 30 min. Proteins in the cell lysates were separated by SDS–PAGE and electrotransferred to PVDF membranes, which were blocked for 1 h with 5% nonfat milk and were then incubated with the indicated antibodies and detected with an Omni-ECL™ Femto Light Chemiluminescence Kit (Epizyme). The following primary antibodies were used: rabbit monoclonal anti-GFP (catalog no. AB0045), rabbit monoclonal anti-ISG15 (catalog no. CY7086), rabbit monoclonal anti-LC3B (catalog no. CY5992) and rabbit monoclonal anti-TOMM20 (catalog no. CY5527), which were obtained from Abways Technology Inc.; mouse monoclonal anti-MAVS (catalog no. sc166583), which was obtained from Santa Cruz Biotechnology; rabbit monoclonal anti-OAS1 (catalog no. 14498), rabbit monoclonal anti-MDA-5 (catalog no. 5321), rabbit monoclonal anti-RIG-I (catalog no. 3743), rabbit monoclonal anti-TBK1 (catalog no. 3504), rabbit monoclonal anti-phospho-TBK1 S172 (catalog no. 5483), rabbit monoclonal anti-IRF3 (catalog no. 4302), rabbit monoclonal anti-phospho-IRF3 S396 (catalog no. 4947), rabbit monoclonal anti-SQSTM1/p62 (catalog no. 8025), rabbit monoclonal anti-BNIP3L/NIX (catalog no. 12396), rabbit monoclonal anti-PINK1 (catalog no. 6946), rabbit monoclonal anti-NDP52 (catalog no. 60732), rabbit monoclonal anti-optineurin (catalog no. 58981), rabbit monoclonal anti-HA tag (catalog no. 3724) and rabbit monoclonal anti-DYKDDDDK tag (catalog no. 14793), which were obtained from Cell Signaling Technology; mouse monoclonal anti-SARS-CoV/SARS-CoV-2 (COVID-19) nucleocapsid antibody (catalog no. GTX632269), which was obtained from GeneTex Inc.; and mouse monoclonal anti-β-actin (catalog no. sc-47778), which was obtained from Santa Cruz Inc. The following secondary antibodies were used: horseradish peroxidase (HRP)-conjugated sheep anti-rabbit IgG and HRP-conjugated ECL sheep anti-mouse IgG (Jackson ImmunoResearch Inc., Baltimore, PA, USA). Protein bands were detected with enhanced chemiluminescence (ECL) reagent. Information on the antibodies used in this study is listed in Table 2.
Gene silencing
Cells were grown to 40% confluence and were then incubated with 50 nM NIX siRNA (5′-AACACGTACCATCCTCATCCT-3′) [37] or pYr-Lvsh-shORF10 (5′- GCACAAGTAGATGTAGTTAAC-3′) and Attractene Transfection Reagent (QIAGEN) for 36 h according to the manufacturer’s instructions.
Generation of NIX-knockout (KO) HeLa cells
NIX-KO cells were created by CRISPR–Cas9 gene editing and purchased from CRISPRdirect (http://crispr.dbcls.jp). The single-guide RNA (sgRNA) sequence targeting the human NIX gene (5′-CTTTGAAGAAAAGTGCGGAC-3′) was cloned into the pSpCas9 (BB)-2A-Puro (PX459) vector (Addgene, Feng Zhang). To generate NIX-KO HeLa cells, the NIX-KO plasmid was transfected into HeLa cells using Attractene Transfection Reagent (QIAGEN). At 36 hpt, transfected cells were selected in medium containing 3.0 μg/ml puromycin, and the medium was changed daily for selection of positive clones. Six days post transfection, the positive clones were subjected to limiting dilution in 96-well plates for selection of monoclonal cells. NIX-KO cell lines were obtained and confirmed by immunoblotting.
Enzyme-linked immunosorbent assay (ELISA)
A human IFN-β Quantikine ELISA Kit (Bio-Techne, Minneapolis, MN, USA, DIFNB0) was utilized to measure the concentrations of IFN-β in culture supernatants according to the manufacturer’s instructions. Assay Diluent RD1–19 was added to each well, and standard substances, including deionized water, control, or sample, were added to each well. Cells were incubated for 2 h at room temperature. Each well was aspirated and washed, and the process was repeated three times for a total of four washes. Human IFN-β Conjugate was added to each well. A fresh adhesive strip was used to cover the plates, and the plates were incubated for 2 h at room temperature on a shaker. The wash step was repeated. Substrate solution was added to each well and incubated for 30 min at room temperature on the bench top. Stop solution was added to each well. The color in the wells changed from blue to yellow. The optical density of each well was determined within 30 min using a microplate reader set to 450 nm. In addition, the absorbance at a wavelength of 570 nm was measured, and this value was used to correct for optical imperfections in the plate.
Dual-luciferase assay
HEK293T cells were seeded in 96-well plates in DMEM supplemented with 10% FBS and were then cotransfected in triplicate with the IFN-β-Luc reporter plasmid and pRL-TK internal control plasmid with or without the indicated expression plasmids using standard calcium phosphate precipitation with Attractene transfection reagent (Qiagen, Valencia, CA, USA). After 24 h of transfection, a dual-luciferase assay was performed using a dual-luciferase assay kit (Promega Corporation, Madison, WI, USA) and a SpectraMax M5 microplate reader (Molecular Devices Instruments Inc., USA).
Immunofluorescence assay (IFA)
HeLa cells plated on coverslips in 24-well plates were transfected with the indicated plasmids for 24 h, after which the original culture was discarded. The cells were then washed with PBS, fixed with room-temperature 4% paraformaldehyde (PFA)/PBS for 10 min, washed, permeabilized with 0.1% Triton X-100/TBS for 10 min, and blocked with 5% BSA. Next, the cells were washed and sequentially incubated with primary antibodies at 4 °C overnight and with secondary antibodies at room temperature for 60 min. Nuclei were stained with DAPI (US Everbright Inc.). The secondary antibodies used in this study were Alexa Fluor 350 goat anti-mouse IgG (H + L) and Alexa Fluor 594 goat anti-rabbit IgG (H + L), which were obtained from Invitrogen. Images were acquired using a Leica DMi8 microscope.
Coimmunoprecipitation (Co-IP)
Co-IP assays were performed using a PierceTM Co-IP Kit (Thermo Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. Whole-cell extracts were collected 48 h after transfection and lysed in lysis buffer supplemented with 1 mM PMSF and complete protease inhibitor cocktail on ice for 30 min. After centrifugation for 10 min at 13,000 × g and 4 °C, the supernatants were collected and incubated with Protein G Sepharose beads coupled to specific antibodies for 2 h or overnight with rotation at 4 °C. The beads were then washed 3× with lysis buffer. The bound proteins were eluted with elution buffer, and the lysates were boiled for 5 min with sample buffer (50 mM Tris-HCl (pH 6.8), 2% SDS, 10% glycerol, 0.1% bromophenol blue and 1% β-mercaptoethanol). The lysates were subjected to immunoblot analysis with the indicated antibodies.
Isolation of mitochondria
Pure mitochondria were isolated using a MinuteTM Mitochondria Isolation Kit (Invent Biotechnologies Inc.). HeLa cells were harvested by low-speed centrifugation (500 – 600 × g for 5 min). The cells were washed once with 1 ml of cold PBS. The supernatant was completely removed, and the cell pellet was resuspended in 250 µl of buffer A by vortexing briefly. The cell suspension was incubated on ice for 5–10 min and vortexed vigorously for 20–30 s. The cell suspension was then transferred to a filter cartridge. The filter cartridge was capped and centrifuged at 16,000 × g for 30 s. The filter was discarded, and the pellet was resuspended by vortexing briefly. The pellet was resuspended by vortexing and centrifuged at 700 × g for 1 min, after which the supernatant was transferred to a fresh 1.5-ml tube and 300 µl of buffer B was added at a supernatant:buffer B ratio of 1:1.2 (250 µl/300 µl). The solution was mixed by vortexing for 10 s. The final supernatant:buffer B ratio ranged from 1:1 to 1:1.6 (250 µl/400 µl). The sample was centrifuged at 16,000 × g for 10 min, after which it was completely removed, and the pellet was resuspended in 200 µl of buffer B by repeated pipetting and subsequent vigorous vortexing for 10–20 s. The tube was centrifuged at 8000 × g for 5 min. The supernatant was then transferred to a fresh 2.0-ml tube to which 1.6 ml of cold PBS was added, and the sample was centrifuged at 16,000 × g for 15–30 min. The supernatant was discarded, and the pellet (isolated mitochondria) was saved.
Statistical analysis
The results are expressed as the mean ± SD values. Comparisons between the different groups were performed by t tests using GraphPad Prism 8. Values of *P < 0.05, **P < 0.01, and ***P < 0.001 were considered significant, and ns indicates a nonsignificant difference.
Results
SARS-CoV-2 ORF10 inhibits the antiviral innate immune response
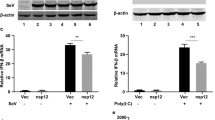
SARS-CoV-2 uses several strategies to block the production of IFN-I to circumvent host antiviral defenses, allowing the virus to undergo rapid replication [16]. To explore the function of ORF10 in the antiviral innate immune response, HeLa-ACE2 cells were transfected with ORF10-HA and empty vector (pCMV-HA), and whole-cell lysates were then examined using quantitative real-time PCR (qRT–PCR) and western blotting. Overexpression of ORF10 markedly suppressed the transcription of the IFN-α1 and IFN-β genes upon stimulation with poly(I:C) or infection with SARS-CoV-2 (Fig. 1a). We further showed that overexpression of ORF10 downregulated the production of IFN-β in HeLa cells upon stimulation with poly(I:C) (Fig. 1b). Next, the effect of ORF10 expression on antiviral ISGs was detected, and it was found that ORF10 expression decreased the mRNA levels of ISG15 and OAS1 compared with those in control cells upon poly(I:C) treatment or SARS-CoV-2 infection (Fig. 1c). Consistent with these results, immunoblot analysis showed that the ISG15 and OAS1 levels were significantly reduced in cells overexpressing ORF10 (Fig. 1d). Taken together, these data suggest that ORF10 suppresses antiviral innate immune responses by abrogating IFN-I signaling.

a HeLa-ACE2 cells were transfected with the ORF10-HA plasmid or empty vector (pCMV-HA) for 24 h and were then treated with poly(I:C) (20 μg/mL) or infected with SARS-CoV-2 (MOI = 0.1), and the transcription levels of IFN-α1 and IFN-β were monitored by qRT–PCR. b HeLa cells transfected with the ORF10-GFP plasmid or pEGFP-N1 were treated with poly(I:C) (20 μg/mL) for 24 h. The relative expression level of IFN-β in the supernatant was measured by ELISA. c qRT–PCR analysis of ISG15 and OAS1 mRNA expression in HeLa-ACE2 cells transfected with the ORF10-HA or pCMV-HA plasmid for 24 h and then treated with poly(I:C) (20 μg/mL) or infected with SARS-CoV-2 (MOI = 0.1). d Immunoblot analysis of ISG15, OAS1, and β-actin expression in HeLa cells transfected with ORF10-HA or pCMV-HA for 24 h and then treated with poly(I:C) (20 μg/mL) or infected with SARS-CoV-2 (MOI = 0.1). Data from three independent experiments were analyzed using t tests (a–c) and are presented as the mean ± SD values (*p < 0.05, **p < 0.01, and ***p < 0.001; ns indicates a nonsignificant difference)
SARS-CoV-2 ORF10 abrogates IFN-I signaling by autophagic degradation of MAVS
To reveal the mechanisms by which ORF10 regulates IFN-I signaling, we investigated the target of ORF10 in the RLR signaling pathway. HEK293T cells were cotransfected with empty vector or the ORF10 expression plasmid along with HA-MDA5, Flag-RIG-I (N), Flag-MAVS, TBK1-HA, the active form of IRF3(5D)-HA, and the IFN-β promoter and were then subjected to dual-luciferase reporter assays to detect IFN-β promoter activity. Overexpression of ORF10 attenuated the induction of IFN-β promoter-mediated luciferase reporter activity through MDA5, RIG-I (N), and MAVS but not through TBK1 or IRF3 (Fig. 2a). Furthermore, overexpression of ORF10 reduced the level of MAVS and significantly reduced the levels of phosphorylated TBK1 and IRF3 but did not affect the expression of RIG-I and MDA5 (Fig. 2b), suggesting that ORF10 specifically targets MAVS. Next, HeLa cells were transfected with the ORF10-GFP plasmid or pEGFP-N1 and were then harvested and subjected to immunoblot analysis. As shown in Fig. 2c, SARS-CoV-2 ORF10 downregulated the expression of MAVS in a dose-dependent manner. These data indicate that ORF10 downregulates RLR-mediated activation of the IFN pathway by inhibiting the expression of MAVS.

a HEK293T cells were cotransfected with the IFN-β promoter plasmid (60 ng), Renilla luciferase reporter plasmid (pRL-TK; 60 ng), HA-MDA5 plasmid (80 ng), Flag-RIG-I(N) plasmid (80 ng), Flag-MAVS plasmid (80 ng), TBK1-HA plasmid (80 ng) or IRF3(5D)-HA plasmid (80 ng) and empty vector or the ORF10-HA plasmid. Luciferase activity in cell lysates was measured 24 h after transfection. b HeLa cells were transfected with ORF10-HA or pCMV-HA for 24 h and were then treated with poly(I:C) (20 μg/mL). The cell lysates were subjected to immunoblot analysis with the indicated antibody. c Immunoblot analysis of the expression of MAVS in HeLa cells transfected with the ORF10-GFP plasmid (0, 400, 800 and 1600 ng) and then treated with poly(I:C) (20 μg/mL). d HeLa cells transfected with the ORF10-GFP plasmid and pEGFP-N1 were stimulated by poly(I:C) transfection and were then treated with MG132 (5 μM), bafilomycin A1 (Baf A1; 5 nM), or chloroquine (CQ; 40 μM) for 24 h. The cell lysates were then analyzed by western blotting. e HeLa cells were transfected with the ORF10-HA or pCMV plasmid for 24 h. The cell lysates were then analyzed by western blotting. f HeLa cells were seeded in 12-well plates and cotransfected with the mCherry-GFP-LC3 plasmid and the ORF10-HA plasmid or transfected with only mCherry-GFP-LC3 (vector). The cells were incubated for 36 h for immunofluorescence analysis. Scale bars: 10 µm. Data from three independent experiments were analyzed using a t test (a) and are presented as the mean ± SD values (*p < 0.05, **p < 0.01, and ***p < 0.001; ns indicates a nonsignificant difference)
The main mechanisms of protein degradation in eukaryotic cells include the ubiquitin (Ub)-proteasome system and the autophagosomal pathway [38]. To explore whether ORF10 regulates MAVS proteostasis via the Ub-proteasome system or the autophagosomal pathway, we expressed ORF10 in HeLa cells and then treated the cells with the protease inhibitor MG132, the autophagy inhibitor bafilomycin A1 (Baf A1), or chloroquine (CQ). ORF10-mediated degradation of the MAVS protein was blocked by the autophagy inhibitors Baf A1 and CQ but not by the proteasome inhibitor MG132 (Fig. 2d). To explore whether autophagy is induced by ORF10, HeLa cells were transfected with ORF10-HA or pCMV-HA, and greater LC3-II accumulation was observed in the presence of ORF10 expression than in the absence of ORF10 expression; furthermore, ORF10 overexpression led to enhanced degradation of P62 compared to that in mock control cells (Fig. 2e), and ORF10-HA expression resulted in increased numbers of mCherry+GFP− puncta (Fig. 2f), suggesting that ORF10 expression induces complete autophagy.
SARS-CoV-2 ORF10 induces mitophagy
Mitochondrial MAVS links mitochondria to antiviral type I IFN signaling. Some viruses induce mitophagy, leading to mitochondrial fragmentation and thereby to detrimental effects on innate immunity [39]. We found that GFP-tagged LC3 was colocalized with DsRed-tagged Mito, a mitochondrial marker protein, in SARS-CoV-2-infected cells (Fig. S1a), indicating that SARS-CoV-2 infection induced mitophagy. Moreover, BafA1 or Mdivi-1 treatment inhibited SARS-CoV-2 infection-induced degradation of MAVS (Fig. S1b), suggesting that SARS-CoV-2 infection results in degradation of MAVS via mitophagy. To verify whether ORF10 inhibits MAVS protein expression by inducing mitophagy, we first assessed the localization of ORF10 in mitochondria. Immunofluorescence analysis showed that GFP-tagged ORF10 was colocalized with DsRed-tagged Mito in ORF10-GFP-transfected HeLa cells (Fig. 3a). Next, we determined whether ORF10 can induce mitophagy. Immunofluorescence analysis showed that GFP-tagged LC3B was colocalized with DsRed-tagged Mito in ORF10-HA-transfected HeLa cells (Fig. 3b), indicating that ORF10 induced LC3 localization in mitochondria. Moreover, we detected the expression of the outer membrane mitochondrial protein TOMM20, a mitochondrial marker protein, and found that ORF10 expression resulted in a decrease in TOMM20 expression (Fig. 3c) and that this ORF10 expression-induced decrease in TOMM20 expression was prevented by Baf A1 treatment (Fig. 3d). Collectively, these data suggest that SARS-CoV-2 ORF10 degrades the MAVS protein via mitophagy.

a HeLa cells were transfected with pDsRed-Mito and the ORF10-GFP plasmid or pEGFP-N1 for 36 h. Fluorescence signals were visualized by confocal immunofluorescence microscopy. Scale bars: 10 μm. b HeLa cells were transfected with pcDNA3.1-GFP-LC3, pDsRed-Mito, the ORF10-HA plasmid or pCMV-HA for 36 h. The cells were then immunostained for ORF10 (blue), and fluorescence signals were visualized by confocal immunofluorescence microscopy. Scale bars: 10 μm. c HeLa cells were transfected with the ORF10-GFP plasmid, and the cell lysates were harvested at the indicated time points and subsequently analyzed by western blotting with anti-P62, anti-LC3B, and anti-TOMM20 antibodies. β-Actin served as the loading control for all analyses. d ORF10-expressing or pEGFP-N1-expressing HeLa cells were treated with or without Baf A1. The cell lysates were analyzed for mitophagy indicator expression by western blotting
SARS-CoV-2 ORF10 induces mitophagy via its interaction with NIX
Mitophagy is triggered by the activation of specific autophagy receptors localized mainly on the outer mitochondrial membrane (OMM) [31]. To determine the autophagy receptor to which ORF10 binds to initiate mitophagy, the expression of the autophagy receptors NIX, OPTN, NDP52, PINK1, and P62 was detected in HeLa cells expressing ORF10. The data revealed that ORF10 expression increased the level of mitochondria-associated NIX but not the levels of the other detected receptors (Fig. 4a). The ORF10-HA and Flag-NIX plasmids were cotransfected into HEK293T cells, and immunoprecipitation with the anti-HA or anti-Flag antibody and immunoblot analysis with the anti-HA or anti-Flag antibody were subsequently performed. The data showed that ORF10 coimmunoprecipitated with NIX (Fig. 4b, c). In addition, endogenous co-IP experiments indicated that ORF10 had a physiological association with NIX (Fig. 4d, e). Furthermore, LC3B was shown by reciprocal co-IP to interact with ORF10 (Fig. 4f). To examine whether ORF10 is colocalized with NIX, immunofluorescence analysis was performed. The results revealed that a significant portion of NIX was colocalized with ORF10 (Fig. 4g). Next, we examined the function of NIX in ORF10-induced mitophagy. Confocal microscopy confirmed that the localization of ORF10 in mitochondria was dependent on the expression of NIX (Fig. 4h). Moreover, we examined autophagy levels in both the cytosolic and mitochondria-enriched fractions of HeLa cells with stable NIX KO and of HeLa cells stably expressing the negative control vector (PX459). As shown in Fig. 4i, there was no obvious difference in the cytosolic LC3B-II level upon ORF10 expression between PX459 and NIX-KO cells; however, the mitochondrial fraction of the lysate of NIX-KO cells expressing ORF10 exhibited a significantly reduced level of LC3B-II compared with the mitochondrial fraction of the lysate of mock control cells. These results suggest that NIX is a specific modulator of ORF10-induced mitophagy.

a Immunoblot analysis of total cell lysates and mitochondrial fractions of HeLa cells transfected with the ORF10-HA or pCMV-HA plasmid for 24 h. The cell lysates were subjected to immunoblot analysis with the indicated antibodies. b, c Immunoblot analysis was performed to detect the interaction between ORF10 and NIX. The cell lysates were immunoprecipitated with an anti-HA or anti-Flag antibody and immunoblotted with an anti-Flag or anti-HA antibody, respectively. d, e Immunoblot analysis was performed to detect the interaction between endogenous NIX and ORF10. The cell lysates were immunoprecipitated with an anti-NIX or anti-HA antibody and immunoblotted with an anti-HA or anti-NIX antibody, respectively. f Immunoblot analysis was performed to detect the interaction between ORF10 and LC3B. The cell lysates were immunoprecipitated with an anti-LC3B antibody and immunoblotted with an anti-HA antibody. g HeLa cells were transfected with plasmids encoding Flag-NIX, DsRed-Mito, or ORF10-HA or with pCMV-HA for 36 h. The cells were then immunostained for ORF10 (blue) and NIX (red), and fluorescence signals were visualized by confocal immunofluorescence microscopy. Scale bars: 10 μm. h Colocalization of ORF10 and mitochondria in NIX-KO HeLa cells and HeLa cells stably expressing the PX459 plasmid and transfected with pDsRed-Mito, the ORF10-GFP plasmid or pEGFP-N1 for 36 h was assessed using IFA. Scale bars: 10 μm. i NIX-KO HeLa cells and HeLa cells stably expressing PX459 were transfected with the ORF10-HA plasmid for 36 h. Cytoplasmic and mitochondrial fractions were purified for western blot analysis
NIX is necessary for ORF10-mediated degradation of MAVS and inhibits the IFN-I response
We hypothesized that ORF10 degrades MAVS and suppresses innate immunity in an NIX-dependent manner. As shown in Fig. 5a, knockdown of NIX expression by transfection of siNIX suppressed the inhibitory effect of ORF10 on IFN-β production triggered by MAVS. Overexpression of NIX increased the inhibitory effect of ORF10 on IFN-β production triggered by MAVS (Fig. 5b). Consistent with these findings, knockout of NIX abolished the inhibitory effect of ORF10 on IFN-β mRNA transcription triggered by MAVS (Fig. 5c). Overexpression of NIX increased the inhibitory effect of ORF10 on IFN-β mRNA transcription triggered by MAVS (Fig. 5d). In addition, our experiments showed that NIX knockout reversed the inhibitory effect of ORF10 on MAVS expression (Fig. 5e). Treatment with the mitochondrial fission inhibitor Mdivi-1 reversed the ORF10-induced inhibition of IFN-β mRNA expression (Fig. 5f). In brief, these results indicate that ORF10 inhibits the innate immune response by regulating MAVS expression in an NIX-dependent manner.

a, b HEK293T cells were cotransfected with the indicated plasmids. Luciferase activity was measured in the cell lysates 24 h after transfection. c qRT–PCR analysis of IFN-β mRNA expression in HeLa cells stably expressing the NIX-KO or PX459 plasmid and transfected with the indicated plasmids for 36 h. d HeLa cells stably expressing Flag-NIX or empty vector were transfected with the indicated plasmids. IFN-β mRNA expression was measured via qRT–PCR. e NIX expression was knocked out in HeLa cells transfected with the ORF10-GFP plasmid or pEGFP-N1, and the cell lysates were then assessed using western blotting. f qRT–PCR analysis of IFN-β mRNA expression in HeLa cells transfected with the ORF10-GFP plasmid or pEGFP-N1 and treated with 50 μM Mdivi-1 or vehicle (DMSO). Data from three independent experiments were analyzed using t tests (a–d and f) and are presented as the mean ± SD values (*p < 0.05, **p < 0.01, and ***p < 0.001; ns indicates a nonsignificant difference)
ORF10 facilitates SARS-CoV-2 replication via degradation of MAVS
Finally, we examined the effects of ORF10 on viral propagation. HeLa-ACE2 cells transfected with the ORF10-HA or pCMV-HA plasmid were infected with SARS-CoV-2 at an MOI of 0.1 and harvested at the indicated time points. The cell lysates were analyzed by western blotting and qRT–PCR. The protein and mRNA levels of the viral N protein were increased in ORF10-HA-transfected cells compared to pCMV-HA-transfected control cells (Fig. 6a, b). Consistent with these observations, we silenced the expression of ORF10 in SARS-CoV-2-infected cells and found that the protein and mRNA levels of the viral N protein were significantly reduced (Fig. 6c–e). Furthermore, ORF10 expression accelerated the SARS-CoV-2 infection-induced degradation of MAVS (Fig. 6a). Consistent with these results, SARS-CoV-2 infection in HeLa-ACE2 cells with inhibition of ORF10 expression no longer downregulated MAVS expression (Fig. 6d). Finally, ORF10 expression was knocked down in HeLa-ACE2 cells, and the cells were infected with SARS-CoV-2. The TCID50 assay showed that inhibition of ORF10 expression reduced the replication of SARS-CoV-2 (Fig. 6f). Collectively, our results indicate that SARS-CoV-2 ORF10 promotes viral replication via degradation of MAVS.

a HeLa-ACE2 cells transfected with the ORF10-HA plasmid or pCMV-HA were infected with SARS-CoV-2 at an MOI of 0.1 for 24 h, after which the cell lysates and culture supernatants were collected to analyze SARS-CoV-2 N protein and MAVS protein expression. b HeLa-ACE2 cells transfected with the HA-tagged ORF10 plasmid or pCMV-HA were infected with SARS-CoV-2 at an MOI of 0.1 for 24 h, and the cell lysates and culture supernatants were then collected to analyze viral RNA levels using qRT–PCR. c–e After cotransfection with the ORF10-GFP plasmid and ORF10-specific shRNA (shORF10) or control shRNA (shNC) for 24 h, cell lysates were subjected to immunoblot analysis or qRT–PCR to determine the knockdown efficiency (c). After transfection with shNC or shORF10, HeLa-ACE2 cells were infected with SARS-CoV-2, and the cell lysates were subjected to immunoblot analysis with the indicated antibodies (d) or to qRT–PCR (e). f HeLa-ACE2 cells were transfected with shNC or shORF10 and were then infected with SARS-CoV-2 at an MOI of 0.1 at 24 h post transfection. Cells were collected at 24 h and subjected to 3 freeze–thaw cycles. The titer of SARS-CoV-2 was determined by a TCID50 assay. Data from three independent experiments were analyzed by t tests (b, c, e, and f) and are presented as the mean ± SD values (*p < 0.05, **p < 0.01, and ***p < 0.001; ns indicates a nonsignificant difference)
Discussion
The SARS-CoV-2 genome encodes eleven accessory proteins, and the functions of several have been identified. ORF3a induces apoptosis [40]; ORF3b is a potent IFN antagonist, and this effect appears to be related to the ability of ORF3b to hinder the nuclear translocation of IRF3 [5, 41]; ORF6 is able to block the transport of the transcription factor STAT from the cytoplasm to the nucleus, in turn blocking IFN activation [42]; ORF8 suppresses the IFN-I signaling pathway by inhibiting binding to the IFN-stimulated response element [43]; ORF9b suppresses IFN-I responses by targeting TOM70 [44]; and ORF9c enables the virus to evade immune surveillance by reducing the HLA abundance and antigen presentation [45]. However, the function of ORF10, which is uniquely expressed in SARS-CoV-2, is unknown. In the present study, we found that SARS-CoV-2 ORF10 induces mitophagy to degrade MAVS by binding to NIX, thus blocking MAVS-mediated antiviral signaling and promoting viral replication.
Viral antagonism of the host innate immune response is critical for viral replication. In this study, we showed for the first time the function of SARS-CoV-2 ORF10 in suppressing the innate immune response. The expression of ORF10 inhibited the expression of IFN-β and ISGs and the transcription of IFN-α1 and IFN-β mRNAs. The RLR signaling pathway plays an important role in identifying infection with RNA viruses and regulating the cellular innate immune response. By screening adapter proteins in the RLR signaling pathway, we identified MAVS as the target via which ORF10 suppresses the IFN-I signaling pathway. At present, the negative posttranslational regulation of MAVS is thought to be achieved mainly by K48-linked ubiquitination [35], blockade of signal transduction, and autophagy [46]. In ORF10-expressing HeLa cells, treatment with CQ or Baf A1 but not MG132 reversed the ORF10-mediated inhibition of MAVS expression, indicating that ORF10 degrades MAVS through the autophagy pathway.
Autophagy is a critical degradation process in all eukaryotes that mediates the elimination of harmful components [47], while the innate immune system is the first line of defense against invading pathogens [48]. Innate immunity and autophagy are inextricably linked and reciprocally regulated. MAVS is predominantly localized at the OMM and performs its functions there, indicating that mitochondria provide a functional platform for innate antiviral signal transduction [49]. Accumulating evidence suggests that the autophagy pathway is also involved in controlling MAVS-mediated antiviral signaling [50]. For example, viral infection activates RLR signaling, and RNF34 binds to MAVS in the mitochondrial compartment to promote the switch from K63-linked to K27-linked polyubiquitination. The Ub chain attached to MAVS is recognized by NDP52, resulting in the recruitment of damaged mitochondria enriched in MAVS aggregates to vacuoles for autophagic degradation [48]. In the present study, ORF10 expression induced complete autophagy. In addition, we found that ORF10 was localized in mitochondria. Overexpression of ORF10 promoted the localization of LC3 in mitochondria, and ORF10 colocalized with LC3 in mitochondria. Moreover, ORF10 expression promoted the degradation of the mitochondrial outer membrane protein TOMM20. Therefore, ORF10 induces mitophagy, ultimately leading to decreased mitochondrial MAVS expression.
In general, mitophagy regulatory pathways are classified as Ub-dependent or Ub-independent (receptor-dependent) [51]. PINK1 phosphorylates Ub chains on mitochondria and recruits autophagy receptors, such as NDP52, OPTN, and P62, to induce mitophagy [52, 53]. Here, we found that ORF10-induced mitophagy was PINK1-independent, since overexpression of ORF10 had no effect on the expression of PINK1, NDP52, OPTN, or P62. In addition to Ub-dependent mitophagy, LC3-interacting region (LIR)-containing mitophagy receptors can directly induce Ub-independent mitophagy [54]. NIX belongs to the BH3-only family and is involved in the initiation of autophagy. NIX is located on the OMM and directly interacts with LC3 via its LIR motif to mediate mitochondrial clearance. In this study, we confirmed that NIX is a specific modulator of ORF10-induced mitophagy. ORF10 interacted directly with NIX and LC3B. Deletion of NIX attenuated the induction of mitophagy by ORF10.
A previous study revealed that in the absence of N-terminally truncated isoforms of MAVS, blockade of NIX-mediated mitophagy stabilized full-length MAVS and induced the subsequent secretion of type I IFNs and other proinflammatory cytokines [55]. Is NIX involved in ORF10-mediated degradation of MAVS? Our data showed that overexpression of NIX significantly enhanced the inhibitory effects of ORF10 on MAVS expression and IFN-β production, while NIX-KO weakened the inhibitory effect of ORF10 on MAVS-mediated antiviral signaling, suggesting that NIX plays a key role in ORF10-mediated blockade of antiviral signaling. However, overexpression of ORF10 reduced MAVS-induced IFN-β mRNA expression even in NIX-KO cells, suggesting that ORF10 degrades MAVS not only through the NIX-mediated autophagy pathway but also possibly through other degradation pathways.
ORF10 transcripts can be detected in patients infected with SARS-CoV-2 [56]. Moreover, Liu et al. found that the expression level of ORF10 in patients with severe disease was much higher than that in patients with moderate disease; in addition, the expression ratio of ORF10 to nucleocapsid (N) in patients with severe disease was significantly higher than that in patients with moderate disease [57]. Therefore, ORF10 plays a vital role at all stages of SARS-CoV-2 infection. In our study, overexpression of ORF10 promoted the degradation of MAVS and the replication of SARS-CoV-2. Consistent with these results, when ORF10 was knocked down by shRNA, MAVS was not degraded and viral replication was weakened, suggesting that ORF10 facilitates SARS-CoV-2 replication via degradation of MAVS. It is possible that silencing ORF10 affects the functions of the total viral RNA genome, leading to attenuated expression of other ORFs. In fact, Sara Åkerstrom et al. found that silencing the viral genes S, 3a, and 7a did not inhibit the expression of the full-length genomic RNA of SARS-CoV [58].
In this study, we report the mechanism by which the SARS-CoV-2 ORF10 protein abrogates antiviral innate immunity and facilitates viral replication (Fig. 7). Our findings indicate that ORF10 interacts with NIX and LC3B to induce mitophagy, subsequently leading to blockade of MAVS-mediated antiviral signaling. The findings of this study provide a new direction for the study of the molecular mechanism by which SARS-CoV-2 resists antiviral innate immunity.

SARS-CoV-2 ORF10 interacts with NIX and LC3B and translocates to mitochondria, where it induces mitophagy, leading to MAVS degradation, thus resists the antiviral natural immune response and facilitate viral infection.
Change history
25 April 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41423-023-01023-y
References
Peck KM, Burch CL, Heise MT, Baric RS. Coronavirus host range expansion and middle east respiratory syndrome coronavirus emergence: biochemical mechanisms and evolutionary perspectives. Annu Rev Virol. 2015;2:95–117.
Su S, Wong G, Shi W, Liu J, Lai A, Zhou J, et al. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016;24:490–502.
Drosten C, Günther S, Preiser W, van der Werf S, Brodt HR, Becker S, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl J Med. 2003;348:1967–76.
Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl J Med. 2012;367:1814–20.
Redondo N, Zaldivar-Lopez S, Garrido JJ, Montoya M. SARS-CoV-2 accessory proteins in viral pathogenesis: knowns and unknowns. Front Immunol. 2021;12:708264.
Li X, Geng M, Peng Y, Meng L, Lu S. Molecular immune pathogenesis and diagnosis of COVID-19. J Pharm Anal. 2020;10:102–8.
Fehr AR, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol. 2015;1282:1–23.
Mousavizadeh L, Ghasemi S. Genotype and phenotype of COVID-19: Their roles in pathogenesis. J MicrobiolImmunol Infect. 2021;54:159–63.
Shang J, Han N, Chen Z, Peng Y, Li L, Zhou H, et al. Compositional diversity and evolutionary pattern of coronavirus accessory proteins. Brief Bioinforma. 2021;22:1267–78.
Wang C, Liu Z, Chen Z, Huang X, Xu M, He T, et al. The establishment of reference sequence for SARS-CoV-2 and variation analysis. J Med Virol. 2020;92:667–74.
Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nat Med. 2020;26:450–2.
Rabaan AA, Al-Ahmed SH, Haque S, Sah R, Tiwari R, Malik YS, et al. SARS-CoV-2, SARS-CoV, and MERS-COV: a comparative overview. Le Infez Med. 2020;28:174–84.
Michel CJ, Mayer C, Poch O, Thompson JD. Characterization of accessory genes in coronavirus genomes. Virol J. 2020;17:131.
Cagliani R, Forni D, Clerici M, Sironi M. Coding potential and sequence conservation of SARS-CoV-2 and related animal viruses. Infect Genet Evol: J Mol Epidemiol Evolut Genet Infect Dis. 2020;83:104353.
Mena EL, Donahue CJ, Vaites LP, Li J, Rona G, O'Leary C, et al. ORF10-Cullin-2-ZYG11B complex is not required for SARS-CoV-2 infection. Proc Natl Acad Sci USA. 2021;118:e2023157118.
Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun. 2020;11:3810.
Sa Ribero M, Jouvenet N, Dreux M, Nisole S. Interplay between SARS-CoV-2 and the type I interferon response. PLoS Pathog. 2020;16:e1008737.
Streicher F, Jouvenet N. Stimulation of Innate Immunity by Host and Viral RNAs. Trends Immunol. 2019;40:1134–48.
Li J, Liu Y, Zhang X. Murine coronavirus induces type I interferon in oligodendrocytes through recognition by RIG-I and MDA5. J Virol. 2010;84:6472–82.
Hou P, Wang H, Zhao G, Hu G, Xia X, He H. MiR-3470b promotes bovine ephemeral fever virus replication via directly targeting mitochondrial antiviral signaling protein (MAVS) in baby hamster Syrian kidney cells. BMC Microbiol. 2018;18:224.
Fu YZ, Wang SY, Zheng ZQ, Yi H, Li WW, Xu ZS, et al. SARS-CoV-2 membrane glycoprotein M antagonizes the MAVS-mediated innate antiviral response. Cell Mol Immunol. 2021;18:613–20.
Du Y, Duan T, Feng Y, Liu Q, Lin M, Cui J, et al. LRRC25 inhibits type I IFN signaling by targeting ISG15-associated RIG-I for autophagic degradation. EMBO J. 2018;37:351–66.
Chen F, Chan KH, Jiang Y, Kao RY, Lu HT, Fan KW, et al. In vitro susceptibility of 10 clinical isolates of SARS coronavirus to selected antiviral compounds. J Clin Virol: Off Publ Pan Am Soc Clin Virol. 2004;31:69–75.
Jin Y, Yang H, Ji W, Wu W, Chen S, Zhang W et al. Virology, Epidemiology, Pathogenesis, and Control of COVID-19. Viruses. 2020;12.
Morgenstern B, Michaelis M, Baer PC, Doerr HW, Cinatl J Jr. Ribavirin and interferon-beta synergistically inhibit SARS-associated coronavirus replication in animal and human cell lines. Biochemical Biophysical Res Commun. 2005;326:905–8.
Yin X, Riva L, Pu Y, Martin-Sancho L, Kanamune J, Yamamoto Y, et al. MDA5 governs the innate immune response to SARS-CoV-2 in lung epithelial cells. Cell Rep. 2021;34:108628.
Hayn M, Hirschenberger M, Koepke L, Nchioua R, Straub JH, Klute S et al. Systematic functional analysis of SARS-CoV-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep. 2021;35:109126.
Gkikas I, Palikaras K, Tavernarakis N. The role of mitophagy in innate immunity. Front Immunol. 2018;9:1283.
Zhang L, Qin Y, Chen M. Viral strategies for triggering and manipulating mitophagy. Autophagy.2018;14:1665–73.
Ding B, Zhang L, Li Z, Zhong Y, Tang Q, Qin Y, et al. The matrix protein of human parainfluenza virus type 3 induces mitophagy that suppresses interferon responses. Cell Host Microbe. 2017;21:538–47 e534.
Vo MT, Smith BJ, Nicholas J, Choi YB. Activation of NIX-mediated mitophagy by an interferon regulatory factor homologue of human herpesvirus. Nat Commun. 2019;10:3203.
Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45–51.
Shi Y, Wu J, Zhong T, Zhu W, She G, Tang H, et al. Upstream ORFs prevent MAVS spontaneous aggregation and regulate innate immune homeostasis. iScience.2020;23:101059.
Ma W, Wang H, He H. Bovine herpesvirus 1 tegument protein UL41 suppresses antiviral innate immune response via directly targeting STAT1. Vet Microbiol. 2019;239:108494.
Zhang Y, Hou P, He DC, Wang H, He H. RACK1 degrades MAVS to promote bovine ephemeral fever virus replication via upregulating E3 ubiquitin ligase STUB1. Vet Microbiol. 2021;257:109096.
Ma W, Wang Y, Yao X, Xu Z, An L, Yin M. The role of Exo70 in vascular smooth muscle cell migration. Cell Mol Biol Lett. 2016;21:20.
Fei P, Wang W, Kim SH, Wang S, Burns TF, Sax JK, et al. Bnip3L is induced by p53 under hypoxia, and its knockdown promotes tumor growth. Cancer Cell. 2004;6:597–609.
Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell.2011;147:728–41.
Wang R, Zhu Y, Ren C, Yang S, Tian S, Chen H, et al. Influenza A virus protein PB1-F2 impairs innate immunity by inducing mitophagy. Autophagy. 2020;17:496–11.
Ren Y, Shu T, Wu D, Mu J, Wang C, Huang M, et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol Immunol. 2020;17:881–3.
Konno Y, Kimura I, Uriu K, Fukushi M, Irie T, Koyanagi Y, et al. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep. 2020;32:108185.
Miorin L, Kehrer T, Sanchez-Aparicio MT, Zhang K, Cohen P, Patel RS, et al. SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc Natl Acad Sci USA. 2020;117:28344–54.
Li JY, Liao CH, Wang Q, Tan YJ, Luo R, Qiu Y, et al. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res. 2020;286:198074.
Jiang HW, Zhang HN, Meng QF, Xie J, Li Y, Chen H, et al. SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell Mol Immunol. 2020;17:998–1000.
Dominguez Andres A, Feng Y, Campos AR, Yin J, Yang CC, James B, et al. SARS-CoV-2 ORF9c is a membrane-associated protein that suppresses antiviral responses in cells. bioRxiv: the preprint server for biology. 2020; https://doi.org/10.1101/2020.08.18.256776.
Ren Z, Ding T, Zuo Z, Xu Z, Deng J, Wei Z. Regulation of MAVS expression and signaling function in the antiviral innate immune response. Front Immunol. 2020;11:1030.
Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;12:1542–52.
He X, Zhu Y, Zhang Y, Geng Y, Gong J, Geng J, et al. RNF34 functions in immunity and selective mitophagy by targeting MAVS for autophagic degradation. EMBO J. 2019;38:e100978.
Sun X, Sun L, Zhao Y, Li Y, Lin W, Chen D, et al. MAVS maintains mitochondrial homeostasis via autophagy. Cell Discov. 2016;2:16024.
Tal MC, Iwasaki A. Autophagic control of RLR signaling. Autophagy. 2009;5:749–50.
Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14.
Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–14.
Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci USA. 2016;113:4039–44.
Song Y, Zhou Y, Zhou X. The role of mitophagy in innate immune responses triggered by mitochondrial stress. Cell Commun Signal: Ccs. 2020;18:186.
Qi N, Shi Y, Zhang R, Zhu W, Yuan B, Li X, et al. Multiple truncated isoforms of MAVS prevent its spontaneous aggregation in antiviral innate immune signalling. Nat Commun. 2017;8:15676.
Davidson AD, Williamson MK, Lewis S, Shoemark D, Carroll MW, Heesom KJ, et al. Characterisation of the transcriptome and proteome of SARS-CoV-2 reveals a cell passage induced in-frame deletion of the furin-like cleavage site from the spike glycoprotein. Genome Med. 2020;12:68.
Liu T, Jia P, Fang B, Zhao Z. Differential expression of viral transcripts from single-cell RNA sequencing of moderate and severe COVID-19 patients and its implications for case severity. Front Microbiol. 2020;11:603509.
Akerstrom S, Mirazimi A, Tan YJ. Inhibition of SARS-CoV replication cycle by small interference RNAs silencing specific SARS proteins, 7a/7b, 3a/3b and S. Antivir Res. 2007;73:219–27.
Acknowledgements
This work was partially supported by grants from the National Natural Science Fund of China (31872490, 31972665, and 32072834) and Taishan Scholar and Distinguished Experts (to H. H.).
Author information
Authors and Affiliations
Contributions
HBH, YWG and HMW developed the concept of the study; HBH, PLH and XYL designed the experiments; XYL performed the experiments; TCW, WQW and YDZ participated in the viral infection experiments. ZPY was involved with the I.F. assay. HSC was involved with the I.P. assay. SJ, XW and YZ helped construct the recombinant plasmids. CQX and XMM provided help with the dual-luciferase assay. XYL, PLH, XFW and WQM collected and analyzed the data; WQM wrote the manuscript. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
The original online version of this article was revised: In the original publication of this article, there was an unintended error during image preparation of Fig. 4d. The blots for β-actin in Fig. 4d and Fig. 4f were generated from the same experiment, but the one in Fig. 4d was reversed.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, X., Hou, P., Ma, W. et al. SARS-CoV-2 ORF10 suppresses the antiviral innate immune response by degrading MAVS through mitophagy. Cell Mol Immunol 19, 67–78 (2022). https://doi.org/10.1038/s41423-021-00807-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41423-021-00807-4
Keywords
This article is cited by
-
Non-cytopathic bovine viral diarrhea virus (BVDV) inhibits innate immune responses via induction of mitophagy
Veterinary Research (2024)
-
Mechanism and role of mitophagy in the development of severe infection
Cell Death Discovery (2024)
-
Apoptosis and pyroptosis in the nasal mucosa of Syrian hamster during SARS-CoV-2 infection and reinfection
Apoptosis (2024)
-
The role of autophagy in viral infections
Journal of Biomedical Science (2023)
-
SARS-CoV-2 and innate immunity: the good, the bad, and the “goldilocks”
Cellular & Molecular Immunology (2023)
