
Abstract
Protein N-myristoylation is an important fatty acylation catalyzed by N-myristoyltransferases (NMTs), which are ubiquitous enzymes in eukaryotes. Specifically, attachment of a myristoyl group is vital for proteins participating in various biological functions, including signal transduction, cellular localization, and oncogenesis. Recent studies have revealed unexpected mechanisms indicating that protein N-myristoylation is involved in host defense against microbial and viral infections. In this review, we describe the current understanding of protein N-myristoylation (mainly focusing on myristoyl switches) and summarize its crucial roles in regulating innate immune responses, including TLR4-dependent inflammatory responses and demyristoylation-induced innate immunosuppression during Shigella flexneri infection. Furthermore, we examine the role of myristoylation in viral assembly, intracellular host interactions, and viral spread during human immunodeficiency virus-1 (HIV-1) infection. Deeper insight into the relationship between protein N-myristoylation and innate immunity might enable us to clarify the pathogenesis of certain infectious diseases and better harness protein N-myristoylation for new therapeutics.
Similar content being viewed by others


Introduction
In eukaryotes, most proteins must undergo alternative splicing and extensive modifications, such as protein glycosylation, phosphorylation, or lipidation, after synthesis to reach their active and functional forms. Indeed, attachment of lipid groups is vital for proteins to achieve their final native structure and to allow intracellular transport for them to reach the appropriate cellular localization.1,2,3 Protein lipidation can be divided into four major types: N-myristoylation, S-palmitoylation, prenylation, and glycosylphosphatidylinositol anchor conjugation.4
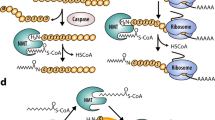
N-myristoylation is a ubiquitous protein lipid modification that occurs cotranslationally in eukaryotes and involves attachment of myristic acid to the N-terminal glycine (Gly) of a wide range of substrate proteins.5,6 Representative N-Gly-myristoylated proteins for which experimental evidence is available are summarized in Table 1. Although N-myristoylation general occurs cotranslationally on newly synthesized polypeptides following the removal of the initiator methionine by methionine aminopeptidase (MetAP2) (Fig. 1A), there is also evidence of posttranslational myristoylation on an internal Gly exposed by caspase cleavage during apoptosis (Fig. 1B).7,8 Myristoylation plays an important role in signal transduction, protein stability, and the localization of proteins to membranes.9,10,11,12,13,14,15,16 Furthermore, attachment of a myristoyl group adds a certain degree of regulation to myristoylated proteins, which is named the myristoyl switch.17 N-myristoylation is catalyzed by the enzyme N-myristoyltransferase (NMT). To date, the presence of NMT has been confirmed in most eukaryotes but not in prokaryotes.18,19 Lower eukaryotes have only one isoform of NMT, whereas most mammals express two isozymes.19 In humans, NMT isozymes, NMT1 and NMT2, are expressed in most tissues and are essential for cell survival, regulation of immune responses, and HIV-1 infection.20,21,22,23,24,25,26

The molecular mechanism of protein N-myristoylation catalyzed by N-myristoyltransferase (NMT). A Cotranslational protein N-myristoylation and the Bi–Bi mechanism. After removal of the initiator methionine by methionine aminopeptidase 2 (MetAP2), NMT catalyzes the delivery of myristic acid from myristoyl-CoA to the N-terminal glycine residue of a protein. The reaction catalyzed by NMT follows an ordered Bi–Bi mechanism. After the recognition and binding of the fatty acid chain of myristoyl-CoA, the substrate-binding pocket is exposed. Subsequently, NMT recognizes and binds to the N-terminal sequence signal of the protein. Finally, NMT catalyzes protein N-myristoylation and releases products. Simultaneously, NMT returns to its previous conformation, in which the substrate-binding pocket is hidden. B Posttranslational protein N-myristoylation during apoptosis. P21-activated kinase 2 (PAK2) is cleaved by caspase 3 to produce caspase-truncated PAK2 (ctPAK2), which has a newly exposed glycine residue at the N-terminus. Then, NMT catalyzes the covalent attachment of myristic acid to the glycine residue of ctPAK2. Posttranslationally myristoylated ctPAK2 translocates to subcellular membrane compartments to induce apoptosis
Innate immunity is an evolutionarily conserved host defense mechanism27,28. In contrast to adaptive immunity, the innate immune system immediately responds to pathogens but does not provide long-lasting immunity to the host.29 The immediate pathogen response of innate immunity is achieved through multiple germline-encoded pattern-recognition receptors (PRRs) that recognize various pathogen-associated molecular patterns (PAMPs), including DNA and RNA from bacteria and viruses, and danger-associated molecular patterns (DAMPs).30,31,32,33,34,35,36,37 Downstream signaling cascades, including adaptors, kinases, and transcription factors, are activated via upstream signaling following the recognition of common pathogen constituents by PRRs. This leads to expression of antimicrobial genes and cytokines, including proinflammatory cytokines, chemokines, and type I interferons (IFNs), to recruit immune cells to infected sites and eliminate pathogens.38,39,40,41 Recent studies showing modulation of the TLR4 pathway by N-myristoylation,42 demyristoylation-induced innate immunosuppression during Shigella flexneri infection,43 and involvement of N-myristoylation in HIV-1 pathogenesis26 have resulted in increasing attention on the interplay between protein N-myristoylation and innate immunity.
In this review, we illustrate the basic principles of protein N-myristoylation and summarize recent important findings on the mechanisms underlying innate immune modulation by N-myristoylation. We also describe the vital role of myristoylation in viral assembly, intracellular host interactions, and spread during HIV-1 infection. Although the field of protein myristoylation is new and rapidly growing, this modification undoubtedly plays essential roles in human physiology as well as in important pathological conditions. Thus, there is a need for an updated overview of recent advances in the field to maximize our knowledge on protein N-myristoylation into therapeutic discovery.
The biology and mechanisms of protein myristoylation
N-myristoyltransferases (NMTs)
Both cotranslational and posttranslational protein myristoylation require the enzymes NMT and myristoyl-CoA to catalyze the transfer of a myristate group to a Gly residue of a protein. NMT belongs to the GCN5-related N-acetyltransferase protein family.6,44,45 Various studies have reported that NMT is a ubiquitous enzyme in eukaryotes, as it can be found in some lower eukaryotic organisms (e.g., Caenorhabditis elegans, Entamoeba histolytica, and Giardia lamblia),46,47,48 various land plants (e.g., Arabidopsis thaliana, Oryza sativa, and Lycopersicon esculentum),49,50 and almost all mammals.51,52 Conversely, prokaryotes do not possess NMTs, and these enzymes appear to have arisen as eukaryotes evolved from the archaeal common ancestor to the eukaryotic common ancestor.19 Some mammalian species (e.g., humans and mice) possess the two isozymes NMT1 and NMT2; lower eukaryotes only have one isoform.53 However, they all share a conserved sequence that constitutes the catalytic domain.
Human NMT1 (hNMT1) and NMT2 (hNMT2), which share only 76%–77% protein sequence identity, contain 496 and 498 amino acids, respectively (Fig. 2A).54 In addition, although hNMT2 is only phosphorylated at Ser38, hNMT1 can be phosphorylated at Ser31, Ser47, and Ser83 (Fig. 2A).55,56,57,58,59,60 hNMT1 and hNMT2 show a similar distribution in different organs, but expression of hNMT1 in the healthy heart, gut, kidney, liver, and other organs is higher than that of hNMT2.61 Studies on NMT1 and NMT2 enzyme kinetics and functions have found different substrate affinities for the two enzymes, with NMT1 playing more substantial roles in embryonic development, apoptosis, and cell proliferation.20,53,62,63 Overall, these findings suggest that NMT1 may exert major functions and require more complex regulation than NMT2.

Human NMTs and the three main myristoyl switch models. A Comparison of the functional domains and phosphorylated residue positions between human NMT1 (hNMT1) and human NMT2 (hNMT2). Both hNMT1 and hNMT2 have three similar myristoyl-CoA binding sites. hNMT2 is only phosphorylated at the Ser38 residue, whereas hNMT1 can be phosphorylated at Ser31, Ser47, and Ser83. B Schematic illustration showing the three main myristoyl switch models: myristoyl-electrostatic switch, myristoyl-ligand switch, and myristoyl-palmitoyl switch. In the myristoyl-electrostatic switch model, when MARCKS is phosphorylated by PKC, the phosphate motif reduces the positive charge on its surface, which results in its dissociation from the membrane to the cytosol. In the myristoyl-ligand switch model, the myristoyl group of recoverin is exposed following the binding of two Ca2+,ions by EF-hand motifs, which induces recoverin binding to the membrane; without Ca2+, the myristoyl moiety is hidden in the hydrophobic pocket. In the myristoyl-palmitoyl switch model, FRS2α requires conjugation to both a myristoyl group and a palmitoyl group for membrane targeting and kinase activity stimulation
The N-terminal amino acid sequences of NMT substrates are similar. In addition to containing a Gly at position one of the N-terminal ends, several myristoylated proteins require a Ser/Thr residue at the fifth position of the N-terminus (G-X-X-X-S/T-X-X-X-).64 NMTs also catalyze the myristoylation of proteins following the Bi–Bi mechanism (Fig. 1A).65 After recognizing myristoyl-CoA, NMT binds myristoyl-CoA to form the NMT-myristoyl-CoA complex, which triggers exposure of the substrate-binding pocket of the enzyme. Subsequently, NMT recognizes and binds to the N-terminal sequence signal of the protein. Finally, NMT catalyzes protein N-myristoylation and releases products; NMT simultaneously returns to its initial conformation, in which the substrate-binding pocket is hidden.
Myristoyl switches
As mentioned earlier, promoting the binding of proteins to the cell membrane is one of the most important functions of protein myristoylation. Hydrophobic insertion of the myristoyl chain into the lipid bilayer is necessary but not sufficient for the membrane binding of myristoylated proteins.66 A fundamental question in the field is as follows: how do cells or organisms regulate stable binding or reversible dissociation between myristoylated proteins and lipid bilayer membranes? Actually, with the help of a second signal, including phosphates, positively charged ions, or another modification, the myristoylated protein can be stably attached to or reversibly dissociate from the cell membrane.67 During this process, the myristoyl motif plays a “switch” role. Three main myristoyl switch models have been proposed: a myristoyl-electrostatic switch, myristoyl-ligand switch, and myristoyl-palmitoyl switch (Fig. 2B).17,67 The myristoyl-electrostatic switch refers to enhancing or preventing the binding of the myristoylated protein to the lipid membrane by affecting the protein’s surface charge.68 One of the best examples of an electrostatic-mediated myristoyl switch protein is myristoylated alanine-rich C kinase substrate (MARCKS). Following phosphorylation by protein kinase C (PKC), the phosphate motif reduces the positive charge on the surface of MARCKS, resulting in its dissociation from the membrane to the cytosol.69 In a few cases, the myristoyl moiety will be sequestered in a hydrophobic pocket following myristoylation. Conformational alterations triggered by specific ligands in such proteins will expose the myristoyl moiety, a mechanism known as the myristoyl-ligand switch. For instance, the myristoyl group of recoverin is exposed following the binding of two Ca2+,ions by EF-hand motifs, which induces recoverin binding to the membrane; without Ca2+, the myristoyl group is hidden in a hydrophobic pocket.70,71 Another example of a myristoyl-ligand switch involves ADP ribosylation factor 1 (ARF1).72,73 In this case, the myristoyl group of GDP-inactive ARF1 is sequestered but liberated by GTP exchange for membrane binding and signaling. Similar to the myristoyl group, the palmitoyl group, which is a 16-carbon fatty chain, can promote attachment of proteins to the cell membrane.74 Regarding the myristoyl-palmitoyl switch, some proteins require both a myristoyl group and palmitoyl group to target the membrane.75,76,77 For example, Barylko et al. demonstrated that stable membrane localization and kinase activity of fibroblast growth factor receptor substrate 2α (FRS2α) are mediated by myristoylation and palmitoylation and proved that N-myristoylation-negative mutants of FRS2α do not incorporate palmitate and therefore cannot attach to the plasma membrane.77
Lys myristoylation
Protein N-myristoylation is thought to occur exclusively at the N-terminal Gly residue. However, protein N-myristoylation also occurs at the epsilon amino groups of internal lysines of proteins.4,78 Recent studies have revealed that the transfer of myristate to the N-terminal ε-amino groups of lysine side chains is also catalyzed by NMT, expanding the range of known myristoylation substrates.79 Moreover, when α- and ε-amino groups are both available on the same peptide, NMT preferentially catalyzes Gly myristoylation rather than Lys myristoylation.79 Interestingly, in contrast to irreversible Gly myristoylation, several acyl hydrolases targeting Lys myristoylation have recently been found, including Sirtuin1/2/3/6 (SIRT1/2/3/6) and histone deacetylase 8/11 (HDAC8/11),78 rendering Lys myristoylation a reversible modification. This suggests that Lys myristoylation, as with other reversible modifications (e.g., palmitoylation, phosphorylation), may be intimately involved in regulating various signaling pathways. Overall, this reversible protein N-myristoylation mechanism provides an exciting field for the exploration of more functions of N-myristoylated proteins in human health and diseases.
Functions of protein myristoylation in human innate immune regulation and responses
Innate immunity
The human immune system is composed of innate immunity and adaptive immunity.80 T and B cells are the main adaptive immune cells, and they have several specific and unique receptors. Numerous B lymphocytes constitute multitudinous B-cell receptors (BCRs), thus increasing the probability of encountering an antigen that binds to a given BCR. The process of clonal expansion of B cells does, however, require 3–5 days, which may allow for enough time for the pathogen to cause damage.29 Compared with adaptive immunity, innate immunity can be activated quickly after pathogen invasion. Indeed, it is the body’s first line of defense against pathogen invasion until a sufficient number of antibodies are produced by B cells.81
When pathogens invade vertebrates, innate lymphocytes, such as macrophages and dendritic cells, recognize and bind to PAMPs82 or DAMPs83 through PRRs, inducing expression of proinflammatory factors, IFNs, and hundreds of interferon-stimulated genes.84,85,86 These effectors help the host to rapidly remove invading pathogens. PAMPs are highly conserved molecular structures unique to a group of specific microbial pathogens and their products. The best-known examples of PAMPs include single- or double-stranded RNA, viral DNA, and lipopolysaccharide (LPS) of gram-negative bacteria. As PAMPs are not produced by the host, they are regarded by innate immune cells as molecular characteristics of infection. A variety of PRRs have been discovered thus far. PRRs can be divided into two categories according to their cellular localization: the first type of PRRs localize to the cytoplasm of most cells, such as RIG-I-like receptors and cyclic GMP-AMP synthase;87 the second type of PRRs localize to the cell membrane, such as Toll-like receptors (TLRs).40,88
TLRs are mainly expressed on the plasma membrane of DCs, macrophages, neutrophils, and lymphocytes.40,88 Early studies of antifungal responses in Drosophila melanogaster were critical to the discovery of the Toll protein.89,90,91 Subsequently, the human homolog of Drosophila Toll was identified as TLR4.92 To date, multiple TLRs have been identified, including at least 13 members in mammals.93 TLRs detect microbial cell surface components; for example, TLR4 detects LPS,94 TLR5 senses flagellin,95,96 and TLRs 1, −2, and −6 recognize bacterial lipoproteins.97,98,99,100 Other TLRs also detect nucleic acids, such as double-stranded RNA (TLR3),101 single-stranded RNA (TLR7 and 8),102,103,104,105 unmethylated CpG containing single-stranded DNA (TLR9),106 and bacterial ribosomal RNA (TLR13).107 After recognizing ligands, TLRs recruit adaptor molecules to induce expression of inflammatory factors or IFNs. This ensemble of inflammatory factors and IFNs promote inflammation and host defense.
Myristoylation regulates TRIF-related adaptor molecule (TRAM) and TLR4-dependent inflammatory responses
TLR4-dependent inflammatory responses are activated by LPS, a complex glycolipid that represents a major constituent of the gram-negative bacterial cell wall.108 Upon binding LPS, TLR4 dimerizes and recruits all four types of Toll/IL-1 resistance (TIR) domain-containing adaptor molecules:109 myeloid differentiation factor 88 (MyD88);110 MyD88 adaptor-like;111,112 TIR domain-containing adaptor-inducing IFN-β (TRIF);113,114,115,116 and TRIF-related adaptor molecule (TRAM).117,118 Among these TIR domain-containing adaptor molecules, TRAM functions exclusively in the TLR4 pathway.118 Recently, Rowe reported that TRAM-mediated regulation of TLR4 signal transduction depends on myristoylation.42 TRAM colocalizes with TLR4 on the plasma membrane through its N-terminal myristoyl group.42 Upon detection of dimerized TLR4, TRAM dissociates from TLR4 and the membrane to interact with TRIF and further activate the NF-κB and IRF3 pathways.42 TRAM relies on two main myristoyl-dependent mechanisms to dissociate from the membrane and regulate TLR4 signaling (Fig. 3). The first mechanism is the myristoyl-electrostatic switch of TRAM. A recent study demonstrated that TRAM is phosphorylated by PKCε during TLR4 signaling. Following LPS stimulation, TRAM is transiently phosphorylated by PKCε on Ser-16 near its N-terminus,119 and this phosphorylation event adjacent to the myristoyl group reduces the electrostatic interaction between multiple residues and the phospholipid head groups, leading to dissociation of TRAM from the membrane. This process is not only essential for the activation of IRF3 and NF-κB but also, more importantly, constitutes a negative feedback mechanism for the TLR4 signaling pathway, avoiding excessive production of inflammatory cytokines. Notably, TRAM also undergoes tyrosine phosphorylation at Tyr167 within its TIR domain following LPS stimulation.120 However, Tyr167 phosphorylation mainly serves to activate downstream signals and is not involved in the myristoyl-electrostatic switch of TRAM.

The role of myristoylation in regulating TRIF-related adaptor molecule (TRAM) during LPS-induced TLR4 inflammatory responses. TRAM is myristoylated and anchored to the plasma membrane. After LPS stimulation, TLR4 dimerizes to recruit the TIR domains of cytoplasmic adaptor molecules, including Mal, TRAM, MyD88, and TRIF, which further activate NF-κB and IRF3 signaling to produce the proinflammatory cytokine IFNβ. Notably, following interaction with TLR4, TRAM is transiently phosphorylated by PKCε on Ser-16, which is near its N-terminus. This phosphorylation event adjacent to the myristoyl group reduces the electrostatic interaction between multiple residues and phospholipid head groups, leading to TRAM dissociation from the membrane. This process is not only essential for the activation of IRF3 and NF-κB but also, more importantly, constitutes a negative feedback mechanism of the TLR4 signaling pathway, avoiding excessive expression of inflammatory cytokines. In addition, LPS-induced activation of the TLR4 pathway promotes the expression and activation of heme oxygenase-2 (HO-2, a myristate-binding protein). HO-2 binds to the myristoyl group and inhibits the function of TRAM, thereby negatively regulating TLR4 signaling. It is not yet known how or whether proteins trapped by HO-2 are released. If they are not released, we speculate that the HO-2-TRAM complex will be degraded via the ubiquitin-mediated protein degradation pathway or delivered to another membrane with the help of an unknown cofactor
The other mechanism by which myristoylated TRAM regulates TLR4 signaling involves the myristate-binding protein heme oxygenase-2 (HO-2) (Fig. 3). Heme oxygenase (HO) catalyzes degradation of heme to generate biliverdin.121 There are two main isoforms of HO: inducible HO-1 and constitutively expressed HO-2.122 Constitutively expressed HO-2 can be found in all cell types, whereas HO-1 is only highly expressed following oxidative stress or induction by inflammatory stimuli.123 Increasing evidence indicates that HO-2 is vital for the timely shutdown of inflammatory responses.124 In contrast to normal mice, HO-2-knockout mice show a defect in wound healing and severe inflammation.125. The LPS-TLR4 inflammatory response in mouse cerebral vascular endothelial cells is inhibited by overexpression of HO-2, though it is enhanced by RNAi-mediated knockdown of HO-2.126 In an exciting new study, Zhu et al. identified HO-2 as a myristate-binding protein that binds to the myristoyl group of TRAM to negatively regulate the LPS-TLR4 pathway. Activation of TLR4 signaling boosts expression of inflammatory cytokines and promotes expression and activation of HO-2. In turn, HO-2 binds to the myristoyl group and inhibits the function of TRAM. Structural analysis has demonstrated that the myristoyl group is inserted almost completely into a deep hydrophobic pocket of HO-2. Moreover, depleting HO-2 or blocking access to its myristate pocket enhances hyperresponsive LPS-TLR4 signaling.127 This work not only illustrates the regulatory mechanism of HO-2 in the LPS-TLR4 pathway but also raises some interesting questions. For example, why is constitutively expressed HO-2, but not inducible HO-1, a myristate-binding protein? Is TRAM trapped within HO-2 released, and how does this occur? If it is not released, is it degraded by the proteasome or delivered to another membrane? These important questions remain to be answered.
Demyristoylation-induced innate immunosuppression in Shigella flexneri infection
To escape host innate and adaptive immune system supervision, microbial pathogens produce virulence proteins (also called effectors) to hijack host cellular signal transduction.128 Bacteria employ specialized needle-like delivery systems to secrete effectors into the host cell, including the type III and IV secretion systems (T3SS and T4SS, respectively).129 Effectors are translocated through either the T3SS or T4SS, whereas DNA is only transferred through the T4SS.130 The effectors are inactive in the bacterium but become activated by eukaryotic activators in host cells following delivery.129 Activated effectors manipulate signal transduction pathways to induce immunosuppression, thereby potentiating their replication.129
For a long time, protein N-terminal Gly myristoylation has been regarded as an irreversible, stable modification. However, in a recent study, Burnaevskiy et al. revealed that invasion plasmid antigen J (IpaJ), a T3SS effector protein secreted by Shigella flexneri, is a demyristoylating enzyme that hydrolyzes the myristoyl group from the N-terminal Gly of human ARF1.43 Functionally, ARF1 couples membrane binding (by the N-terminal Gly-myristoyl group) to regulate cargo transport through the Golgi apparatus, which is essential for the secretion of cytokines and chemokines.131 As mentioned earlier, ARF1 has a myristoyl-ligand switch.72,131 In GDP-bound states, the myristoyl moiety is sequestered in a hydrophobic pocket of ARF1, and the protein is inactive.131 After exchange from the GDP- to GTP-bound state, the myristoyl group is exposed from the hydrophobic pocket, allowing ARF1 to bind to the membrane and recruit target proteins, thereby triggering transport vesicle formation.131 During Shigella flexneri infection, IpaJ is translocated through the T3SS into host cells (Fig. 4). IpaJ will recognize and remove the myristoyl group of ARF1, which leads to ARF1 disassociation from Golgi membranes, thus inhibiting the secretion of cytokines and chemokines.43 Hence, infected host cells cannot release these signaling molecules to warn other cells or recruit immune cells to clear the pathogen Shigella flexneri. In addition to ARF1, IpaJ removes the myristoyl moiety from numerous N-terminal Gly-myristoylated proteins in vitro, including c-Src, MARCKS, hVPS15, and GRASP65,43 suggesting that IpaJ might also target multiple myristoylated proteins during Shigella infection. However, whole-cell myristoylome profiling to identify physiological substrates of IpaJ during Shigella infection has revealed that IpaJ targets only some members of the ARF family (ARF1-5 but not ARF6) and members of the related ARF-like (ARL) family.132 Further analysis showed that to cleave the substrate, IpaJ not only requires the presence of myristoyl but also must interact with structural elements and residues utilized by ARF effector proteins.132 In addition, IpaJ displays selectivity toward Golgi-associated ARFs.132 ARF1-5 localizes to the Golgi and functions to regulate trafficking between the Golgi and endoplasmic reticulum.133 ARF6, however, resides at the plasma membrane and is the only member of the ARF family that is not demyristoylated by IpaJ during Shigella infection.132 A recent study may explain why IpaJ cannot demyristoylate ARF6. Unlike other proteins that are myristoylated at N-terminal Gly residues, ARF6 is myristoylated at an N-terminal ε lysine residue by NMTs.134 As IpaJ can only cleave the peptide bond between N-myristoylated Gly and the N-terminal second amino acid residue,132 ARF6 is not efficiently cleaved by IpaJ. Interestingly, the myristoyl group of ARF6 can be removed by the NAD+-dependent deacylase SIRT2. Moreover, SIRT2 binds preferentially to GDP-bound ARF6, while NMTs prefer to bind GTP-bound ARF6. Therefore, the Lys-myristoylation–demyristoylation cycle can couple to and promote the GTPase cycle of ARF6.134 Nevertheless, whether bacterial Lys demyristoylases exist and the relationship between Lys myristoylation and innate immunity remain unknown. Although an initial framework has been formed from these findings to explore the multiple functions of protein N-myristoylation in human innate immunity, additional studies in this field are necessary to develop more effective and safer antimicrobial drugs.

Demyristoylation-induced innate immunosuppression in Shigella flexneri pathogenesis. During infection, Shigella flexneri secretes various molecules into host cells to manipulate and control host cellular processes. These effectors (virulence factors, including IpaJ, IpgD, etc.) are translocated through a T3SS or T4SS, whereas DNA is only transferred through the T4SS. The host cell also senses Shigella flexneri invasion and triggers expression of immune genes, including various cytokines and chemokines. Golgi-associated ARF family GTPases (ARF1-5) are vital for the secretion of these cytokines and chemokines. Moreover, these ARFs all have the myristoyl-ligand switch. IpaJ recognizes GTP-ARFs and inactivates them by irreversibly releasing them from Golgi membranes via demyristoylation, thereby inhibiting the secretion of cytokines and chemokines
Myristoylation and HIV-1 pathogenesis
In addition to its indispensable roles in protein stability, cellular signal transduction, and targeting of proteins to plasma membrane systems, myristoylation is vital for viral invasion, assembly, intracellular host interactions, and budding.135 Viruses lack the enzyme NMT, which is required for N-myristoylation; therefore, their proteins are myristoylated by eukaryotic NMTs. Apart from a few exceptions,136,137 the Gag proteins of nearly all retroviruses are cotranslationally myristoylated at the amino-terminal Gly of the matrix domain (MA).138,139 Pharmacological inhibition of NMTs or mutation of the Gag MA domain to prevent myristoylation suppresses the assembly and budding of HIV-1 in host cells.140,141 Thus, N-myristoylation of Gag proteins is vital for HIV-1 replication. During the late stages of infection, Gag, which consists of the four domains p17 matrix (MA), p24 capsid (CA), p7 nucleocapsid (NC), and p6 starts to be synthesized in host cells142 (Fig. 5A). However, the myristoyl group of newly synthesized Gag is partially (~60%) sequestered in the MA domain, preventing the interaction between Gag and membranes.143 In the late stages of infection, the myristate groups of Gag need to be exposed to be able to bind to the plasma membrane and promote assembly. A recent study showed that Gag multimerization triggers a conformational switch from the myristoyl-sequestered state to the myristoyl-exposed state.143 The interaction between the viral RNA template and the NC domain promotes Gag multimerization, thereby exposing their myristoyl moieties,143 and Gag oligomers will then be able to bind to the plasma membrane via their myristoyl groups and direct particle assembly. During or shortly after budding, the viral protease is activated to cleave Gag, which liberates the mature MA, CA, and NC proteins and the unstructured peptides p2, p1, and p6 for viral maturation.144 In addition to TRAM, HO-2, a newly discovered cellular myristate-binding protein, can specifically bind to the myristoyl moiety of HIV-1 Gag, thereby inhibiting the association between HIV-1 Gag and the membrane.127 Inhibition of HO-2 myristate-binding activity by mutation or with the use of a noncleavable substrate analog significantly enhances the production of HIV-1.127. These findings indicate that negative regulation of HIV-1 Gag by HO-2 may be a host cell innate defense mechanism during virus infection. Regardless, it is unclear how intracellular HO-2 is activated and binding specificity is achieved during HIV-1 infection. Answering these important questions is necessary to develop new anti-HIV-1 drugs.

The myristoylated proteins Gag and Nef promote the invasion and spread of human immunodeficiency virus-1 (HIV-1). A Upon invading a host cell, HIV integrates its genome into the host genome, enabling viral protein expression. The viral proteins Nef and Gag are cotranslationally myristoylated by the host’s NMT. The N-terminal myristate of newly synthesized Gag is partially (≈60%) trapped in the MA domain, limiting contact between Gag and membranes. The interaction between the viral RNA template and the NC domain promotes Gag multimerization and exposes myristoyl groups, thus rendering Gag oligomers able to bind to the plasma membrane and direct particle assembly. During viral maturation, the viral protease is activated to cleave Gag. In addition, myristoylated Nef mediates downregulation of CD4 and MHC-I receptors on the host cell surface. To prevent premature apoptosis of infected cells, Nef inhibits the activity of ASK1. HO-2 can bind the myristate moiety of Gag and inhibit its activity, though it is not clear whether HO-2 is able to bind the myristate of Nef and inhibit its activity. B Nef inhibits the endocytosis of dendritic cells (DCs) to upregulate the surface level of DC-SIGN, which promotes efficient spread of HIV-1 in uninfected DCs and T cells. Nef also induces the production of the CC chemokines macrophage inflammatory protein 1α and 1β and other soluble factors (soluble CD23 and soluble ICAM) in macrophages to recruit T cells and facilitate productive transmission of HIV-1 infection
A previous study described a protein that negatively regulates virus replication: Nef.145 Interestingly, subsequent research found that the Nef protein can promote virus replication and infection.146,147 After being N-terminal Gly-myristoylated by host NMTs, HIV-1 Nef is found in the cytoplasm and plasma membrane.148,149 HIV-1 Nef promotes HIV-1 invasion and spread by several mechanisms (Fig. 5A): (1) by activating CD4+ T lymphocytes, Nef promotes viral spread among CD4+ T lymphocytes;150 (2) to prevent premature apoptosis of infected cells, Nef inhibits the activity of ASK1151,152; and (3) Nef downregulates major histocompatibility complex class I (MHC-I), which is essential for cytotoxic T lymphocyte (CTL) recognition, to prevent the death of HIV-1-infected cells by CTLs.153 Moreover, Nef is involved in the downregulation of CD4+ receptors from the cell surface154. NMT1 has been shown to preferentially associate with HIV-1 Nef; moreover, the myristoyl group of Nef is essential for its activity, and mutation of the N-terminal myristoylation site on Nef cripples its activities. Overall, the myristoylation of Nef may play a vital role in HIV-1 activities.
Nef is also critical for the spread of HIV-1. Following recognition and binding of HIV-1 by a DC-specific receptor (DC-SIGN), DCs deliver viral particles to target cells without becoming effectively infected. Nonetheless, DCs may be infected by viral particles, further promoting the efficient spread of HIV to other uninfected DCs and T cells by Nef (Fig. 5B).155 In addition to DCs, Nef facilitates the productive transmission of HIV-1 infection from macrophages to T cells.156,157 Nef induces the production of the CC chemokines macrophage inflammatory protein 1α and 1β as well as other soluble factors (soluble CD23 and soluble ICAM) in macrophages to recruit T cells and facilitate productive transmission of HIV-1 infection (Fig. 5B). However, it is unclear whether the myristoyl moiety of Nef is essential for the induction of these chemokines and factors. Moreover, the role of myristoylation in the Nef-mediated spread of HIV-1 infection remains to be fully elucidated.
Conclusions and perspectives
Over the past decade, N-myristoylation has become an attractive area of protein modification research. The exciting evidence obtained from structural, genetic, and biomedical studies has unquestionably revealed that protein N-myristoylation plays nonnegligible functions in broad biological processes, such as apoptosis, hematopoiesis, carcinogenesis, immune response, and viral infections.26,158,159 Despite some breakthroughs, an increasing number of unknown areas have emerged, including protein Lys myristoylation, demyristoylation, and novel cellular myristate-binding proteins.
Protein N-myristoylation is well established as Gly myristoylation, but the new area of Lys myristoylation remains mostly unexplored. Although a large number of Gly-myristoylated proteins involved in various biological processes have been identified, Lys-myristoylated proteins have rarely been found. As Lys myristoylation is reversible, several important proteins in various signaling pathways may undergo this modification. With the improvement of mass spectrometry, not only will more Lys-myristoylated proteins be identified, but our knowledge about the functions of protein myristoylation in cellular activities will also increase. Furthermore, both Gly myristoylation and Lys myristoylation are catalyzed by NMTs, but the catalytic mechanism and substrate specificity of NMTs require further investigation.
Recent discoveries regarding the regulation of Gly myristoylation during infection have revealed IpaJ as the first Gly-demyristoylating enzyme.43,132 These findings point to novel directions with regard to the dynamic regulation of Gly myristoylation and pathogen interference of host protein myristoylation. However, our understanding of the regulation of Gly myristoylation is still in its infancy, and it remains to be determined whether other microbial proteases cleave Gly myristoylation as a pathogenic mechanism. A better understanding of the dynamic cellular regulation of Gly myristoylation by pathogens would deepen our knowledge of the mechanisms of pathogen invasion. In addition, evidence has identified HO-2 as a negative regulator of virus replication and demonstrated that the LPS-TLR4 signaling pathway is involved in binding of N-myristoylated Gag and TRAM.127 Nevertheless, how the binding specificity and activities of HO-2 are regulated in a timely manner is still largely unknown. More in-depth studies on the regulation of HO-2 may reveal new perspectives on how the host defends itself against pathogen invasion.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a novel coronavirus, has caused the coronavirus disease 2019 (COVID-19) global pandemic.160 Following the identification of the first case in China, many studies have been conducted to understand this novel virus.161,162,163,164,165 SARS-CoV-2 has four main structural proteins: membrane (M), envelope (E), nucleocapsid (N), and spike (S).166 Among them, the S protein is the most important for the virus to successfully invade cells. S proteins decorate the surface of the virus, and viral entry into cells is initiated when these proteins bind to the host angiotensin-converting enzyme 2 receptor.162,167 Accordingly, many antibody therapies in development target the S proteins.168,169,170,171,172 However, there are few studies on other therapeutic targets of SARS-CoV-2. Although there is no known protein motif for Gly myristoylation of coronavirus proteins,173 the recently discovered lysine myristoylation protein indicates potential SARS-CoV-2 protein myristoylation. This is limited to speculation, and there is no report confirming it. Therefore, it is necessary to determine the relationship between the pathogenesis of SARS-CoV-2 and protein N-myristoylation, which may constitute a target in the development of COVID-19 therapeutic drugs.
Finally, as protein N-myristoylation is crucial for virus assembly and spread, host NMT may be an exciting target for developing antiviral drugs.
References
Casey, P. Protein lipidation in cell signaling. Science 268, 221–225 (1995).
Chen, B., Sun, Y., Niu, J., Jarugumilli, G. K. & Wu, X. Protein lipidation in cell signaling and diseases: function, regulation, and therapeutic opportunities. Cell Chem. Biol. 25, 817–831 (2018).
Nadolski, M. J. & Linder, M. E. Protein lipidation. FEBS J. 274, 5202–5210 (2007).
Jiang, H. et al. Protein lipidation: occurrence, mechanisms, biological functions, and enabling technologies. Chem. Rev. 118, 919–988 (2018).
Boutin, J. A. Myristoylation. Cell. Signal. 9, 15–35 (1997).
Farazi, T. A., Waksman, G. & Gordon, J. I. The biology and enzymology of protein N-myristoylation. J. Biol. Chem. 276, 39501–39504 (2001).
Zha, J., Weiler, S., Oh, K. J., Wei, M. C. & Korsmeyer, S. J. Posttranslational N-myristoylation of BID as a molecular switch for targeting mitochondria and apoptosis. Science 290, 1761–1765 (2000).
Martin, D. D. O., Beauchamp, E. & Berthiaume, L. G. Post-translational myristoylation: fat matters in cellular life and death. Biochimie 93, 18–31 (2011).
Adam, R. M. et al. Cholesterol sensitivity of endogenous and myristoylated Akt. Cancer Res. 67, 6238–6246 (2007).
Hu, T. et al. Myristoylated Naked2 antagonizes Wnt-β-Catenin activity by degrading Dishevelled-1 at the plasma membrane. J. Biol. Chem. 285, 13561–13568 (2010).
Schwertassek, U. et al. Myristoylation of the dual-specificity phosphatase c-JUN N-terminal kinase (JNK) stimulatory phosphatase 1 is necessary for its activation of JNK signaling and apoptosis. FEBS J. 277, 2463–2473 (2010).
Tan, Y. W., Hong, W. J. & Chu, J. J. H. Inhibition of enterovirus VP4 myristoylation is a potential antiviral strategy for hand, foot and mouth disease. Antivir. Res. 133, 191–195 (2016).
Wright, M. H., Heal, W. P., Mann, D. J. & Tate, E. W. Protein myristoylation in health and disease. J. Chem. Biol. 3, 19–35 (2010).
McIlhinney, R. A. J. Protein Targeting Protocols (ed. Clegg, R. A.) 211–225 (Humana Press, 1998).
Maurer-Stroh, S. et al. MYRbase: analysis of genome-wide glycine myristoylation enlarges the functional spectrum of eukaryotic myristoylated proteins. Genome Biol. 5, R21 (2004).
Timms, R. T. et al. A glycine-specific N-degron pathway mediates the quality control of protein N-myristoylation. Science 365, eaaw4912 (2019).
Ames, J. B., Tanaka, T., Stryer, L. & Ikura, M. Portrait of a myristoyl switch protein. Curr. Opin. Struct. Biol. 6, 432–438 (1996).
Towler, D. A. et al. Purification and characterization of yeast myristoyl CoA:protein N-myristoyltransferase. Proc. Natl Acad. Sci. 84, 2708–2712 (1987).
Meinnel, T., Dian, C. & Giglione, C. Myristoylation, an ancient protein modification mirroring eukaryogenesis and evolution. Trends Biochem. Sci. 45, 619–632 (2020).
Ducker, C. E., Upson, J. J., French, K. J. & Smith, C. D. Two N-myristoyltransferase isozymes play unique roles in protein myristoylation, proliferation, and apoptosis. Mol. Cancer Res. 3, 463–476 (2005).
Bouamr, F., Scarlata, S. & Carter, C. Role of myristylation in HIV-1 Gag assembly. Biochemistry 42, 6408–6417 (2003).
Resh, M. D. A myristoyl switch regulates membrane binding of HIV-1 Gag. Proc. Natl Acad. Sci. U. S. A. 101, 417–418 (2004).
Li, H., Dou, J., Ding, L. & Spearman, P. Myristoylation is required for human immunodeficiency virus type 1 Gag-Gag multimerization in mammalian cells. J. Virol. 81, 12899–12910 (2007).
Takamune, N., Tanaka, T., Takeuchi, H., Misumi, S. & Shoji, S. Down-regulation of N-myristoyl transferase expression in human T-cell line CEM by human immunodeficiency virus type-1 infection. FEBS Lett. 506, 81–84 (2001).
Takamune, N., Hamada, H., Misumi, S. & Shoji, S. Novel strategy for anti-HIV-1 action: selective cytotoxic effect of N-myristoyltransferase inhibitor on HIV-1-infected cells. FEBS Lett. 527, 138–142 (2002).
Udenwobele, D. I. et al. Myristoylation: an important protein modification in the immune response. Front Immunol. 8, 751 (2017).
Muthamilarasan, M. & Prasad, M. Plant innate immunity: an updated insight into defense mechanism. J. Biosci. 38, 433–449 (2013).
Riera Romo, M., Pérez-Martínez, D. & Castillo Ferrer, C. Innate immunity in vertebrates: an overview. Immunology 148, 125–139 (2016).
Kaur, B. P. & Secord, E. Innate immunity. Pediatr. Clin. North Am. 66, 905–911 (2019).
Akira, S., Uematsu, S. & Takeuchi, O. Pathogen recognition and innate immunity. Cell 124, 783–801 (2006).
Kato, H. et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105 (2006).
Hornung, V. et al. 5’-Triphosphate RNA is the ligand for RIG-I. Science 314, 994–997 (2006).
Pichlmair, A. et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science 314, 997–1001 (2006).
Yoneyama, M. et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5, 730–737 (2004).
Alexopoulou, L., Holt, A. C., Medzhitov, R. & Flavell, R. A. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 413, 732–738 (2001).
Barrat, F. J., Elkon, K. B. & Fitzgerald, K. A. Importance of nucleic acid recognition in inflammation and autoimmunity. Annu. Rev. Med. 67, 323–336 (2016).
Hornung, V., Hartmann, R., Ablasser, A. & Hopfner, K.-P. OAS proteins and cGAS: unifying concepts in sensing and responding to cytosolic nucleic acids. Nat. Rev. Immunol. 14, 521–528 (2014).
Kawai, T. & Akira, S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384 (2010).
Paludan, S. R., Bowie, A. G., Horan, K. A. & Fitzgerald, K. A. Recognition of herpesviruses by the innate immune system. Nat. Rev. Immunol. 11, 143–154 (2011).
Takeuchi, O. & Akira, S. Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010).
Takeuchi, O. & Akira, S. Innate immunity to virus infection. Immunol. Rev. 227, 75–86 (2009).
Rowe, D. C. et al. The myristoylation of TRIF-related adaptor molecule is essential for Toll-like receptor 4 signal transduction. Proc. Natl Acad. Sci. U. S. A. 103, 6299–6304 (2006).
Burnaevskiy, N. et al. Proteolytic elimination of N-myristoyl modifications by the Shigella virulence factor IpaJ. Nature 496, 106–109 (2013).
Vetting, M. W. et al. Structure and functions of the GNAT superfamily of acetyltransferases. Arch. Biochem. Biophys. 433, 212–226 (2005).
Raju, R. V. S., Magnuson, B. A. & Sharma, R. K. Mammalian myristoyl CoA: protein N-myristoyltransferase. Mol. Cell. Biochem. 149, 191–202 (1995).
The C. elegans Sequencing Consortium. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282, 2012–2018 (1998).
Maurer-Stroh, S., Eisenhaber, B. & Eisenhaber, F. N-terminal N-myristoylation of proteins: refinement of the sequence motif and its taxon-specific differences11Edited by J. Thornton. J. Mol. Biol. 317, 523–540 (2002).
Šarić, M. et al. Dual acylation accounts for the localization of α19-Giardin in the ventral Flagellum pair of Giardia lamblia. Eukaryot. Cell 8, 1567–1574 (2009).
Qi, Q. et al. Molecular cloning, genomic organization, and biochemical characterization of myristoyl-CoA:ProteinN-Myristoyltransferase from Arabidopsis thaliana. J. Biol. Chem. 275, 9673–9683 (2000).
Podell, S. & Gribskov, M. Predicting N-terminal myristoylation sites in plant proteins. BMC Genom. 5, 37 (2004).
King, M. J. & Sharma, R. K. Demonstration of multiple forms of bovine brain myristoyl CoA: protein N-myristoyl transferase. Mol. Cell. Biochem. 113, 77–81 (1992).
Glover, C. J., Goddard, C. & Felsted, R. L. N-myristoylation of p60src. Identification of a myristoyl-CoA:glycylpeptide N-myristoyltransferase in rat tissues. Biochem. J. 250, 485–491 (1988).
Giang, D. K. & Cravatt, B. F. A second mammalian N-Myristoyltransferase. J. Biol. Chem. 273, 6595–6598 (1998).
Selvakumar, P. et al. Potential role of N-myristoyltransferase in cancer. Prog. Lipid Res. 46, 1–36 (2007).
Zhou, H. et al. Toward a comprehensive characterization of a human cancer cell phosphoproteome. J. Proteome Res. 12, 260–271 (2013).
Bian, Y. et al. An enzyme assisted RP-RPLC approach for in-depth analysis of human liver phosphoproteome. J. Proteom. 96, 253–262 (2014).
Beausoleil, S. A., Villén, J., Gerber, S. A., Rush, J. & Gygi, S. P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 (2006).
Dephoure, N. et al. A quantitative atlas of mitotic phosphorylation. Proc. Natl Acad. Sci. 105, 10762–10767 (2008).
Rigbolt, K. T. G. et al. System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci. Signal. 4, rs3 (2011).
Olsen, J. V. et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3 (2010).
Wang, D. et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 15, e8503 (2019).
Yang, S. H. et al. N-myristoyltransferase 1 is essential in early mouse development. J. Biol. Chem. 280, 18990–18995 (2005).
Shrivastav, A. et al. Requirement of N-myristoyltransferase 1 in the development of monocytic lineage. J. Immunol. 180, 1019–1028 (2008).
Johnson, D. R., Bhatnagar, R. S., Knoll, L. J. & Gordon, J. I. Genetic and biochemical studies of protein N-myristoylation. Annu. Rev. Biochem. 63, 869–914 (1994).
Rudnick, D. A. et al. Kinetic and structural evidence for a sequential ordered Bi Bi mechanism of catalysis by Saccharomyces cerevisiae myristoyl-CoA:protein N-myristoyltransferase. J. Biol. Chem. 266, 9732–9739 (1991).
Peitzsch, R. M. & McLaughlin, S. Binding of acylated peptides and fatty acids to phospholipid vesicles: pertinence to myristoylated proteins. Biochemistry 32, 10436–10443 (1993).
Resh, M. D. Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim. Biophys. Acta 1451, 1–16 (1999).
McLaughlin, S. & Aderem, A. The myristoyl-electrostatic switch: a modulator of reversible protein-membrane interactions. Trends Biochem. Sci. 20, 272–276 (1995).
Braun, T., McIlhinney, R. A. & Vergères, G. Myristoylation-dependent N-terminal cleavage of the myristoylated alanine-rich C kinase substrate (MARCKS) by cellular extracts. Biochimie 82, 705–715 (2000).
Tanaka, T., Ames, J. B., Harvey, T. S., Stryer, L. & Ikura, M. Sequestration of the membrane-targeting myristoyl group of recoverin in the calcium-free state. Nature 376, 444–447 (1995).
Ames, J. B., Porumb, T., Tanaka, T., Ikura, M. & Stryer, L. Amino-terminal myristoylation induces cooperative calcium binding to recoverin. J. Biol. Chem. 270, 4526–4533 (1995).
Goldberg, J. Structural basis for activation of ARF GTPase: mechanisms of guanine nucleotide exchange and GTP-myristoyl switching. Cell 95, 237–248 (1998).
Liu, Y., Kahn, R. A. & Prestegard, J. H. Structure and membrane interaction of myristoylated ARF1. Structure 17, 79–87 (2009).
Linder, M. E. & Deschenes, R. J. Palmitoylation: policing protein stability and traffic. Nat. Rev. Mol. Cell Biol. 8, 74–84 (2007).
Cadwallader, K. A., Paterson, H., Macdonald, S. G. & Hancock, J. F. N-terminally myristoylated Ras proteins require palmitoylation or a polybasic domain for plasma membrane localization. Mol. Cell Biol. 14, 4722–4730 (1994).
Martín, M. L. & Busconi, L. Membrane localization of a rice calcium-dependent protein kinase (CDPK) is mediated by myristoylation and palmitoylation. Plant J. 24, 429–435 (2000).
Barylko, B. et al. Myristoylation-dependent palmitoylation of the receptor tyrosine kinase adaptor FRS2α. Biochemistry 58, 2809–2813 (2019).
Cao, J. et al. HDAC11 regulates type I interferon signaling through defatty-acylation of SHMT2. Proc. Natl Acad. Sci. U. S. A. 116, 5487–5492 (2019).
Dian, C. et al. High-resolution snapshots of human N-myristoyltransferase in action illuminate a mechanism promoting N-terminal Lys and Gly myristoylation. Nat. Commun. 11, 1132 (2020).
Iwasaki, A. & Medzhitov, R. Regulation of adaptive immunity by the innate immune system. Science 327, 291–295 (2010).
Adams, N. M., Grassmann, S. & Sun, J. C. Clonal expansion of innate and adaptive lymphocytes. Nat. Rev. Immunol. 20, 694–707 (2020).
Medzhitov, R. & Janeway, C. A. Jr Decoding the patterns of self and nonself by the innate immune system. Science 296, 298–300 (2002).
Roers, A., Hiller, B. & Hornung, V. Recognition of endogenous nucleic acids by the innate immune system. Immunity 44, 739–754 (2016).
Chen, W. et al. Induction of Siglec-G by RNA viruses inhibits the innate immune response by promoting RIG-I degradation. Cell 152, 467–478 (2013).
Ran, Y. et al. SENP2 negatively regulates cellular antiviral response by deSUMOylating IRF3 and conditioning it for ubiquitination and degradation. J. Mol. Cell Biol. 3, 283–292 (2011).
Levy, D. E., Marié, I. J. & Durbin, J. E. Induction and function of type I and III interferon in response to viral infection. Curr. Opin. Virol. 1, 476–486 (2011).
Goubau, D., Deddouche, S. & Reis e Sousa, C. Cytosolic sensing of viruses. Immunity 38, 855–869 (2013).
Dambuza, I. M. & Brown, G. D. C-type lectins in immunity: recent developments. Curr. Opin. Immunol. 32, 21–27 (2015).
Gay, N. J. & Keith, F. J. Drosophila Toll and IL-1 receptor. Nature 351, 355–356 (1991).
Steward, R. Dorsal, an embryonic polarity gene in Drosophila, is homologous to the vertebrate proto-oncogene, c-rel. Science 238, 692–694 (1987).
Schneider, D. S., Hudson, K. L., Lin, T. Y. & Anderson, K. V. Dominant and recessive mutations define functional domains of Toll, a transmembrane protein required for dorsal-ventral polarity in the Drosophila embryo. Genes Dev. 5, 797–807 (1991).
Medzhitov, R., Preston-Hurlburt, P. & Janeway, C. A. Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388, 394–397 (1997).
Brubaker, S. W., Bonham, K. S., Zanoni, I. & Kagan, J. C. Innate immune pattern recognition: a cell biological perspective. Annu. Rev. Immunol. 33, 257–290 (2015).
Poltorak, A. et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088 (1998).
Gewirtz, A. T., Navas, T. A., Lyons, S., Godowski, P. J. & Madara, J. L. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J. Immunol. 167, 1882–1885 (2001).
Hayashi, F. et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410, 1099–1103 (2001).
Kang, J. Y. et al. Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity 31, 873–884 (2009).
Takeuchi, O. et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11, 443–451 (1999).
Takeuchi, O. et al. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int Immunol. 13, 933–940 (2001).
Takeuchi, O. et al. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J. Immunol. 169, 10–14 (2002).
Alexopoulou, L., Holt, A. C., Medzhitov, R. & Flavell, R. A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413, 732–738 (2001).
Diebold, S. S., Kaisho, T., Hemmi, H., Akira, S. & Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303, 1529–1531 (2004).
Greulich, W. et al. TLR8 is a sensor of RNase T2 degradation products. Cell 179, 1264–1275.e13 (2019).
Heil, F. et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 303, 1526–1529 (2004).
Hemmi, H. et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat. Immunol. 3, 196–200 (2002).
Hemmi, H. et al. A Toll-like receptor recognizes bacterial DNA. Nature 408, 740–745 (2000).
Hidmark, A., von Saint Paul, A. & Dalpke, A. H. Cutting edge: TLR13 is a receptor for bacterial RNA. J. Immunol. 189, 2717–2721 (2012).
Raetz, C. R. H. & Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71, 635–700 (2002).
Vogel, S. N. & Fenton, M. Toll-like receptor 4 signalling: new perspectives on a complex signal-transduction problem. Biochem Soc. Trans. 31, 664–668 (2003).
Muzio, M., Ni, J., Feng, P. & Dixit, V. M. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science 278, 1612–1615 (1997).
Fitzgerald, K. A. et al. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature 413, 78–83 (2001).
Horng, T., Barton, G. M., Flavell, R. A. & Medzhitov, R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature 420, 329–333 (2002).
Yamamoto, M. et al. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J. Immunol. 169, 6668–6672 (2002).
Yamamoto, M. et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301, 640–643 (2003).
Hoebe, K. et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 424, 743–748 (2003).
Oshiumi, H., Matsumoto, M., Funami, K., Akazawa, T. & Seya, T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat. Immunol. 4, 161–167 (2003).
Fitzgerald, K. A. et al. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J. Exp. Med. 198, 1043–1055 (2003).
Yamamoto, M. et al. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat. Immunol. 4, 1144–1150 (2003).
McGettrick, A. F. et al. Trif-related adapter molecule is phosphorylated by PKC{epsilon} during Toll-like receptor 4 signaling. Proc. Natl Acad. Sci. U. S. A. 103, 9196–9201 (2006).
Huai, W. et al. Phosphatase PTPN4 preferentially inhibits TRIF-dependent TLR4 pathway by dephosphorylating TRAM. J. Immunol. 194, 4458–4465 (2015).
Noguchi, M., Yoshida, T. & Kikuchi, G. Specific requirement of NADPH-cytochrome c reductase for the microsomal heme oxygenase reaction yielding biliverdin IX alpha. FEBS Lett. 98, 281–284 (1979).
Maines, M. D., Trakshel, G. M. & Kutty, R. K. Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J. Biol. Chem. 261, 411–419 (1986).
Prawan, A., Kundu, J. K. & Surh, Y. J. Molecular basis of heme oxygenase-1 induction: implications for chemoprevention and chemoprotection. Antioxid. Redox Signal 7, 1688–1703 (2005).
Seta, F. et al. Heme oxygenase-2 is a critical determinant for execution of an acute inflammatory and reparative response. Am. J. Pathol. 169, 1612–1623 (2006).
Bellner, L. et al. Heme oxygenase-2 deletion impairs macrophage function: implication in wound healing. Faseb j. 29, 105–115 (2015).
Chen, R. J. et al. Heme oxygenase-2 suppress TNF-α and IL6 expression via TLR4/MyD88-dependent signaling pathway in mouse cerebral vascular endothelial cells. Mol. Neurobiol. 50, 971–978 (2014).
Zhu, Y. et al. Heme oxygenase 2 binds myristate to regulate retrovirus assembly and TLR4 signaling. Cell Host Microbe 21, 220–230 (2017).
Alto, N. M. & Orth, K. Subversion of cell signaling by pathogens. Cold Spring Harb. Perspect. Biol. 4, a006114 (2012).
Hayes, C. S., Aoki, S. K. & Low, D. A. Bacterial contact-dependent delivery systems. Annu. Rev. Genet. 44, 71–90 (2010).
Cabezón, E., Ripoll-Rozada, J., Peña, A., de la Cruz, F. & Arechaga, I. Towards an integrated model of bacterial conjugation. FEMS Microbiol. Rev. 39, 81–95 (2014).
Kahn, R. A. Toward a model for Arf GTPases as regulators of traffic at the Golgi. FEBS Lett. 583, 3872–3879 (2009).
Burnaevskiy, N., Peng, T., Reddick, L. E., Hang, H. C. & Alto, N. M. Myristoylome profiling reveals a concerted mechanism of ARF GTPase deacylation by the bacterial protease IpaJ. Mol. Cell 58, 110–122 (2015).
Donaldson, J. G. & Jackson, C. L. ARF family G proteins and their regulators: roles in membrane transport, development and disease. Nat. Rev. Mol. Cell Biol. 12, 362–375 (2011).
Kosciuk, T. et al. NMT1 and NMT2 are lysine myristoyltransferases regulating the ARF6 GTPase cycle. Nat. Commun. 11, 1067 (2020).
Maurer-Stroh, S. & Eisenhaber, F. Myristoylation of viral and bacterial proteins. Trends Microbiol. 12, 178–185 (2004).
Palmiter, R. D., Gagnon, J., Vogt, V. M., Ripley, S. & Eisenman, R. N. The NH2-terminal sequence of the avian oncovirus gag precursor polyprotein (Pr76gag). Virology 91, 423–433 (1978).
Pepinsky, R. B., Papayannopoulos, I. A., Campbell, S. & Vogt, V. M. Analysis of Rous sarcoma virus Gag protein by mass spectrometry indicates trimming by host exopeptidase. J. Virol. 70, 3313–3318 (1996).
Henderson, L. E., Krutzsch, H. C. & Oroszlan, S. Myristyl amino-terminal acylation of murine retrovirus proteins: an unusual post-translational proteins modification. Proc. Natl Acad. Sci. U. S. A. 80, 339–343 (1983).
Pal, R. et al. Myristoylation of gag proteins of HIV-1 plays an important role in virus assembly. AIDS Res Hum. Retroviruses 6, 721–730 (1990).
Göttlinger, H. G., Sodroski, J. G. & Haseltine, W. A. Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proc. Natl Acad. Sci. U. S. A. 86, 5781–5785 (1989).
Bryant, M. & Ratner, L. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc. Natl Acad. Sci. U. S. A. 87, 523–527 (1990).
Freed, E. O. HIV-1 assembly, release and maturation. Nat. Rev. Microbiol. 13, 484–496 (2015).
Tang, C. et al. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc. Natl Acad. Sci. U. S. A. 101, 517–522 (2004).
Hermida-Matsumoto, L. & Resh, M. D. Human immunodeficiency virus type 1 protease triggers a myristoyl switch that modulates membrane binding of Pr55(gag) and p17MA. J. Virol. 73, 1902–1908 (1999).
Cheng-Mayer, C., Iannello, P., Shaw, K., Luciw, P. A. & Levy, J. A. Differential effects of nef on HIV replication: implications for viral pathogenesis in the host. Science 246, 1629–1632 (1989).
Miller, M. D., Feinberg, M. B. & Greene, W. C. The HIV-1 nef gene acts as a positive viral infectivity factor. Trends Microbiol. 2, 294–298 (1994).
Miller, M. D., Warmerdam, M. T., Gaston, I., Greene, W. C. & Feinberg, M. B. The human immunodeficiency virus-1 nef gene product: a positive factor for viral infection and replication in primary lymphocytes and macrophages. J. Exp. Med. 179, 101–113 (1994).
Fackler, O. T. et al. Association of human immunodeficiency virus Nef protein with actin is myristoylation dependent and influences its subcellular localization. Eur. J. Biochem. 247, 843–851 (1997).
Bentham, M., Mazaleyrat, S. & Harris, M. Role of myristoylation and N-terminal basic residues in membrane association of the human immunodeficiency virus type 1 Nef protein. J. Gen. Virol. 87, 563–571 (2006).
Skowronski, J., Parks, D. & Mariani, R. Altered T cell activation and development in transgenic mice expressing the HIV-1 nef gene. EMBO J. 12, 703–713 (1993).
Badley, A. D. et al. Macrophage-dependent apoptosis of CD4+ T lymphocytes from HIV-infected individuals is mediated by FasL and tumor necrosis factor. J. Exp. Med. 185, 55–64 (1997).
Geleziunas, R., Xu, W., Takeda, K., Ichijo, H. & Greene, W. C. HIV-1 Nef inhibits ASK1-dependent death signalling providing a potential mechanism for protecting the infected host cell. Nature 410, 834–838 (2001).
Hung, C. H. et al. HIV-1 Nef assembles a Src family kinase-ZAP-70/Syk-PI3K cascade to downregulate cell-surface MHC-I. Cell Host Microbe 1, 121–133 (2007).
Goldsmith, M. A., Warmerdam, M. T., Atchison, R. E., Miller, M. D. & Greene, W. C. Dissociation of the CD4 downregulation and viral infectivity enhancement functions of human immunodeficiency virus type 1 Nef. J. Virol. 69, 4112–4121 (1995).
Sol-Foulon, N. et al. HIV-1 Nef-induced upregulation of DC-SIGN in dendritic cells promotes lymphocyte clustering and viral spread. Immunity 16, 145–155 (2002).
Swingler, S. et al. HIV-1 Nef mediates lymphocyte chemotaxis and activation by infected macrophages. Nat. Med. 5, 997–103 (1999).
Swingler, S. et al. HIV-1 Nef intersects the macrophage CD40L signalling pathway to promote resting-cell infection. Nature 424, 213–219 (2003).
Yuan, M. et al. N-myristoylation: from cell biology to translational medicine. Acta Pharmacol. Sin. 41, 1005–1015 (2020).
Shrivastav, A. et al. Expression and activity of N-myristoyltransferase in lung inflammation of cattle and its role in neutrophil apoptosis. Vet. Res 41, 9 (2010).
Romagnoli, S., Peris, A., De Gaudio, A. R. & Geppetti, P. SARS-CoV-2 and COVID-19: from the bench to the bedside. Physiol. Rev. 100, 1455–1466 (2020).
Nkengasong, J. China’s response to a novel coronavirus stands in stark contrast to the 2002 SARS outbreak response. Nat. Med. 26, 310–311 (2020).
Lan, J. et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581, 215–220 (2020).
Cai, Y. et al. Distinct conformational states of SARS-CoV-2 spike protein. Science 369, 1586–1592 (2020).
Turoňová, B. et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 370, 203–208 (2020).
Plante, J. A. et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature (2020). https://doi.org/10.1038/s41586-020-2895-3.
Wu, A. et al. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe 27, 325–328 (2020).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8 (2020).
Cao, L. et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science 370, 426–431 (2020).
Rogers, T. F. et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science 369, 956–963 (2020).
Chi, X. et al. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science 369, 650–655 (2020).
Barnes, C. O. et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 588, 682–687 (2020).
Monteil, V. et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 181, 905–913.e7 (2020).
Schoeman, D. & Fielding, B. C. Coronavirus envelope protein: current knowledge. Virol. J. 16, 69 (2019).
Ono, A. & Freed, E. O. Binding of human immunodeficiency virus type 1 Gag to membrane: role of the matrix amino terminus. J. Virol. 73, 4136–4144 (1999).
Yu, G. & Felsted, R. L. Effect of myristoylation on p27 nef subcellular distribution and suppression of HIV-LTR transcription. Virology 187, 46–55 (1992).
Resh, M. D. Myristylation and palmitylation of Src family members: the fats of the matter. Cell 76, 411–413 (1994).
Streuli, C. H. & Griffin, B. E. Myristic acid is coupled to a structural protein of polyoma virus and SV40. Nature 326, 619–622 (1987).
Moscufo, N., Simons, J. & Chow, M. Myristoylation is important at multiple stages in poliovirus assembly. J. Virol. 65, 2372–2380 (1991).
Gripon, P., Le Seyec, J., Rumin, S. & Guguen-Guillouzo, C. Myristylation of the hepatitis B virus large surface protein is essential for viral infectivity. Virology 213, 292–299 (1995).
Tillotson, L. & Shatkin, A. J. Reovirus polypeptide sigma 3 and N-terminal myristoylation of polypeptide mu 1 are required for site-specific cleavage to mu 1C in transfected cells. J. Virol. 66, 2180–2186 (1992).
Martin, K. H., Grosenbach, D. W., Franke, C. A. & Hruby, D. E. Identification and analysis of three myristylated vaccinia virus late proteins. J. Virol. 71, 5218–5226 (1997).
Lang, M. L. et al. IgA Fc receptor (FcalphaR) cross-linking recruits tyrosine kinases, phosphoinositide kinases and serine/threonine kinases to glycolipid rafts. Biochem. J. 364, 517–525 (2002).
O’Callaghan, D. W. et al. Differential use of myristoyl groups on neuronal calcium sensor proteins as a determinant of spatio-temporal aspects of Ca2+ signal transduction. J. Biol. Chem. 277, 14227–14237 (2002).
Spilker, C., Dresbach, T. & Braunewell, K. H. Reversible translocation and activity-dependent localization of the calcium-myristoyl switch protein VILIP-1 to different membrane compartments in living hippocampal neurons. J. Neurosci. 22, 7331–7339 (2002).
Perrino, B. A. & Martin, B. A. Ca(2+)- and myristoylation-dependent association of calcineurin with phosphatidylserine. J. Biochem. 129, 835–841 (2001).
Timm, S., Titus, B., Bernd, K. & Barroso, M. The EF-hand Ca(2+)-binding protein p22 associates with microtubules in an N-myristoylation-dependent manner. Mol. Biol. Cell 10, 3473–3488 (1999).
Hwang, J. Y. & Koch, K. W. Calcium- and myristoyl-dependent properties of guanylate cyclase-activating protein-1 and protein-2. Biochemistry 41, 13021–13028 (2002).
Ueda, T., Yamaguchi, M., Uchimiya, H. & Nakano, A. Ara6, a plant-unique novel type Rab GTPase, functions in the endocytic pathway of Arabidopsis thaliana. EMBO J. 20, 4730–4741 (2001).
Chen, C. A. & Manning, D. R. Regulation of G proteins by covalent modification. Oncogene 20, 1643–1652 (2001).
Moffett, S., Brown, D. A. & Linder, M. E. Lipid-dependent targeting of G proteins into rafts. J. Biol. Chem. 275, 2191–2198 (2000).
Martin, G. B., Bogdanove, A. J. & Sessa, G. Understanding the functions of plant disease resistance proteins. Annu. Rev. Plant Biol. 54, 23–61 (2003).
Pepperkok, R. et al. Intracellular distribution of mammalian protein kinase A catalytic subunit altered by conserved Asn2 deamidation. J. Cell Biol. 148, 715–726 (2000).
Fraser, I. D. et al. A novel lipid-anchored A-kinase Anchoring Protein facilitates cAMP-responsive membrane events. EMBO J. 17, 2261–2272 (1998).
Vaandrager, A. B. et al. Membrane targeting of cGMP-dependent protein kinase is required for cystic fibrosis transmembrane conductance regulator Cl- channel activation. Proc. Natl Acad. Sci. U. S. A. 95, 1466–1471 (1998).
Lu, S. X. & Hrabak, E. M. An Arabidopsis calcium-dependent protein kinase is associated with the endoplasmic reticulum. Plant Physiol. 128, 1008–1021 (2002).
Michel, T. Targeting and translocation of endothelial nitric oxide synthase. Braz. J. Med. Biol. Res. 32, 1361–1366 (1999).
Acknowledgements
We would like to apologize to those researchers whose related work we were not able to cite in this review. This work was supported by a special program from the Ministry of Science and Technology of China (2016YFA0502500 to L.Z.), the Chinese National Natural Science Funds (U20A20393, 82041009, 31925013, 31671457, and 91753139 to L.Z.; 31871405 and 31571460 to F.Z.), Jiangsu National Science Foundation (BK20180043 and 19KJA550003 to F.Z.), the Zhejiang Natural Science Fund (LD19C070001 to L.Z.), the Key Project of University Natural Science Foundation of Jiangsu Province (19KJA550003), and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Contributions
B.W., T.D., W.S., and Y.W. contributed equally to this work. B.W., T.D., W.S., and Y.W. conceived and drafted the manuscript. W.B. prepared the figures. M.Z. and R.J. discussed the concepts of the manuscript. F.Z. and L.Z. provided valuable discussion and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Wang, B., Dai, T., Sun, W. et al. Protein N-myristoylation: functions and mechanisms in control of innate immunity. Cell Mol Immunol 18, 878–888 (2021). https://doi.org/10.1038/s41423-021-00663-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41423-021-00663-2
Keywords
This article is cited by
-
Protein lipidation in health and disease: molecular basis, physiological function and pathological implication
Signal Transduction and Targeted Therapy (2024)
-
Protein N-myristoylation plays a critical role in the mitochondrial localization of human mitochondrial complex I accessory subunit NDUFB7
Scientific Reports (2023)
-
Myristic acid as a checkpoint to regulate STING-dependent autophagy and interferon responses by promoting N-myristoylation
Nature Communications (2023)
-
Inflammatory crosstalk between saturated fatty acids and gut microbiota–white adipose tissue axis
European Journal of Nutrition (2023)
-
Functional characterization of FBXL7 as a novel player in human cancers
Cell Death Discovery (2022)
