
Abstract
The aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor initially identified as the receptor for dioxin. Almost half a century after its discovery, AHR is now recognized as a receptor for multiple physiological ligands, with important roles in health and disease. In this review, we discuss the role of AHR in the gut–brain axis and its potential value as a therapeutic target for immune-mediated diseases.
Similar content being viewed by others

Introduction
In the 1970s, Poland et al.1,2 identified a potential receptor for the anthropogenic compound 2,3,7,8-tetraclorodibenzo-p-dioxin (TCDD).3 Two years later, the same group demonstrated that TCDD binds to this unknown receptor in hepatic cells, inducing expression of the aryl hydrocarbon hydroxylase enzyme encoded by CYP1A1.4 Those and other seminal studies by Nebert and Poland led to the identification and characterization of the aryl hydrocarbon receptor (AHR),3,4,5,6 a ligand-activated transcription factor with important physiological roles in health and disease.7
Indeed, although initial studies focused on ligands such as polycyclic aromatic hydrocarbons (PAHs), polychlorinated biphenyls (PCBs) and halogenated aromatic hydrocarbons (HAHs), it is now clear that a broad range of dietary, commensal and endogenous, ligands activate AHR.8,9,10,11 To date, multiple physiological and dietary AHR ligands (Table 1) have been identified, including tryptophan metabolites such as 6-formylindolo[3,2-b]carbazole (FICZ), kynurenine, indigo, indirubin, the pigment curcumin,12 carotenoids,13 flavonoids, bilirubin and biliverdin,14 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE),15 indoxyl-3-sulfate (I3S), indole-3-carbinol (I3C), gallic acid,16 prostaglandins and eicosanoids.17 Additional AHR agonists are produced by the metabolism of commensal microorganisms.9,18,19 Moreover, some medications are reported to activate AHR, including omeprazole,20 sulindac,21 laquinimod,22 tapinarof,23 and diclofenac.24 The ability of AHR to interact with multiple molecules and its broad expression enables it to modulate diverse physiological processes in response to environmental, microbial and metabolic cues. In this review, we discuss the role of AHR in the immune response, with a focus on the gut–brain axis.25
The AHR
In mice and humans, AHR is an 848-amino acid-long protein. It is encoded by a gene located on chromosomes 7 and 12 in humans26 and mice,27 respectively. The Ahr promoter harbors several transcription initiation sites rich in GC-rich regions but without a TATA or a CCAAT box.28 These GC-rich regions contain binding sites for ubiquitously expressed zinc-finger transcription factors, including Sp1 and Sp3, which seem to be required for basal AHR expression.29 AHR is a member of the basic-helix/loop/helix per-Arnt-sim (bHLH/PAS) family of transcription factors. The bHLH domain of AHR is responsible for DNA binding and dimerization, stabilizing protein–protein interactions. The PAS domain contains two subdomains: PAS-A, which is essential for dimerization with other proteins, and PAS-B, which harbors ligand- and heat shock protein (HSP) 90-binding motifs (Fig. 1A). The AHR transcriptional activation domain is located in the N terminal region and encompasses a region rich in glutamine (Q-rich region) that also harbors a nuclear translocation signal30,31 (Fig. 1A).

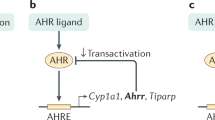
AHR and its signaling pathway. A Schematic representation of AHR protein domains. B The AHR signaling pathway. The inactive form of AHR is localized in the cytosol in a complex composed of HSP90, AIP, p23, and c-SRC. AHR agonists induce conformational changes in AHR that result in its translocation to the nucleus. In the nucleus, AHR interacts with ARNT, and the heterodimer is responsible for the transcription of XRE-containing genes. Notes: (AHR) aryl hydrocarbon receptor, (N) N terminal motif, (C) C terminal motif, (NLS) nuclear localization signal, (bHLH) basic-helix loop helix, (PAS) Per-Arnt-Sim, (Q-rich) glutamine rich, (HSP90) heat shock protein 90, (AIP) AHR-interacting protein, (XRE) xenobiotic responsive elements, (AHRR) AHR repressor, (CYP) cytochrome P450, (IDO) indoleamine 2,3-dioxygenase
Inactive AHR is located in the cytoplasm complexed with several chaperones that stabilize it. The cytoplasmic AHR complex contains the following: (1) an HSP90 dimer that maintains AHR in a conformation that maximizes its affinity for ligands;32 (2) p23 as a cochaperone; (3) AHR-interacting protein (AIP, also known as Ara9 or XAP2), which stabilizes AHR in the cytoplasm, preventing its ubiquitination and degradation;33 and (4) the c-SRC protein kinase.34 AHR genomic signaling is triggered by ligand binding, which induces a conformational change in AHR, releasing AIP35 and exposing the nuclear localization signal36 and a protein kinase C target site that when phosphorylated promotes AHR nuclear translocation37 (Fig. 1B). HSP90 is reported to translocate to the nucleus together with AHR,38 but knowledge of the cofactors that translocate to the nucleus together with AHR and their function remains limited.
In the nucleus, AHR exchanges its chaperones with the AHR nuclear translocator (ARNT, also known as HIF-1β)39 to interact with DNA sequences known as xenobiotic responsive elements (XRE, also known as DRE) in the regulatory regions of target genes (e.g., CYP1A1, CYP1A2, CYP1B1, and AHRR). AHR-targeted components of cytochrome P450 (CYP) affected by AHR catalyze the degradation of AHR ligands40 and hence participate in a negative feedback loop that limits AHR activation. AHR also induces expression of the AHR repressor (AHRR), which limits AHR activation.41
In addition to its direct effects mediated via XREs, AHR controls transcriptional responses through interaction with other transcription factors and coactivators, including nuclear factor-kappaB (NF-κB), estrogen receptor (ER), retinoic acid receptor, and members of the signal transducers and activators of transcription family.42 For instance, NF-kB/RelA-dependent AHR expression following LPS stimulation in dendritic cells (DCs) has been described.43 AHR has also been shown to interact with c-Maf to control the expression of IL-10 and IL-21.44,45,46,47
AHR has been shown to modulate the epigenetic status of the cell via the control of noncoding RNAs,48 microRNAs,49 and histone acetylation/methylation mechanisms that regulate chromatin conformation and accessibility.50
AHR signaling also involves nongenomic pathways. For example, once released from its complex with AHR, c-SRC can phosphorylate enzymes involved in the arachidonic acid and leukotriene signaling pathways.51 These nongenomic mechanisms are important for the induction of endotoxin tolerance in DCs via c-SRC-driven phosphorylation and stabilization of indoleamine 2,3-dioxygenase 1 (IDO1).52 Finally, AHR has been reported to act as an E3 ubiquitin protein ligase, inducing proteasomal degradation of protein targets such as p53, FOS, hypoxia-inducible factor (HIF)-1α, MYC, and ER.53,54,55 Altogether, these data demonstrate that almost half a century after the cloning of AHR, the mechanisms mediating the control of cellular responses by AHR still need to be fully elucidated.
AHR in the control of the immune response
The purification of AHR and the generation of knockout mice (AHR KO) led to the identification of multiple physiologic roles for AHR.56,57 One of those roles is the regulation of the immune response.
Modulation of inflammation by AHR expressed in mucosal tissues and skin
The intestinal epithelium interacts with a myriad of microbial metabolites, pollutants, and dietary molecules. AHR acts as a sensor for many of these environmental stimuli, mediating some of their effects on the immune response. AHR conditional knockout mice generated using an intestinal epithelial cell (IEC)-specific Cre recombinase (Vil1-Cre) show increased susceptibility to Citrobacter rodentium infection.58 In addition, these mice display defective differentiation of intestinal stem cells, resulting in the malignant transformation of IECs.58 Conversely, AHR activation by dietary ligands limits intestinal stem cell proliferation by regulating the E3 ubiquitin ligases Rnf43 and Znrf3, suppressing Wnt/β-Catenin signaling. Within the context of inflammation, IFN-γ induces the expression of IDO1, which produces the AHR agonist L-kynurenine, thereby triggering AHR-driven upregulation of IL-10 receptor 1 and consequently amplifying the anti-inflammatory effects of IL-10.59 These findings highlight the anti-inflammatory role of AHR in IECs and its contribution to the integrity of the intestinal barrier.
AHR also controls the expression of antimicrobial peptides that fight pathogens in the gut. For instance, regenerating islet-derived protein III (REGIII)β and REGIIIγ are upregulated following administration of the probiotic-derived AHR agonist 1,4-dihydroxy-2-naphthoic acid (DHNA), altering the microbiome and ameliorating dextran sodium sulfate-induced colitis in mice.60 In support of a role for AHR in therapeutic interventions already in use, the increase in Th22 cell differentiation and IL-22 production induced by TNF blockade was abrogated by AHR inhibition in Crohn’s disease (CD) patients treated with antitumor necrosis factor (TNF)-blocking antibodies. These data suggest a new role for AHR agonists as potential adjuvants for anti-TNF therapy in CD patients.61
Although these and additional studies40,47,58,62,63,64,65,66,67,68,69 support a protective role for AHR in intestinal inflammation, some studies have challenged this notion. In particular, it was recently reported that environmental oxazoles induce the production of AHR agonists by IDO1 expressed in IECs and other cell types,70 which surprisingly leads to increased intestinal inflammation via the suppression of IL-10 secretion, modulation of CD1d-dependent antigen presentation and production of IFNγ/IL-13 by NKT cells.70 These provocative findings should be further investigated, particularly within the context of alternative interpretations, such as the expansion of AHR-driven nonpathogenic Th17 cells, which may acquire full pathogenic activity following exposure to additional factors in the inflamed gut, such as IL-23.71
Innate lymphoid cells (ILCs) are tissue-resident innate immune cells that participate in the response to infection and contribute to tissue homeostasis and chronic inflammation.72,73 ILCs are classified into five subsets: NK cells, lymphoid tissue inducer cells, group 1 ILCs (ILC1s), group 2 ILCs (ILC2s), and group 3 ILCs (ILC3s).74 Each of these subsets is controlled by different transcription factors,75 and AHR controls IL-22 expression in ILC3s.76 Indeed, AHR-deficient mice exhibit expansion of segmented filamentous bacteria in the small intestine due to reduced IL-22 production by ILC3s, which in turn promotes Th17 cell expansion in the gut and the development of spontaneous colitis.72,73 Of note, polymorphisms in caspase recruitment domain family member 9 (CARD9) have been associated with intestinal inflammation.77 Interestingly, CARD9 risk alleles associated with inflammatory bowel disease promote a decrease in the abundance of intestinal commensals that produce AHR agonists, leading to decreased AHR activation and intestinal inflammation.77 These findings highlight how AHR’s role as a sensor of commensal products contributes to mechanisms of intestinal pathogenesis.
DCs play central roles in the maintenance of tolerance and the generation of protective immune responses against pathogens in the gut as well as in other tissues.78,79 AHR is highly expressed in DCs,80 affecting their differentiation and function.9,81 AHR-driven cytokine, kynurenine,82,83,84 and retinoic acid85 production in DCs boosts the differentiation of regulatory T cells that suppress the development of experimental autoimmune encephalomyelitis (EAE), the model of multiple sclerosis (MS).86
Different subsets of DCs sense the lumen microenvironment, and following their migration to mesenteric lymph nodes (MLNs) via CCR7 signaling, they control peripheral differentiation regulatory cells and prime effector T cells.87 For example, AHR activation by the commensal metabolite indole-3-pyruvic acid reduces the ability of DCs in MLNs to promote the differentiation of IFN-γ-producing T cells, thus preventing chronic inflammation during colitis.88 AHR signaling in DCs is also reported to affect nonimmune cells in unexpected ways, as demonstrated by reports of increased numbers of small intestinal epithelial stem cells and atypical differentiation of epithelial precursors following AHR deletion from CD11c+ DCs.89
AHR signaling controls T-cell responses not only via the modulation of APC function but also through intrinsic effects in T cells. For example, AHR modulates the expansion and differentiation of Th17 cells,90,91 though AHR appears to be more relevant for the control of the transcriptional program of nonpathogenic Th17 cells.71 Indeed, AHR signaling promotes the conversion of Th17 cells to type 1 regulatory T cells (Tr1 cells).92 Moreover, AHR has been linked to the control of regulatory T cells through multiple mechanisms involving their differentiation and stability as well as effector mechanisms.44,46,63,85,90,93 Overall, the effects of AHR on T cells are likely to have consequences for inflammation in other tissues in addition to the gut.
Intraepithelial lymphocytes (IELs) constitute a population of T cells localized in the epithelial layer of mammalian mucosal linings such as the intestine. IELs are antigen-experienced T cells of both T-cell receptor γδ (TCRγδ)+ and TCRαβ+ lineages.94 AHR modulates IEL survival and response to nutritional and microbial stimuli.95 For example, administration of the AHR agonist FICZ ameliorates DDS-induced colitis by reducing the apoptotic rate of CD8αα+TCRαβ+ IELs, while decreasing and increasing their production of IFN-γ and IL-10, respectively.96 Furthermore, Colonna and collaborators established that CD8αα+TCRαβ+ IELs in the small intestine are supported by the activation of AHR signaling by tryptophan metabolites produced by Lactobacillus reuteri.65 Interestingly, Kadowaki et al. reported that microbial AHR agonists promote the differentiation of regulatory CD4+ IELs, which can migrate to the CNS and suppress inflammation through LAG3-dependent mechanisms.97 These important findings suggest that AHR-dependent immunoregulatory mechanisms operating in the gut can affect inflammatory processes in other tissues.
Modulation of inflammation in the central nervous system (CNS)
The gut and brain axis is now recognized as a key factor in the pathology of multiple neurological disorders, including MS and its experimental model EAE.25 In EAE and MS, CD4+ effector T cells primed in the periphery migrate to the CNS, where they are reactivated by cDCs and other cells to cause myelin destruction.98,99 In addition, recruited T cells secrete cytokines that modulate the activity of CNS-resident immune cells, such as microglia and astrocytes.100,101,102
In pioneering studies, Wekerle and coworkers demonstrated that the commensal gut flora controls autoreactive T cells that migrate to the CNS and cause inflammation and tissue pathology.103 Follow-up studies defined alterations in the gut microbiota associated with MS104 and identified specific components of the microbiome involved in the regulation of effector and regulatory T cells.105,106,107 Similarly, it was recently reported that anti-inflammatory B cells controlled by the commensal flora migrate the gut to the CNS to limit tissue pathology in MS.108 Interestingly, AHR controls B-cell anti-inflammatory activities.109,110 Moreover, AHR controls the differentiation and stability of intestinal Tregs,47,85 and oral administration of the AHR agonist ITE increases the myelin-reactive Treg/Teff ratio and suppresses EAE.85 These findings suggest that AHR signaling contributes to the anti-inflammatory effects of the commensal flora not only in the gut but also in other tissues, such as the CNS.
Astrocytes are the most abundant glial cells in the CNS and have essential roles associated with the support of neurons and synapses, the control of neurotransmitters and the regulation of blood–brain barrier development and function.111,112,113,114,115,116,117,118 Astrocytes also play important roles in CNS inflammation and neurodegeneration via their own neurotoxic activities as well as the recruitment and activation of other cells involved in CNS pathogenesis.19,100,119,120,121,122,123,124,125,126 Transcriptional analyses of astrocytes revealed AHR upregulation in the context of EAE and MS.119,121 Indeed, specific inactivation of AHR in astrocytes via conditional knockout mice and cell-specific shRNA knockdown identified AHR as a negative regulator of the NF-κB transcriptional responses that promote microglial activation, neurotoxic peripheral monocyte recruitment to the CNS, and astrocyte-intrinsic neurotoxic activities.19 Moreover, in-depth molecular studies have established that AHR inhibits NF-κB activation in astrocytes through a SOCS2-dependent mechanism19 that also operates in DCs.127 Interestingly, microbiota perturbation studies showed that metabolites produced from the degradation of tryptophan by the intestinal commensal flora reach the CNS and activate AHR in astrocytes to limit CNS inflammation,19 describing for the first time a mechanism mediating the control of astrocytes by the gut flora.
Microglia are CNS-resident macrophages with multiple functions in health and disease,128 playing important roles in the control of astrocyte responses.129 Interestingly, microglia express AHR,130,131 and conditional knockout mice revealed that AHR limits NF-κB activation in microglia.19 In addition, AHR controls microglial production of TGF-α and VEGF-B: AHR transactivates the Tgfa promoter, interfering with NF-κB-driven VEGF-B expression.19 Microglial TGF-α and VEGF-B suppress and induce astrocyte responses, respectively, that promote CNS pathogenesis.19 In fact, deletion of microglial AHR worsens EAE, increasing demyelination and monocyte recruitment to the CNS.19 As microbial agonists can also activate microglial AHR, these findings provide a molecular mechanism by which the gut microbiome modulates microglial and astrocyte responses as well as interactions between these CNS-resident cells.
AHR is expressed in CNS endothelial cells,132 neurons,133 and oligodendrocytes.134 Endothelial cell AHR is suggested to contribute to detoxification processes132,135,136 and studies in fish suggest that AHR hyperactivation in endothelial cells trigger apoptosis and vascular defects, resulting in hemorrhage, edema, and embryonic mortality.132 Metabolites of the pesticide DDT induce AHR-dependent neurotoxicity.137 Finally, AHR has been proposed to participate in oligodendrocyte differentiation.138 These findings indicate that AHR participates in the regulation of endothelial cells, neurons and oligodendrocytes in health and disease, though further studies are needed to identify the specific mechanisms involved.
As mentioned above, AHR signaling can modulate peripheral T-cell differentiation.9,81 Moreover, peripheral T cells recruited to the CNS control astrocyte100,101,120 and microglial102 responses. Hence, these findings suggest that AHR signaling participates in the gut–brain axis through multiple mechanisms ranging from activation of AHR in CNS-resident cells by microbial metabolites to AHR-mediated peripheral modulation of immune cells that migrate to the CNS.
Role of AHR in infections targeting the CNS
The microbiota establishes multiple types of relationships with the host, ranging from mutualism to parasitism: in the former, the interaction is beneficial for both organisms; in the latter, this interaction is only beneficial for the parasite and harmful for the host.139 We discussed AHR-mediated microbiota–host relationships beneficial for the host above; below, we describe the role of AHR in relationships detrimental to the host (Fig. 2).

Role of AHR in infections targeting the CNS. AHR can affect the outcome of infectious diseases that target the CNS. Notes: (NP) nanoparticles, (ROS) reactive oxygen species, (NO) nitric oxide
Lysteria monocitogenesis targets the gastrointestinal tract and can also cause meningitis. In a murine model of listeriosis, AHR-deficient mice displayed higher mortality than their WT counterparts, concomitant with higher levels of pro-inflammatory cytokines, decreased ROS production and macrophage survival.140 Zika virus (ZIKV) infection has been associated with severe outcomes, including fetal brain abnormalities141 and Guillain–Barré syndrome.142 Similarly, it was recently reported that ZIKV infection triggers the production of AHR agonists by the host.126 AHR activation interferes with IFN-I-dependent mechanisms of anti-ZIKV immunity,126 in agreement with previous reports.143 AHR also interferes with mechanisms of intrinsic immunity mediated by the protein PML. Most importantly, AHR inhibition with clinical antagonists suppresses ZIKV replication in vitro and in vivo and ameliorates CNS abnormalities associated with ZIKV.126 Similar mechanisms appear to operate within the context of infection by dengue virus.126
Trypanosoma cruzi is the etiological agent of Chagas’s disease, a chronic illness endemic to Central and South America with long-term consequences for the heart, esophagus, colon and nervous system.144 In the experimental model of Chagas disease, AHR activation expands the Treg compartment, increasing parasite replication.145 In agreement with these findings, AHR-deficient mice show reduced T. cruzi parasitemia and a heightened immune response characterized by the production of proinflammatory cytokines, increased NO in serum, and downregulation of SOCS2.145,146 Conversely, within the context of infection by Toxoplasma gondii, AHR deficiency results in higher mortality as a result of increased pro-inflammatory responses and decreased IL-10 production.147 Taken together, these findings highlight the complex roles played by AHR in infection: AHR can limit immunopathology but can also be exploited by pathogens to evade the immune response.
AHR and CNS tumors
Based on its multiple physiological roles, it is not surprising that AHR contributes to tumor pathogenesis. Glioblastoma is the most common primary malignant brain tumor in adults148 and one of the most aggressive cancers, with a median survival of 15–18 months despite standard of care therapy.148,149 Opitz et al. reported that tryptophan 2,3-dioxygenase in glioblastoma leads to the production of kynurenine, which acts in an autocrine manner to enhance tumor invasiveness and replication.150,151 In addition, Gramarzki et al. reported that AHR in glioma cells drives expression of TGF-β, suggesting that AHR signaling promotes an immune suppressive microenvironment in glioma.152 Indeed, AHR expression has been detected in tumor-associated macrophages (TAMs), which constitute more than 30% of infiltrating cells in glioblastoma. Takenaka et al. recently showed that AHR activation induces an anti-inflammatory phenotype in glioblastoma TAMs.153 Moreover, AHR drives expression of CD39 in TAMs, which promotes CD8+ cell dysfunction153 (Fig. 3). These findings suggest a role for AHR in tumor immunoevasion and highlight the intrinsic tumor cell functions, emphasizing its potential as a therapeutic target.151,154,155

Role of AHR in glioblastoma. Kynurenine in the tumor microenvironment activates AHR in TAMs, promoting expression of CCR2, CD39 and KLF4. CCR2 contributes to the recruitment of TAMs to the tumor microenvironment, CD39 promotes CD8+ T-cell dysfunction, and KLF4 together with SOCS2 influences TAM polarization. Notes: (Kyn) kynurenine, (TAM) tumor-associated macrophages
AHR as a target for therapeutic immunomodulation
As briefly discussed in this manuscript, AHR signaling has multiple effects on the immune response. AHR constitutes a potential target for therapeutic intervention based on the ability of small molecules to control its activity (Fig. 4).

AHR sensor and immunomodulatory roles. AHR senses diverse environmental cues provided by the diet, microbiome, and anthropogenic compounds. AHR signaling participates in physiological and pathological processes, making it a potential target for therapeutic intervention
With regard to autoimmune diseases, laquinimod22 and tapinarof23,156,157 have been developed as AHR-targeting drugs for the treatment of MS, psoriasis and atopic dermatitis. Furthermore, codelivery of tolerogenic AHR agonists and antigens to DCs with nanoparticles provides an attractive approach. This nanoparticle-based approach is based on the induction of a tolerogenic phenotype in DCs, which are concomitantly loaded with disease-relevant antigens,158 thereby boosting antigen-specific tolerance with minimal effects on nonrelated immune responses. This approach leads to expansion of Tregs (both FoxP3 + Tregs and Tr1 cells) that suppress inflammation in EAE.159 Similar observations have been made in other autoimmune diseases, such as type 1 diabetes.127 Within the context of infection or tumors, AHR inhibitors may offer a novel pathway to limit immune evasion,151 with the caveat that AHR may also play a role in limiting immunopathology. Nonetheless, in considering the therapeutic targeting of AHR, it should be kept in mind that AHR participates in multiple physiological processes in addition to immune regulation. Moreover, AHR signaling is regulated by microbial metabolites, with important effects on the immune response. Thus, therapeutic targeting of AHR should consider not only its effects on the immune response but also its important roles in the host–microbiome relationship and the multiple effects of the microbiome in autoimmunity, cancer, and infections.
Concluding remarks
Five decades after its identification, AHR has emerged as an important immune regulator. It is therefore important to characterize the physiological AHR agonists involved in immune regulation, as they may provide lead molecules for the development of novel immunomodulators. In addition, they may contribute to the identification of ligand-specific downstream effects of AHR signaling of therapeutic interest. Within this context, there remains an important need to characterize the cell-specific effects of AHR signaling and the mechanisms involved.
Finally, the participation of AHR in the gut–brain axis prompts new research questions, as follows: (1) Which microbial AHR agonists reach the CNS? (2) Which components of the commensal flora produce AHR ligands, and how are they regulated in health and disease? (3) Which peripheral cells are educated by the commensal flora in the periphery to then migrate to the CNS and control the activity of resident cells? These questions will guide future research efforts and reveal novel opportunities for AHR-targeted therapeutics.
References
Nebert, D. W., Winker, J. & Gelboin, H. V. Aryl hydrocarbon hydroxylase activity in human placenta from cigarette smoking and nonsmoking women. Cancer Res. 29, 1763–1769 (1969).
Nebert, D. W. & Gelboin, H. V. The in vivo and in vitro induction of aryl hydrocarbon hydroxylase in mammalian cells of different species, tissues, strains, and developmental and hormonal states. Arch. Biochem. Biophys. 134, 76–89 (1969).
Poland, A. P. et al. Genetic expression of aryl hydrocarbon hydroxylase activity. Induction of monooxygenase activities and cytochrome P1-450 formation by 2,3,7,8-tetrachlorodibenzo-p-dioxin in mice genetically “nonresponsive” to other aromatic hydrocarbons. J. Biol. Chem. 249, 5599–5606 (1974).
Poland, A., Glover, E. & Kende, A. S. Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J. Biol. Chem. 251, 4936–4946 (1976).
Nebert, D. W. The Ah locus. A gene with possible importance in cancer predictability. Arch. Toxicol. Suppl. 3, 195–207 (1980).
Nebert, D. W. et al. The Ah phenotype. Survey of forty-eight rat strains and twenty inbred mouse strains. Genetics 100, 79–87 (1982).
Hahn, M. E. et al. Molecular evolution of two vertebrate aryl hydrocarbon (dioxin) receptors (AHR1 and AHR2) and the PAS family. Proc. Natl Acad. Sci. USA 94, 13743–13748 (1997).
Denison, M. S. et al. Exactly the same but different: promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol. Sci. 124, 1–22 (2011).
Gutierrez-Vazquez, C. & Quintana, F. J. Regulation of the immune response by the Aryl hydrocarbon receptor. Immunity 48, 19–33 (2018).
Quintana, F. J. & Sherr, D. H. Aryl hydrocarbon receptor control of adaptive immunity. Pharmacol. Rev. 65, 1148–1161 (2013).
Lee, H. U. et al. Host-microbiome interactions: the aryl hydrocarbon receptor and the central nervous system. J. Mol. Med. 95, 29–39 (2017).
Ciolino, H. P. et al. Effect of curcumin on the aryl hydrocarbon receptor and cytochrome P450 1A1 in MCF-7 human breast carcinoma cells. Biochem. Pharmacol. 56, 197–206 (1998).
Wanner, R. et al. The differentiation-related upregulation of aryl hydrocarbon receptor transcript levels is suppressed by retinoic acid. Biochem. Biophys. Res. Commun. 209, 706–711 (1995).
Phelan, D. et al. Activation of the Ah receptor signal transduction pathway by bilirubin and biliverdin. Arch. Biochem. Biophys. 357, 155–163 (1998).
Abron, J. D. et al. An endogenous aryl hydrocarbon receptor ligand, ITE, induces regulatory T cells and ameliorates experimental colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 315, G220–G230 (2018).
Abdullah, A. et al. Activation of aryl hydrocarbon receptor signaling by a novel agonist ameliorates autoimmune encephalomyelitis. PLoS ONE 14, e0215981 (2019).
Nebert, D. W. Aryl hydrocarbon receptor (AHR): “pioneer member” of the basic-helix/loop/helix per-Arnt-sim (bHLH/PAS) family of “sensors” of foreign and endogenous signals. Prog. Lipid Res. 67, 38–57 (2017).
Roager, H. M. & Licht, T. R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 9, 3294 (2018).
Rothhammer, V. et al. Microglial control of astrocytes in response to microbial metabolites. Nature 557, 724–728 (2018).
Quattrochi, L. C. & Tukey, R. H. Nuclear uptake of the Ah (dioxin) receptor in response to omeprazole: transcriptional activation of the human CYP1A1 gene. Mol. Pharmacol. 43, 504–508 (1993).
Ciolino, H. P. et al. Sulindac regulates the aryl hydrocarbon receptor-mediated expression of Phase 1 metabolic enzymes in vivo and in vitro. Carcinogenesis 27, 1586–1592 (2006).
Kaye, J. et al. Laquinimod arrests experimental autoimmune encephalomyelitis by activating the aryl hydrocarbon receptor. Proc. Natl Acad. Sci. USA 113, E6145–E6152 (2016).
Smith, S. H. et al. Tapinarof is a natural AhR agonist that resolves skin inflammation in mice and humans. J. Investig. Dermatol. 137, 2110–2119 (2017).
Bass, S. E. et al. Novel dithiolethione-modified nonsteroidal anti-inflammatory drugs in human hepatoma HepG2 and colon LS180 cells. Clin. Cancer Res. 15, 1964–1972 (2009).
Kadowaki, A. & Quintana, F. J. The Gut-CNS axis in multiple sclerosis. Trends Neurosci. 43, 622–634 (2020).
Le Beau, M. M. et al. Chromosomal localization of the human AHR locus encoding the structural gene for the Ah receptor to 7p21–>p15. Cytogenet Cell Genet. 66, 172–176 (1994).
Seldin, M. F., Howard, T. A. & D’Eustachio, P. Comparison of linkage maps of mouse chromosome 12 derived from laboratory strain intraspecific and Mus spretus interspecific backcrosses. Genomics 5, 24–28 (1989).
Eguchi, H. et al. Molecular cloning of the human AH receptor gene promoter. Biochem. Biophys. Res. Commun. 203, 615–622 (1994).
Fitzgerald, C. T., Nebert, D. W. & Puga, A. Regulation of mouse Ah receptor (Ahr) gene basal expression by members of the Sp family of transcription factors. DNA Cell Biol. 17, 811–822 (1998).
Fukunaga, B. N. et al. Identification of functional domains of the aryl hydrocarbon receptor. J. Biol. Chem. 270, 29270–29278 (1995).
Wu, D. et al. Structure and dimerization properties of the aryl hydrocarbon receptor PAS-A domain. Mol. Cell Biol. 33, 4346–4356. (2013).
Denis, M. et al. Association of the dioxin receptor with the Mr 90,000 heat shock protein: a structural kinship with the glucocorticoid receptor. Biochem. Biophys. Res. Commun. 155, 801–807 (1988).
Lees, M. J., Peet, D. J. & Whitelaw, M. L. Defining the role for XAP2 in stabilization of the dioxin receptor. J. Biol. Chem. 278, 35878–35888 (2003).
Mulero-Navarro, S. & Fernandez-Salguero, P. M. New trends in Aryl hydrocarbon receptor biology. Front Cell Dev. Biol. 4, 45 (2016).
Ikuta, T. et al. Nuclear localization and export signals of the human aryl hydrocarbon receptor. J. Biol. Chem. 273, 2895–2904 (1998).
Ikuta, T., Kobayashi, Y. & Kawajiri, K. Phosphorylation of nuclear localization signal inhibits the ligand-dependent nuclear import of aryl hydrocarbon receptor. Biochem. Biophys. Res Commun. 317, 545–550 (2004).
Kudo, I. et al. The regulation mechanisms of AhR by molecular chaperone complex. J. Biochem. 163, 223–232 (2018).
Tsuji, N. et al. The activation mechanism of the aryl hydrocarbon receptor (AhR) by molecular chaperone HSP90. FEBS Open Bio 4, 796–803 (2014).
Reyes, H., Reisz-Porszasz, S. & Hankinson, O. Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science 256, 1193–1195 (1992).
Schiering, C. et al. Feedback control of AHR signalling regulates intestinal immunity. Nature 542, 242–245 (2017).
Sakurai, S., Shimizu, T. & Ohto, U. The crystal structure of the AhRR-ARNT heterodimer reveals the structural basis of the repression of AhR-mediated transcription. J. Biol. Chem. 292, 17609–17616 (2017).
Quintana, F. J. The aryl hydrocarbon receptor: a molecular pathway for the environmental control of the immune response. Immunology 138, 183–189 (2013).
Vogel, C. F. et al. Cross-talk between aryl hydrocarbon receptor and the inflammatory response: a role for nuclear factor-kappaB. J. Biol. Chem. 289, 1866–1875 (2014).
Apetoh, L. et al. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat. Immunol. 11, 854–861 (2010).
Farez, M. F. et al. Melatonin contributes to the seasonality of multiple sclerosis relapses. Cell 162, 1338–1352 (2015).
Gandhi, R. et al. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells. Nat. Immunol. 11, 846–853 (2010).
Mascanfroni, I. D. et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-alpha. Nat. Med. 21, 638–646 (2015).
Wu, Y. et al. Benzo(a)pyrene regulated A549 cell migration, invasion and epithelial-mesenchymal transition by up-regulating long non-coding RNA linc00673. Toxicol. Lett. 320, 37–45 (2020).
Mobini, K. et al. 6-Formylindolo[3,2-b]carbazole (FICZ) enhances the expression of tumor suppressor miRNAs, miR-22, miR-515-5p, and miR-124-3p in MCF-7 cells. Cell J. 22, 115–120 (2020).
Wang, S. & Hankinson, O. Functional involvement of the Brahma/SWI2-related gene 1 protein in cytochrome P4501A1 transcription mediated by the aryl hydrocarbon receptor complex. J. Biol. Chem. 277, 11821–11827 (2002).
Matsumura, F. The significance of the nongenomic pathway in mediating inflammatory signaling of the dioxin-activated Ah receptor to cause toxic effects. Biochem. Pharmacol. 77, 608–626 (2009).
Bessede, A. et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature 511, 184–190 (2014).
Bunaciu, R. P. & Yen, A. Activation of the aryl hydrocarbon receptor AhR Promotes retinoic acid-induced differentiation of myeloblastic leukemia cells by restricting expression of the stem cell transcription factor Oct4. Cancer Res. 71, 2371–2380 (2011).
Mejia-Garcia, A. et al. Activation of AHR mediates the ubiquitination and proteasome degradation of c-Fos through the induction of Ubcm4 gene expression. Toxicology 337, 47–57 (2015).
Ohtake, F. et al. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature 446, 562–566 (2007).
Fernandez-Salguero, P. et al. Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science 268, 722–726 (1995).
Lahvis, G. P. & Bradfield, C. A. Ahr null alleles: distinctive or different? Biochem. Pharmacol. 56, 781–787 (1998).
Metidji, A. et al. The environmental sensor AHR protects from inflammatory damage by maintaining intestinal stem cell homeostasis and barrier integrity. Immunity 49, 353–362 (2018).
Lanis, J. M. et al. Tryptophan metabolite activation of the aryl hydrocarbon receptor regulates IL-10 receptor expression on intestinal epithelia. Mucosal Immunol. 10, 1133–1144 (2017).
Fukumoto, S. et al. Identification of a probiotic bacteria-derived activator of the aryl hydrocarbon receptor that inhibits colitis. Immunol. Cell Biol. 92, 460–465 (2014).
Fang, L. et al. Anti-TNF therapy induces CD4+ T-cell production of IL-22 and promotes epithelial repairs in patients With Crohn’s disease. Inflamm. Bowel Dis. 24, 1733–1744 (2018).
Goettel, J. A. et al. AHR activation is protective against colitis driven by T cells in humanized mice. Cell Rep. 17, 1318–1329 (2016).
Wu, H. Y. et al. In vivo induction of Tr1 cells via mucosal dendritic cells and AHR signaling. PLoS One 6, e23618 (2011).
Yeste, A. et al. IL-21 induces IL-22 production in CD4+ T cells. Nat. Commun. 5, 3753 (2014).
Cervantes-Barragan, L. et al. Lactobacillus reuteri induces gut intraepithelial CD4(+)CD8αα(+) T cells. Science 357, 806–810 (2017).
Lee, J. S. et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat. Immunol. 13, 144–151. (2011).
Longhi, M. S. et al. Bilirubin suppresses Th17 immunity in colitis by upregulating CD39. JCI Insight. 2, e92791 (2017).
Obata, Y. et al. Neuronal programming by microbiota regulates intestinal physiology. Nature 578, 284–289 (2020).
Lamas, B., Natividad, J. M. & Sokol, H. Aryl hydrocarbon receptor and intestinal immunity. Mucosal Immunol. 11, 1024–1038 (2018).
Iyer, S. S. et al. Dietary and microbial oxazoles induce intestinal inflammation by modulating aryl hydrocarbon receptor responses. Cell 173, 1123–1134.e11 (2018).
Lee, Y. et al. Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol. 13, 991–999 (2012).
Qiu, J. et al. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity 39, 386–399 (2013).
Qiu, J. et al. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity 36, 92–104 (2012).
Vivier, E. et al. Innate lymphoid cells: 10 Years On. Cell 174, 1054–1066 (2018).
Klose, C. S. & Artis, D. Neuronal regulation of innate lymphoid cells. Curr. Opin. Immunol. 56, 94–99 (2019).
Melo-Gonzalez, F. & Hepworth, M. R. Functional and phenotypic heterogeneity of group 3 innate lymphoid cells. Immunology 150, 265–275 (2017).
Lamas, B. et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 22, 598–605 (2016).
Banchereau, J. & Steinman, R. M. Dendritic cells and the control of immunity. Nature 392, 245–252 (1998).
Quintana, F. J., Yeste, A. & Mascanfroni, I. D. Role and therapeutic value of dendritic cells in central nervous system autoimmunity. Cell Death Differ. 22, 215–224 (2015).
Esser, C. et al. Functions of the aryl hydrocarbon receptor in the skin. Semin. Immunopathol. 35, 677–691 (2013).
Rothhammer, V. & Quintana, F. J. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 19, 184–197 (2019).
Vogel, C. F. et al. Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase. Biochem. Biophys. Res. Commun. 375, 331–335 (2008).
Nguyen, N. T. et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl Acad. Sci. USA 107, 19961–19966 (2010).
Mezrich, J. D. et al. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 185, 3190–3198 (2010).
Quintana, F. J. et al. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc. Natl Acad. Sci. USA 107, 20768–20773 (2010).
Wheeler, M. A., Rothhammer, V. & Quintana, F. J. Control of immune-mediated pathology via the aryl hydrocarbon receptor. J. Biol. Chem. 292, 12383–12389 (2017).
Cerovic, V. et al. Intestinal macrophages and dendritic cells: what’s the difference? Trends Immunol. 35, 270–277 (2014).
Aoki, R. et al. Indole-3-pyruvic acid, an aryl hydrocarbon receptor activator, suppresses experimental colitis in mice. J. Immunol. 201, 3683–3693 (2018).
Chng, S. H. et al. Ablating the aryl hydrocarbon receptor (AhR) in CD11c+ cells perturbs intestinal epithelium development and intestinal immunity. Sci. Rep. 6, 23820 (2016).
Quintana, F. J. et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453, 65–71 (2008).
Veldhoen, M. et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453, 106–109 (2008).
Gagliani, N. et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 523, 221–225 (2015).
Mascanfroni, I. D. et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-α. Nat. Med 21, 638–646 (2015).
Cheroutre, H., Lambolez, F. & Mucida, D. The light and dark sides of intestinal intraepithelial lymphocytes. Nat. Rev. Immunol. 11, 445–456 (2011).
Li, Y. et al. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell 147, 629–640 (2011).
Chen, W. et al. Aryl hydrocarbon receptor activation modulates CD8alphaalpha(+)TCRalphabeta(+) IELs and suppression of colitis manifestations in mice. Biomed. Pharmacother. 87, 127–134 (2017).
Kadowaki, A. et al. Gut environment-induced intraepithelial autoreactive CD4(+) T cells suppress central nervous system autoimmunity via LAG-3. Nat. Commun. 7, 11639 (2016).
Mundt, S. et al. Conventional DCs sample and present myelin antigens in the healthy CNS and allow parenchymal T cell entry to initiate neuroinflammation. Sci Immunol. 4, eaau8380 (2019).
Jordao, M. J. C. et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 363, eaat7554 (2019).
Wheeler, M. A. et al. MAFG-driven astrocytes promote CNS inflammation. Nature 578, 593–599 (2020).
Ito, M. et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 565, 246–250 (2019).
Pasciuto, E. et al. Microglia require CD4 T cells to complete the fetal-to-adult transition. Cell 182, 625–640.e24 (2020).
Berer, K. et al. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 479, 538–541 (2011).
Jangi, S. et al. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 7, 12015 (2016).
Berer, K. et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc. Natl Acad. Sci. USA 114, 10719–10724 (2017).
Cekanaviciute, E. et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc. Natl Acad. Sci. USA 114, 10713–10718 (2017).
Quintana, F. J. & Prinz, M. A gut feeling about multiple sclerosis. Proc. Natl Acad. Sci. USA 114, 10528–10529 (2017).
Rojas, O. L. et al. Recirculating intestinal IgA-producing cells regulate neuroinflammation via IL-10. Cell 176, 610–624.e18 (2019).
Piper, C. J. M. et al. Aryl hydrocarbon receptor contributes to the transcriptional program of IL-10-producing regulatory b cells. Cell Rep. 29, 1878–1892 (2019).
Xiao, S. et al. Checkpoint receptor TIGIT expressed on Tim-1(+) B cells regulates tissue inflammation. Cell Rep. 32, 107892 (2020).
Allen, N. J. et al. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 486, 410–414 (2012).
Anderson, M. A. et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200 (2016).
Anderson, M. A. et al. Required growth facilitators propel axon regeneration across complete spinal cord injury. Nature 561, 396–400 (2018).
Chung, W. S. et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504, 394–400 (2013).
Molofsky, A. V. et al. Astrocyte-encoded positional cues maintain sensorimotor circuit integrity. Nature 509, 189–194 (2014).
Singh, S. K. et al. Astrocytes assemble thalamocortical synapses by bridging NRX1alpha and NL1 via Hevin. Cell 164, 183–196 (2016).
Stogsdill, J. A. et al. Astrocytic neuroligins control astrocyte morphogenesis and synaptogenesis. Nature 551, 192–197 (2017).
Vainchtein, I. D. et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 359, 1269–1273 (2018).
Mayo, L. et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat. Med. 20, 1147–1156 (2014).
Mayo, L. et al. IL-10-dependent Tr1 cells attenuate astrocyte activation and ameliorate chronic central nervous system inflammation. Brain 139, 1939–1957 (2016).
Rothhammer, V. et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 22, 586–597 (2016).
Rothhammer, V. et al. Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proc. Natl Acad. Sci. USA 114, 2012–2017 (2017).
Chao, C. C. et al. Metabolic control of astrocyte pathogenic activity via cPLA2-MAVS. Cell 179, 1483–1498. e22 (2019).
Wheeler, M. A. et al. Environmental control of astrocyte pathogenic activities in CNS Inflammation. Cell 176, 581–596.e18 (2019).
Alaamery, M. et al. Role of sphingolipid metabolism in neurodegeneration. J. Neurochem. 2020.
Giovannoni, F. et al. AHR is a Zika virus host factor and a candidate target for antiviral therapy. Nat. Neurosci. 23, 939–951 (2020).
Yeste, A. et al. Tolerogenic nanoparticles inhibit T cell-mediated autoimmunity through SOCS2. Sci. Signal 9, ra61 (2016).
Prinz, M., Jung, S. & Priller, J. Microglia biology: one century of evolving concepts. Cell 179, 292–311 (2019).
Linnerbauer, M., Wheeler, M.A. & Quintana, F.J. Astrocyte crosstalk in CNS inflammation. Neuron (2020). In press.
Ayata, P. et al. Epigenetic regulation of brain region-specific microglia clearance activity. Nat. Neurosci. 21, 1049–1060 (2018).
Buttgereit, A. et al. Sall1 is a transcriptional regulator defining microglia identity and function. Nat. Immunol. 17, 1397–1406 (2016).
Filbrandt, C. R. et al. Presence and functional activity of the aryl hydrocarbon receptor in isolated murine cerebral vascular endothelial cells and astrocytes. Neurotoxicology 25, 605–616 (2004).
Kim, M. D., Jan, L. Y. & Jan, Y. N. The bHLH-PAS protein Spineless is necessary for the diversification of dendrite morphology of Drosophila dendritic arborization neurons. Genes Dev. 20, 2806–2819 (2006).
RNAseq, B. Brain RNAseq. 2020. https://www.brainrnaseq.org/.
Dauchy, S. et al. Expression and transcriptional regulation of ABC transporters and cytochromes P450 in hCMEC/D3 human cerebral microvascular endothelial cells. Biochem. Pharmacol. 77, 897–909 (2009).
Jacob, A. et al. Aryl hydrocarbon receptor regulates CYP1B1 but not ABCB1 and ABCG2 in hCMEC/D3 human cerebral microvascular endothelial cells after TCDD exposure. Brain Res. 1613, 27–36 (2015).
Wnuk, A. et al. Autophagy-related neurotoxicity is mediated via AHR and CAR in mouse neurons exposed to DDE. Sci. Total Environ. 742, 140599 (2020).
Wu, P. Y. et al. Novel Endogenous Ligands of Aryl Hydrocarbon Receptor Mediate Neural Development and Differentiation of Neuroblastoma. ACS Chem. Neurosci. 10, 4031–4042 (2019).
Paracer, S. & Ahmadjian V. Symbiosis: an introduction to biological associations. 2nd ed. (Oxford University Press, New York, 2000). 291 p.
Kimura, A. et al. Aryl hydrocarbon receptor protects against bacterial infection by promoting macrophage survival and reactive oxygen species production. Int. Immunol. 26, 209–220 (2014).
França, G. V. A. et al. Congenital Zika virus syndrome in Brazil: a case series of the first 1501 livebirths with complete investigation. Lancet 388, 891–897 (2016).
Cao-Lormeau, V. M. et al. Guillain-Barre Syndrome outbreak associated with Zika virus infection in French Polynesia: a case-control study. Lancet 387, 1531–1539 (2016).
Yamada, T. et al. Constitutive aryl hydrocarbon receptor signaling constrains type I interferon–mediated antiviral innate defense. Nat. Immunol. 17, 687–694 (2016).
Machado, F. S. et al. Current understanding of immunity to Trypanosoma cruzi infection and pathogenesis of Chagas disease. Semin. Immunopathol. 34, 753–770 (2012).
Ambrosio, L. F. et al. Role of Aryl hydrocarbon receptor (AhR) in the regulation of immunity and immunopathology during trypanosoma cruzi infection. Front. Immunol. 10, 631 (2019).
Barroso, A. et al. The Aryl hydrocarbon receptor modulates production of cytokines and reactive oxygen species and development of myocarditis during trypanosoma cruzi infection. Infect. Immun. 84, 3071–3082 (2016).
Sanchez, Y. et al. The unexpected role for the aryl hydrocarbon receptor on susceptibility to experimental toxoplasmosis. J. Biomed. Biotechnol. 2010, 505694 (2010).
Wen, P. Y. & Reardon, D. A. Neuro-oncology in 2015: progress in glioma diagnosis, classification and treatment. Nat. Rev. Neurol. 12, 69–70 (2016).
Stupp, R. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 (2005).
Opitz, C. A. et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478, 197–203 (2011).
Platten, M. et al. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 18, 379–401 (2019).
Gramatzki, D. et al. Aryl hydrocarbon receptor inhibition downregulates the TGF-beta/Smad pathway in human glioblastoma cells. Oncogene 28, 2593–2605. (2009).
Takenaka, M. C. et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci. 22, 729–740 (2019).
Gabriely, G. & Quintana, F. J. Role of AHR in the control of GBM-associated myeloid cells. Semin Cancer Biol. 64, 13–18 (2020).
Gabriely, G. et al. Role of AHR and HIF-1α in glioblastoma metabolism. Trends Endocrinol. Metab. 28, 428–436 (2017).
Gold, L. S. et al. A phase IIb, randomized clinical trial of tapinarof cream for the treatment of plaque psoriasis: secondary efficacy and patient-reported outcomes. J. Am. Acad. Dermatol. 9622, 30957–30959 (2020).
Robbins, K. et al. Phase 2, randomized dose-finding study of tapinarof (GSK2894512 cream) for the treatment of plaque psoriasis. J. Am. Acad. Dermatol. 80, 714–721 (2019).
Rothhammer, V. & Quintana, F. J. Environmental control of autoimmune inflammation in the central nervous system. Curr. Opin. Immunol. 43, 46–53 (2016).
Yeste, A. et al. Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proc. Natl Acad. Sci. USA 109, 11270–11275 (2012).
Acknowledgements
This work was supported by grants NS102807, ES02530, ES029136, AI126880 from the NIH and RG-1902-33606 from the NMSS and PA-1604-08459 from the International Progressive MS Alliance. J.V.M. and P.H.F.C. were supported by Santander Universidades and by Fundação Faculdade de Medicina (FFM), São Paulo, SP, Brazil.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Barroso, A., Mahler, J.V., Fonseca-Castro, P.H. et al. The aryl hydrocarbon receptor and the gut–brain axis. Cell Mol Immunol 18, 259–268 (2021). https://doi.org/10.1038/s41423-020-00585-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41423-020-00585-5
Keywords
This article is cited by
-
Molecular Evolution of Aryl Hydrocarbon Receptor Signaling Pathway Genes
Journal of Molecular Evolution (2023)
-
Lactobacillus reuteri tryptophan metabolism promotes host susceptibility to CNS autoimmunity
Microbiome (2022)
-
Honeybee gut Lactobacillus modulates host learning and memory behaviors via regulating tryptophan metabolism
Nature Communications (2022)
-
Elastin-Derived Peptides in the Central Nervous System: Friend or Foe
Cellular and Molecular Neurobiology (2022)
-
The aryl hydrocarbon receptor regulates expression of mucosal trafficking receptor GPR15
Mucosal Immunology (2021)
