
Abstract
Glucagon-like peptides (GLP-1 and GLP-2) are two proglucagon-derived intestinal hormones that mediate distinct physiological functions through two related receptors (GLP-1R and GLP-2R) which are important drug targets for metabolic disorders and Crohn’s disease, respectively. Despite great progress in GLP-1R structure determination, our understanding on the differences of peptide binding and signal transduction between these two receptors remains elusive. Here we report the electron microscopy structure of the human GLP-2R in complex with GLP-2 and a Gs heterotrimer. To accommodate GLP-2 rather than GLP-1, GLP-2R fine-tunes the conformations of the extracellular parts of transmembrane helices (TMs) 1, 5, 7 and extracellular loop 1 (ECL1). In contrast to GLP-1, the N-terminal histidine of GLP-2 penetrates into the receptor core with a unique orientation. The middle region of GLP-2 engages with TM1 and TM7 more extensively than with ECL2, and the GLP-2 C-terminus closely attaches to ECL1, which is the most protruded among 9 class B G protein-coupled receptors (GPCRs). Functional studies revealed that the above three segments of GLP-2 are essential for GLP-2 recognition and receptor activation, especially the middle region. These results provide new insights into the molecular basis of ligand specificity in class B GPCRs and may facilitate the development of more specific therapeutics.
Similar content being viewed by others


Introduction
GLP-2R belongs to class B GPCR subfamily, which is mainly expressed in the gut, pancreas and brain.1 Its endogenous ligand is GLP-2 (GLP-2(1–33)), a member of the glucagon-like peptide family that also includes GLP-1 and glucagon. GLP-2 is encoded by the proglucagon gene and becomes active after post-translational processing by prohormone convertases. As a gastrointestinal hormone, it mainly regulates intestinal epithelial cell growth and functions that are crucial for digestion and absorption of nutrients.2 Similar to GLP-1, GLP-2 is rapidly inactivated by dipeptidyl peptidase 4 in vivo.3 Clinically, GLP-2 analogue, teduglutide, is used to treat short bowel syndrome and Crohn’s disease. Other therapeutic indications, including colitis, pediatric gastrointestinal disorders and inflammation of the intestinal mucosa, are currently under development.4 Recent studies in animal models have shown that GLP-2 also exerts beneficial effects on glucose homeostasis,5,6 spontaneous hypertension7,8,9 and depression.10,11,12 This prompts a renewed interest in examining the role of this peptide beyond the gastrointestinal tract.
The cryo-electron microscopy (cryo-EM) emerges as a primary methodology to determine the structure of GPCR–G protein complexes starting with the GLP-1R13 and the calcitonin receptor,14 followed by eight other class B GPCRs in complex with various G proteins.15,16,17,18,19,20 These structures suggest that class B GPCRs have a common activation pattern which basically coincides with the previously proposed two-domain binding model.21 Although class B receptors have roughly the same activation mode, the molecular details are receptor specific, especially in the ligand-binding region. Therefore, exploring the recognition mechanism between GLP-2 and GLP-2R is of interest for further understanding the mechanism of ligand recognition and receptor activation among class B GPCRs. In this study, we employed cryo-EM to determine the high-resolution structure of the human GLP-2R in complex with Gs protein. Based on our prior experience,22 NanoBiT strategy23,24 was used in this study to stabilize the GLP-2–GLP-2R–Gs complex and strengthen the interactions between GLP-2R and Gβ, resulting in a cryo-EM structure at a global resolution of 3.0 Å (Supplementary information, Table S1). Our data provide a rational template to facilitate the design of better GLP-2 analogues for therapeutic use and expand our knowledge on the biology of this receptor family.
Results and discussion
Structure determination
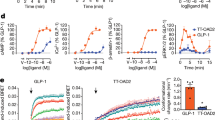
We used NanoBiT strategy22,23,24 to stabilize the complex, in which GLP-2R was directly attached to LgBiT and rat Gβ was linked to HiBiT with a 15-amino acid linker in between. Transiently transfected HEK 293T cells showed that the C-terminus-truncated GLP-2R(1–490)-LgBiT had a 6-fold stronger interaction with Gβ-SmBiT than the wild-type (WT) receptor(1–553)-LgBiT (Supplementary information, Fig. S1a, b). When SmBiT was replaced by HiBiT, the luminescence signal detected between GLP-2R and Gβ was 10-fold stronger (Supplementary information, Fig. S1c). A haemagglutinin (HA) signal peptide was added to the N-terminus of GLP-2R to increase the expression level. In addition to linking with LgBiT, a TEV protease site and an OMBP (optimized maltose binding protein)-MBP tag were attached to the C-terminus (Supplementary information, Fig. S2a). The OMBP tag is codon-optimized on the basis of MBP to increase its expression in insect cells. At the same time, using OMBP-MBP tag can enhance affinity between the protein complex and amylose resin in order to obtain more complexes. Rat Gβ1 was attached to HiBiT with a 15-amino acid linker between them (Supplementary information, Fig. S2b). To form an active G protein-coupled complex, HA-GLP-2R(1–490)-LgBiT-TEV-OMBP-MBP was co-expressed with a dominant-negative bovine Gαs (DNGαs), rat Gβ1-HiBiT and bovine Gγ2 in Sf9 insect cells. Compared with the protein expressed without NanoBiT strategy, the yield decreased but the stability increased (Supplementary information, Fig. S1d, e). Negative-staining EM visualization displayed more particles with similar size (Supplementary information, Fig. S1f).
A large-scale purification was subsequently performed using this approach. Cells were stimulated by 20 μM GLP-2 together with the camelid-derived nanobody Nb35 to stabilize the interface between Gαs and Gβ.25 The complex was solubilized in lauryl maltose neopentyl glycol (LMNG) and cholesteryl hemi succinate (CHS), treated with TEV enzyme to remove tags from GLP-2R and then isolated by amylose resin. It was further purified by size-exclusion chromatography for cryo-EM specimen preparation (Supplementary information, Fig. S3a, b). The production of cAMP and ligand-binding ability of our modified hGLP-2R construct were reduced presumably caused by its lower expression level (~73% of WT; Supplementary information, Fig. S3c, d and Table S2).
Frozen-hydrated GLP-2–GLP-2R–Gs complexes were imaged using a Titan Krios microscope (Supplementary information, Fig. S4a). 2D classification showed clear secondary structure features and random distribution of particles in different directions enabling high-resolution cryo-EM map reconstruction (Supplementary information, Fig. S4b). A total of 284,669 particles were selected after consecutive 3D classifications, which yielded a cryo-EM density map with a global resolution of 3.0 Å (Supplementary information, Fig. S4c, d).
Overall architecture
In the cryo-EM structure of the GLP-2–GLP-2R–Gs complex, most of the receptor core, G protein subunits and the first 31 amino acids of GLP-2 were observed with high-resolution features, thereby allowing for accurate modeling of side-chain rotamers (Fig. 1; Supplementary information, Fig. S5). The receptor extracellular domain (ECD) was omitted from the final reported structure because of the relatively low resolution owning to its intrinsic flexibility, which is a general feature in most reported class B GPCR–Gs complex structures.

a Cut-through view of cryo-EM density map illustrating the GLP-2–GLP-2R–Gs complex and the transparent disc-shaped micelle. Receptor density is shown in orange red, GLP-2 in forest green, Gαs in yellow, Gβ in light blue, Gγ in medium blue and Nb35 in light gray. b Model of the complex as a cartoon, with GLP-2 as helix in forest green. The receptor is shown in orange red, Gαs in yellow, Gβ in light blue, Gγ in medium blue and Nb35 in light gray.
The overall structure of active GLP-2R–Gs complex is similar to other class B GPCR–Gs complexes such as GLP-1–GLP-1R–Gs,13 ExP5–GLP-1R–Gs,17 glucagon–GCGR–Gs,18 LA-PTH–PTH1R–Gs19 and GHRH–GHRHR–Gs,26 with root mean square deviation (RMSD) values of 1.76, 1.05, 1.55, 1.09 and 0.95 Å for the whole complex, respectively. When pointing toward the glucagon receptor subfamily, notable conformational differences were observed in the extracellular half of GLP-2R where the peptide hormone binds, especially in TM1, ECL1 and TM7, indicating that GLP-2R utilizes a distinct peptide-recognition mode specific for GLP-2, but not for GLP-1, glucagon or GHRH.
Molecular recognition
The activated GLP-2R structure shows that GLP-2 is stably anchored in its position through an extensive network of interactions that involves TMs 1, 2, 3, 5 and 7, and ECLs 1, 2 and 3 (Fig. 2). The N-terminus of GLP-2, His1P (P indicates that the residue belongs to GLP-2), which is located above the conserved central polar network of class B GPCRs, is oriented toward TM3 and forms a hydrogen bond with H2683.37b (Wootten numbering in superscript27) and hydrophobic contacts with V2713.40b, W3405.36b and R3445.40b (Fig. 2a). Consistently, the truncated metabolite GLP-2(3–33) acts as a partial agonist with great reduction in binding affinity and potency.28,29 Meanwhile, mutants H268A, W340A and R344A decreased GLP-2 potency by 457-, 34- and 16-fold, respectively (Fig. 2d; Supplementary information, Table S3). Despite an identical N-terminal histidine in GLP-2, GLP-1 and glucagon, the side-chain orientations of histidine in the cognate receptors are different: the GLP-2 His1P is more distant from TM5 than that of GLP-1 and closer to TM3 than that of glucagon (Figs. 2, 3). Such difference may arise from the subtle changes in pocket residues at 3.37b (H268 for GLP-2R, Q234 for GLP-1R and Q232 for GCGR), 3.40b (V271 for GLP-2R, V237 for GLP-1R and I235 for GCGR) and ECL2 (N334 for GLP-2R, R299 for GLP-1R and N298 for GCGR). Another key residue in the N-terminus of GLP-2 is Asp3P, whose side-chain forms a salt bridge with K2312.67b and a hydrogen bond with Y1861.47b. Alanine substitutions in Y1861.47b and K2312.67b had a moderate effect (4–16 fold) on GLP-2 potency (Fig. 2d).

a Extensive contacts between GLP-2R (sticks with orange red carbons) and GLP-2 (sticks with forest green carbons), showing that the N-terminal residue His1P can interact with H2683.37b, V2713.40b and W3405.36b, together with other interactions between agonist and receptor (left). The interactions focus on the ECL1 of GLP-2R and GLP-2 (right). b, c Structural comparison of the TMD binding pockets among GLP-2–GLP-2R–Gs, GLP-1–GLP-1R–Gs,13 and glucagon–GCGR–Gs.19 GLP-2 in forest green, GLP-2R in orange red; GLP-1 in blue, GLP-1R in light blue; glucagon in magenta and GCGR in light magenta. Receptor ECD and G protein are omitted for clarity. d Effects of mutation in the peptide-binding pocket on cAMP accumulation. cAMP levels were measured in WT receptor and alanine-mutated TMs and ECLs. Data shown are means ± SEM of at least three independent experiments.

The middle region of GLP-2 forms both a polar network with several charged and polar amino acids in TM7 (K4147.35b and R4177.38b) and ECL2 (T332ECL2 and N334ECL2) via Gly4P, Ser5P, Asp8P, Glu9P and Asn11P and a complementary nonpolar network with TM1 (R1711.32b, Y1721.33b, L1751.36b, L1781.39b and Q1791.40b) via Phe6P, Met10P, Ile13P and Leu14P (Fig. 2a). Intriguingly, we observed one water molecule bridging Ser7P and D2322.68b, which is absent in the GLP-1–GLP-1R structure.13 When comparing the active GLP-2R structure with the GLP-1-bound GLP-1R structure,13 the extracellular end of TM1 in GLP-2R is extended by six additional residues, thereby bending toward GLP-2 with the formation of additional hydrogen bond and hydrophobic contacts. These interactions further stabilize the peptide conformation in the bound state and may contribute to specific recognition of GLP-2.
Partial unwinding of the short α-helix elevates ECL1 to an extended conformation that stands upwards in line with TM2 and TM3, thereby engaging the C-terminal half of GLP-2 through an extensive interface spanning from Leu14P to Trp25P (Figs. 2a, 3). Specifically, Asp21P forms a salt bridge with R242ECL1, while Trp25P forms a hydrogen bond with D244ECL1 and pi-pi stacking with W249ECL1, which was stabilized by Phe22P. Besides, Leu14P and Ala18P pack against Y239ECL1, R242ECL1 and Y252ECL1. These interactions strengthen the binding and function of GLP-2, consistent with the decreased potencies of R242A (by 5-fold), R242E (by 20-fold), W249A (by 1552-fold) and Y252A (by 5-fold) (Fig. 2d). Similar effects of mutations were observed with the binding of teduglutide (Table 1). Together, these results highlight the importance of ECL1 in GLP-2 recognition and receptor activation.
G protein coupling
In our model of the GLP-2R–Gs complex, Gs protein is anchored by the α5 helix of Gαs (GαH5), thereby fitting to the cytoplasmic cavity of the transmembrane domain (TMD) involving TMs 2, 3, 5, 6, and 7 (Supplementary information, Fig. S6). Structural comparison of the GLP-2R–Gs complex with that of other class B GPCRs reveals substantial similarity in the G protein-binding interface, consistent with a common mechanism of Gs protein engagement. However, many receptor-specific interactions were observed. Y391GαH5 is an essential residue in determining G protein coupling specificity by forming H-bond in class A GPCRs,30 however, this interaction was not observed among available class B GPCR–G protein complexes. Most strikingly, Y391GαH5 formed a tight hydrogen bond with a water molecule, which connects to E2813.50b and H2142.50b in the GLP-2R–Gs complex (Supplementary information, Fig. S6), a phenomenon that was not observed among other class B GPCRs. Polar interactions also occurred between K3685.64b and Q384GαH5, H372ICL3 and R385GαH5 as well as R3826.37b and E392GαH5. The ICL2 of GLP-2R formed additional hydrophobic interactions with GαH5. There were also limited interactions between GαH5 subunit and helix 8 of the receptor (E392GαH5, N4408.47b and G4418.48b). These receptor-specific interactions may contribute to the G protein specificity of GLP-2R.
Ligand specificity
GLP-2 and GLP-1 share high sequence similarity (69.7%) and have almost identical residues in two regions: the N-terminus (first six residues in GLP-2) that penetrates into the TMD core, and the C-terminus (residues 18–33) that is recognized by the ECD and ECL1 of the receptor. On the contrary, the central region of the peptide mainly interacts with TMs 1, 2, 7, ECL1 and ECL2 of the receptor and differs to that of GLP-1 (Fig. 4; Supplementary information, Fig. S7a, b).

a Structural comparison of GLP-2–GLP-2R–Gs and GLP-1–GLP-1R–Gs.13 Receptor ECD and G protein are omitted for clarity. b Schematic diagram of interactions between peptide and receptor. Conserved residues in GLP-2 and GLP-1 are highlighted in pink, while those similar are shown in light pink. Length of gray shading box illustrates the interface area between peptide and corresponding segment. c Signaling profiles of the chimeric GLP-2/GLP-1 peptides. Replacement of the N-terminus (residues 1–6), middle region (residues 7–19) and C-terminus (residues 20–33) of GLP-2 by corresponding segment of GLP-1 was named as chimeric GLP-2/GLP-1(1–6), GLP-2/GLP-1(7–19) and GLP-2/GLP-1(20–33), respectively. Dose-response curves of peptide-induced cAMP accumulation for GLP-2R were generated and graphed as means ± SEM from three independent experiments.
Structural comparison of GLP-2–GLP-2R–Gs and GLP-1–GLP-1R–Gs13 complexes reveals distinct features of their ligand-binding pockets (Figs. 3, 4; Supplementary information, Fig. S7c–e). Superimposing the TMD of GLP-2R with that of GLP-1R shows that the intracellular portions of both receptors, except ICL2 and ICL3, overlap very well, whereas the extracellular parts of TM1, TM5 and TM7 as well as all ECLs have different conformations and side-chain orientations (Fig. 4a; Supplementary information, Fig. S7c–e). Consistently, the N-terminal part of GLP-2 is reoriented relative to GLP-1. Furthermore, the rest of GLP-2 is elevated vertically (~0.5 Å) and moves toward TM1–TM2 by 1–3 Å, allowing the extensive interaction with ECL1. Overall, the interface area measured by buried solvent-accessible surface of GLP-2–GLP-2R TMD is 3050 Å2, significantly larger than that of GLP-1–GLP-1R TMD (2297 Å2).
The orientation of the N-terminal histidine between GLP-1 and GLP-2 is rather different (Fig. 2b; Supplementary information, Fig. S7c). GLP-1 faces toward TM5 to form cation-pi stacking and hydrogen bond with the rotated down side-chain of R299ECL2, while that of W3065.36b and R3105.40b switch out from the receptor core to accommodate the approaching of histidine. However, by replacing arginine with threonine, the ECL2 of GLP-2R is no longer able to dip into the receptor core and becomes distant from the N-terminal histidine of GLP-2. Therefore, W3405.36b and R3445.40b of GLP-2R rotate inwards. Together with the inward movement of the extracellular part of TM5, they push the N-terminus of GLP-2 away from TM5 to reside closely with TM3, where H2683.37b contributes a hydrogen bond to stabilize the conformation (Supplementary information, Fig. S7c). Compared to GLP-1, the middle region of GLP-2 has more extensive contacts with the ligand-binding pocket (Fig. 4b). The interface areas between TMD and GLP-2 contributed by TM1 and TM7 are 864 Å2 and 432 Å2, respectively, markedly larger than those between TMD and GLP-1 (330 Å2 and 303 Å2, respectively). Specifically, in the extracellular half of GLP-2R, TM1 forms at least one additional helical turn, thereby moving toward TM2 (5 Å, measured by Cα of the residue at 1.33b) and bending down toward GLP-2. This ultimately leads to a much larger interaction pattern for GLP-2 (ranging from Asp3P to Asn16P) than for GLP-1, where only two residues of GLP-1 interact with TM1.
Meanwhile, side-chain orientations of TM1 residues are reorganized (Supplementary information, Fig. S7d). For example, Y1821.43b is reoriented ~90° from an outside facing position to a position pointing to TM2 and engages T-shaped stacking with Phe6P of GLP-2. Accompanying TM1 movement to TM2, the extracellular tip of TM7 moves toward TM1 and the side-chain of K4147.35b points to GLP-2 with formation of a salt bridge with Glu9P. In the C-terminus of GLP-2, we observed that it forms a more extensive interaction with the ECL1 of GLP-2R compared to the GLP-1–GLP-1R ECL1 interaction (interface area = 791 Å2 vs 424 Å2). Different from a 2.5-turn α-helix of ECL1 in both activated GLP-1R and GCGR, the ECL1 of GLP-2R partially unwinds vertically in line with TM2 and TM3, providing additional interactions not seen in its counterparts (Figs. 3, 4a; Supplementary information, Fig. S7e). Alternatively, the α1-helix of GLP-1R ECD is in close contact with the C-terminus of GLP-1. Because the ECD structure of GLP-2R is unavailable, its impact on the structure and dynamics remains elusive.
Considering the uneven sequence conservation and distinct binding modes of GLP-2 and GLP-1, we designed chimeric GLP-2/GLP-1 peptides to explore the molecular basis of ligand specificity. As shown in Fig. 4c, replacement of the first six residues of GLP-2 by those of GLP-1 (i.e., double mutant D3PE and S5PT) decreased cAMP accumulation by 12-fold compared to the native peptide, suggesting that subtle substitution may directly alter receptor activation. In addition, chimeric GLP-2 whose C-terminal fragment (residues 20–33) was replaced with that of GLP-1 displayed a 17-fold potency reduction in GLP-2R signaling but successfully elicited GLP-1R-mediated cAMP accumulation (Fig. 4c). Replacement of the GLP-2 middle region (residues 7–19) that mainly interacts with TM1, TM7 and ECL1 by the corresponding part of GLP-1, almost abolished GLP-2R-mediated cAMP accumulation but fully rescued that induced by GLP-1R (Fig. 4c), indicative of a fundamental role of this region in determining the specificity for ligand recognition.
Conformational changes
Triggered by GLP-2 binding and G protein coupling, GLP-2R would presumably undergo significant conformational changes. It shares a high degree of sequence similarity in the TMD region with GLP-1R (61%) that has been studied extensively both structurally and functionally.31 Thus, inactive, intermediate and active GLP-1R structures published previously13,17,32,33,34,35 provide good references for the present study. Superimposing the TMD of GLP-2R with that of GLP-1R reveals that activated GLP-2R shows a similar conformation to that of GLP-1-bound or ExP5-bound GLP-1R–Gs complexes13,17 (Cα RMSD = 1.41 and 0.91 Å, respectively), distinct from inactive GLP-1R33 (Cα RMSD = 1.79 Å). Further comparison demonstrates that diversified peptide–receptor interactions converge to induce conserved and extensive conformational transition of the TMD of GLP-2R and GLP-1R (Fig. 5).

In the extracellular half of GLP-2R, accompanying the unwinding of the last two turns of TM6, TM7 moves outward and bends toward TM6 via a conserved pivot point G4297.50b, which expands the TMD-binding pocket to accommodate peptide entrance and connection. Meanwhile, TM1 moves inwards and folds down toward GLP-2, which contributes both polar and hydrophobic contacts to stabilize the binding. Furthermore, different from the ordered α-helical ECL1 in inactive GLP-1R33 and β-hairpin in inactive GCGR36 that cover the binding pocket and probably block peptide insertion, ECL1 of active GLP-2R, GLP-1R and GCGR simultaneously move outward and stand upwards in line with TM2 and TM3. Together with inward movement of α-helical extended TM2, raised ECL1 of GLP-2R forms extensive interaction with the C-terminal half of GLP-2 (Fig. 4a, b) and was shown to be essential for peptide binding and receptor activation (Fig. 4c). As far as the intracellular half is concerned, the positions of TM1–2–3 of active GLP-2R resemble that of active GLP-1R (Fig. 5). The most profound conformational change is the sharp kink in the middle of TM6, leading to an outward movement of the intracellular portion of TM6 as measured by Cα carbon of Lys6.35b (19.0 Å, similar to that of other Gs-coupled class B receptors). In contrast, in the absence of G protein coupling, the binding of peptide 5 promotes a rigidly outward shift of the intracellular tip of TM6 of GLP-1R by 12.1 Å without a sharp kink,34 thereby creating an intracellular crevice for G protein coupling. For PTH1R37 and GCGR38 (whose crystal structures were determined with PGS domain fusion in ICL3 and T4L fusion at ICL2, respectively), agonist binding failed to promote the outward movement of the intracellular half of TM6 as seen in the inactive state, probably due to the fusion. With more structural and dynamics studies on class B GPCRs, the activation process from inactive to active states via an intermediate state, i.e., allosteric communication between extracellular peptide binding and intracellular G protein coupling, will be better understood.
Role of ECL1
Relative to ECL2 and ECL3, ECL1 in class B GPCRs is the most divergent extracellular loop with flexible lengths, ranging from 8 (CRF2R) to 27 (PTH1R) residues. Inspired by the intriguing ECL1 conformation of GLP-2R, we performed structural comparison of ECL1 across class B GPCRs to unveil its functionality (Fig. 6). Among 20 agonist-bound structures from 9 receptors, we calculated the height of ECL1 (defined as the maximum vertical distance of Cα carbon relative to the membrane layer) and the ECL1–agonist interface area. We found that GLP-2R has the most protruded ECL1 with a height of 22.6 Å and this ECL1 closely attaches to GLP-2 with the largest interface area of 791 Å2. The ECL1 of all activated GLP-1R and GCGR structures consistently adopt α-helical conformation bridging extended TM2 and TM3 helices by two short linkers to form extensive interactions with bound agonists. Remarkably, significant conformational changes of ECL1 upon receptor activation were observed relative to their inactive conformations: (i) in the inactive GCGR, ECL1 forms a β-hairpin conformation and further compacts β-sheet with the stalk region; and (ii) in the inactive GLP-1R, ECL1 moves toward TM1 and connects ECD in its closed TMD-interacting conformation. As far as CRF1R, CRF2R, AM1R, AM2R and CLR are concerned, their ECL1 are relatively short (consisting of 8–11 residues) and can only form one-turn α-helix to connect the adjacent transmembrane helices TM2 and TM3, contributing limited contacts with the peptides. PTH1R has the longest ECL1, but it is unstructured in both LA-PTH–PTH1R–Gs19 and ePTH–PTH1R37 structures, probably owing to the great mobility of PTH1R’s ECD and specific binding mode, consistent with the mutagenesis studies.37,39,40 To date, four PAC1R structures were reported, namely, PACAP38–PAC1R–Gs (3.0 Å),20 PACAP–PAC1R–mini-Gs (3.9 Å),41 PACAP38–PAC1R–Gs (3.5 Å)42 and maxadilan–PAC1R–Gs (3.6 Å).42 The ECL1 was not resolved in the first two structures, and the third map showed that the ECL1 closely interacted with PACAP38 (pituitary adenylate cyclase activating polypeptide-38) without detailed residue contact information due to limited resolution.42 Interestingly, maxadilan, a peptidic PAC1R agonist, reorganized the ECD orientation and reshaped the ligand-binding pocket conformation, including ECL142 (Fig. 6b). Collectively, these data demonstrate the diversity and flexibility of ECL1 among class B GPCRs.

a Scatter plot of ECL1 from 9 class B GPCRs. x axis shows the interface area between ECL1 and agonist, and y axis is the height of ECL1. b Structural comparison of diversified ECL1 conformations among class B GPCRs. All structures are superimposed on the GLP-1-bound GLP-1R (PDB: 5VAI)13 using the Cα carbons of the residues in the TM2–3–4. The locations of the membrane planes predicted by OPM (Orientations of Proteins in Membranes)54 are shown as black dashed lines. The height of ECL1 is defined as the maximum vertical distance of Cα carbon relative to the membrane layer, while the interface areas were calculated using freeSASA.53
To further explore the functional importance of ECL1 in ligand binding and receptor activation, we performed mutagenesis studies on this part of the receptor (Table 1; Supplementary information, Fig. S8 and Table S4). Replacement of GLP-2R ECL1 by poly-alanine abolished the ability of GLP-2 to elicit cAMP response, while replacement by that of GLP-1R diminished its potency by 196-fold. Meanwhile, single-point mutations on deeply buried residues (R242, W249 and Y252) in the interface between GLP-2 and ECL1 also demonstrated negative impact on ligand binding (Supplementary information, Table S5).
In conclusion, the structure of the GLP-2–GLP-2R–Gs complex delineates detailed interactions between GLP-2 and GLP-2R that account for the peptide binding specificity, and provides new insights into the structural reorganization of class B GPCRs upon activation. Combined with previous research on the GLP-1R, our results reveal that subtle differences in the sequence of TMD binding pocket and the consequent diversified conformational changes of the extracellular half accommodate peptide binding and empower the receptor to be specifically trigged by the cognate hormone. This complex structure points to an essential role of ECL1 in peptide recognition and receptor activation, in addition to that of ECD. This work also highlights the applicability of the NanoBiT tethering strategy to stabilize GPCR–G protein complexes for structural studies.
Materials and methods
Constructs
WT human GLP-2R was modified to include a HA signal peptide at the N-terminus. LgBiT and an OMBP-MBP tag were added to the C-terminus of the receptor with a TEV site between them. A bovine Gαs was used with G226A and A366S mutations to stabilize the complex;43 rat Gβ1 was attached to HiBiT with a 15-amino acid linker between them; bovine Gγ2 was also used in cloning. All of the above constructs were generated in pFastBac and pcDNA3.1 vectors for protein expression in insect cells and functional assays in mammalian cells, respectively.
Protein expression
The complex was co-expressed in Sf9 cells (Expression System, Davis, USA) grown in ESF-921 serum-free medium (Expression System) at 27 °C, 120 rpm. Baculovirus was added at a proportion of 1:1:1:1 for HA-GLP-2R(1–490)-LgBiT-TEV-OMBP-MBP, Gαs, Gβ-HiBit and Gγ when the cell density reached 3 × 106/mL. After 48 h incubation, the pellet was harvested by PBS using centrifugation at 2000 rpm for 20 min.
Complex purification
Sf9 cells were suspended in 20 mM HEPES, pH 7.4, 100 mM NaCl and 10% (v/v) glycerol in the presence of Protease Inhibitor Cocktail (Topscience, Shanghai, China). Complex was formed by adding 10 mM CaCl2, 10 mM MgCl2, 1 mM MnCl2, 50 mU/mL apyrase (Sigma-Aldrich, Darmstadt, Germany), 20 μM GLP-2, 100 μM TCEP (Sigma-Aldrich) and 10 μg/mL Nb35 into the cell lysate and incubated at room temperature (RT) for 1.5 h. Cell membranes were solubilized by 0.5% (w/v) LMNG (Anatrace, Maumee, USA) supplemented with 0.1% (w/v) CHS (Anatrace) with 5 μM GLP-2 and Protease Inhibitor Cocktail (Topscience) at 4 °C for 2 h, followed by centrifugation at 65,000× g for 30 min at 4 °C. The supernatant was taken to bind with amylose resin (NEB, Ipswich, USA) for 2 h at 4 °C. After packing, the column was washed with buffer containing 20 mM HEPES, pH 7.4, 100 mM NaCl, 10% (v/v) glycerol, 5 μM GLP-2, 25 μM TCEP, 5 mM MgCl2, 1 mM MnCl2, 0.1% (w/v) LMNG and 0.02% (w/v) CHS first (10 column volumes), and then with decreased concentrations of detergents 0.03% (w/v) LMNG, 0.01% (w/v) GDN and 0.008% (w/v) CHS (20 column volumes). TEV enzyme was added to the resin and kept at 4 °C overnight to remove OMBP-MBP tag. The complex was eluted from the resin and concentrated to 500 μL using a 100 kDa MWCO Amicon Ultra Centrifugal Filter (Merck Millipore, Darmstadt, Germany). Size-exclusion chromatography was carried out by loading the protein sample to Superdex 200 Increase 10/30GL (GE Healthcare, Boston, USA) to obtain the monomer complex. The column was pre-equilibrated with 20 mM HEPES, pH 7.4, 100 mM NaCl, 5 μM GLP-2, 100 μM TCEP, 2 mM MgCl2, 0.00075% (w/v) LMNG, 0.00025% (w/v) GDN and 0.0002% (w/v) CHS.
Nb35 production
E. coli BL21 cells were transformed with the plasmid containing Nb35 target gene and were grown in TB medium with 100 μg/mL ampicillin, 1 M MgCl2, 2% (w/v) glucose at 37 °C, 180 rpm for 3 h. IPTG (1 M) was added to induce the expression when OD600 reached 0.7–1.2. After 8 h expression, the cell pellet was collected and stored at −80 °C until use. Nb35 was purified by nickel affinity chromatography as previously described,25 followed by size-exclusion chromatography using a HiLoad 16/600 Superdex 75 column (GE Healthcare). The column was pre-equilibrated with 20 mM HEPES, pH 7.4 and 100 mM NaCl. Glycerin 30% (v/v) was added to collect Nb35 which was frozen in liquid nitrogen and stored at −80 °C.
Negative-staining electron microscopy
Purified GLP-2R–Gs complex was diluted to 0.012 mg/mL with 20 mM HEPES, pH 7.4, 100 mM NaCl, 5 μM GLP-2, 100 μM TCEP, 2 mM MgCl2, 0.00075% (w/v) LMNG, 0.00025% (w/v) GDN and 0.0002% (w/v) CHS. Eight microliters of protein complex and 8 μL uranium formate were applied to a glow-discharged 300-mesh cooper grid (EMCN, Beijing, China) for 1 min and then sucked with filter paper. After grilling the grid with a lamp, the samples were imaged at RT with a Talos L120C electron microscope (FEI, Hillsboro, USA) operated at 120 kV. Images were recorded at magnification of 57,000× and a defocus value of −1 μm.
Cryo-EM
Three microliters of the purified GLP-2–GLP-2R–Gs complex at about 20 mg/mL was applied onto a glow-discharged holey carbon grid (Quantifoil R1.2/1.3), blotted, and subsequently sample-coated grids were vitrified by plunging into liquid ethane using a Vitrobot Mark IV (ThermoFisher Scientific, Waltham, USA). Automatic data collection was performed on a Titan Krios equipped with a Gatan K2 Summit direct electron detector in the Center of Cryo-Electron Microscopy, Zhejiang University (Hangzhou, China). The microscope was operated at 300 kV accelerating voltage, at a nominal magnification of 29,000× in counting mode, corresponding to a pixel size of 1.014 Å. A total of 2296 movies were obtained at a dose rate of about 8 electrons per Å2 per second with a defocus ranging from −0.5 to −2.5 μm. The total exposure time was 8 s and intermediate frames were recorded in 0.2 s intervals, resulting in an accumulated dose of 64 electrons per Å2 and a total of 40 frames per micrograph.
Data processing and 3D reconstructions
Dose-fractionated image stacks were subjected to beam-induced motion correction using MotionCor2.1.44 A sum of all frames, filtered according to the exposure dose, in each image stack was used for further processing. Contrast transfer function parameters for each micrograph were determined by Gctf v1.06.45 Particle selection, 2D and 3D classifications were performed on a binned dataset with a pixel size of 2.028 Å using RELION-3.0-beta2.46 Auto-picking yielded 1,498,893 particle projections were subjected to reference-free 2D classification to discard false positive particles or particles categorized in poorly defined classes, producing 485,548 particle projections for further processing. This subset of particle projections was subjected to a round of maximum-likelihood-based three dimensional classifications with a pixel size of 2.028 Å, resulting in one well-defined subsets with 162,932 projections. Further 3D classifications with mask on the complex produced two good subsets accounting for 332,946 particles, which were subsequently subjected to a round of 3D classifications with mask on the receptor. A selected subset containing 284,669 projections was then subjected to 3D refinement and Bayesian polishing with a pixel size of 1.014 Å. After last round of refinement, the final map has an indicated global resolution of 3.0 Å at a Fourier shell correlation (FSC) of 0.143. Local resolution was determined using the Bsoft package with half maps as input maps.47
Model building
The initial template of the TMDs in GLP-2R was generated using SWISS-MODEL.48 The models of Gs protein and Nb35 from PTH1R complex structure (PDB: 6NBF)19 were used as the relevant components of the GLP-2R–Gs complex models. While the whole TMDs, Gs protein, and Nb35 showed decent density, the ECDs of GLP-2R complex structures displayed very poor density in the whole density maps and were thus omitted in the final structural models. All the models were docked into the EM density map using UCSF Chimera.49 Model adjustment and rebuilding were performed in COOT.50 Real space and Rosetta refinements were carried out using Phenix software package.51 All residues were checked for fitting in electron density, and Ramachandran and rotamer restraints. The refined models exhibited good model-density fitting and model geometry (see Supplementary information, Table S1 for data and model refinement statistics).
cAMP accumulation
HEK 293T cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco, Waltham, USA) supplemented with 10% (v/v) fetal bovine serum (FBS) (Gibco) and 1% (v/v) sodium pyruvate (Gibco) at 37 °C, 5% CO2 in a humidified incubator. They were seeded onto 6-well cell culture plates and transiently transfected with different GLP-2R constructs using Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA, USA). After 24 h, the transfected cells were seeded onto 384-well microtiter plates (3000 cells per well) in HBSS supplemented with 5 mM HEPES, 0.1% (w/v) bovine serum albumin (BSA) and 0.5 mM 3-isobutyl-1- methylxanthine, and stimulated with increasing concentrations of GLP-2 for 30 min at RT. Eu and Ulight were diluted by cAMP detection buffer and added to the plates separately to terminate the reaction. Plates were incubated at RT for 60 min before measuring the fluorescence intensity at 620 nm and 650 nm by an EnVision multilabel plate reader (PerkinElmer, Boston, USA).
NanoBiT assay
HEK 293T cells were seeded onto 6-well cell culture plates in DMEM containing 10% FBS and 1% sodium pyruvate at 37 °C, 5% CO2 before transfection. They were transiently transfected with truncated GLP-2 receptors attached to LgBiT and Gβ-SmBiT (or Gβ-HiBiT) in a ratio of 1:1 using Lipofectamine 2000 transfection reagent (Invitrogen) and incubated for 24 h. After seeding onto 96-well microtiter plates (48,000 cells per well), the medium was replaced by HBSS followed by 30 min incubation at 37 °C. Coelenterazine H (5 μM) was then added to the plates and luminescence signals measured 1 h thereafter using an EnVision multilabel plate reader (PerkinElmer) at 18 s intervals before and after ligand addition (25 °C).
Oxidative iodination
The GLP-2 radioligand was created by oxidative iodination using ChloramineT to incorporate the iodine isotope [125I] at position Tyr10 of human GLP-2(1–33, M10Y). A stepwise oxidation reaction was performed by addition of 6 aliquots of 5 µL ChloramineT (30 µL/mL) with a 1-min interval during constant stirring. The reaction was terminated by the addition of 400 µL phase A (0.1% trifluoracetic acid) before being fractionated into 1 mL volume by reverse-phase high-performance liquid chromatography (RP-HPLC, Waters 600 SimiPrep system) on a C18 column. The iodinated protein was assembled by increasing concentrations of phase B (Acetonitrile + 0.1% TFA) and tested for competitive binding ability.
Receptor binding
One day before transfection, the COS-7 cells were seeded in 75 cm2 culture flasks (3 million cells/flask) in DMEM, containing 3.9 g/L NaHCO3 and supplemented with 10% FBS, 1% l-glutamine, 180 units/mL penicillin and 45 µg/mL streptomycin. After an overnight culture at 37 °C, 10% CO2 and 95% air humidity, the cells were transiently transfected with different GLP-2R constructs or pcDNA3.1 vector as previously described.52 Following 24-h incubation, the cells were transferred to Costa® microtiter plates (150,000 cells/well, PerkinElmer) to achieve a 5%–10% specific binding. Forty-eight hours after transfection, the cells were washed twice in binding buffer (50 mM HEPES, pH 7.4, supplemented with 1 mM CaCl2, 5 mM MgCl2 and 0.5% (w/v) BSA) and incubated for 15 min at 4 °C. An increasing concentration of unlabeled GLP-2(1–33) or teduglutide (ranging from 0.1 nM to 1 μM) followed by a fixed concentration of [125I]GLP-2(1–33, M10Y) (10–15 pM) was added to the cells and incubated for 4 h at 4 °C. The cells were then washed twice in binding buffer (4 °C), lysed and counted for radioactivity using a Wallac Gamma Counter (PerkinElmer).
Structural analysis
The interface area was calculated by FreeSASA53 using the Sharke-Rupley algorithm with a probe radius of 1.2 Å. Structural figures were prepared using UCSF Chimera49 and PyMOL (The PyMOL Molecular Graphics System v.2.1). RMSD analysis was performed in PyMOL.
Data availability
The atomic coordinates and the electron microscopy maps have been deposited in the Protein Data Bank (PDB) under accession number 7D68 and Electron Microscopy Data Bank (EMDB) under accession number EMD-30590, respectively. All relevant data are available from the authors upon request and/or included in the manuscript or Supplementary information.
References
Drucker, D. J. & Yusta, B. Physiology and pharmacology of the enteroendocrine hormone glucagon-like peptide-2. Annu. Rev. Physiol. 76, 561–583 (2014).
Drucker, D. J., Erlich, P., Asa, S. L. & Brubaker, P. L. Induction of intestinal epithelial proliferation by glucagon-like peptide 2. Proc. Natl. Acad. Sci. USA 93, 7911–7916 (1996).
Drucker, D. J. et al. Regulation of the biological activity of glucagon-like peptide 2 in vivo by dipeptidyl peptidase IV. Nat. Biotechnol. 15, 673–677 (1997).
Austin, K., Markovic, M. A. & Brubaker, P. L. Current and potential therapeutic targets of glucagon-like peptide-2. Curr. Opin. Pharmacol. 31, 13–18 (2016).
Baldassano, S., Amato, A. & Mule, F. Influence of glucagon-like peptide 2 on energy homeostasis. Peptides 86, 1–5 (2016).
Amato, A., Baldassano, S. & Mule, F. GLP2: an underestimated signal for improving glycaemic control and insulin sensitivity. J. Endocrinol. 229, R57–R66 (2016).
Sasaki-Hamada, S., Ito, K. & Oka, J. Neuronal Fos-like immunoreactivity associated with dexamethasone-induced hypertension in rats and effects of glucagon-like peptide-2. Life Sci. 93, 889–896 (2013).
Sasaki-Hamada, S., Narusawa, K., Nakamura, R., Ishibashi, H. & Oka, J. I. Effects of centrally administered glucagon-like peptide-2 on blood pressure and barosensitive neurons in spontaneously hypertensive rats. Neuropeptides 69, 66–75 (2018).
Sasaki-Hamada, S., Okada, S., Ito, K., Iwai, T. & Oka, J. I. Immunohistochemical determination of the site of hypotensive effects of glucagon-like peptide-2 in the rat brain. Neuroscience 212, 140–148 (2012).
Sasaki-Hamada, S., Nakamura, Y., Koizumi, K., Nabeta, R. & Oka, J. I. Pharmacological evidence for the relationship between the NMDA receptor and nitric oxide pathway and the antidepressant-like effects of glucagon-like peptide-2 in the mouse forced-swim test. Behav. Brain Res. 364, 162–166 (2019).
Sasaki-Hamada, S. et al. Antidepressant-like effects exerted by the intranasal administration of a glucagon-like peptide-2 derivative containing cell-penetrating peptides and a penetration-accelerating sequence in mice. Peptides 87, 64–70 (2017).
Sasaki-Hamada, S., Yuri, Y., Hoshi, M. & Oka, J. I. Immunohistochemical determination of the site of antidepressant-like effects of glucagon-like peptide-2 in ACTH-treated mice. Neuroscience 294, 156–165 (2015).
Zhang, Y. et al. Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature 546, 248–253 (2017).
Liang, Y. L. et al. Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature 546, 118–123 (2017).
Liang, Y. L. et al. Cryo-EM structure of the active, Gs-protein complexed, human CGRP receptor. Nature 561, 492–497 (2018).
Ma, S. et al. Molecular basis for hormone recognition and activation of corticotropin-releasing factor receptors. Mol. Cell 77, 669–680 (2020).
Liang, Y. L. et al. Phase-plate cryo-EM structure of a biased agonist-bound human GLP-1 receptor-Gs complex. Nature 555, 121–125 (2018).
Qiao, A. et al. Structural basis of Gs and Gi recognition by the human glucagon receptor. Science 367, 1346–1352 (2020).
Zhao, L. H. et al. Structure and dynamics of the active human parathyroid hormone receptor-1. Science 364, 148–153 (2019).
Liang, Y. L. et al. Toward a structural understanding of class B GPCR peptide binding and activation. Mol. Cell 77, 656–668 (2020).
Hoare, S. R. Mechanisms of peptide and nonpeptide ligand binding to Class B G-protein-coupled receptors. Drug Discov. Today 10, 417–427 (2005).
Duan, J. et al. Cryo-EM structure of an activated VIP1 receptor-G protein complex revealed by a NanoBiT tethering strategy. Nat. Commun. 11, 4121 (2020).
Dixon, A. S. et al. NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem. Biol. 11, 400–408 (2016).
Inoue, A. et al. Illuminating G-protein-coupling selectivity of GPCRs. Cell 177, 1933–1947 (2019).
Rasmussen, S. G. et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 (2011).
Zhou, F. L. et al. Structural basis for activation of the growth hormone-releasing hormone receptor. Nat. Commun. 11, 5205 (2020).
Wootten, D., Simms, J., Miller, L. J., Christopoulos, A. & Sexton, P. M. Polar transmembrane interactions drive formation of ligand-specific and signal pathway-biased family B G protein-coupled receptor conformations. Proc. Natl. Acad. Sci. USA 110, 5211–5216 (2013).
Thulesen, J. et al. The truncated metabolite GLP-2(3-33) interacts with the GLP-2 receptor as a partial agonist. Regul. Pept. 103, 9–15 (2002).
Munroe, D. G. et al. Prototypic G protein-coupled receptor for the intestinotrophic factor glucagon-like peptide 2. Proc. Natl. Acad. Sci. USA 96, 1569–1573 (1999).
Liu, X. et al. Structural insights into the process of GPCR-G Protein complex formation. Cell 177, 1243–1251 (2019).
Graaf, C. et al. Glucagon-like peptide-1 and its class B G protein-coupled receptors: a long march to therapeutic successes. Pharmacol. Rev. 68, 954–1013 (2016).
Ma H., et al. Structural insights into the activation of GLP-1R by a small molecule agonist. Cell Res. https://doi.org/10.1038/s41422-020-0384-8 (2020).
Wu, F. et al. Full-length human GLP-1 receptor structure without orthosteric ligands. Nat. Commun. 11, 1272 (2020).
Jazayeri, A. et al. Crystal structure of the GLP-1 receptor bound to a peptide agonist. Nature 546, 254–258 (2017).
Song, G. et al. Human GLP-1 receptor transmembrane domain structure in complex with allosteric modulators. Nature 546, 312–315 (2017).
Zhang, H. et al. Structure of the full-length glucagon class B G-protein-coupled receptor. Nature 546, 259–264 (2017).
Ehrenmann, J. et al. High-resolution crystal structure of parathyroid hormone 1 receptor in complex with a peptide agonist. Nat. Struct. Mol. Biol. 25, 1086–1092 (2018).
Zhang, H. et al. Structure of the glucagon receptor in complex with a glucagon analogue. Nature 553, 106–110 (2018).
Zhao, L. H. et al. Differential requirement of the extracellular domain in activation of class B G protein-coupled receptors. J. Biol. Chem. 291, 15119–15130 (2016).
Lee, C. et al. Role of the extracellular regions of the parathyroid hormone (PTH)/PTH-related peptide receptor in hormone binding. Endocrinology 135, 1488–1495 (1994).
Kobayashi, K. et al. Cryo-EM structure of the human PAC1 receptor coupled to an engineered heterotrimeric G protein. Nat. Struct. Mol. Biol. 27, 274–280 (2020).
Wang, J. et al. Cryo-EM structures of PAC1 receptor reveal ligand binding mechanism. Cell Res. 30, 436–445 (2020).
Liu, P. et al. The structural basis of the dominant negative phenotype of the Galphai1beta1gamma2 G203A/A326S heterotrimer. Acta Pharmacol. Sin. 37, 1259–1272 (2016).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods. 14, 331–332 (2017).
Zhang, K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Scheres, S. H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Heymann, J. B. Guidelines for using Bsoft for high resolution reconstruction and validation of biomolecular structures from electron micrographs. Protein Sci. 27, 159–171 (2018).
Waterhouse, A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018).
Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Jensen, P. C., Thiele, S., Ulven, T., Schwartz, T. W. & Rosenkilde, M. M. Positive versus negative modulation of different endogenous chemokines for CC-chemokine receptor 1 by small molecule agonists through allosteric versus orthosteric binding. J. Biol. Chem. 283, 23121–23128 (2008).
Mitternacht, S. FreeSASA: an open source C library for solvent accessible surface area calculations. F1000Res. 5, 189 (2016).
Lomize, M. A., Lomize, A. L., Pogozheva, I. D. & Mosberg, H. I. OPM: orientations of proteins in membranes database. Bioinformatics 22, 623–625 (2006).
Acknowledgements
We thank C. Li for construct screening, A. Dragan, M.S. Baggesen and A. Dai for technical assistance, S. Chang for data collection at the Center of Cryo-Electron Microscopy, Zhejiang University and C. Ma for sample preparation at the Protein Facilities, Zhejiang University. This work was partially supported by the National Natural Science Foundation of China (81872915 (M.W.W.), 81922071 (Y. Zhang), 81773792 (D.Y.), 81973373 (D.Y.), 21704064 (Q.Z.), 31770796 (Y.J.) and 31971178 (S.Z.)); the National Science & Technology Major Project of China — Key New Drug Creation and Manufacturing Program (2018ZX09735–001 (M.W.W.), 2018ZX09711002–002–005 (D.Y.) and 2018ZX09711002–002–002 (Y.J.)); the National Key Basic Research Program of China (2018YFA0507000 (M.W.W. and S.Z.) and 2019YFA0508800 (Y. Zhang)); the Shanghai Municipal Science and Technology Major Project (2019SHZDZX02 (H.E.X.)); the Ministry of Science and Technology of China Major Project (XDB08020303 (H.E.X.)); Zhejiang Province Science Fund for Distinguished Young Scholars (LR19H310001 (Y. Zhang)); Fundamental Research Funds for Central Universities (2019XZZX001-01-06 (Y. Zhang)); Shanghai Science and Technology Development Fund (18ZR1447800 (L.H.Z.)); Young Innovator Association of CAS (2018325 (L.H.Z.)); SA-SIBS Scholarship Program (L.H.Z. and D.Y.); Fudan-SIMM Joint Research Fund (FU-SIMM-20174003 (L.H.Z. and M.W.W.)) and Novo Nordisk-CAS Research Fund grant (NNCAS-2017–1-CC (D.Y.)).
Author information
Authors and Affiliations
Contributions
W.S., Z.C., F. Zhao and F. Zhou designed the expression constructs, purified the GLP-2–GLP-2R–Gs complex, prepared the final samples for negative staining/data collection towards the structure and participated in manuscript preparation; L.N.C. and D.-D.S. performed specimen screening, cryo-EM grid preparation/data collection, map calculation, structure analysis, bulk figure preparation and participated in manuscript writing; Q.Z. performed structural analysis, figure preparation, and participated in manuscript writing; S.G., W.J.C.v.d.V., X.C., Y.C. and Y. Zhou conducted ligand binding and signaling experiments; L.H.Z. supervised construct preparation, protein expression and purification; S.Z. and H.Z. assisted in structural analysis; Y.J. participated in data analysis and manuscript editing; M.M.R. directed receptor binding studies and edited the manuscript; D.Y. supervised mutagenesis and signaling experiments, participated in data analysis and manuscript preparation; H.E.X., Y. Zhang and M.W.W. initiated the project, supervised the studies, analyzed the data and wrote the manuscript with inputs from all co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, W., Chen, LN., Zhou, Q. et al. A unique hormonal recognition feature of the human glucagon-like peptide-2 receptor. Cell Res 30, 1098–1108 (2020). https://doi.org/10.1038/s41422-020-00442-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41422-020-00442-0
This article is cited by
-
The Potential Mechanism of Remission in Type 2 Diabetes Mellitus After Vertical Sleeve Gastrectomy
Obesity Surgery (2024)
-
Insights into the structure and activation mechanism of some class B1 GPCR family members
Molecular Biology Reports (2024)
-
Structure, function and drug discovery of GPCR signaling
Molecular Biomedicine (2023)
-
Structural basis of hydroxycarboxylic acid receptor signaling mechanisms through ligand binding
Nature Communications (2023)
-
Structure insights into selective coupling of G protein subtypes by a class B G protein-coupled receptor
Nature Communications (2022)
