
Abstract
In vivo genome editing represents a powerful strategy for both understanding basic biology and treating inherited diseases. However, it remains a challenge to develop universal and efficient in vivo genome-editing tools for tissues that comprise diverse cell types in either a dividing or non-dividing state. Here, we describe a versatile in vivo gene knock-in methodology that enables the targeting of a broad range of mutations and cell types through the insertion of a minigene at an intron of the target gene locus using an intracellularly linearized single homology arm donor. As a proof-of-concept, we focused on a mouse model of premature-aging caused by a dominant point mutation, which is difficult to repair using existing in vivo genome-editing tools. Systemic treatment using our new method ameliorated aging-associated phenotypes and extended animal lifespan, thus highlighting the potential of this methodology for a broad range of in vivo genome-editing applications.
Similar content being viewed by others

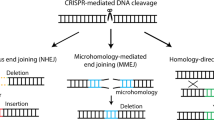
Introduction
Direct gene modification in living organisms by in vivo targeted genome-editing is a powerful tool for many fields of life science, including animal science and developmental biology. Furthermore, this technology could potentially be used to correct inherited diseases by eliminating disease-causing mutations, offering the possibility of a permanent cure.1 In particular, in the presence of an ectopic donor that possesses two stretches of homologous sequences to the target genome, homology-directed repair (HDR) can replace endogenous genomic sequences with exogenously supplied donor sequences, thus enabling site-specific integration of a transgene or the correction of a disease-causing mutation (either recessive or dominant). However, these conventional HDR-based targeted gene knock-in strategies have practical limitations because HDR is mainly active in dividing cells.2 Thus, adult tissues comprised of non-dividing cells are inaccessible. In vivo, tissues consist of many cell types whose status can be either dividing or non-dividing and which can change during development and regeneration. HDR-mediated gene correction strategies have shown promise in curing inherited diseases in mice, but the targets are currently limited to tissues with cells that are able to divide in vivo (Supplementary information, Fig. S1a).1,3,4,5
To overcome the limitations of HDR-mediated genome editing, we previously developed CRISPR/Cas9-based homology-independent targeted integration (HITI), which allows for efficient targeted knock-ins in both dividing and non-dividing cells in vitro and in vivo.6 Rather than utilizing HDR, HITI relies instead on the other major DNA double-strand break (DSB) repair pathway, the non-homologous end joining (NHEJ) pathway. In the case of HITI, the donor DNA lacks a homology arm and is designed to include a Cas9 cleavage site that flanks the donor sequence (Supplementary information, Fig. S1b). Cas9-mediated DSBs are created simultaneously in both genomic target sequences and the exogenously provided donor DNA, generating blunt ends. The linearized donor DNA can be used for repair by the NHEJ pathway, allowing for its integration into the genomic DSB site. Once incorporated into the genome, donor DNA inserted in the desired orientation disrupts the Cas9 target sequence and prevents further Cas9 cutting. If the donor DNA is inserted in the undesired orientation, the Cas9 target sequence will remain intact and a second round of Cas9 cutting will remove the integrated donor DNA. Thus, HITI inserts the donor DNA into the targeted chromosome in a predetermined direction (Supplementary information, Fig. S1c).
Since NHEJ is active throughout the cell cycle in a variety of adult cell types (including proliferating and post-mitotic cells) and its activity far exceeds HDR,7 the HITI strategy has enabled the targeted integration of transgene cassettes in many organs, including non-dividing tissues such as the brain.6 Notably, HITI was used to restore visual function in a rat model of retinitis pigmentosa by targeted insertion of a functional copy of exon 2 of the Mertk gene to correct the gene’s loss-of-function due to a 1.9 kb deletion, whereas conventional HDR was not able to restore it.6 These results suggest that HITI-based treatments could be used to ameliorate a variety of genetic diseases and be target to many target tissues. However, HITI is not without limitations. For example, although HITI can insert DNA at a precise location within the genome, it cannot repair point or frameshift mutations due to the fact that HITI cannot remove pre-existing mutations.8 Thus, HITI-mediated gene-correction strategies are effective for targeting loss-of-function mutations caused by large deletions, but not all mutations, such as gain-of-function dominant mutations (Supplementary information, Fig. S1b). This severely limits the types of diseases that could be potentially treated. Therefore, improved technologies for the in vivo manipulation of the genome are still needed.
Recent studies have suggested that elements of DNA-repair complexes are more promiscuous than previously thought, and are not restricted to NHEJ or HDR pathways, and even operate in post-mitotic cells.9,10,11,12 This endows cells with flexibility to overcome DNA damage and provides new opportunities for correcting the genome. Previously, we attempted to combine NHEJ-mediated HITI and canonical HDR by constructing a HITI donor with two homology arms for conventional HDR.6 This donor structure is similar to the one used in homology-mediated end joining (HMEJ) strategy as previously reported (Supplementary information, Fig. S2a).13 However, the targeted integration efficiency of the HMEJ-like HITI–HDR combined donor was lower than the HITI donor in HEK293 cells, suggesting that the addition of the traditional two-homology arms does not increase targeted gene knock-in efficiency in dividing cells.6
Here we describe a unique NHEJ and HDR mediated targeted gene knock-in method that requires a DSB induction site within a single stretch of homologous sequence on the donor (Supplementary information, Fig. S2b, c). We termed this “intercellular linearized Single homology Arm donor mediated intron-Targeting Integration (SATI)”. SATI allows DNA knock-in via single homology arm mediated HDR or homology independent NHEJ-based HITI, enabling the targeting of a broad range of mutations and cell types. We demonstrate the utility of this system as a potential therapy through the in vivo correction of a dominant point mutation that causes premature aging in mice. Our data indicate that SATI, due to its target flexibility and versatility, is a powerful genetic tool for in vivo genome editing.
Results
Development of single homology arm donor mediated gene knock-ins in post-mitotic neurons
The HITI system takes advantage of the intrinsic NHEJ pathway, a relatively mutagenic form of DNA repair compared to HDR.6 With NHEJ, small insertions/deletions (indels) are often created at the junction between the inserted DNA and the targeted genomic locus. This can cause an out-of-frame mutation when targeting an exon, leading to gene inactivation (Supplementary information, Fig. S3a). To overcome this limitation, we targeted intronic sequences upstream of a relevant exon (or mutation) and included a splice acceptor, relevant downstream exon(s), the 3′UTR, and genetic elements, such as GFP, within the donor DNA. In theory, this would result in transcription of the donor exon(s), rather than the endogenous exon(s) downstream of the insertion site, thereby enabling production of a normal transcript, thus correcting the mutation, or a fusion transcript in the case of knock-in genetic elements, such as GFP (Supplementary information, Fig. S2b). Importantly, small indels introduced into the intron have reduced probability of affecting target gene function.
To evaluate the efficacy of this new approach, we targeted the Tubulin beta-3 chain, Tubb3 gene in non-dividing cultured mouse primary neurons using a series of donor DNAs, gRNAs, and Cas9 from Streptococcus pyogenes (SpCas9) (Fig. 1a–d). Protospacer adjacent motif (PAM) sequences (5′-NGG-3′) are commonly recognized by wild-type SpCas9 and are abundant throughout the mammalian genome, though they are not always found at the exact position required to target all genes using HITI. Recently, some novel Cas9s that can target flexible PAM sequences (5′-NG-3′) have been developed by protein engineering.14,15 Using these newly developed Cas9s, the target region can be expanded, owing to its flexibility (Supplementary information, Fig. S3b). However, because the activity of these novel Cas9s is not higher than that seen with the original SpCas9, and as we are targeting introns (providing more flexibility in designing gRNAs), we decided to keep using wild-type SpCas9 (hereafter termed Cas9) for further experiments (Supplementary information, Fig. S3b–d). For experiments on neurons, most of the donor DNA was in the form of minicircles (MC). A MC is double-stranded DNA devoid of the bacterial backbone that enhances the stability of the integrated transgene.6,16 We targeted intron 3 of the Tubb3 gene using a donor DNA, Tubb3int3-SATI. This donor included sequence identical to the target genome, including exon 4, GFP, and the Tubb3 3′UTR, thus possessing one homology arm for the target site. In addition, we included a Cas9 cleavage site to flank the donor sequence in order to give HITI the capacity for target integration. Therefore, the intracellularly linearized donor DNA plasmid can then be used for repair by the NHEJ pathway, enabling its unidirectional integration into the genomic DSB site via HITI (Fig. 1a; Supplementary information, Fig. S2b). A series of donors, including previously developed exon-targeting HITI, conventional HDR, and HMEJ, which is a combination vector that carries two homology arms and cutting sites13 (Tubb3ex4-HITI, Tubb3ex4-HDR, and Tubb3int3-HMEJ, respectively), were also constructed for comparison (Fig. 1b–d; Supplementary information, Figs. S1a, b and S2a).6

Single homology arm donor-mediated gene knock-in in non-dividing primary neurons. a Schematic representation of targeted GFP knock-in at the Tubb3 locus by a SATI (intercellular linearized Single homology Arm donor-mediated intron-Targeting Integration) donor harboring a single homology arm for targeting in intron 3. Pink pentagons, Intron 3 gRNA target sequences. Yellow scissors or Black lines within gRNA target sequence, Cas9 cleavage site. Light blue trapezoid, homologous sequence between target and donor. b Schematic representation of targeted GFP knock-in at Tubb3 locus by no homology HITI donor targeting in exon 4. Light blue pentagons, Exon 4 gRNA target sequences. Black lines within pentagon, Cas9 cleavage site. c Schematic representation of targeted GFP knock-in at Tubb3 locus by a conventional HDR donor harboring two homology arms targeting in exon 4. Light blue pentagons, Exon 4 gRNA target sequences. Light blue parallelograms, homologous sequence between target and donor. d Schematic representation of targeted GFP knock-in at Tubb3 locus by an HMEJ donor harboring two homology arms targeting in intron 3. Red bars (splicing acceptor and downstream sequence from rat Tubb3 gene) and inserting cassette (i.e. exon 4, GFP and 3′UTR) lack any homology sequences, in order to avoid undesired recombination. Pink pentagons, Intron 3 gRNA target sequences. Light blue parallelograms, homologous sequence between target and donor. e Experimental scheme for GFP knock-in in cultured primary neurons. f Representative immunofluorescence images of neurons transfected with Cas9, one-armed SATI donor and int3gRNA-mCherry detected by anti-β-III tubulin antibody (magenta), mCherry signal (red), anti-GFP antibody (green), DAPI signal (blue), and EdU signal (white). Scale bar: 10 μm. g Percentage of knock-in cells (GFP+) per transfected cells (mCherry+) with different combinations of gRNAs and donors. Each value indicates percentage of GFP-positive cells among transfected cells. Data are represented as box with whisker with all the input data points as green dots, the average is the line inside the box. One-way ANOVA with Bonferroni’s multiple comparison test for analysis, ****P < 0.0001. h Ratio of HITI- and oaHDR-mediated GFP knock-in after transfected with one-armed SATI donor into primary neurons. The following combinations of donor and gRNA were transfected (Donor cut: MC-Tubb3int3-scramble and mScramblegRNA-mCherry; Ch cut: MC-Tubb3int3-scramble and int3gRNA-mCherry; Donor+Ch cut (SATI): MC-Tubb3int3-SATI and int3gRNA-mCherry). Analyzed number is indicated on top
We co-transfected mouse primary neurons with sets of donor DNAs, gRNAs with mCherry expression vector (gRNA-mCherry), and Cas9. To ensure that gene editing was occurring in post-mitotic neurons, we incubated the cells in EdU, allowing us to verify the timing whereby neurons in culture become post-mitotic and which cell populations were transfected. Five days post-transfection, the occurrence of correct gene knock-in was confirmed by immunocytochemistry (Fig. 1e, f; Supplementary information, Fig. S4a). Using the intron 3 targeting donor (Tubb3int3-SATI), we detected the expected Tubb3-GFP fusion protein in the cytoplasm. Tubb3-GFP co-localized with β-III-tubulin/Tuj1, the product of the Tubb3 gene. Moreover, GFP-positive (GFP+) cells were negative for EdU (EdU−), demonstrating that the intronic gene knock-in approach worked in non-dividing neurons (Fig. 1f; Supplementary information, Fig. S4b).
Next, we compared the GFP knock-in efficiency and importance of the donor sequence at the integration site for different combinations of donors and gRNAs. Similar to our previous report,6 GFP knock-in efficiency was very low (~0.07% of transfected cell) using a conventional HDR donor (Tubb3ex4-HDR) that harbored two homology arms for the cutting site on the genome (Fig. 1c, g). A non-homology HITI donor (Tubb3ex4-HITI) achieved efficient NHEJ-mediated GFP knock-in by HITI (36.25% of transfected cells) (Fig. 1b, g), in agreement with previous data.6 Using Tubb3int3-SATI, we observed knock-in events at a low GFP knock-in efficiency when either only the target was cut at intron 3 in the genome, or only the Tubb3int3-SATI donor was cut (6.3% and 2.7% per transfected cells) (Fig. 1a, g). Surprisingly, the junction site of the donor with GFP inserted at the targeted locus remained intact, like the targeted genome sequence. In other words, the sequence of the junction site of the gRNA-targeting sequence showed no features of HITI (Fig. 1h; Supplementary information, Fig. S4c). Therefore, we speculated that an unknown, non-canonical HDR pathway was responsible for the insertion of donor DNA when a single homology arm was used. We refer to the utilization of this non-canonical HDR as one-armed HDR (oaHDR), distinguishing it from conventional HDR which utilizes two homology arms for the chromosomal cutting site (Fig. 1a, c). By simultaneously cutting the genome and one-homology arm donor DNA (Tubb3int3-SATI), we observed efficient GFP knock-in (~37% of transfected cells) (Fig. 1g). The efficiency was equivalent for exon-targeted non-homology HITI donor (Tubb3ex4-HITI, ~36%),6 and also comparable to the efficiency seen for the HMEJ donor (Tubb3int3-HMEJ, ~40%) (Fig. 1g).13 In addition, when Cas9 was replaced with Cas9 nickase (Cas9D10A), which introduces a single-strand break (SSB),17 the GFP knock-in efficiency was extremely low, suggesting that HITI and oaHDR need DSBs, not SSBs (Supplementary information, Fig. S4d). While analyzing the gene-editing events after GFP integration with double digestion of donor Tubb3int3-SATI and chromosomal target, we found that ~95% of gRNA target sites showed a feature of oaHDR, which shows no difference in genomic sequence except for the GFP insertion (Fig. 1h; Supplementary information, Fig. S4c). Only 5% of GFP knock-in events were mediated by HITI, suggesting that the donor DNA was inserted mainly via oaHDR, which is expected to require the participation of elements of both NHEJ- and HDR-related pathways.
Together, these results suggest that a non-canonical HDR process occurs in neurons when the cut is in either the single-homology arm donor or the chromosomal target sequence. Knock-in efficiency is significantly increased by cutting both the donor and chromosomal target (Fig. 1g). In summary, we successfully developed a genome targeting system that we termed “intercellular linearized Single homology Arm donor mediated intron-Targeting Integration (SATI),” which induces a DSB at both the donor and chromosomal target and utilizes features of both HITI and oaHDR. Using this system to target introns provides flexibility in designing gRNAs specific for a wider range of genome sequences and minimizes the effects of NHEJ-created indels (Supplementary information, Figs. S2b, c and S3a, b).
Measurement of oaHDR and HITI based knock-in efficiency in dividing cells
DNA repair by canonical HDR can only efficiently occur during the S-G2 phase of the cell cycle, making it inaccessible to non-dividing cells.2 To test the range of potential applications for SATI, we next asked whether oaHDR takes place in dividing cells in vitro. We used genetically modified human HEK293 cells and human embryonic stem (hES) cell lines that harbored a mutated GFP transgene expressed under the EF1α promoter.6,18 We compared knock-in efficiencies via HDR- or oaHDR-mediated targeted integration using three functional gRNAs: gRNA1, gRNA2, and gRNA3 (Supplementary information, Fig. S5a, b). The conventional two homology arm donor-mediated HDR is active in these cells in accord with our previous reports.6,18 Interestingly, we observed very few knock-in events when both genomic and donor DNAs were cut simultaneously, suggesting that the oaHDR-mediated integration is rare in dividing HEK293 and hES cells (Supplementary information, Fig. S5c, d). To potentially increase oaHDR efficiency in dividing cells, we performed knock-ins during different phases of the cell cycle. Non-dividing cells, such as neurons, are arrested in the G0/G1 stage and exhibit high levels of oaHDR. We therefore speculated that arresting proliferating cells in G0/G1 may boost the oaHDR-mediated integration. To examine this possibility, we arrested cells in G1 (using Lovastatin or by expressing a G1-phase specific Cas9, Cas9-Cdt1). However, we did not observe an increase in oaHDR activity in G1-phase specific genome editing, suggesting that G1 arrest does not boost oaHDR-mediated integration (Supplementary information, Fig. S6).
In contrast, in actively dividing cells, the activity of HITI was one order of magnitude higher than for conventional HDR (18.2% vs. 1.4% in HEK293 cells; 111.6 vs. 11.4 per 106 hESCs), as demonstrated by the knock-in of an mCherry reporter into HEK293 and hES cells (Supplementary information, Fig. S7a–c). Thus, when a SATI construct is used the integration can predominantly occur either via the non-canonical one-armed HDR (in non-dividing cells) or via HITI (in activly dividing cells), with a higher efficiency compared with HDR (Supplementary information, Fig. S7d).
Gene correction of a dominant mutation using SATI
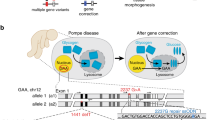
To demonstrate the versatility of the SATI strategy for gene targeting, we used it to correct a dominant mutation in exon 11 of the Lamin A/C, Lmna gene (c.1827C>T; p.Gly609Gly) using a progeria model mouse.19 This mutation results in the production of an abnormal form of Lamin A protein called progerin, whose accumulation causes pathological changes in multiple tissues.19,20,21 To correct this dominant mutation, we constructed Adeno-associated virus (AAV) and minicircle (MC) vectors that contained the SATI-mediated gene-correction donor (AAV-Progeria-SATI and MC-Progeria-SATI, respectively) (Fig. 2a; Supplementary information, Fig. S8a). These Progeria-SATI donors contained one 1.9-kb homology arm (including the wild-type exon 11, exon 12, and the 3′UTR of the Lmna gene) sandwiched by the intron 10 gRNA target sequence and AAV-Progeria-SATI that included the intron 10 gRNA expression cassette. We hypothesized that both HITI- and oaHDR-mediated targeted gene knock-ins would result in production of the wild type Lmna gene transcript (Fig. 2a).

oaHDR- or HITI-mediated gene knock-in profile after SATI mediated gene-correction of progeria mice in vitro and in vivo. a Schematic representation of the LmnaG609G (c.1827C>T) gene correction with SATI-mediated gene-correction donor. Red box indicates exon 11 with single point mutant. After gene correction mediated by NHEJ-mediated HITI, targeted sequence including corrected mutation are inserted in intron 10, just upstream of mutated exon 11 (left). After gene correction mediated by oaHDR, the mutation is corrected with no change of other genomic sequence except for point mutation (right). The expression level of Lamin C transcribed from exon 1–10 is not affected by Lmna c.1827C>T mutation. After gene correction, Lamin A protein is expressed instead of Progerin expression. Pink pentagon, Lmna intron 10 gRNA target sequence. Yellow scissors or Black line within gRNA target sequence, Cas9 cleavage site (see also Supplementary information, Fig. S8a). b The ratio of HITI, oaHDR, and undetermined (due to large deletion) in targeted sequence after SATI mediated gene correction from progeria MEF (top panel, n = 48), primary neuron (middle panel, n = 47), and brain (lower panel, n = 19). Actual knock-in ratio is indicated in the graph (%). c The ratio of HITI, oaHDR, and undetermined (due to large deletion) with or without indel at targeting site after gene correction by Cas9/Lmna-gRNA-mCherry/MC-Progeria-SATI transfection with shRNA gene knock down for progeria MEFs. Actual targeting ratio is indicated in the graph (%). Each target of shRNA knockdown is indicated at bottom. Scramble control, n = 48; Ku80, n = 19; Lig3, n = 32; Rad51, n = 17. d Experimental scheme for in vivo gene correction by AAV-Progeria-SATI via intravenous (IV) AAV injections to LmnaG609G/G609G progeria mouse model. AAV-Progeria-SATI is injected into newborn (postnatal day 1, P1) mouse together with AAV-Cas9. The phenotypes are analyzed in the indicated date in each experiment. e Gene correction efficiency at Lmna c.1827C>T dominant point mutation site from the indicated tissues in SATI-treated (Pro+SATI) or only donor-treated without Cas9 (Pro+donor) progeria mice at day 100. f Indel percentages at Lmna intron 10 gRNA target site from the indicated tissues in SATI-treated (Pro+SATI) or only donor-treated without Cas9 (Pro+donor) progeria mice at day 100. g The ratio of HITI, oaHDR and undetermined (due to large deletion) with or without indel at targeting site after gene correction by systemic AAV-Progeria-SATI injection for progeria mice. Deep sequencing was performed using the extracted DNA from liver (top) and heart (bottom), respectively. Actual knock-in ratio is indicated in the graph (%)
To determine whether gene correction of the c.1827C>T mutation was successful and determine the ratio of oaHDR- and HITI-mediated knock-ins, we isolated mouse embryonic fibroblasts (MEFs) and primary neurons from progeria mice (Supplementary information, Fig. S8b). It is notable that MEFs exhibit low HDR activity, even though they are highly proliferative.22 Progeria-SATI donors were delivered to these cells by transfection or infection. We also injected AAV-Progeria-SATI and AAV-Cas9 into the adult brains of progeria mice. Genomic DNA was extracted from the edited progeria cells or brain tissue. Since the DNA delivery efficiency is low for these cells and tissue, we first enriched the corrected sequence by cutting with the restriction endonuclease BstXI, which specifically recognizes the non-corrected allele, and analyzed the sample by Sanger sequencing. We observed gene-corrected events, and both oaHDR (80–90%) and HITI (10–20%) were evident in the gene-corrected cells, suggesting that SATI-mediated gene correction had been achieved for the dominant point mutation causing progeria and that the oaHDR-mediated integration of the SATI donor was the predominant pathway in these cell types (Fig. 2b).
To determine the pathway responsible for oaHDR- and HITI-mediated gene knock-ins, we explored wild-type primary neurons transfected with the Tubb3-GFP knock-in SATI system (Tubb3int3-SATI donor; Cas9; and dual cut gRNA) together with shRNAs against genes involved in DSB repair pathways (Supplementary information, Fig. S9a, b). GFP knock-in efficiency of the SATI donor was affected by shRNAs targeting DSB repair related genes including genes for the canonical NHEJ (cNHEJ) (Ku70 and Ku80), alternative NHEJ (altNHEJ) (Lig3 and Xrcc1) and HDR (Rad50 and Rad51) pathways. We then examined changes in the ratio of oaHDR and HITI in progeria MEFs (Fig. 2c). The Ku80 knockdown prevented HITI-mediated knock-in. This is consistent with our previous results, where we demonstrated that HITI involves the canonical NHEJ mediated knock-in machinery.6 In contrast, Lig3 knockdown moderately increased HITI (21.9% from 12.6% in control), suggesting that alternative end-joining (altNHEJ) is involved in oaHDR-mediated gene knock-in. Interestingly, Rad51 knockdown resulted in large deletions, suggesting that Rad51 may stabilize the genomic structure during SATI-mediated gene modification. These results indicate that gene knock-in by the SATI system is mediated by multiple DSB repair pathways (Supplementary information, Fig. S9c).
SATI-mediated systemic gene correction of a dominant mutation in vivo
To test the ability of SATI to correct a dominant mutation in vivo, we systemically delivered AAV-Progeria-SATI, together with an AAV expressing Cas9,6 via intravenous (IV) injection into neonatal LmnaG609G/G609G progeria mice at postnatal day 1 (P1) (Fig. 2d). We packaged the SATI donor in serotype 9 AAV, based on their ability to infect a wide range of tissues.23 Genomic PCR and Sanger sequence analyses at day 100 revealed that SATI-mediated targeted gene knock-in occurred in several tissues, including the liver, heart, muscle, kidney, and aorta even though the efficiency varied (Supplementary information, Fig. S10). We determined the frequency and sequence of indels at the gRNA target site in intron 10, as well as the efficiency of SATI-mediated gene correction (2.06% in the liver and 0.34% in the heart) using next-generation sequencing (NGS) in several organs at day 100 (Supplementary information, Fig. S11a). To exclude the possibility that the observed events were due to a PCR artifact, we included control progeria mice, which were injected with only donor AAV (labeled as “Pro+donor”) for NGS experiments. It is notable that the gRNA target site was in intron 10 of the Lmna gene, and the sizes of the indels were small, and not expected to affect the splicing of the Lmna transcript (Supplementary information, Fig. S11b–d).
To study off-target effects of SATI in vivo, we examined mutation rates associated with the ten highest-ranked off-target sites for the Lmna intron 10 gRNA. Liver tissues treated with SATI were analyzed via NGS, revealing only minimal indels at computationally predicted off-target sites (Supplementary information, Fig. S11e). Next, we decided to test the potential off-target integration of donor DNA in other regions of the genome using 5′RACE and sequencing to identify the sequence upstream of exon 11 of Lmna mRNA transcribed from the integrated donor DNA in liver and heart (Supplementary information, Fig. S12a). We detected on-target integration at the Lmna locus in the liver and heart of treated progeria mice (Supplementary information, Fig. S12b). However, several exons of Alb and Myh6 genes were captured in the liver and heart, respectively, suggesting a possibility for donor DNA to become trapped in the open-chromatin regions (Supplementary information, Fig. S12c, d). Importantly, the expression level of the Alb gene is more than 10,000-fold higher than the Lmna gene in liver, suggesting that the trapped donor derived fusion transcript is significantly less than the wild type endogenous Alb gene transcript, and that this minimal off-target integration should not affect the tissues, unless the fusion protein initiates tumorigenesis (Supplementary information, Fig. S12e).
To evaluate SATI-mediated oaHDR and HITI efficiency in vivo at day 100, we amplified ~600 bp that included the gRNA target sites and the c.1827C>T mutation site and determined the efficiency by paired-end sequencing. We estimated that the percentage of gene correction was 2.07% in the liver and 0.14% in the heart, similar to the above NGS results (Fig. 2e, f; Supplementary information, Fig. S11a). Moreover, oaHDR events were observed in liver and heart analyses by paired-end sequencing after in vivo systemic SATI treatment (Fig. 2g). Although this number may seem low, it is important to note that gene-corrected cells are still present in some organs even 100 days after treatment and that the correction efficiency was sufficient to elicit SATI-mediated phenotypic rescue in several tissues and organs (see below).
Phenotypic rescue of progeroid syndrome by SATI
Progeria mice typically exhibit progressive weight loss and a shortened lifespan. These phenotypes were delayed by SATI treatment (Fig. 3a; Supplementary information, Fig. S13a and Movie S1) resulting in a slowdown of progressive weight loss and an extension of median survival time by 1.45-fold (untreated and SATI-treated animals showed 105 and 152 days median survival time, respectively). The Lmna gene encodes both Lamin A and Lamin C proteins, and the LmnaG609G/G609G mutation results in abnormal splicing of just the Lamin A transcript (Fig. 2a).24 Quantitative RT-PCR analysis of SATI treated progeria mice revealed an increase in wild-type Lamin A transcript relative to total Lamin C transcript (up to around 3.5-fold) and a decrease in Progerin transcripts in total Lamin A transcripts (up to around 5.4-fold) in the liver, heart, and aorta on day 100 (Fig. 3b).

Prevention of aging phenotypes and molecular analyses in the SATI-treated progeria mice. a Survival plots of Lmna+/+ (WT), SATI treated Lmna+/+ (WT+SATI), LmnaG609G/G609G (Pro), SATI treated LmnaG609G/G609G (Pro+SATI), Lmna+/G609G heterozygous (Het), SATI treated Lmna+/G609G heterozygous (Het+SATI) mice. WT, n = 72; WT+SATI, n = 8; Het, n = 33; Het+SATI, n = 11; Progeria, n = 25; Progeria+SATI, n = 15. P < 0.0001 according to log-rank (Mantel-Cox) test. Median survival and maximum survival date of each group are indicated at bottom. b RT-qPCR analysis for the expression ratio of Lamin A to Lamin C (left) and Progerin to Lamin A (right) from represented tissues (n = 3). Expression level of each gene is normalized by Gapdh first, and then ratio is calculated. Relative values after SATI treated are indicated. Data are represented as mean ± s.e.m. Each P-value is indicated according to unpaired Student’s t-test. N.S., not significant. Relative ratios are indicated at top of each graph. c Representative photographs of WT, Progeria (Pro), and Progeria+SATI (Pro+SATI) mice at 17 weeks old. d–g Histological analysis of skin d, spleen e, kidney f, and aorta g at 17 weeks old. Left: representative pictures of hematoxylin and eosin (H&E) staining. Middle and right: quantitative analyses represented as mean ± s.e.m. d–g Skin, n = 39; spleen, n = 20; kidney glomerulus, n = 20; kidney renal tubules, n = 50; aorta, n = 9. Scale bars: skin, kidney, and aorta 100 μm, spleen 250 μm. Black arrowheads indicate decreased epidermal thickness and increased keratinization d, and small lymphoid nodules in the splenic white pulp e. The thickness of epidermis is significantly decreased in untreated mice and restored in SATI treated mice d. The area of germinal center is significantly decreased in untreated mice and restored in SATI-treated mice e. The area of glomerulus (middle panel) and diameter of renal tubules (right panel) are significantly decreased in untreated mice and restored in SATI treated mice f. The density of aortic nuclei is significantly decreased in untreated mice and restored in SATI-treated mice g. P-values are indicated in each graph, one-way ANOVA with Tukey’s multiple comparisons test d–g. h Electrocardiogram (ECG) analysis in WT, Pro, and Pro+SATI mice between day 92 and day 110. Heart rate represented as beats per minute (bpm), n = 7. P-values are indicated in each graph, one-way ANOVA with Tukey’s multiple comparisons test
In 3-month old progeria mice, age-associated pathological changes are typically observed in multiple organs, including skin, spleen, and kidneys.25,26,27,28 These aging phenotypes were diminished in 17-week-old progeria mice that received the SATI treatment (Fig. 3c–f; Supplementary information, Fig. S13b). SATI-treated mice showed increased epidermal thickness, a rescue of germinal centers in the spleen, and decreased tubular atrophy in the kidney. We checked for a knock-in event and altered protein levels in established tail tip fibroblasts (TTFs) from SATI treated mice at day 70 but we were unable to detect any knock-in by PCR (Supplementary information, Fig. S13c). Instead, SATI treatment slightly decreased Progerin/LaminA protein levels and partially rescued the nuclear envelope abnormalities typically observed in progeria (Supplementary information, Fig. S13d–f). Progeria mice carry the mutant allele (the c.1827C>T; p.Gly609Gly mutation), which is equivalent to the Hutchinson-Gilford progeria syndrome (HGPS) c.1824C>T; p.Gly608Gly mutation in the human LMNA gene. Complications related to atherosclerosis, including cardiovascular problems or stroke, are the eventual causes of death for most patients with HGPS (or progeria).29 Progeria mice present histological and transcriptional alterations characteristic of progeroid symptoms, reminiscent of the main clinical manifestations of human HGPS, including shortened life span and cardiovascular aberrations.19 Therefore, we analyzed the aorta and heart rate of progeria mice. SATI treatment increased the number of nuclei in the smooth muscle layer of the aortic arch, compared with untreated controls (Fig. 3g). Electrocardiogram (ECG) recordings revealed that SATI treatment prevented the progressive development of bradycardia, which is usually observed in progeria mice (Fig. 3h).
Since almost all patients with HGPS are heterozygous for the same dominant c.1824C>T mutation,20 we also treated heterozygous progeria mice (Lmna+/G609G) with SATI. Median survival of these heterozygous mice was also improved following SATI treatment (untreated and SATI-treated animals showed 323 and 403 days median survival, respectively) (Fig. 3a). Importantly, we did not observe any morphological/histological alterations in wild-type mice treated with SATI for over 500 days (Supplementary information, Fig. S13g), suggesting that the deleterious effects of the observed off-target integrations are of little consequence. Collectively, these data demonstrate that SATI can be used to correct dominant mutations in vivo to prevent the development of pathological phenotypes.
In vivo correction in adult tissues using SATI
Patients with HGPS are diagnosed at a median age of 19 months (range, 3.5 months-4.0 years).30 Similarly, many other diseases caused by dominant mutations are diagnosed well beyond the neonatal stage. We therefore asked whether delivering SATI later in life could provide therapeutic benefits. We delivered the SATI system to 10-week old progeria mice through local intramuscular (IM) injection. Skeletal muscle is one of the affected tissues in progeria mice (Fig. 4a).19 We found that fiber size distribution of the injected tibialis anterior muscle was improved three weeks after injection of the SATI-treated progeria mice (Fig. 4b, c). Together with the successful gene knock-in by SATI in the adult post-mitotic mouse brain (Fig. 2b), these results suggest that local gene repair in specific tissues at juvenile or adult stages could provide a complementary treatment option for patients with dominant mutations.

Intramuscular treatment of the SATI in adult progeria tibialis anterior muscle. a Experimental scheme for in vivo gene repair by AAV-Progeria-SATI via Intramuscular (IM) AAV injections into the tibialis anterior (TA) muscles of adult LmnaG609G/G609G progeria. TA muscle of 10-weeks-old progeria mouse was injected AAV(s) and analyzed at three weeks later. b Representative pictures of H&E staining of TA muscle at 13-weeks-old. Top: wild type with PBS injection as control (WT+PBS), middle: AAV-Progeria-SATI only treated without AAV-Cas9 (Pro-Cas9), bottom: AAV-Progeria-SATI and AAV-Cas9 treated (Pro+Cas9). Scale bars: 100 μm. c Muscle fiber cross-sectional area distribution of TA muscles in progeria mice at 13-weeks-old. Each color of bar shows representative muscle from independent mouse. WT+PBS, n = 6; Pro-Cas9, n = 6; Pro+Cas9, n = 8. Average of % fibers is indicated at right upper corner. Each trendline is indicated as broken line. Data are represented as mean ± s.e.m. Each P-value is indicated according to unpaired Student’s t-test
Discussion
Here we describe a versatile in vivo gene knock-in method, termed SATI. SATI is a unique strategy combining intron-targeting gene knock-in with a specific donor vector possessing a single homology arm and Cas9 cleavage site. The unique vector structure for SATI has a bipotential capacity to achieve efficient gene knock-in by choosing the predominant DSB repair machinery (i.e. non-canonical HDR mediated by single homology arm or NHEJ) in the target cell. SATI differs from HMEJ because the HMEJ donor contains two homology arms in addition to cutting sites and so allows the exogenous cassette to be integrated at a target site through either the canonical HDR or NHEJ pathway.13 Previously, we attempted to make the same donor structure by constructing a HITI donor with two homology arms for conventional HDR.6 However, the targeted integration efficiency of this combined HMEJ-like donor was lower than the HITI donor, suggesting that the addition of the traditional two-homology arms does not increase targeted gene knock-in efficiency in dividing cells and that the canonical HDR and NHEJ pathways are competing with each other as previously described.31,32 In addition, the in vivo applications of HDR are limited to tissues that have dividing cells. In this study, HMEJ is equally effective with HITI and SATI in primary neuron cultures. This result suggests that canonical HDR and NHEJ pathways do not compete in this cell type because canonical HDR is not active in neurons. Thus, the efficiency of HMEJ might be affected by canonical HDR activity in the target cell types. Since in vivo tissues consist of a mixture of cell types of both either dividing or non-dividing status, it is still unclear whether HMEJ can target a wide range of cell types in vivo. By contrast, SATI-mediated knock-in has been achieved in both dividing and non-dividing cells, as is the case with HITI. To clarify the details of how HMEJ and SATI differ, a further side-by side comparison is needed in many different cell types. Regarding its applicability, SATI is a versatile in vivo genome-editing method that can target a broad range of mutations and cell types (Supplementary information, Fig. S2c). In addition, the design of the HMEJ donor is less flexible than SATI because of the need to include two homology arms without the possibility of including the splicing acceptor on the left homology arm, in order to avoid undesired splicing. Furthermore, two homology arms reduce the size of the insert cassette that can be packaged in AAV, thus limiting its in vivo application.
The proof of concept of SATI enabling targeted transgene knock-in in neurons in vitro and in vivo, will help to advance both basic and translational neuroscience research. For example, this system could be used to insert optogenetic activators downstream of a relevant genetic locus to gain precise cell type-specific control of neuronal activity.33 SATI-mediated genome editing in the adult mouse brain and muscle in vivo brings about the possibility to generate knock-in reporters to trace cell lineages in non-dividing tissues other species. This would be particularly useful in animal models where transgenic tools are limited (e.g., non-human primates).34,35 Current viral vector-mediated transgene-complementation approaches can be used to effectively treat diseases caused by recessive mutations, specifically those for which the mutant allele produces no (or very little) functional protein.36 For inherited disorders such as these, gene therapy has provided remarkable therapeutic benefits in clinical trials. However, this gene-complementation strategy cannot be used to treat gain-of-function genetic mutations that produce proteins with increased or aberrant function37 such as achondroplasia,38 Huntington’s disease,39 and progeria syndrome.29 The SATI system allowed us to achieve targeted gene knock-in in multiple tissues, thus providing a first in vivo proof-of-concept for in vivo gene correction.
Although the SATI-mediated in vivo gene correction efficiency achieved in a premature aging mouse model caused by a dominant point mutation in this study is mild (2% in liver), we could observe diminished aging phenotypes in several tissues as well as an extension of lifespan. This is similar to the HITI-mediated gene-correction efficiency (4% in retina), which was sufficient to alleviate some of the phenotypes associated with retinitis pigmentosa in rats.6 Other genome-editing methodologies with similar or even less efficiency, have been shown to improve phenotypic outcomes like hearing loss in mouse models of hereditary tyrosinemia type I and deafness.40,41 Additionally, we could observe diminished aging phenotypes in the skin and spleen as well as in tail-tip fibroblasts, even though SATI-mediated gene knock-in could not be detected by PCR and NGS at later stages (around postnatal day 90) in these tissues. Together, these observations beg the question of how the mild to low correction efficiencies achieved can explain the extent of phenotypic improvements observed. This might be accounted for by an indirect non-cell-autonomous effect from the small number of gene-corrected cells in the same or other tissues. Indeed, aging is a complex and multifactorial process, where non-autonomous signals play key roles. For example, clearance of senescent cells in some tissues can systemically delay aging-associated disorders.42 Similarly, we have observed in our previous43 studies that even a small percentage of edited cells in one tissue can have non-cell-autonomous beneficial aging effects systemically on other parts of the organism. These indirect improvements of aging phenotypes are also seen in other in vivo progeria studies such as methionine restriction in the diet.44 Notwithstanding these observations, the development of efficient gene-delivery tools as well as elucidation of the detailed mechanisms of oaHDR, are needed to increase the efficiency of SATI and to clarify the extent of the phenotypic improvement as well as the relationship between corrected cells and non-cell-autonomous effects.
In this regard, two recent reports have shown that classical HDR occurs efficiently in cultured cardiomyocytes and in the mouse brain in vivo. In these experiments, transgenic mice were used having constitutively active Cas9 or AAVs were used to deliver donor DNA with two homology arms.9,45 However, we have previously shown that a similar HDR strategy did not rescue visual function in a rat model of retinitis pigmentosa, whereas a HITI-based strategy was effective.6 These contradictory results may be explained by the promiscuity of DNA repair complexes, differences in cell types, delivery methods, target loci, and/or experimental design. In this study, we have compared DNA knock-in efficiencies using HDR-, HITI-, HMEJ- and SATI-mediated systems in HEK293 cells, hES cells, MEFs, and primary neurons. These comparisons revealed that each knock-in method has different levels of efficiency in different cell types in agreement with recent discoveries11,12 suggesting that different cell-types might utilize preferred DSB repair pathways. These observations highlight the importance of a detailed study of the most appropriate genome-editing method for targeting specific cell types particularly as these technologies progress towards the clinic.
Our DSB related gene knockdown experiments have revealed that SATI requires the participation of components of multiple DSB repair sub-pathways. There is evidence that coupling of the HDR and NHEJ pathways repairs DSB by recombination with a homologous sequence on a heterologous chromosome in mouse ES cells,46 and a model was proposed in which repair is initiated by HDR and completed by NHEJ-mediated ligation. More recently, linear single-strand oligonucleotides (ssODNs) with only a single homology arm have been inserted into target loci in HEK293T cells. Here, the authors speculated that the donor ssODNs were integrated by synthesis-dependent strand annealing (SDSA) coupled with DNA synthesis.47 Although the activities of basal HDR and DNA synthesis are thought to be quite low in non-dividing cells, a similar combined HDR/NHEJ repair process, or SDSA, may be activated when both donor and target sequences are cut. Another possibility is that oaHDR may be a potential consequence of the break induced replication (BIR) mechanism. However, the ability of SATI to proceed in non-dividing neurons and its activity, despite knockdown of Rad52, may refute this interpretation.48 Additional studies are warranted to elucidate the detailed molecular mechanisms of the oaHDR-mediated gene knock-in process. Once better understood, this approach may prove instrumental in developing strategies for in vivo target-gene replacement.
Taken together, our results indicate that SATI could potentially be used to generate knock-in animals and correct dominant mutations in vivo, even in adult tissues, through the targeting of multiple tissues via systemic delivery. Importantly, it should be noted that over 90% of human RefSeq genes have open reading frames that are <4 kb,49 which is within the capacity of current AAV-based delivery methods. Once better understood, this advanced gene-repair approach may prove instrumental in developing effective strategies for in vivo target-gene replacement of a broad range of mutation types, including dominant mutations, as well as devastating genetic multi-organ and systemic pathologies.
Materials and methods
Plasmids and Minicircle DNA
To construct gRNA expression vectors, each 20 bp target sequence was sub-cloned into pCAGmCherry-gRNA (Addgene 87110) or gRNA_Cloning Vector (Addgene 41824). The CRISPR-Cas9 target sequences (20 bp target and 3 bp PAM sequence) used in this study are shown as following: Tubb3 intron 3 targeting gRNA (int3gRNA-mCherry: GAAGGCTGACCTATTTATCCAGG), gRNA2 (GGTCGCCACCATGGTGAGCAAGG), gRNA3 (CAGCTCGACCAGGATGGGCACGG), and Lmna intron 10 targeting gRNA (Lmna-gRNA-mCherry: CCCATAAGTGTCTAAGATTCAGG). The Scramble-gRNA (mScramblegRNA-mCherry; GCTTAGTTACGCGTGGACGAAGG), gRNA1 (CAGGGTAATCTCGAGAGCTTAGG), and Tubb3 exon4 targeting gRNA (ex4gRNA-mCherry; GCTTAGTTACGCGTGGACGAAGG) expression plasmids have been established in a previous publication.6 hCas9 (Addgene 41815) and tGFP (Addgene 26864) were purchased from Addgene. In our previous paper, we established the enhanced version of Cas9 (pCAG-1BPNLS-Cas9-1BPNLS (Addgene 87108) and pCAG-1BPNLS-Cas9-1BPNLS-2AGFP (Addgene 87109), IRESmCherry-HDR-0c, IRESmCherry-MC and Tubb3ex4-HDR, Tubb3ex4-HITI (pTubb3-MC: Addgene 87112).6 Minicircles (MCs) are double stranded DNAs devoid of the bacterial backbone that enhance the stability of the integrated transgene.6,16 To construct the SATI donor for mouse Tubb3 (pMC-Tubb3int3-SATI and pMC-Tubb3int3-scramble), gRNA target sequence and one-side homology arm including GFP was amplified from pTubb3-HR as established in the previous paper6 and subcloned into the ApaI (NEB #R0114S) and SmaI (NEB #R0141S) sites of the minicircle producer plasmid (pMC.BESPX from System Biosciences #MN100B-1) using the In-Fusion HD Cloning kit (Clontech #639650). To construct a HMEJ donor for mouse Tubb3 (pTubb3int3-HMEJ), we removed unnecessary homologous sequence from the inserting cassette by inserting a codon optimized exon 4 and non-translated sequence derived from the rat genome. The mouse Tubb3 exon 4 was codon optimized and synthesized in IDT. Part of intron 3 including the splicing acceptor site, 3′UTR and downstream sequences were amplified from rat genomic DNA isolated from a Brown Norway rat. Two homology arms (left arm: 1.0 kb, right arm: 1.2 kb) were amplified from mouse genomic DNA and then assembled with the inserting cassette. The assembled fragment was sandwiched by two gRNA target sequence and subcloned into pCAG-floxSTOP plasmid following the above strategy. To construct the SATI donor for progeria gene correction (pMC-progeria-SATI), the gRNA target sequence and one side homology arm including c.1827C was amplified from wild type C57BL/6 mouse genomic DNA and then subcloned into pMC.BESPX following the above strategy. These parental pre-minicircle DNAs were removed from backbone DNA to generate minicircle DNA vector as described in the previous paper.6 To construct NG PAM xCas9 (pCAG-1BPNLS-xCas9-1BPNLS) (xCas9 3.7 (Addgene 108379) was a gift from David Liu (Addgene plasmid # 108379; http://n2t.net/addgene:108379; RRID:Addgene_108379),14 the xCas9 3.7 was amplified by PCR, then inserted in pCAG-1BPNLS-Cas9-1BPNLS using In-Fusion HD Cloning kit. To construct NG PAM SpCas9-NG (pCAG-1BPNLS-SpCas9NG-1BPNLS), SpCas9-NG15 was synthesized in IDT and then inserted into pCAG-1BPNLS-Cas9-1BPNLS using the In-Fusion HD Cloning kit. To construct cell-cycle specific Cas9,50 Cdt1 and Geminin were synthesized in IDT, then inserted in pCAG-1BPNLS-Cas9-1BPNLS (Addgene 87108) using the In-Fusion HD Cloning kit. This resulted in pCAG-1BPNLS-Cas9-Cdt1 and pCAG-1BPNLS-Cas9-Geminin are G1- or S/G2/M-phase specific Cas9 expression plasmid, respectively. To construct nickase Cas9 (pCAG-1BPNLS-Cas9D10A-1BPNLS), the D10A point mutation was inserted into pCAG-1BPNLS-Cas9-1BPNLS (Addgene 87108) using the In-Fusion HD Cloning kit. shRNA expression vectors (pLKO-shRNA) were purchased from Sigma (Supplementary information, Fig. S9a). The control, pLKO-shRNA-Scramble, was established in a previous study.51 To construct donor/gRNA AAVs for SATI-mediated progeria gene correction, the one side homology arm including c.1827C was amplified from wild type C57BL/6 mouse genomic DNA. The homology arm was sandwiched by the Cas9/gRNA target sequence, Lmna intron 10 gRNA expression cassette and mCherryKASH expression cassettes, subcloned between ITRs of PX552 purchased from Addgene (Addgene 60958) to generate the pAAV-Progeria-SATI. pAAV-nEFCas9 (Addgene 87115) were established in the previous paper.6
AAV production
All of AAVs (AAV-progeria-SATI and AAV-nEFCas9) were packaged with serotype 9 and were generated by the Gene Transfer Targeting and Therapeutics Core (GT3) at the Salk institute for biological studies or ViGene Biosciences.
Animals
ICR and C57BL/6 mice were purchased from the Jackson laboratory. The mouse model of Hutchinson–Gilford progeria syndrome (HGPS) carrying the Lmna G609G (c.1827C>T) mutation (Progeria) was generated by Carlos López-Otín at the University of Oviedo, Spain and kindly donated by Brian Kennedy at the Buck Institute.19,52 All mice were bred in Animal Resource Department in Salk Institute. All mouse procedures were performed according to NIH guidelines and approved by the Committee on Animal Care at the Salk Institute. All mice used in this study were from mixed gender, mixed strains and age from E12.5 to 17 months and later.
Primary culture of mouse neurons
Mouse neurons were obtained from the cortex of E14.5 ICR mice brains or P0.5 progeria mice brains. Brain dissection was performed in a cold solution of 2% glucose in PBS. Then tissue was dissociated with Accutase (Innovative Cell Technologies #AT104), and the suspension was transferred across a 40 μm cell strainer to get a single cell suspension. Cells were plated in a ratio of 200,000 cells per each 12 mm poly-d-lysine coverslip (Neuvitro #H-12-1.5-PDL) with Neurobasal media (Gibco #21103-049) supplemented with 5 mM taurine (Sigma #T8691-25G), 2% B27 (Gibco #17504-044) and 1x GlutaMAX (Gibco #35050-061). Cultures were maintained on standard conditions (37 °C in humidified 5% CO2/95% air). Half the volume of culture media was replaced every other day. We tracked the disappearance of the proliferative neuronal progenitors present in the primary culture by 10 μM EdU-pulses every day after plating (using EdU from the Invitrogen #C10640 kit). In agreement with our previous study,6 5 days after culture, the percentage of EdU+ cells was reduced until basal levels. We could then proceed with such a post-mitotic cell population for subsequent experiments.
Transfection and AAV infection of in vitro cultured primary neurons
For transfection of minicircles or plasmids, CombiMag (OZBiosciences #CM20200) reagent in combination with Lipofectamine 2000 (Invitrogen #P-N52758) was used for transfection of mouse primary neurons according to the manufacturer’s instructions. Plasmids of Cas9, gRNA, and shRNAs were transfected in a ratio of 1 μg each per 1 mL, while donors at ratio of 2 μg per mL of culture media after 5 days in culture. The following combinations of donor and gRNA were transfected in primary neurons (single homology arm/chromosome cut: MC-Tubb3int3-scramble and int3gRNA-mCherry; single homology arm/donor cut: MC-Tubb3int3-scramble and mScramblegRNA-mCherry; single homology arm/donor- chromosome dual cut (SATI): MC-Tubb3int3-SATI and int3gRNA-mCherry; Exon 4 targeting HITI: Tubb3ex4-HITI and ex4gRNA-mCherry; Exon 4 targeting HDR: Tubb3ex4-HDR and ex4gRNA-mCherry); and Intron 3 targeting HMEJ: Tubb3int3-HMEJ. For AAV infection, the AAV mixtures (AAV9-nEFCas9 [2 × 1011 genome copy (GC)] and AAV9-Progeria-SATI [2 × 1011 GC]) were infected into primary culture in 6-well scale after 5 days in culture. Cells were analyzed by following methods after 5 days of transfection or infection.
Immunocytochemistry of primary neurons
Fixation was performed for 15 min in 4% paraformaldehyde solution. Blocking and permeabilization was carried out for 1 h at room temperature with 5% Bovine Serum Albumin (BSA, Sigma #A1470-100) and 0.1% Triton-X100 (EMD #TX1568-1) in PBS. Primary antibodies were diluted in PBS and incubated overnight at 4 °C in a humid chamber at the following concentrations [1:1000] anti-GFP (Aves #GFP-1020) or [1:250] anti-βIII-tubulin (Sigma #T2200-200UL). On the following day, cells were incubated with secondary antibodies: [1:1000] Alexa-Fluor 488 or 647 (Thermo Fisher, #A11039 and #A21244). Five washing steps with 0.2% of Tween 20 (Fisher #BP337-500) in PBS were performed to remove the excess of primary and secondary antibodies after respective incubations. Then, cells were mounted using DAPI-Vector Shield mounting media (Vector #H-1200). To determine the proliferation status, EdU was detected by Click-iT EdU kit according manufacturer instructions (Invitrogen #C10640).
Image capture and processing of primary neurons
Immunocytochemistry samples of neuronal primary culture were visualized by confocal microscopy using a Zeiss LSM 710 Laser Scanning Confocal Microscope (Zeiss) for detection and quantification of GFP knock-in efficiency. For quantification purposes, the proportion of GFP+ cells was calculated with reference to total transfected mCherry+ cells per coverslip by direct counting. Representative pictures were acquired with an Airyscan LSM880 microscope (Zeiss). For imaging purposes, at least five pictures were obtained from each sample. Cultures were derived at least from 30 different litters, the exact n values are described in each figure. Images were processed by ZEN2 Black edition software (Zeiss), ICY software for bio-imaging version 1.9.5.1 (http://icy.bioimageanalysis.org/), and NIH ImageJ (Fiji) software53 according the experimental requirements.
Genotyping of cultured primary neurons
To determine GFP knock-in and distinguish between one-armed HDR (oaHDR) and HITI events in the cultured primary neurons, genomic DNA was extracted using a Pico Pure DNA Extraction Kit (Thermo Fisher Scientific #KIT0103) or a Blood & Tissue kit (QIAGEN #69506) according to the manufacturer’s instructions. The GFP knock-in sequence including gRNA target site was first amplified with PrimeSTAR GXL DNA polymerase (Takara #R050A) with the following primers: mTubb3GFP-F1: 5′-GCAGAACTCCCAGCACCACAATTTTCAACCATGNNNNNNNNACAGCCCTCATCTGACATCACAGTCTCAGC-3′ and mTubb3GFP-R1: 5′-GTTGCTTCTTTAACTTATGTGACTCCAGACAGTTGTTTCCTATGAAGGCTCCGTTTACGTCGCCGTCCAGCTCGACCAG-3′. Then, the PCR product was nested using the following primers and 1st PCR product as a template. mTubb3GFP-F2: 5′-GCAGAACTCCCAGCACCACAATTTTCAACCATG-3′ and mTubb3GFP-R2: 5′-GTTGCTTCTTTAACTTATGTGACTCCAGACAGTTGTTTCCTATGAAGGCT-3′. PCR products were cloned into the pCR-Blunt II-TOPO vector with Zero Blunt TOPO cloning kit (Invitrogen #450245). Amplicons were sequenced using an ABI 3730xl sequencer (Applied Biosystems) and the ratio of oaHDR and HITI was determined from the gRNA target sequence. Of note, the NNNNNNNN in the mTubb3GFP-F1 primer is barcode sequence to distinguish each origin. To avoid inaccuracy by PCR bias, we counted as one if the PCR products contain same barcode sequence.
Generation and culture of GFP-correction HEK293, HeLa and hES cell lines
We used the mutated GFP gene-based reporter system to assess the knock-in efficiency in dividing cells and to optimize the SATI method in HEK293 and hES cells as established in a previous report.6,18 The mutated GFP gene-based reporter line in HeLa cell was established in a previous study.6 hES cells were cultured as previously described.54 HEK293 and HeLa cells were cultured with HEK293 medium containing DMEM (Gibco #11995-040), 10% heat-inactivated Fetal Bovine Serum (FBS, Gibco #16000-044), 1x GlutaMAX, 1x MEM Non-Essential Amino Acids (Gibco #11140-050) and 1x Penicillin Streptomycin (Gibco #15140-122).
Measurement of targeted gene knock-in efficiency in GFP-correction HEK293, HeLa, and hES cell lines
To measure the targeted gene knock-in efficiency of HDR, oaHDR and HITI in GFP-correction HEK293, hES and HeLa cell lines, Lipofectamine 3000 (Invitrogen #L3000008) and FuGENE HD (Promega #E2311) were used for transfection of HEK293/HeLa-derived cell lines and human ES-derived cell line, respectively. Transfection complexes were prepared following the manufacturers’ instructions. Cas9 expression plasmid (hCas9 [HEK293 and HeLa cell] or pCAG-1BPNLS-Cas9-1BPNLS [hESCs]), gRNA (gRNA1, gRNA2, and/or gRNA3) and donor DNA (tGFP) were used for transfection. gRNA1 was used to measure HDR efficiency. Co-transfection of gRNA2 and gRNA3 was used to measure the oaHDR efficiency. gRNA2 or gRNA3 single transfections were used as controls to cut only genomic DNA and only DNA donor, respectively. For GFP-correction of the HEK293 cell line, plasmids of Cas9, gRNA and donor were transfected in a ratio of 1 μg each per reaction for 12-wells. For GFP-correction of the hES cell line, 0.5 μg of Cas9 expression vector, each 0.5 μg of gRNA expression plasmids vector and 1 μg of donor vector were co-transfected for 6 wells. For GFP-correction HeLa cell line, plasmids of Cas9, gRNA, and donor were transfected in a ratio of 0.5 μg each per reaction for 12-wells. To compare the HDR and HITI efficiency in HEK293 and hESC cells, we co-transfected pCAG-1BPNLS-Cas9-1BPNLS, gRNA1, and donor DNAs (IRESmCherry-HDR-0c or IRESmCherry-MC). A promoterless IRESmCherry minicircle DNA (IRESmCherry-MC) was used to measure HITI efficiency. A promoterless IRESmCherry with a two-homology arm plasmid (IRESmCherry-HDR-0c) was used to measure HDR efficiency. The efficiencies of targeted gene knock-in via HDR, oaHDR, and HITI were determined 6 days after transfection by determining the number of GFP+ or mCherry+ cells by FACS LSR Fortessa (BD) or CytoFLEX S (Beckman coulter). To arrest HeLa cells in G1 phase, they were treated with 20 μM Lovastatin (Sigma #1370600) as in a previous report.55 To examine the effect of cell-cycle-specific genome editing, pCAG-1BPNLS-Cas9-1BPNLS, pCAG-1BPNLS-xCas9-1BPNLS, pCAG-1BPNLS-SpCas9NG-1BPNLS, pCAG-1BPNLS-Cas9-Cdt1and pCAG-1BPNLS-Cas9-Geminin were also transfected into HEK293 or HeLa cells instead of hCas9. The cell cycle phase was determined by propidium iodide (PI) (Sigma #P4170) staining and FACS analysis following a previous report.55
Surveyor assay
To examine the efficacy of the generated gRNA1, gRNA2 and gRNA3, we performed a Surveyor assay in HEK293 cells as described previously.56
Establishment and maintenance of progeria mouse embryonic fibroblasts (MEFs)
Mouse embryonic fibroblasts (MEFs) were isolated from Progeria (LmnaG609G/G609G) embryos at E12.5 and maintained on standard conditions (37 °C in humidified 5% CO2/95% air) in DMEM, 10% heat-inactivated FBS, 1x GlutaMAX, 1x MEM non-essential amino acids and 1x Penicillin Streptomycin. Progeria MEFs (passage 5) were transfected with pCAG-1BPNLS-Cas9-1BPNLS-2AGFP, pCAGmCherry-Lmna-gRNA, MC-progeria-SATI, and pLKO-shRNAs using Nucleofection P4 Kit (Lonza #V4XP-4024). Two days later, the transfected cells were treated with Puromycin (final 1 μg/mL, Gibco #A11138-03) to select shRNA transfected cells and harvested two days later for genomic DNA extraction using the PicoPure DNA Extraction Kit.
Stereotaxic AAV injection in the adult brain
The 8-week-old Progeria (LmnaG609G/G609G) mice received AAV injections with a 1:1 mixture of AAV9-nEFCas9 (5.33 × 1013 genome copy (GC)/mL) and AAV9-Progeria-SATI (2.26 × 1013 GC/mL). Mice were anesthetized with 100 mg/kg of ketamine (Putney) and 10 mg/kg of xylazine (AnaSed Injection) cocktail via intra-peritoneal injections and mounted in a stereotaxis (David Kopf Instruments Model 940 series) for surgery and stereotaxic injections. Virus was injected into the center of V1, using the following coordinates: 3.4 mm rostral, 2.6 mm lateral relative to bregma, and 0.5–0.7 mm ventral from the pia. We injected 3 μL of AAVs using a 33 Gauge neuros syringe (Hamilton #65460-06). To prevent virus backflow, the injection needle was left in the brain for 5–10 min after completion of injection. After injection, the skull and skin were closed, and mice were allowed to recover on a 37 °C warm pad. Mice were housed for two weeks to permit gene knock-in. After three weeks, the injected site was harvested and genomic DNA extracted using a Blood & Tissue kit for subsequent experiments.
Evaluation of oaHDR/HITI events in progeria mice by sanger sequencing
Genomic DNA is extracted from progeria MEF, primary neuron, and brain tissue, respectively. To enrich the corrected sequence, the junction site of gene knock-in sequence including gRNA target site was first amplified with PrimeSTAR GXL DNA polymerase with following primers: LMNAex11NGS1-F: 5′-TGCATGCTTCTCCTCAGATTTCCCTGCAACAA-3′ and LMNAex11NGS1-R: 5′-GATGAGGGTAAAGCCAAGGCAGCAGGACAAA-3′. Then, the PCR product was nested using the following primers and the 1st PCR product as a template. mLMNAex11-F4: 5′-TCCTCAGATTTCCCTGCAACAATGTTCTCTTTCCTTCCTGT-3′ and mLMNAex11-R4: 5′-TGTGACACTGGAGGCAGAAGAGCCAGAGGAGA-3′. Using these PCR products, BstXI enzyme (NEB #R0113S) digestion, which can only recognize the uncorrected mutation, was performed at 37 °C. Using the BstXI-digested products, the junction site of only the knock-in gene sequence including gRNA target site was amplified with following primers: LMNAenrich2-F: 5′-AACAATGTTCTCTTTCCTTCCTGTCCCC-3′ and LMNA enrich2-R: 5′-CAGAAGAGCCAGAGGAGATGGAT-3′. Final PCR products were cloned into the pCR-Blunt II-TOPO vector with Zero Blunt TOPO cloning kit. Amplicons were sequenced using an ABI 3730xl sequencer (Applied Biosystems).
Intravenous (IV) AAV injection for gene delivery of targeting vectors
The newborn (P1) LmnaG609G/G609G (Progeria), Lmna+/G609G (Heterozygous Progeria) and Lmna+/+ (WT) mice were subjected to IV AAV9 injection as in a previous report.57 Briefly, P1 mice were anesthetized and a total 30 μL of AAV mixtures (AAV9-nEFCas9 (2 × 1011 genome copy (GC)) and AAV9-Progeria-SATI (2 × 1011 GC) was injected via the temporal vein using a 30 G insulin syringe (Simple Diagnostics #SY139319). After injection, bleeding was stopped by applying pressure using a cotton swab and mice were allowed to recover on a 37 °C warm pad.
Genotyping of SATI correction in the progeria tissues
To examine the SATI-mediated knock-in event by Sanger sequencing, genomic DNA was extracted using a Blood & Tissue kit according to the manufacturer’s instructions. The HITI-mediated gene knock-in locus was amplified with PrimeSTAR GXL DNA polymerase with following HITI-specific primers: mLmnaHITI-F1: 5′-CTGCCTTACCTTCTTCCTGCCCTTCCCTAGCCT-3′ and mLmnaHITI-R1: 5′-ATGATGGGGGAAATAGCCAGGAAGCCTTCGAAA-3′. For the internal control, Fanca gene was amplified with following primers: mFA-3F: 5′-CGGCCTTCCACCATTGCAGAC-3′ and mFA-3R: 5′-CCATGATCTCGCTGACAAGGACTG-3′. To determine the efficiency of indels at target site and gene correction of mutation, Lmna intron 10 gRNA target site was amplified with PrimeSTAR GXL DNA polymerase with the following primers: mLmna-F1: 5′-TGCATGCTTCTCCTCAGATTTCCCTGCAACAA-3′ and mLmna-R1: 5′-GATGAGGGTAAAGCCAAGGCAGCAGGACAAA-3′. PCR products were cloned into the pCR-Blunt II-TOPO vector with the Zero Blunt TOPO cloning kit. Amplicons were sequenced using an ABI 3730xl sequencer (Applied Biosystems).
Measurement of the gene-correction frequency by targeted deep sequencing
To determine the gene-correction efficiency, indel efficiency and large deletion, relatively large fragments (1. 4 kb) including on-gRNA cutting and mutation sites, were amplified using PrimeSTAR GXL DNA polymerase from the indicated organs in AAV infected mice (Progeria (Pro)+donor, AAV-progeria-SATI only; Pro+SATI, AAV-Cas9 and AAV-progeria-SATI) 100 days following injection. The following primers were used: LMNAex11NGS1-F: 5′- TGCATGCTTCTCCTCAGATTTCCCTGCAACAA-3′ and LMNAex11NGS1-R: 5′- GATGAGGGTAAAGCCAAGGCAGCAGGACAAA-3′. For library construction, 2 μg PCR product was treated with dsDNA fragmentase (NEB #M0348) for 18 min, purified by AxyPrep Mag FragmentSelect Kits (Axygen # 14-223-160) and then prepared according to the instructions for BGISeq Whole Genome Sequencing library preparation. Sequencing was carried out on a BGISeq-500 platform with pair-end 100 (PE100) strategy. Raw data were filtered by SOAPnuke58 v1.5.6 using the following criteria: N rate threshold 0.05, low quality threshold 20, low quality rate 0.2. 10 million clean reads of each sample were mapped to house mouse reference sequences (GRCm38.p6) using BWA59 v 0.7.15 with standard settings. For the editing status, the alignment result is counted for the base composition of target site c.1827C>T in exon 11. All the insertion and deletion around the gRNA cutting site was counted. Sequencing data were also analyzed by split-read methods to detect large deletions. Briefly, reads were split to pairwise ends (split-reads) base by base with a minimum length of 30 bp. These pairwise ends were aligned to the reference using Bowtie260 v2.2.5 with the parameter –k 100. If the pairwise ends from a same read individually mapped back to the sequences of PCR region, the distance of the two mapped regions is calculated and referred to as a deletion. All the samples went through the pairwise ends analysis, but no large deletion (>42 bp) was found.
Measurement of off-target mutation and the ratio of oaHDR and HITI by targeted deep sequencing
The on-target site was amplified using PrimeSTAR GXL DNA polymerase from the indicated organs in AAV infected mice (Pro+donor, AAV-progeria-SATI only; Pro+SATI, AAV-Cas9 and AAV-progeria-SATI) after 100 days injection. To determine off-target effects, the top 10 predicted off-target sites61 were also amplified using PrimeSTAR GXL DNA polymerase. Then, PCR amplicons were purified using Agencourt AMPure XP (Beckman coulter #A63380) and 2nd round PCR to attach Illumina P5 adapters and sample-specific barcodes. The purified PCR products were pooled at equal ratios for single and/or pair-end sequencing using Illumina MiSeq at the Zhang laboratory (UCSD). High-quality reads (score > 23) were analyzed for insertion and deletion (indel) events and Maximum Likelihood Estimate (MLE) calculation made as with previously described methods.6,61 Briefly, for off-target site analysis, raw reads with an average Phred quality score of 23 were locally aligned to their respective on or off-target sites. All reads were required to match 85% of the genomic reference region, and also to span the entire 20 base-pair target regions along with 5 base-pair flanking regions in both directions. Then such 30 base-pair regions were analyzed for indels, with the final indel rate calculated by using maximum likelihood estimate method similar to previously described methods6,61 that correct for background errors. On-target sites were analyzed using a similar approach. High quality reads were analyzed for insertions and deletions within the gRNA target ±5 base-pair by matching the expected surrounding 10 base-pair flanking regions. Correction efficiency was determined using a similar exact match approach to determine SNP identity within reads that contained an indel event within the expected target region. As next generation sequencing analysis of indels cannot detect large size deletion and insertion events, CRISPR-Cas9 targeting efficiency and activity shown above is underestimated. To distinguish oaHDR and HITI events, we examined the sequence of gRNA target and mutation sites on the same read and separated the read in 6 categories (i.e. no mutation, indels, correction by oaHDR with indels, correction by oaHDR without indels, correction by HITI with indels, correction by HITI without indels and correction by undetermined event) based on the sequence feature of gRNA target as well as the linkage of gRNA target and mutation sites.
Data availability of target deep sequencing
Raw Illumina sequencing reads for this study have been deposited in the National Center for Biotechnology Information Short Read Archive and accessible through SRA accession number SRP126448. BGISeq-500 sequencing reads for this study have been deposited in the CNGB Nucleotide Sequence Archive (https://db.cngb.org/cnsa/) of CNGBdb with accession code CNP0000221.
5′-rapid amplification of cDNA ends (RACE)-based genome-wide off-target analysis
SMARTer RACE 5′/3′ Kit (Takara Bio USA, Inc. #634858) was used for performing the 5′- rapid amplification of cDNA ends (RACE) according manufacturer’s instructions. 1 μg total RNA was used for this reaction. Lmna exon 11-specific primers used in this experiment were 5′-GATTACGCCAAGCTTCCCACACTGCGGAAGCTTCGAGT-3′ for 1st PCR and 5′-GATTACGCCAAGCTTACACTGGAGGCAGAAGAGCCAGAGGAGATGGA-3′ for nested PCR. PCR products were cloned into the In-Fusion HD Cloning Kit. RACE fragments were sequenced using an ABI 3730xl sequencer (Eton Bioscience, Inc.). The captured exons which are located to upstream of Lmna exon 11 were mapped on UCSC mouse genome browser (NCBI37/mm9) (https://genome.ucsc.edu/cgi-bin/hgGateway?db=mm9). The chromatin and expression status of the mapped Alb and Myh6 genes loci were analyzed using H3K27ac ChIPSeq and RNASeq from Encode/LICR and DNase I hypersensitive sites (DHSs) from Encode/University of Washington. These data were obtained from liver or heart tissues at adult 8-week-old mice.
RNA analysis
Total RNA was extracted using an RNeasy Protect Mini Kit (QIAGEN #74124) or an RNeasy Fibrous Tissue Mini Kit (QIAGEN #74704) according to the manufacturer’s instructions, followed by cDNA synthesis using the Maxima H Minus cDNA Synthesis Master Mix (Thermo Fisher Scientific #M1681). TaqMan or SYBR green Gene Expression Assays were performed with the CFX384 Real-Time System C1000 Touch Thermal Cycler (Bio-Rad). TaqMan probes (Thermo Fisher Scientific) used in this experiment were Gapdh [Mm99999915_g1], LaminA [Forward primer: 5′-GTGGCAGCTTCGGGGACAAC-3′, Reverse primer: 5′-AGCAGACAGGAGGTGGCATGTG-3′ and Probe: 5′-CCCAGGAGGTAGGAGCGGGTGACT-3′], LaminC [Forward primer: 5′-GCCTTCGCACCGCTCTCATCAAC-3′, Reverse primer: 5′-ATGGAGGTGGGAGAGCTGCCCTAG-3′ and Probe: 5′-CACCAGCTTGCGCATGGCCACTTCT-3′] and Progerin [Forward primer: 5′-TGAGTACAACCTGCGCTCAC-3′, Reverse primer: 5′-TGGCAGGTCCCAGATTACAT-3′ and Probe: 5′-CGGGAGCCCAGAGCTCCCAGAA-3′]. For Alb gene expression analysis, SsoAdvanced SYBR Green Super mix (Bio-Rad # 1725274) was used with following primers [Forward primer: 5′-CTGTCTGCAATCCTGAACCGTGTG-3′ and Reverse primer: 5′-AAGCATGGCCGCCTTTCC-3′]. The datasets of the RT-qPCR were first normalized by a housekeeping gene, Gapdh and followed by the ratio of LaminA/LaminC and Progerin/LaminA. Because the endogenous expression level of Lmna gene itself is affected by the physiological aging,62 the same Lmna gene transcripts were compared. After replacement of the mutant exon with the wildtype exon without affecting the endogenous short form Lamin C transcript, the ratio of normalized LaminA/LaminC should be increased with SATI treatment. Similarly, replacement of the mutant exon with wildtype exon, the ratio of normalized Progerin/LaminA should be decreased.
Histological analysis of mouse tissues
For hematoxylin and eosin (H&E) staining, mice were harvested after transcardial perfusion using phosphate-buffered saline (PBS (-)) followed by 4% paraformaldehyde (PFA, Sigma #P6148). Subsequently, each organ was removed by dissection, post-fixed with 4% PFA at 4 °C, and embedded in paraffin. Paraffin sections were used for H&E staining in standard protocols.
Heart rate analysis
For analysis of heart rate, mice were anesthetized with 2.5% isoflurane (HENRY SCHEIN #NDC11695-6776-1), and heart rate was monitored using Power Lab data acquisition instrument with Chat5 for Windows (AD Instruments). Data were processed and analyzed using LabChart 8 (AD Instruments).
Intramuscular (IM) AAV injection
The 10-week-old Progeria (LmnaG609G/G609G) mice were anesthetized with intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg). A small portion of the quadriceps muscle was surgically exposed in front of the hind limb. The AAV mixture (Pro-Cas9, AAV-progeria-SATI (1.5 × 1010 GC) only; Pro+Cas9, AAV-Cas9 (1.5 × 1010 GC) and AAV-progeria-SATI (1.5 × 1010 GC)) was injected into the tibialis anterior (TA) muscle using a 29 Gauge insulin syringe. As a control, the same volume of PBS was injected into wild type B6 TA muscles. After injection, the skin was closed, and mice were recovered on a 37 °C warm pad. Three weeks later, the injection site was harvested for histological analysis.
Muscle fiber analysis
Three weeks after TA muscle injection, mice were euthanized, and the TA muscles were dissected and processed for histological analysis. The muscle fiber area was manually analyzed using NIH ImageJ (Fiji) software53 and processed by Microsoft Excel. 300 muscle fibers were measured for each muscle.
Establishment of tail-tip fibroblasts (TTFs) and their maintenance
TTFs were isolated from Lmna+/+ (WT), LmnaG609G/G609G (Progeria), and AAV-Progeria-SATI-treated LmnaG609G/G609G (Progeria+SATI) mice at day 70 and established as previously described.52 TTFs were maintained at 37 °C in DMEM, 10% heat-inactivated FBS, 1x GlutaMAX, 1x MEM non-essential amino acids and 1x Penicillin–Streptomycin.
Western blot analysis of TTFs
Western blotting was performed as previously described.52 Briefly, protein samples were harvested with RIPA buffer from confluent TTFs. Protein concentration was measured by Bradford reagent (Sigma #B6916-500ML). A total 10 μg of protein were loaded on 4–12% bis–tris gel (Invitrogen #NP0321BOX). Following protein transfer, PVDF membranes (EMD Millipore #IPVH00010) were blocked with 3% skimmed milk (RPI #M17200) and incubated overnight at 4 °C with primary antibody of anti-laminA/C [1:1000] (E-1, Santa Cruz #sc-376248). HRP-anti-mouse IgG antibody [1:4000] (Cell signaling #7076S) was used as secondary antibody. The blots were incubated for 1 h at room temperature and developed by ECL (GE healthcare #RPN2232). For internal controls, anti-actin antibody [1:4000] (Santa Cruz #sc-47778) and HRP-anti-mouse IgG secondary antibody [1:4,000] (Cell signaling #7076S) were used.
Immunocytochemistry of TTFs
1 × 104 TTFs (passage 5) were plated onto coverslips (Fisherbrand #12-545-82 12CIR-1D) on a 12-well plate. After 2 days incubation, coverslips were washed two times with PBS (-) and fixed with 4% paraformaldehyde (PFA) at room temperature for 30 min and then treated with blocking buffer (0.2% TritonX-100 in PBS (-), pH 7.4) for 1 h at room temperature, followed by incubation with primary antibodies diluted in PBS (-) and incubated overnight at 4 °C. The primary antibodies used in this study were [1:150] Anti-laminA/C (E-1, Santa Cruz #sc-376248). Sections were washed three times in PBS (-) and treated with secondary antibodies conjugated to [1:500] Alexa Fluor 488 goat anti-Mouse (Life technology #11001) with [1:2000] Hoechst 33342 (Thermo Fisher #H3570) for 30 min at room temperature. After three sequential washes with PBS (-), the sections were mounted with ProLong Diamond Antifade Mountant (Invitrogen #P36970).
Image capture and processing for TTFs and tissues
Representative pictures for H&E staining of each tissue were acquired with an Olympus IX51 microscope. Representative pictures for immunocytochemistry and samples of TTFs were acquired using a Zeiss LSM 710 laser scanning confocal microscope. At least five pictures were obtained from each sample. For quantification, the exact n values are described in each figure. Images were processed by ZEN2 Black edition software (Zeiss), and NIH ImageJ (FIJI) software53 according to the experimental requirements. Western blotting bands were analyzed by NIH ImageJ (FIJI) software.53
Statistical analyses
Average (mean), standard deviation (s.d.), standard error of the mean (s.e.m.), and statistical significance based on unpaired Student’s t-test for absolute values using Microsoft Excel or GraphPad Prism version 7.03 for Windows (GraphPad Software, www.graphpad.com). One-way ANOVA followed by Bonferroni’s multiple comparisons test, Tukey’s multiple comparisons test, and log-rank (Mantel-Cox) test were performed using GraphPad Prism version 7.03 for Windows.
References
Cox, D. B. T., Platt, R. J. & Zhang, F. Therapeutic genome editing: prospects and challenges. Nat. Med. 21, 121–131 (2015).
Orthwein, A. et al. A mechanism for the suppression of homologous recombination in G1 cells. Nature 528, 422–426 (2015).
Genovese, P. et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature 510, 235–240 (2014).
Li, H. et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature 475, 217–221 (2011).
Lombardo, A. et al. Site-specific integration and tailoring of cassette design for sustainable gene transfer. Nat. Methods 8, 861–869 (2011).
Suzuki, K. et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540, 144–149 (2016).
Lieber, M. R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 79, 181–211 (2010).
Suzuki, K. & Izpisua Belmonte, J. C. In vivo genome editing via the HITI method as a tool for gene therapy. J. Hum. Genet. 63, 157–164 (2018).
Nishiyama, J., Mikuni, T. & Yasuda, R. Virus-mediated genome editing via homology-directed repair in mitotic and postmitotic cells in mammalian brain. Neuron 96, 755–768.e5 (2017).
Verma, P. & Greenberg, R. A. Noncanonical views of homology-directed DNA repair. Genes Dev. 30, 1138–1154 (2016).
Welty, S. et al. RAD52 is required for RNA-templated recombination repair in post-mitotic neurons. J. Biol. Chem. 293, 1353–1362 (2018).
Yasuhara, T. et al. Human Rad52 promotes XPG-mediated R-loop processing to initiate transcription-associated homologous recombination repair. Cell 175, 558–570.e11 (2018).
Yao, X. et al. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Res. 27, 801–814 (2017).
Hu, J. H. et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556, 57–63 (2018).
Nishimasu, H. et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 361, 1259–1262 (2018).
Chen, Z.-Y., He, C.-Y., Ehrhardt, A. & Kay, M. A. Minicircle DNA vectors devoid of bacterial DNA result in persistent and high-level transgene expression in vivo. Mol. Ther. J. Am. Soc. Gene Ther. 8, 495–500 (2003).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Wu, J. et al. An alternative pluripotent state confers interspecies chimaeric competency. Nature 521, 316–321 (2015).
Osorio, F. G. et al. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci. Transl. Med. 3, 106ra107 (2011).
Eriksson, M. et al. Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome. Nature 423, 293–298 (2003).
Lopez-Mejia, I. C. et al. Antagonistic functions of LMNA isoforms in energy expenditure and lifespan. EMBO Rep. 15, 529–539 (2014).
Tichy, E. D. et al. The abundance of Rad51 protein in mouse embryonic stem cells is regulated at multiple levels. Stem Cell Res. 9, 124–134 (2012).
Saraiva, J., Nobre, R. J. & Pereira de Almeida, L. Gene therapy for the CNS using AAVs: the impact of systemic delivery by AAV9. J. Control. Release Soc. 241, 94–109 (2016).
Coutinho, H. D. M., Falcão-Silva, V. S., Gonçalves, G. F. & da Nóbrega, R. B. Molecular ageing in progeroid syndromes: Hutchinson–Gilford progeria syndrome as a model. Immun. Ageing A 6, 4 (2009).
Cesta, M. F. Normal structure, function, and histology of the spleen. Toxicol. Pathol. 34, 455–465 (2006).
Khanna, P. B., Davies, I. & Faragher, E. B. Age-related changes in the stomach of the laboratory mouse: a quantitative morphological study. Age Ageing 17, 257–264 (1988).
Kurban, R. S. & Bhawan, J. Histologic changes in skin associated with aging. J. Dermatol. Surg. Oncol. 16, 908–914 (1990).
Zhou, Q., Brown, J., Kanarek, A., Rajagopal, J. & Melton, D. A. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 455, 627–632 (2008).
Gonzalez, J. M., Pla, D., Perez-Sala, D. & Andres, V. A-type lamins and Hutchinson–Gilford progeria syndrome: pathogenesis and therapy. Front. Biosci. Sch. Ed. 3, 1133–1146 (2011).
Merideth, M. A. et al. Phenotype and course of Hutchinson–Gilford progeria syndrome. N. Engl. J. Med. 358, 592–604 (2008).
Chu, V. T. et al. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 33, 543–548 (2015).
Maruyama, T. et al. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 33, 538–542 (2015).
Boyden, E. S., Zhang, F., Bamberg, E., Nagel, G. & Deisseroth, K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci. 8, 1263–1268 (2005).
Izpisua Belmonte, J. C. et al. Brains, genes, and primates. Neuron 86, 617–631 (2015).
Sasaki, E. et al. Generation of transgenic non-human primates with germline transmission. Nature 459, 523–527 (2009).
Naldini, L. Gene therapy returns to centre stage. Nature 526, 351–360 (2015).
Lewin, A. S., Glazer, P. M. & Milstone, L. M. Gene therapy for autosomal dominant disorders of keratin. J. Investig. Dermatol. Symp. Proc. 10, 47–61 (2005).
Horton, W. A., Hall, J. G. & Hecht, J. T. Achondroplasia. Lancet Lond. Engl. 370, 162–172 (2007).
Walker, F. O. Huntington’s disease. Lancet Lond. Engl. 369, 218–228 (2007).
Gao, X. et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 553, 217–221 (2018).
Yin, H. et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 32, 551–553 (2014).
Baker, D. J. et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236 (2011).
Beyret, E. et al. Single-dose CRISPR-Cas9 therapy extends lifespan of mice with Hutchinson–Gilford progeria syndrome. Nat. Med. 25, 419–422 (2019).
Bárcena, C. et al. Methionine restriction extends lifespan in progeroid mice and alters lipid and bile acid metabolism. Cell Rep. 24, 2392–2403 (2018).
Ishizu, T. et al. Targeted genome replacement via homology-directed repair in non-dividing cardiomyocytes. Sci. Rep. 7, 9363 (2017).
Richardson, C. & Jasin, M. Coupled homologous and nonhomologous repair of a double-strand break preserves genomic integrity in mammalian cells. Mol. Cell Biol. 20, 9068–9075 (2000).
Paix, A. et al. Precision genome editing using synthesis-dependent repair of Cas9-induced DNA breaks. Proc. Natl Acad. Sci. USA 114, E10745–E10754 (2017).
Kramara, J., Osia, B. & Malkova, A. Break-induced replication: the where, the why, and the how. Trends Genet. TIG 34, 518–531 (2018).
Strausberg, R. L. et al. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc. Natl Acad. Sci. USA 99, 16899–16903 (2002).
Gutschner, T., Haemmerle, M., Genovese, G., Draetta, G. F. & Chin, L. Post-translational regulation of Cas9 during G1 enhances homology-directed repair. Cell Rep. 14, 1555–1566 (2016).
Hishida, T. et al. Sirt1, p53, and p38(MAPK) are crucial regulators of detrimental phenotypes of embryonic stem cells with Max expression ablation. Stem Cells Dayt. Ohio 30, 1634–1644 (2012).
Ocampo, A. et al. In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell 167, 1719–1733.e12 (2016).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Liu, G.-H. et al. Recapitulation of premature ageing with iPSCs from Hutchinson–Gilford progeria syndrome. Nature 472, 221–225 (2011).
Ma, H. T. & Poon, R. Y. C. Synchronization of HeLa cells. Methods Mol. Biol. 1524, 189–201 (2017).
Sanjana, N. E. et al. A transcription activator-like effector toolbox for genome engineering. Nat. Protoc. 7, 171–192 (2012).
Gombash Lampe, S. E., Kaspar, B. K. & Foust, K. D. Intravenous injections in neonatal mice. J. Vis. Exp. e52037 (2014). https://doi.org/10.3791/52037.
Chen, Y. et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience 7, 1–6 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Hsu, P. D. et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832 (2013).
Luo, Y.-B. et al. Investigation of splicing changes and post-translational processing of LMNA in sporadic inclusion body myositis. Int. J. Clin. Exp. Pathol. 6, 1723–1733 (2013).
Acknowledgements
We are grateful to M. Schwarz and P. Schwarz for administrative help; D. O’Keefe and S. Tsuji for help with manuscript preparation; M. Kay and Z.Y. Chen for sharing experimental materials; J. Naughton and J. Marlett for AAV production; K. Peterson and Y. Gu for helping the measurement of heart rate; U. Manor and K. Diffenderfer for imaging; K. McIntyre for mouse histology processing and J. Li for helping the molecular work; K. Sumiyama for data analysis. M.Y. was partially supported by 2016 Salk Women & Science Special Award. K.S. was supported by JSPS KAKENHI (15K21762 and 18H04036), Takeda Science Foundation, The Uehara Memorial Foundation, National Institutes of Natural Sciences (BS291007), The Sumitomo Foundation (170220), The Naito Foundation, The Kurata Grants (1350), Mochida Memorial Foundation, and The Inamori Foundation. This research was supported by Guangdong Provincial Key Laboratory of Genome Read and Write (No. 2017B030301011), Guangdong Provincial Academician Workstation of BGI Synthetic Genomics (No. 2017B090904014) and Shenzhen Peacock Plan (No. KQTD20150330171505310). J.C.I.B. was supported by The Leona M. and Harry B. Helmsley Charitable Trust (2012-PG-MED002), the G. Harold and Leila Y. Mathers Charitable Foundation, NIH (R01HL123755 and 5 DP1 DK113616), The Progeria Research Foundation, The Glenn Foundation, KAUST, The Moxie Foundation, Fundación Dr. Pedro Guillen, AFE and Universidad Católica San Antonio de Murcia (UCAM).
Author information
Authors and Affiliations
Contributions
K.S., M.Y., R.H.-B., and J.C.I.B. conceived the project and designed experiments. K.S., M.Y., R.H.-B. and E.A. constructed plasmids and minicircles. M.Y. and R.H.-B. performed work on primary neurons. K.S., M.Y., R.H.-B., R.D.S., E.A., M.S., and A.N.N. performed genomic DNA and RNA analyses. M.Y., F.H., M.K., T.H., and C.R.E. performed phenotypic analyses of progeria mice. M.Y. performed AAV IV injection in neonatal mice. Z.L., C.W., J.G., Y.Y., Y.G. and K.Z. performed deep sequencing and analyzed data. K.S., M.Y., P.R. and A.O. performed ECG recordings. K.S., E.N.D., J.M.C., P.M., P.G., C.L.-O., G.-H.L., and J.C.I.B. supervised and contributed to the direction of the project. K.S., M.Y., R.H.-B., P.R., J.W. and J.C.I.B. wrote the manuscript with input from all the authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
41422_2019_213_MOESM14_ESM.mov
Supplementary Movie S1 | Movement activity of SATI-treated progeria mouse at 17-weeks-old. Representative movie of WT, Progeria (-SATI), and Progeria with SATI treatment (+SATI) mice at 17-weeks-old
Rights and permissions
About this article

Cite this article
Suzuki, K., Yamamoto, M., Hernandez-Benitez, R. et al. Precise in vivo genome editing via single homology arm donor mediated intron-targeting gene integration for genetic disease correction. Cell Res 29, 804–819 (2019). https://doi.org/10.1038/s41422-019-0213-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41422-019-0213-0
This article is cited by
-
REMOVER-PITCh: microhomology-assisted long-range gene replacement with highly multiplexed CRISPR-Cas9
In Vitro Cellular & Developmental Biology - Animal (2024)
-
Site-specific transgene integration in chimeric antigen receptor (CAR) T cell therapies
Biomarker Research (2023)
-
Efficient targeted insertion of large DNA fragments without DNA donors
Nature Methods (2022)
-
Establishment of mouse model of inherited PIGO deficiency and therapeutic potential of AAV-based gene therapy
Nature Communications (2022)
-
Improved and Flexible HDR Editing by Targeting Introns in iPSCs
Stem Cell Reviews and Reports (2022)
