
Abstract
Cells may die from accidental cell death (ACD) or regulated cell death (RCD). ACD is a biologically uncontrolled process, whereas RCD involves tightly structured signaling cascades and molecularly defined effector mechanisms. A growing number of novel non-apoptotic forms of RCD have been identified and are increasingly being implicated in various human pathologies. Here, we critically review the current state of the art regarding non-apoptotic types of RCD, including necroptosis, pyroptosis, ferroptosis, entotic cell death, netotic cell death, parthanatos, lysosome-dependent cell death, autophagy-dependent cell death, alkaliptosis and oxeiptosis. The in-depth comprehension of each of these lethal subroutines and their intercellular consequences may uncover novel therapeutic targets for the avoidance of pathogenic cell loss.
Similar content being viewed by others


Introduction
The scientific observation of regulated cell death (RCD) historically began in 1842 when Karl Vogt noticed dying cells in toads. However, the surge in RCD research only started when the term “apoptosis” was coined in 1972 by John Kerr, Andrew Wyllie, and Alastair Currie1 (Fig. 1). Kerr et al. defined apoptosis as a form of programmed cell death (PCD) with morphological changes that differ from necrosis.1 Apoptosis and its dysregulation underlies various pathological and physiological processes, including cell homeostasis, tissue remodelling, and tumorigenesis.2 The identification of CED9 (also known as BCL2 in mammalian cells) and CED4 (also known as apoptotic peptidase-activating factor 1 [APAF1] in mammalian cells) from the studies of Caenorhabditis elegans development in the 1990s3,4,5 marks the beginning of an era of molecular apoptosis research that triggered the rapid expansion of RCD research. The molecular mechanisms regulating apoptosis have been extensively investigated in multiple organisms over the last 30 years. It is now established that apoptosis consists of two major subtypes, namely extrinsic and intrinsic apoptosis (Fig. 2). Extrinsic apoptosis is mediated by membrane receptors, especially by death receptors (e.g., fas cell surface death receptor [FAS, also known as CD95] and TNF receptor superfamily member 1A [TNFRSF1A, also known as TNFR1]), and is driven by initiator caspases CASP8 (also known as caspase 8) and CASP10 (also known as caspase 10).6 Alternatively, dependence receptors (e.g., unc-5 netrin receptor B [UNC5B, also known as UNC5H2] and DCC netrin 1 receptor [DCC]) may ignite extrinsic apoptosis via the activation of the initiator caspase CASP9 or dephosphorylation of death-associated protein kinase 1 (DAPK1, also known as DAPK) following the withdrawal of their ligands.7 In contrast, intrinsic apoptosis is ignited by mitochondrial outer membrane permeabilization (MOMP) that leads to the release of the mitochondrial proteins (e.g., cytochrome C, somatic [CYCS], diablo IAP-binding mitochondrial protein [DIABLO, also known as Smac], and HtrA serine peptidase 2 [HTRA2]) and subsequent activation of initiator caspase CASP9.8 MOMP is tightly controlled by the BCL2 family, including pro-apoptotic (e.g., BCL2 associated X, apoptosis regulator [BAX], BCL2 antagonist/killer 1 [BAK1, also known as BAK]), and anti-apoptotic (e.g., BCL2 and BCL2 like 1 [BCL2L1, also known as BCL-XL]) members.2,9 Although caspase activation does not guarantee cell death, CASP3, CASP6, and CASP7 are considered as important executioners due to their function in substrate cleavage and the destruction of subcellular structures10,11 (Box 1), culminating in the acquisition of the apoptotic morphotype.

Timeline of the terms used in cell death research

Extrinsic and intrinsic apoptosis. Extrinsic apoptosis is induced by the addition of death receptor ligands or by the withdrawal of dependence receptor ligands. CASP8 and CASP10 initiate death receptor-mediated extrinsic apoptosis, whereas CASP9 initiates the withdrawal of dependence receptor ligand-mediated extrinsic apoptosis. Pro-CASP8 and pro-CASP10 are enzymatically inactive until they interact with FADD (Fas-associated via death domain), which is activated upon binding to cell death receptors responding to their ligands. DNA damage, hypoxia, metabolic stress, and other factors can induce intrinsic apoptosis, which begins with MOMP and leads to the release of mitochondrial proteins (e.g., CYCS) into the cytosol. Cytosolic CYCS interacts with APAF1, which recruits pro-CASP9 to form the apoptosome. MOMP is tightly controlled by the BCL2 family, including its pro-apoptotic and anti-apoptotic members. CASP3, CASP6, and CASP7 are considered the common effector caspases for both extrinsic and intrinsic apoptosis. In addition, the extrinsic pathway can trigger intrinsic mitochondrial apoptosis through the generation of truncated BID (tBID) by activated CASP8. tBID can further translocate to mitochondria and cause MOMP through the activation of BAX and BAK1
Cell death may occur in multiple forms in response to different stresses, especially oxidative stress (Box 2). The loss of control over single or mixed types of cell death contributes to human diseases such as cancer, neurodegeneration, autoimmune diseases, and infectious diseases.12,13 During the past few decades, many novel forms of non-apoptotic RCD have been identified. In this review, we discuss our current understanding of the molecular machinery of each of the main types of non-apoptotic RCD, including necroptosis, pyroptosis, ferroptosis, entotic cell death, netotic cell death, parthanatos, lysosome-dependent cell death, autophagy-dependent cell death, alkaliptosis, and oxeiptosis, all of which can be inhibited by small-molecule compounds or drugs (Table 1). Finally, we describe the immunogenicity of cell death, which affects immune surveillance, inflammatory responses, tissue regeneration, and tumor therapy.
Classification of cell death
Early classifications of cell death modalities depended on the morphological and structural details of individual tissues and cells. Accordingly, Schweichel and Merker published in 1973 a morphological hallmark system for classifying cell death into types I, II, and III in prenatal tissues treated with various embryotoxic substances.14 Type I cell death corresponds to apoptosis, and is characterized by cell shrinkage (pyknosis), membrane blebbing, apoptotic body formation, DNA fragmentation (karyorrhexis), and chromatin condensation. Apoptosis was also termed “shrinkage necrosis,” a form of nonpathologic cell death, by John Kerr in 1971.15 Type II cell death is often referred to as autophagy-dependent cell death, with the formation of large-scale autophagic vacuolization-containing cytosolic materials and organelles. Although there is no doubt that autophagy promotes cell survival in most cases,16 autophagy can also cause cell death, namely autophagy-dependent cell death, in specific circumstances.17,18 Type III cell death, namely necrosis, is characterized by the loss of membrane integrity and swelling of subcellular organelles (oncosis). Necrosis has long been considered as an uncontrolled type of cell death. In contrast, regulated types of necrosis such as necroptosis occur in a controlled manner.12,13,19
The current classification system of cell death has been updated by the Nomenclature Committee on Cell Death (NCCD), which formulates guidelines for the definition and interpretation of all aspects of cell death since 2005.20 The NCCD has released five position papers dealing with the classification of cell death (2005 and 2009),20,21 the molecular definitions of cell death subroutines (2012),22 essential versus accessory aspects of cell death (2015),23 and molecular mechanisms of cell death (2018).24 Currently, cell death can be fundamentally divided into accidental cell death (ACD) and RCD, based on functional aspects.23 ACD can be triggered by unexpected attack and injury that overwhelms any possible control mechanisms. In contrast, RCD involves precise signaling cascades, is executed by a set of defined effector molecules and has unique biochemical, functional, and immunological consequences (Table 1). RCD is also known as PCD when it occurs in physiological conditions.23 Based on its molecular characteristics, RCD can be classified into multiple subroutines, a few of which have clear physiological bearing (like necroptosis and pyropotosis, which are observed during development and/or in the context of viral infections) while others (like ferroptosis, entotic cell death, netotic cell death, parthanatos, lysosome-dependent cell death, autophagy-dependent cell death, alkaliptosis, and oxeiptosis) are less well-studied and may actually be limited to cellular responses to specific toxins that do not reflect normal physiology. Here, we adopt the viewpoint that cell death involves some kind of “regulation” (hence “RCD”) as long as specific genetic or pharmacological manipulations are able to interrupt the lethal cascade-causing cellular dismantling in response to external stimuli.
Necroptosis
Necroptosis, a programmed form of necrosis showing morphological features similar to necrosis,25 was first observed in 1996 in pig kidney cells infected by the cowpox virus that expresses cytokine response modifier A (CrmA), a viral CASP1 and CASP8 inhibitor.26 In 1998, this observation was extended when L-M cells (a mouse fibroblast cell line) were found to be strongly sensitized to tumor necrosis factor (TNF, also known as TNFα)-induced necrotic cell death, suggesting that CASP8 negatively controls this type of cell death.27 Today it is known that necroptosis can be triggered by multiple stimuli, including the activation of death receptors (e.g., FAS and TNFRSF1A),28 toll-like receptors (e.g., toll-like receptor 3 [TLR3] and TLR4),29 nucleic acid sensors (e.g., Z-DNA–binding protein 1 [ZBP1, also known as DAI],30 retinoic acid receptor responder 3 [RARRES3, also known as RIG1],31 transmembrane protein 173 [TMEM173, also known as STING]32,33), and adhesion receptors.34 The same ligands (e.g., TNF, TNF superfamily member 10 [TNFSF10, also known as TRAIL], and Fas ligand [FASLG, also known as FasL or CD95L]) that ignite the extrinsic apoptosis pathway can trigger necroptosis when CASP8 activation at the death-inducing signaling complex (DISC) is prevented by means of caspase inhibitors (such as Z-VAD-FMK) or by the depletion of fas-associated via death domain (FADD).35
The era of molecular necroptosis research began in 2000 with the discovery of receptor-interacting serine/threonine kinase 1 (RIPK1) as a regulator of FASLG-induced necroptosis in T cells.28 RIPK1 is indeed a multifunctional signal kinase at the crossroads between inflammation, immunity, cell stress, cell survival, and cell death (Box 3). Subsequently, the identification of the pharmacological RIPK1 inhibitor necrostatin-1 led to the coining of the term “necroptosis.”36,37 Later, receptor-interacting serine/threonine kinase 3 (RIPK3) was unravelled as a downstream mediator of RIPK1 in death receptor-induced necroptosis.38,39,40 The subsequent discovery of mixed lineage kinase domain-like pseudokinase (MLKL) as the effector of necroptosis has largely enhanced our understanding of the molecular process of necroptosis.41,42
An array of signaling pathways facilitate RIPK3 activation in several distinct ways (Fig. 3a) involving the homotypic interaction of RIP homotypic interaction motif (RHIM) domain-containing receptors, adaptors and kinases (ZBP1, toll-like receptor adaptor molecule 1 [TICAM1, also known as TRIF], RIPK1, and RIPK3). These RHIM domains of RIPK1 and RIPK3 mediate the formation of large hetero-amyloid signaling complexes that are initiated by different ligands.43,44 First, death receptor ligands induce the RHIM-mediated binding of RIPK1 to RIPK3, triggering the formation of specific signaling complexes, the “necrosomes,” ultimately resulting in MLKL activation.38,39,40 This process requires protein posttranslational modifications that are regulated by the ubiquitin ligase STIP1 homology and u-box containing protein 1 (STUB1, also known as CHIP),45 the aurora kinase A (AURKA),46 the protein phosphatase Mg2+/Mn2+-dependent 1B (PPM1B, also known as PP2CB),47 and the deubiquitinase TNF alpha-induced protein 3 (TNFAIP3, also known as A20).48 Second, TICAM1, but not RIPK1, is required for RIPK3-MLKL–dependent necroptosis in response to TLR ligands.29 Third, certain viruses can directly bind to RIPK349 or promote the binding of the host protein ZBP1 to RIPK3 and subsequent MLKL activation.30 Fourth, RIPK3 activation by interferon alpha receptor or adhesion receptor occurs through an alternative pathway that does not require RIPK1, TICAM1 nor ZBP1.34,50,51

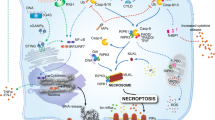
Core molecular mechanism of non-apoptotic regulated cell death. a RIPK3-stimulated MLKL is necessary for membrane rupture formation in necroptosis. Upstream elicitors include DR, TLR, and viruses, which induce RIPK3 activation through RIPK1, TICAM1, and ZBP1, respectively. In addition, RIPK3 is activated by AR via an unknown adaptor protein or kinases. b Pyroptosis is mostly driven by GSDMD after cleavage of this protein by CASP1 and CASP11 in response to PAMPs and DAMPs, or cytosolic LPS. c Ferroptosis is a form of cell death that relies on the balance between iron accumulation-induced ROS production and the antioxidant system during lipid peroxidation. The ACSL4-LPCAT3-ALOX15 pathway mediates lipid peroxidation. In contrast, several antioxidant systems, especially system xc- that includes the core components SLC7A11, GPX4, and NFE2L2, inhibit this process. d Parthanatos is a PARP1-dependent form of cell death that relies on the AIFM1-MIF pathway. e Entotic cell death is a form of cellular cannibalism through the activation of entosis followed by the engulfing and killing of cells through LAP and the lysosomal degradation pathway. RHOA, ROCK, myosin, and CDC42 are required for entosis. f Netotic cell death is driven by NET release, which is regulated by NADPH oxidase-mediated ROS production and histone citrullination. g Lysosome-dependent cell death is mediated by releasing hydrolytic enzymes (cathepsins) or iron upon LMP. h Autophagy-dependent cell death is driven by the molecular machinery of autophagy. i Alkaliptosis is driven by intracellular alkalinization after IKBKB-NF-κB pathway-dependent downregulation of CA9. j Oxeiptosis is an oxygen radical-induced form of cell death driven by the activation of the KEAP1-PGAM5-AIFM1 pathway
The phosphorylation of MLKL by RIPK3 at different residues in the C-terminal pseudokinase domain (S345/S347/T349 in mouse and S357/T358 in human) results in a conformational change and binding of inositolhexaphosphate (IP6) with positively charged patches in the N-terminal part of MLKL, followed by its recruitment to phosphatidylinosites, and insertion and multimerization in the plasma membrane, resulting in plasma membrane permeabilization.41,42,52,53,54,55,56 MLKL oligomerization and translocation to the plasma membrane can be enhanced by interactions with the molecular chaperone heat shock protein 90 alpha family class A member 1 (HSP90AA1, also known as HSP90)57,58,59,60 or by the local accumulation of inositol phosphates resulting from the activation of inositol phosphate kinase (e.g., inositol polyphosphate multikinase [IPMK] and inositol-tetrakisphosphate 1-kinase [ITPK1]).61 Strikingly, the endosomal sorting complexes required for transport (ESCRT)-III complex, a membrane scission machine, limits MLKL-mediated necroptosis and promotes membrane repair.62 MLKL has also been shown to regulate endosomal trafficking and extracellular vesicle generation.63 Thus, a fine balance between membrane injury and repair ultimately decides cell fate in necroptosis.
Early studies have revealed that mitochondrial events such as the production of mitochondrial reactive oxygen species (ROS),64 the activation of the mitochondrial phosphatase PGAM family member 5 (PGAM5, mitochondrial serine/threonine protein phosphatase), or the presence of a mitochondrial permeability transition may trigger necroptosis.65 How exactly mitochondrial ROS production contributes to necroptosis induction is still unsolved, but it may involve a redox sensing upstream of RIPK1 activation and RIPK3 recruitment.66 A connection between aerobic metabolism and necroptosis sensitivity might exist, as evidenced by RIPK3-mediated positive regulation of glutaminolysis and pyruvate dehydrogenase activity.38,67 However, other studies demonstrate that mitochondria are dispensable for necroptosis induced by death receptor signaling using PGAM5 knockdown cells.55,68 It has also been suggested that the formation of necrosomes with RIPK1, RIPK3, and MLKL in the nucleus may increase MLKL activity in plasma membranes.69,70 Moreover, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-derived ROS have been implicated in necroptosis in neutrophils.71 The functional significance of different ROS sources in necroptosis and how they impact the signal transduction remain to be further investigated.
In conclusion, RIPK3 and its substrate MLKL are necessary for necroptosis, whereas upstream RIPK1 contributes to this process in some cases (e.g., death receptor activation). RIPK3, independent of its kinase activity and independent of MLKL, also plays a regulatory role in apoptosis72 and in NLRP3-inflammasome activation and pyroptosis.73 Neither RIPK3 nor MLKL knockout mice show deficiency in embryogenesis, development, and homeostasis, suggesting no major role of necroptosis in nonchallenged conditions.74,75 A role for necroptosis in development and homeostasis is only revealed in conditions of FADD or CASP8 deficiency, demonstrating the important checkpoint function of CASP8 in controlling necroptosis in vivo.74,75 In contrast to the apparent absence of function during development and homeostasis, necroptosis is implicated in neurodegenerative diseases, chemotherapy responses, and tissue injury.76 Of note, data obtained from conditional knockout mice should be favoured over the use of systemic knockout mice that were generated using different sources of ES cells (129, C57BL/6J, or C57BL/6N) to avoid phenotypic interference of passenger mutations.77
Pyroptosis
Pyroptosis is a form of RCD driven by the activation of inflammasome, a cytosolic multiprotein complex responsible for the release of interleukin (IL) 1 family members (e.g., interleukin-1β [IL1B] and IL18), the formation of ASC (apoptosis-associated speck-like protein containing a CARD, also called PYCARD or PYRIN and CARD domain-containing) specks, and the activation of pro-inflammatory caspases. The term pyroptosis was coined by Brad Cookson and coworkers78 to describe CASP1-dependent PCD in macrophages infected by Salmonella or Shigella and associated with the release of IL1B (IL1 was historically called leukocytic pyrogen, inspiring the name pyroptosis).79,80 CASP1 mediates the proteolytic processing of pro-IL1B and pro-IL18 into mature IL1B and IL18, respectively. This type of inflammatory cell death can be triggered by the activation of CASP1 or CASP11 in mice (the latter corresponding to CASP4 and CASP5 in humans) in macrophages, monocytes and other cells81 (Fig. 3b). Pyroptosis is morphologically distinct from apoptosis. Pyroptosis is characterized by the absence of DNA fragmentation in vitro, but by the presence of nuclear condensation coupled to cell swelling and the formation of large bubbles at the plasma membrane that eventually ruptures.82,83
The activation of inflammasomes in macrophages or monocytes requires two signals: a priming signal (that may be mediated by TLR ligands and IFN signaling) that induces the transcriptional upregulation of inflammasome components through nuclear factor of κB (NF-κB), and then a sensing signal (e.g., adenosine triphosphate [ATP] and lipopolysaccharide [LPS]) that triggers pro-inflammatory caspase-mediated pyroptosis. Inflammasomes that include canonical and noncanonical types can be activated in the context of infection, tissue injury, or metabolic imbalances.81 Canonical CASP1-dependent inflammasomes are divided into two subtypes, Nod-like receptors (NLR, e.g., NLR family pyrin domain-containing 1 [NLRP1], NLRP2, NLRP3, NLRP6, NLRP7, NLR family CARD domain-containing 4 [NLRC4]) and non-NLR (e.g., absent in melanoma 2 [AIM2]). They can be selectively activated by pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs), or other immune challenges. For example, NLRP3, the most intensively studied inflammasome, can be activated by a wide range of inflammatory stimuli such as bacterial peptidoglycans, extracellular ATP, and uric acid crystals, facilitated by the kinase NIMA-related kinase 7 (NEK7).84 The non-NLR inflammasome involving AIM2 is activated by cytosolic double-stranded DNA from bacteria or host cells.85,86 The CASP11-dependent noncanonical inflammasome is activated by cytosolic LPS from invading Gram-negative bacteria in macrophages, monocytes, or other cells.87 Lipid A moiety is required for cytosolic LPS binding to CASP11’s CARD domain, which causes CASP11 oligomerization.88 TLR4, a cell membrane receptor for LPS, is not required for cytosolic LPS-induced CASP11 activation.89,90 The cytoplasmic delivery of LPS requires the release of bacterial outer membrane vesicles (OMVs) by Gram-negative bacteria91 or the binding of LPS with high-mobility group Box 1 (HMGB1).92 The interplay between canonical (e.g., NLRP3- and AIM2-dependent) and noncanonical inflammasome pathways can amplify the inflammatory response and pyroptosis.93,94 Although eukaryotic translation initiation factor 2 alpha kinase 2 (EIF2AK2)/PKR and glycolysis may participate in CASP1-dependent inflammasome activation under certain conditions,95,96,97,98 their roles in CASP11 inflammasome remain unclear.
Several recent breakthroughs indicate that gasdermin D (GSDMD) is the key effector of pyroptosis82,99,100,101,102,103 (Fig. 3b). GSDMD is cleaved by CASP11 or CASP1 to produce a 22 kDa C- (GSDMD-C) and a 3l kDa N-terminal fragment (GSDMD-N).101,102,103 CASP11 auto-cleavage at the inter-subunit linker is essential for optimal catalytic activity and subsequent GSDMD cleavage.104 Once formed, GSDMD-N translocates to the inner leaflet of the plasma membrane and binds phospholipids, thus inducing the formation of pores that ultimately cause membrane lysis.99 In contrast, GSDMD-C inhibits GSDMD-N activity.99,100 While deficiency of the phospholipid hydroperoxidase glutathione peroxidase 4 (GPX4) in myeloid-derived cells increases CASP1- or CASP11-mediated GSDMD-N production, pyroptosis, and lethality after cecal ligation and puncture (CLP)-induced sepsis, the pharmacological inhibition of phospholipid hydrolysing enzyme phospholipase C gamma 1 (PLCG1) strongly protects against pyroptosis and CLP-induced septic death,105 indicating that lipid peroxidation promotes pyroptosis. Protein kinase A (PKA) is a major cyclic adenosine monophosphate (cAMP) effector to directly block CASP11-mediated GSDMD-N production in macrophages.106 Like necroptosis, ESCRT-III is also recruited to the plasma membrane to trigger membrane repair upon GSDMD activation.107 Other members of the gasdermin family (GSDMA, GSDMB, GSDMC, GSDME/DFNA5, and GSDMA3) have similar functions in membrane-disrupting cytotoxicity.99 It has been shown that following the blockage or deficiency of the bona fide pyroptosis pathway (ASC-CASP1/4), the induction of pyroptosis can be engaged through mechanisms such as CASP8-GSDMD108,109 and CASP3-GSDME,110 although the contribution of these alternative pathways to pyroptosis elicited by different triggers remains to be established in vivo.
Neutrophil elastase (ELANE), one of the antibacterial serine proteases, triggers GSDMD cleavage at a site that is closer to the N-terminus than the caspase cleavage site.111 Elastase-mediated GSDMD-N production induces neutrophil death as well as the formation of neutrophil extracellular traps (NETs) to intercept invading microorganisms.112,113 In addition, GSDMD-N can directly lyse bacteria (such as Escherichia coli, Staphylococcus aureus, and Listeria monocytogenes) after binding to cardiolipin and forming pores in their membranes.100 CASP1 and CASP11 also play a pyroptosis-independent role in antibacterial host defence. The formation of GSDMD pores can directly trigger IL1B secretion by macrophages before the cells undergo pyroptosis,114 indicating that distinct activation thresholds may control the active IL1B release by live cells and its passive shedding from dead cells once the cell explodes. The dynamics of pore formation and interaction with ion channels allow the existence of different stages and extents of plasma membrane permeabilization, resulting in the release of IL1B prior to spilling of DAMPs following full permeabilization.115
In summary, pyroptosis is a form of GSDMD-mediated RCD that plays a cell type-dependent role in inflammation and immunity. Of note, the first Casp1-/- mice were established from 129 embryonic stem cells carrying an inactivating passenger mutation of the Casp11 locus.87 Thus, the phenotype reported for Casp1-/- mice actually results from deficiencies in both CASP1 and CASP11. Novel individual or combined transgenic mice are required to distinguish the contributions of CASP1 and CASP11 to pyroptotic signaling in a variety of different diseases that were studied in the past using the unintended double knockout.
Ferroptosis
Ferroptosis is an iron- and lipotoxicity-dependent form of RCD. It was originally observed in 2003 using erastin (a cell-permeable compound from high-content screening) to selectively kill genetically-engineered cells with an oncogenic RAS mutation, but not normal cells116 In 2012, the term ferroptosis was formally used by Brent Stockwell to describe an iron-dependent form of non-apoptotic RCD induced by erastin.117 The morphology of erastin-induced ferroptotic cells is characterized by dysmorphic small mitochondria with decreased crista, as well as condensed, ruptured outer membranes,117,118 which might be under control of the pro-apoptotic BCL2 family members such as BH3-interacting domain death agonist (BID)119 and BCL2-binding component 3 (BBC3, also known as PUMA),120 but not BAX or BAK1.117 Mechanistically, these dying cells do not display any hallmarks of apoptosis or necroptosis.117,118 Instead, ferroptosis occurs via an iron-catalyzed process of lipid peroxidation initiated through non-enzymatic (Fenton reactions) and enzymatic mechanisms (lipoxygenases) (Fig. 3c). Polyunsaturated fatty acids (PUFAs) are the prime targets of lipid peroxidation of membranes.121 The deleterious effects of lipid peroxidation in ferroptosis execution can be neutralized by lipophilic radical traps such as vitamin E, ferrostatin-1, and liproxstatin-1.117,118 The mechanistic consequences of uncontrolled lipid peroxidation leading to ferroptotic cell death are still elusive. Using molecular dynamics models, it is hypothesized that membrane thinning and increased curvature drives a vicious cycle of access by oxidants, which ultimately destabilizes the membrane leading to pore and micelle formation.122 Additionally, lipid hydroperoxides decompose to reactive toxic aldehydes such as 4-hydroxy-2-nonenals or malondialdehydes, which may inactivate proteins through crosslinking122.
Essentially, ferroptosis can be induced in a canonical way by either inactivating GPX4, the major protective mechanism of biomembranes against peroxidation damage, or in a noncanonical way by increasing the labile iron pool. Two mechanisms have been described to inactivate GPX4: (1) an indirect way by the deprivation of the cofactor glutathione (GSH) through the depletion of the precursor Cys, as a result of the inhibition of the cystine/glutamate antiporter system xc-117 or the transsulfuration pathway,123 and (2) a direct way by binding and inactivating GPX4 by compounds such as RSL3, ML162, FINO2, withaferin A, or the FDA-approved anticancer agent altretamine.121,124,125,126,127,128,129 In addition, recent findings have proposed a noncanonical ferroptosis induction pathway upon iron overload using, for example, iron chloride, hemoglobin, hemin, or ferrous ammonium sulfate, which suffices to induce ferroptosis.129,130 CDGSH iron sulphur domain 1 (CISD1), a mitochondrial iron export protein, also inhibits ferroptosis by preventing mitochondrial iron accumulation and ROS production.131 The mitochondrial outer membrane proteins voltage-dependent anion channel 2 (VDAC2) and VDAC3 have been identified as direct targets of erastin that modulate mitochondrial function and contribute to ferroptosis.132 However, the contribution of mitochondria to ferroptosis remains controversial and may be context-dependent.133
System xc- is composed of a regulatory subunit solute carrier family 3 member 2 (SLC3A2) and a catalytic subunit solute carrier family 7 member 11 (SLC7A11). This complex promotes the exchange of extracellular cystine and intracellular glutamate across the plasma membrane. Cystine in the cell is reduced to cysteine, which is required for the production of GSH. GPX4 uses GSH to eliminate the production of phospholipid hydroperoxides (PLOOH), the major mediator of chain reactions in lipoxygenases (Fig. 3c). System xc- inhibitors (e.g., erastin, sulfasalazine, sorafenib, and glutamate) are considered as class I ferroptosis inducers (FINs), whereas direct GPX4 inhibitors are referred to class II FINs.134 Of note, GPX4 depletion was also shown to confer sensitivity to apoptosis,135,136 necroptosis,137 and pyroptosis.105 These findings suggest that lipid peroxidation can accelerate an array of distinct RCD modalities.
The likelihood of ferroptosis is determined by the balance between iron accumulation-induced ROS production and the antioxidant system that avoids lipid peroxidation (Fig. 3c). Increased iron uptake by transferrin receptor (TFRC, also known as TFR1) and reduced iron export by ferroportin favor oxidative damage and ferroptosis.138 Lipid peroxidation is influenced by several lipids and enzymes. Thus, the oxidation of PUFAs, including arachidonic acid (AA), by a catalytic pathway involving acyl-CoA synthetase long chain family member 4 (ACSL4), lysophosphatidylcholine acyltransferase 3 (LPCAT3), and arachidonate lipoxygenases (ALOXs, especially ALOX15) is required for lipotoxicity in ferroptosis.121,139,140,141,142 Phosphatidylethanolamine binding protein 1 (PEBP1, also known as RKIP), a scaffold protein inhibitor of protein kinase cascades, is required for the enzymatic activity of ALOX15 in ferroptosis.142 The upregulation of ACSL4, but not other ACSL members, seems to be a marker of ferroptosis.141 In addition to system xc-and GPX4, several integrated antioxidant and pro-survival proteins such as the transcription factor nuclear factor, erythroid 2 like 2 (NFE2L2, also known as NRF2)143 and certain heat shock proteins (HSPs),144,145 can inhibit lipid peroxidation in ferroptosis. In contrast, ROS generated during glutaminase 2 (GLS2)-mediated glutaminolysis may promote ferroptosis.146
NFE2L2 is a key transcription factor that regulates antioxidant defence or detoxification in the context of various stressors. NFE2L2-mediated transactivation of metallothionein 1G (MT1G, a cysteine-rich protein with a high affinity for divalent heavy metal ions), SLC7A11, and heme oxygenase 1 (HMOX1) limits ferroptosis.143,147,148 However, upon excessive activation of NRF2, HMOX1 gets hyperactivated and induces ferroptosis through increasing the labile iron pool upon metabolizing heme.129,149,150 Thus, the protective effect of HMOX1 is attributed to its antioxidant activity, while its toxic effect is mediated through the generation of ferrous iron that might boost Fenton-mediated decomposition of peroxides in case of insufficient buffering capacity by ferritin.
The tumor suppressor tumor protein p53 (TP53)151 and BRCA1-associated protein 1 (BAP1)152 can promote ferroptosis through the downregulation of SLC7A11 via transcriptional and epigenetic mechanisms, respectively. TP53 may also suppress ferroptosis by directly inhibiting the enzymatic activity of membrane-bound glycoprotein dipeptidyl peptidase 4 (DPP4, also known as CD26)153 or by increasing the expression of cell-cycle regulator cyclin-dependent kinase inhibitor 1A (CDKN1A, also known as p21).154 This has been observed in some cancers, in particular colorectal carcinoma, suggesting a context-dependent role of TP53 in the regulation of ferroptosis.155 An African-specific coding region variant of TP53, namely Pro47Ser, also affects ferroptosis senstivity and tumor supression.156
HSPs are a family of highly conserved molecular chaperones that are expressed in response to environmental stresses and render cells resistant to different types of cell death, including ferroptosis. In particular, heat shock protein family B [small] member (HSPB1, also known as HSP25 or HSP27)-mediated actin cytoskeleton protection inhibits ferroptosis via reducing iron uptake and subsequent oxidative injury.144 Heat shock protein family A [Hsp70] member 5 (HSPA5, also known as BIP or GRP78), an endoplasmic reticulum (ER)-sessile chaperone, binds and stabilizes GPX4, thus indirectly counteracting lipid peroxidation in ferroptosis.145 However, 2-amino-5-chloro-N,3-dimethylbenzamide (CDDO), an HSP90 inhibitor, can inhibit ferroptosis in cancer cells, indicating that HSP90 may play a different role in ferroptosis.157
The term “autophagy-dependent cell death” was originally used to describe cell death associated with autophagy based on morphological observation.14 It is now defined by the NCCD as a type of RCD that can be blocked by the suppression of autophagy.24 Recent findings indicate that ferroptosis induction is coupled to an increase in the turnover of lipidated microtubule-associated protein 1 light chain 3 beta (MAP1LC3B, also known as LC3, a marker of autophagosome) as well as the fusion of the autophagosome with lysosomes (namely, autolysosome formation, an important stage of autophagic flux), consistent with the notion that lipid oxidation stimulates autophagy.158,159 The genetic depletion of core autophagy effector molecules such as autophagy-related 5 (ATG5) and ATG7 block cell death by ferroptosis.158,159 Tat-Beclin 1, a strong direct inducer of autophagy, also enhances ferroptosis in cancer cells.160 The molecular mechanisms through which autophagy may contribute to ferroptotic demise may involve multiple pathways,161 such as the degradation of ferritin via nuclear receptor coactivator 4 (NCOA4)-dependent ferritinophagy (e.g., ferritin-specific autophagy),158,159 the inhibition of system xc- activity via the formation of a BECN1-SLC7A11 protein complex,160 and the degradation of lipid droplets via ras-associated protein RAB7 (RAB7A)-dependent lipophagy.162 In addition, chaperone-mediated autophagy promotes GPX4 degradation and subsequent ferroptosis.157
In summary, ferroptosis is an non-apoptotic form of RCD driven by iron accumulation and lipid peroxidation, which can also involve autophagic processes, depending on the trigger.163 Excessive ferroptosis is likely to occur in certain human diseases, especially neurodegenerative and iron overload disorders, calling for its therapeutic suppression.164 In contrast, the induction of ferroptosis constitutes a potential strategy in cancer therapy.164 Note that almost 30 years ago a calcium-dependent non-apoptotic form of neuronal cell death, glutamate-induced toxicity, was coined as oxytosis that could be initiated by system xc- inhibition and GSH depletion,165,166 and it was recently suggested that oxytosis and ferroptosis should be regarded as the same or at least a highly overlapping cell death pathway.167 That said, it remains to be determined whether ferroptosis is involved in “normal” physiology (e.g., development) or whether it only occurs in the context of pathologogical distortions (e.g., tissue injury) or pharmacological manipulations (e.g., anticancer therapy). Further evidence is required to understand this point. This general caveat applies to all modalities of RCD that are discussed below.
Parthanatos
Parthanatos is a poly [ADP-Ribose] polymerase 1 (PARP1)-dependent RCD that is activated by oxidative stress-induced DNA damage and chromatinolysis (Fig. 3d). The term was coined by Valina and Ted Dawson in 2009.168 Unlike apoptosis, parthanatotic cell death occurs without the formation of an apoptotic body and small-size DNA fragments.169 Parthanatos also occurs in the absence of cell swelling, but is accompanied by plasma membrane rupture.170 PARP1 is a chromatin-associated nuclear protein that plays a critical role in the repair of DNA single-strand or double-strand breaks. PARP1 can recognize DNA breaks and use nicotinamide adenine dinucleotide (NAD+) and ATP to trigger poly (ADP-ribose)-sylation. The cleavage-mediated inactivation of PARP1 by caspases has been considered as a marker of apoptotic cell death.171,172 In contrast, 8-oxo-7,8-dihydroguanine, the common DNA base modification resulting from oxidative injury (e.g., ROS, ultraviolet light, and alkylating agents), triggers PARP1 hyperactivation.173 Hyperactivated PARP1 mediates parthanatos through at least two mechanisms, namely, the depletion of NAD+ and ATP (as it occurs during necrosis) and the dissipation of the mitochondrial inner transmembrane potential (an event commonly associated with apoptosis).174
Mechanistically, apoptosis-inducing factor mitochondria-associated 1 (AIFM1, also known as AIF),175 but not caspases and apoptotic DNase endonuclease G (ENDOG), is required for parthanatos execution.176 Hyperactive PARP1 binds AIFM1, which leads to AIFM1 release from mitochondria into the nucleus to produce parthanatotic chromatinolysis.177 This process can be negatively controlled by blocking PARP1 activity via the poly (ADP-ribose)-degrading protein ADP-ribosylhydrolase-like 2 (ADPRHL2, also known as ARH3)178 and the poly (ADP-ribose)-binding protein ring finger protein 146 (RNF146, also known as IDUNA),179 whereas it is positively regulated by enhancing PARP1 activity by the DNA glycosylase enzyme 8-oxoguanine DNA glycosylase (OGG1).173 More recently, macrophage migration inhibitory factor (MIF) has been identified as an AIFM1-binding protein with nuclease activity to produce large DNA fragments in the induction of parthanatos.180 AIFM1-independent parthanatos may also occur in some conditions such as dry macular degeneration.181 The interplay between AIFM1-dependent and -independent parthanatos with other RCDs such as autophagy-dependent cell death182 and necroptosis183 may be involved in various types of oxidative DNA damage-associated diseases, including neurodegenerative disease, myocardial infarction, and diabetes.
Entotic cell death
Entotic cell death is a form of cell cannibalism in which one cell engulfs and kills another cell. The term entosis was coined in 2007 by Joan Brugge.184 Entosis and entotic cell death occur mostly in epithelial tumor cells in the contexts of aberrant proliferation,184 glucose starvation,185 matrix deadhesion,184 or mitotic stress.186 Entosis is characterized by the formation of cell-in-cell structures,184 which have also been observed in the urine and ascites from tumor patients.187 Entosis plays an ambiguous role in tumorigenesis, since it may trigger aneuploidy in engulfing cells188 and provide nutritional support for tumor growth,185 but may also mediate the removal of cancer cells by healthy neighbouring cells.186
Although their underlying mechanisms are not well-understood, cell adhesion and cytoskeletal rearrangement pathways play a central role in the control of entosis induction.189,190 The invasion of a live cell into a neighbouring cell during entosis requires the formation of adherent junctions, which is mediated by adhesion proteins cadherin 1 (CDH1, also known as E-cadherin) and catenin alpha 1 (CTNNA1), but not integrin receptors.184,191,192 Both intact actin and microtubules are required for cytoskeletal rearrangement during entosis. In particular, actomyosin, the actin-myosin complex in the cytoskeleton, is essential for the formation of cell-in-cell structures in entosis. The generation and activity of actomyosin is spatiotemporally controlled by ras homolog family member A (RHOA), rho-associated coiled-coil containing protein kinase (ROCK), and the myosin pathway184,185,192,193,194,195,196 (Fig. 3e). Consequently, pharmacologically targeting these core pathways by inhibitors such as C3-toxin, Y-27632, and blebbistatin diminishes entosis in vitro and in vivo.189
In addition to cell adhesion and cytoskeletal rearrangement pathways, other signaling molecules and regulators are also involved in the regulation of entosis through different mechanisms.189 For example, cell division cycle 42 (CDC42) depletion enhances changes in mitotic morphology and subsequent entosis.186 AURKA197 and the AMP-activated protein kinase (AMPK)185 promote entosis through the control of microtubule plasticity or energy metabolism, respectively. The chromatin-binding protein nuclear protein 1 (NUPR1, also known as P8), a transcriptional regulator, negatively regulates entosis through modulating AURKA activity or autophagy.198
The possible fates of the engulfed cells include cell division, release, or death. Entotic cell death involves the digestion of the engulfed cells by the host cells, which requires LC3-associated phagocytosis (LAP) and the cathepsin B (CTSB)-mediated lysosomal degradation pathway189,190 (Fig. 3e). However, entosis does not involve apoptosis effector caspases and is not regulated by proteins of the BCL2 family.189,199 LAP bridges phagocytosis and autophagy; this process is regulated by the core LC3 lipidation machinery (e.g., ATG5, ATG7, class III phosphatidylinositol 3-kinase complex [e.g., phosphatidylinositol 3-kinase catalytic subunit type 3 (PI3KC3), also known as VPS34], phosphoinositide-3-kinase regulatory subunit 4 [PIK3R4, also known as VPS15], and BECN1), cytochrome B-245 beta chain (CYBB, also known as NOX2)-mediated ROS production, other autophagy regulators (e.g., UV radiation resistance-associated [UVRAG] and RUN domain and cysteine-rich domain-containing beclin 1 interacting protein [RUBCN, also known as Rubicon]).200 Entosis is often observed in neoplasia and its frequency correlates with tumor stage, calling for a further in-depth evaluation of the possibility of targeting this phenomenon. However, at this stage, there are no reagents available that would allow us to inhibit or induce entosis in a selective fashion, i.e., without influencing other cell death modalities.
Netotic cell death
Netotic cell death is a form of RCD driven by NET release. NETs are extracellular net-like DNA-protein structures released by cells in response to infection or injury. NET formation and release, or NETosis, was first observed in neutrophils upon exposure to phorbol myristate acetate or IL8 by Arturo Zychlinsky’s lab in 2004.201 NETs can also be generated by other leukocyte populations (e.g., mast cells, eosinophils, and basophils), epithelial cells, and cancer cells in response to various stresses.202,203,204 Elevated NETosis not only acts against the spread of infection by trapping pathogenic microorganisms (e.g., bacteria and viruses), but also promotes DAMP release, thus possibly contributing to the pathogenesis of autoimmune disorders (e.g., systemic lupus erythematosus, rheumatoid arthritis, asthma, vessel vasculitis, and psoriasis), ischemia-reperfusion injury, and tumor development.205 A recent study indicates that inflammation-associated NET production can awaken nearby dormant cancer cells to redivide.206 This effect may rely on the degradation of laminins, a major adhesive component of basement membranes206; however, this needs further mechanistic exploration.
NETosis is a dynamic process and relies on multiple signals and steps including NADPH oxidase-mediated ROS production, autophagy, the release and translocation of granular enzymes (e.g., elastase, neutrophil expressed [ELANE], matrix metalloproteinase [MMP], and myeloperoxidase [MPO]) and peptides from the cathelecidin family (e.g., cathelicidin antimicrobial peptide [CAMP, also known as LL37]) from the cytosol to the nucleus.207,208 This is followed by histone citrullination, favoring chromatin decondensation, the destruction of the nuclear envelope, and the release of chromatin fibres209 (Fig. 3f). Peptidyl arginine deiminase 4 (PADI4, also known as PAD4) is the enzyme responsible for catalyzing the conversion of arginine to citrullin residues in histones.210 A recently discovered pathway of PADI4-independent NETosis211 may occur downstream of death signals that are normally involved in other types of RCD such as pyroptosis, necroptosis, and autophagy-dependent cell death.212 The alterations of cell surface associated to netotic cell death are initiated by entropic swelling of chromatin through a yet elusive mechanism.213 Of note, lactoferrin, a component of neutrophil granules, can block netotic cell death and inflammation both in vitro and in vivo.214 In addition to the key role of GSDMD in pyroptosis, GSDMD is also involved in the induction of NETosis to digest the pathogen,112,113 indicating a crosstalk between pyroptosis and NETosis pathways in the innate immunity.
Lysosome-dependent cell death
Lysosome-dependent cell death (LCD), also known as lysosomal cell death, is a type of RCD mediated by hydrolytic enzymes that are released into the cytosol after lysosomal membrane permeabilization (Fig. 3g).215 The idea of LCD was first expressed by Christian de Duve, who discovered lysosomes as the cellular degradation machinery in 1955, and the term “lysosomal cell death” was coined in 2000.216 Lysosomes are acidic cellular organelles that can degrade a variety of heterophagic and autophagic cargos, including unused intracellular macromolecules (nucleic acids, proteins, lipids, and carbohydrates), entire organelles (e.g., mitochondria), and invading pathogens.
Lysosomes become leaky when cells are exposed to lysosomotropic detergents (e.g., O-methyl-serine dodecylamide hydrochloride), dipeptide methyl esters (e.g., Leu-Leu-OMe), lipid metabolites (e.g., sphingosine and phosphatidic acid), and ROS.215 Lysosomal membrane permeabilization may also amplify or initiate cell death signaling in the context of apoptosis, autophagy-dependent cell death, and ferroptosis.217,218
Among lysosomal hydrolases, cathepsins (a large family of cysteine peptidases) play a major role in LCD. Different cathepsins are responsible for the initiation and execution of LCD, depending on the context of lysosomal membrane permeabilization.219 The transcription factors signal transducer and activator of transcription 3 (STAT3)220 and TP53221 may favor LCD induction through the selective upregulation of cathepsins (e.g., CTSB, cathepsin L [CTSL] and cathepsin D [CTSD]) expression. In contrast, the NF-κB–elicited expression of serpin family A member 3 (SERPINA3) results in the inhibition of CTSB and CTSL.222 Moreover, the suppression of mucolipin 1, an ion channel in the lysosome (MCOLN1, also known as TRPML1) results in a lysosomal trafficking defect, which promotes CTSB release and consequent LCD.223
Blocking cathepsin expression or activity can block LCD. However, cathepsins are not the sole effectors of LCD because lysosomes store abundant iron, meaning that lysosomal membrane permeabilization can result in the release of this toxic metal into the cytosol,224 thus contributing to ferroptosis.218,225 Impaired lysosomal degradation and the LCD pathway are associated with increased oxidative injury and contribute to lysosomal storage disorders and age-associated diseases.226,227 Based on the cellular context, LCD can adopt necrotic, apoptotic, autophagic, or ferroptotic-like features, adding complexity to this cell death pathway.215,217,218
Autophagy-dependent cell death
Autophagy-dependent cell death is a type of RCD driven by the molecular machinery of autophagy (Fig. 3h). Macroautophagy (hereafter called “autophagy”) is an evolutionarily conserved degradation pathway and has been implicated in human disease and aging.228,229 The process of autophagy involves the sequential formation of three unique membrane structures, namely the phagophore, autophagosome, and autolysosome. Over 40 autophagy-related genes/proteins (ATGs) play key roles in autophagic membrane dynamics and processes.230
As a dynamic recycling system, the bulk and nonselective autophagy process is generally considered as a pro-survival mechanism in response to multiple types of cellular stresses.16 Nevertheless, autophagy can selectively degrade pro-survival proteins related to other types of RCD, thereby tipping the balance from life to death.231,232,233,234 Ferritinophagy causes ferroptosis due to the selective degradation of ferritin (an iron storage protein), consequently causing iron release and oxidative injury.158,159 The degradation of protein tyrosine phosphatase, nonreceptor type 13 (PTPN13, a negative regulator of extrinsic apoptosis)235 favors apoptosis, while autophagic digestion of baculoregulator repeat containing 2 (BIRC2, also known as cIAP1, a negative regulator of necroptosis)236 facilitates the ignition of necroptosis.
In 2013, Beth Levine described “autosis” as a subtype of autophagy-dependent cell death induced by nutrient deprivation or by Tat-Beclin 1, an autophagy-inducing peptide fusing amino acids from BECN1 and HIV Tat protein.17 Autosis is morphologically characterized by enhanced cell-substrate adherence, fragmented or vanished ER structure, focal swelling of the perinuclear space, and mild chromatin condensation.17 At the molecular level, Tat-Beclin 1–induced autosis can be inhibited by blocking upstream Na+/K+-ATPase, a plasma pump linking ion homeostasis and ER stress.17 Interestingly, iron overload stimulates Na+/K+-ATPase activity in the human erythrocyte membrane,237 which may lead to ferroptosis. However, the exact relationship between autosis and ferroptosis remains to be determined.
Autophagy-dependent cell death probably plays a pathogenic role in neurotoxicity and hypoxia-ischemia-induced neuronal death,232,233,234 indicating that this type of RCD can possibly be targeted for neuroprotection.
Alkaliptosis
Alkaliptosis is a novel type of RCD driven by intracellular alkalinisation.238 The word “alkaliptosis” was termed in 2018 by our group.238 A screen of small-molecule compound library targeting G-protein coupled receptors (GPCR) for cytotoxic activity on a human pancreatic cancer cell line led to the idenfication of JTC801. The latter is an opioid analgesic drug that efficiently kills a panel of human pancreas, kidney, prostate, skin, and brain cancer cell lines,238 and these cytotoxic effects were not related to apoptosis, necroptosis, autophagy, or ferroptosis, because genetically or pharmacologically blocking these forms of RCD failed to reverse JTC801-induced cell death.238 In contrast, the inhibition of intracellular alkalinization by N-acetyl cysteine, N-acetyl alanine acid, and acidic culture media blocked JTC801-induced cell death.238 At the molecular levels, alkaliptosis requires inhibitor of nuclear factor kappa B kinase subunit beta (IKBKB, also known as IKKβ)-NF-κB pathway-dependent downregulation of carbonic anhydrase 9 (CA9), an enzyme participating in pH regulation (Fig. 3i). Opioid-related nociceptin receptor 1 (OPRL1), the target that accounts for the analgesic activity of JTC801, is dispensable for alkaliptosis.238 Of note, JTC801 has also been reported to induce apoptosis in human osteosarcoma cells,239 suggesting that the type of RCD triggered by JTC801 depends on the cellular context.
It should be noted that the pathological significance of alkaliptosis in human disease remains fully elusive although metabolic alkalosis is a unique acid-base disorder with kidney or lung injury.240 The significance of the core effector molecules of alkaliptosis also remains unclear.
Oxeiptosis
Oxeiptosis is a novel oxygen radical-induced caspase-independent RCD driven by the activation of the KEAP1-PGAM5-AIFM1 pathway (Fig. 3j).241 This term was introduced in 2018 by Andreas Pichlmair’s lab in a study reporting on the response of mice to ozone and that of cultured fibroblasts and epithelial cells to hydrogen peroxide (H2O2).241 Ozone- or H2O2-induced oxeiptosis is independent of apoptotic or pyroptotic caspases, necroptosis, autophagy, and ferroptosis.241 The KEAP1-NFE2L2 pathway has been known to mediate cytoprotective responses to oxidative injury.241 However, hyperactivated KEAP1 can mediate H2O2-induced oxeiptosis in an NFE2L2-independent manner,241 through a pathway that involves KEAP1 interaction partner PGAM5, a mitochondrial serine-threonine phosphatase that dephosphorylates AIFM1 at Ser116.241 Unlike AIFM1-mediated caspase-independent apoptosis and parthanatos, dephosphorylated AIFM1-mediated oxeiptosis does not require the translocation of AIFM1 from mitochondria to the nucleus.241 In vivo, Pgam5–/– mice are more sensitive to inflammation and injury following ozone treatment or viral infection, indicating that oxeiptosis may suppress inflammation.241 However, it remains an open conundrum how H2O2 may induce so many different cell death modalities including oxeiptosis,241 apoptosis,242 necrosis,242 and ferroptosis.243 Understanding the location- and modification-dependent role of AIFM1 may help us to distinguish these different types of RCD. The role of oxeiptosis in pathological cell death in human diseases also remains largely unknown.
Immunological consequences of cell death
Cell death induced by stimuli may occur in a way that the immune system is alerted, triggering immunity against dead-cell antigens. This “immunogenic cell death” (ICD), a term coined in 2005,244 contrasts with silent efferocytosis, in which dying and dead cells are cleared by phagocytosis without any inflammatory or immune reaction, as well as with tolerogenic cell death (TCD) that actively inhibits immune responses.245 Although apoptosis has generally been considered as a TCD, accumulating evidence suggests that apoptosis can be an ICD when induced under certain conditions.246 An acute or chronic inflammatory response elicited by dying cells not only promotes tissue regeneration and limits infection, but may also cause tissue injury and disease.247 Given the fundamental role of inflammation in a variety of human diseases, it is important to understand the key mediators and pathways that drive this response.
There is no doubt that the release of DAMPs by dead or dying cells is an important factor regulating the balance between ICD and TCD. The immune system can recognize two types of danger signals.248 PAMPs from microbes are recognized by pattern recognition receptors (PRRs). Endogenous DAMPs, which act on the same PRRs as the PAMPs, can be proteins (e.g., HMGB1, histones, and transcription factor A, mitochondrial [TFAM]) and nonproteaceous entities (e.g., DNA, RNA, and extracellular ATP). The release of DAMPs is a hallmark of various types of cell death, although they may exhibit distinct expression profiles in response to different stimuli.249 DAMPs activate different PRRs, such as TLRs, advanced glycosylation end-product specific receptor (AGER, also known as RAGE), and DNA sensors (Box 4) that are widely expressed in leukocytes and other cell types.248 A number of inflammation-related pathways, involving for example the RIPK1-NF-κB,250 DNA-TMEM173 (Fig. 4),251 and IL-17A–IL-17R252 pathways have been documented to mediate the ICD-associated immune response. However, another study suggests that the immunogenicity of necroptotic cells does not correlate with the activation of the RIPK3-RIPK1-NF-κB pathway.253

Central role of TMEM173 in inflammation, immunity, and cell death. TMEM173 can be activated by cytosolic DNA sensors (e.g., CGAS, DDX41, MRE11, and IFI16), or cell surface receptors (e.g., ALK and EGFR) in response to various DNAs from the pathogen and host. The activation of STING not only promotes inflammation and the immune response through TBK1-mediated transcription factor activation, but also ignites various cell death pathways, including apoptosis, necroptosis, pyroptosis, and lysosome-dependent cell death
The pathophysiological role of ICD has amply been documented in the context of chemotherapy-induced anticancer immune responses.244 Several cytotoxic antineoplastics stimulate the immune system by stressing and killing cancer cells in a way that results in the exposure of DAMPs such as calreticulin246 at the surface or the release of DAMPs such as ATP,254 annexin A1,255 HMGB1,256 and TFAM257 into the extracellular space. ICD may occur in the context of apoptosis and necroptosis, meaning that the immunological consequences are not tied to the cell death modality itself but rather to the exposure/release of DAMPs that may occur as a consequence of premortem stress responses.247 Thus, calreticulin exposure is tied to a partial endoplasmic reticulum stress response,246 while ATP release occurs through an autophagy-dependent mechanism.254 ICD of malignant cells favors the cross-presentation of tumor-associated antigens by dendritic cells, resulting in the induction of cytotoxic T lymphocytes that then play an essential role in keeping tumors in check.247 In addition to cancer, ICD is also implicated in infectious disease.247
It is interesting to note that the redox status of DAMPs may affect their immune activity as this has been exemplified for HMGB1, a protein that is usually present in the nucleus yet can translocate to the cytoplasm and undergo cell death-associated release. Extracellular HMGB1 remarkably initiates, amplifies, and perpetuates the inflammatory response if it is nonoxidized.258 However, the oxidized form of HMGB1 favors the induction of immune tolerance in antigen-presenting cells259 and may also promote the expression of immune checkpoint molecules (such as CD274, also known as PD-L1) to limit anticancer immunity.260 The proteolytic cleavage or degradation of HMGB1 also limits its immunostimulatory activity under some conditions.261,262 Thus, HMGB1 may act as a tightly controlled universal DAMP that mediates both ICD and TCD depending on its abundance and oxidation status.263
In spite of the wealth of information on the rules governing the immunological consequences of cell death, a systematic exploration of non-apoptotic RCD subroutines with respect to their immunogenicity is still elusive.
Conclusions and perspectives
RCD occurs through a variety of subroutines that cause cells to be dismantled in different ways, hence producing distinct morphological changes and immunological consequences. In spite of this “biodiversity,” the evolutionary relationship between distinct RCD pathways remains unknown. Oxidative stress can lead to various types of RCD, while the sources of ROS as well as the efficacy of antioxidant defences are context-dependent. It will be important to assemble a standard panel of biomarkers and functional tests including genetic and pharmacological inhibition studies to accurately distinguish between different forms of RCD that may occur in a “pure” form or in “mixed” variants, in which distinct lethal subroutines come into action in a parallel and sometimes hierarchized fashion. It is well known that the suppression of apoptosis by caspase inhibition may reveal necroptotic pathways, and similar backup systems might come into action when other cell death modalities are inhibited. It is plausible that RCD does not only play a housekeeping role in maintaining organismal homeostasis, and that it may play a major role in unwarranted cellular demise. The release of DAMPs during RCD provides potent signals to stimulate local inflammatory or systemic immune responses. The development of novel drugs for selectively intercepting (or, in sharp contrast, activating) the RCD pathway holds great promise for preventing and treating human diseases in which cell loss must be avoided (or when the elimination of malignant cells is a therapeutic goal). More research on RCD is needed to define the interplay between distinct cell death signaling pathways, identify unique molecular effectors for each type of RCD, and evaluate pro-survival or reprogramming mechanisms against RCD. In addition, more research is needed to define the critical point of no return of each RCD subroutine and to investigate the role of excessive or deficient RCD in human disease.
References
Kerr, J. F., Wyllie, A. H. & Currie, A. R. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257 (1972).
Singh, R., Letai, A. & Sarosiek, K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 20, 175–193 (2019).
Hengartner, M. O., Ellis, R. E. & Horvitz, H. R. Caenorhabditis elegans gene ced-9 protects cells from programmed cell death. Nature 356, 494–499 (1992).
Hengartner, M. O. & Horvitz, H. R. C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell 76, 665–676 (1994).
Yuan, J. & Horvitz, H. R. The Caenorhabditis elegans cell death gene ced-4 encodes a novel protein and is expressed during the period of extensive programmed cell death. Development 116, 309–320 (1992).
Schulze-Osthoff, K., Ferrari, D., Los, M., Wesselborg, S. & Peter, M. E. Apoptosis signaling by death receptors. Eur. J. Biochem. 254, 439–459 (1998).
Bredesen, D. E., Mehlen, P. & Rabizadeh, S. Apoptosis and dependence receptors: a molecular basis for cellular addiction. Physiol. Rev. 84, 411–430 (2004).
Chipuk, J. E., Bouchier-Hayes, L. & Green, D. R. Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ. 13, 1396–1402 (2006).
Czabotar, P. E., Lessene, G., Strasser, A. & Adams, J. M. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 15, 49–63 (2014).
McIlwain, D. R., Berger, T. & Mak, T. W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. https://doi.org/10.1101/cshperspect.a026716 (2015).
Galluzzi, L., Lopez-Soto, A., Kumar, S. & Kroemer, G. Caspases connect cell-death signaling to organismal homeostasis. Immunity 44, 221–231 (2016).
Linkermann, A., Stockwell, B. R., Krautwald, S. & Anders, H. J. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat. Rev. Immunol. 14, 759–767 (2014).
Vanden Berghe, T., Linkermann, A., Jouan-Lanhouet, S., Walczak, H. & Vandenabeele, P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 15, 135–147 (2014).
Schweichel, J. U. & Merker, H. J. The morphology of various types of cell death in prenatal tissues. Teratology 7, 253–266 (1973).
Kerr, J. F. Shrinkage necrosis: a distinct mode of cellular death. J. Pathol. 105, 13–20 (1971).
Kroemer, G., Marino, G. & Levine, B. Autophagy and the integrated stress response. Mol. Cell 40, 280–293 (2010).
Liu, Y. et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl Acad. Sci. USA 110, 20364–20371 (2013).
Nassour, J. et al. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature https://doi.org/10.1038/s41586-019-0885-0 (2019).
Weinlich, R., Oberst, A., Beere, H. M. & Green, D. R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 18, 127–136 (2017).
Kroemer, G. et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 12, 1463–1467 (2005). Suppl 2.
Kroemer, G. et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 16, 3–11 (2009).
Galluzzi, L. et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 19, 107–120 (2012).
Galluzzi, L. et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 22, 58–73 (2015).
Galluzzi, L. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25, 486–541 (2018).
Pasparakis, M. & Vandenabeele, P. Necroptosis and its role in inflammation. Nature 517, 311–320 (2015).
Ray, C. A. & Pickup, D. J. The mode of death of pig kidney cells infected with cowpox virus is governed by the expression of the crmA gene. Virology 217, 384–391 (1996).
Laster, S. M., Wood, J. G. & Gooding, L. R. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J. Immunol. 141, 2629–2634 (1988).
Holler, N. et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1, 489–495 (2000).
He, S., Liang, Y., Shao, F. & Wang, X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl Acad. Sci. USA 108, 20054–20059 (2011).
Upton, J. W., Kaiser, W. J. & Mocarski, E. S. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host. Microbe. 11, 290–297 (2012).
Schock, S. N. et al. Induction of necroptotic cell death by viral activation of the RIG-I or STING pathway. Cell Death Differ. 24, 615–625 (2017).
Brault, M., Olsen, T. M., Martinez, J., Stetson, D. B. & Oberst, A. Intracellular nucleic acid sensing triggers necroptosis through synergistic type I IFN and TNF signaling. J. Immunol. 200, 2748–2756 (2018).
Chen, D. et al. PUMA amplifies necroptosis signaling by activating cytosolic DNA sensors. Proc. Natl Acad. Sci. USA 115, 3930–3935 (2018).
Wang, X., He, Z., Liu, H., Yousefi, S. & Simon, H. U. Neutrophil necroptosis is triggered by ligation of adhesion molecules following GM-CSF priming. J. Immunol. 197, 4090–4100 (2016).
Vercammen, D. et al. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 187, 1477–1485 (1998).
Degterev, A. et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119 (2005).
Degterev, A. et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321 (2008).
Zhang, D. W. et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336 (2009).
He, S. et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137, 1100–1111 (2009).
Cho, Y. S. et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123 (2009).
Sun, L. et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 (2012).
Zhao, J. et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl Acad. Sci. USA 109, 5322–5327 (2012).
Mompean, M. et al. The structure of the necrosome RIPK1-RIPK3 core, a human hetero-amyloid signaling complex. Cell 173, 1244–1253 e1210 (2018).
Li, J. et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150, 339–350 (2012).
Seo, J. et al. CHIP controls necroptosis through ubiquitylation- and lysosome-dependent degradation of RIPK3. Nat. Cell Biol. 18, 291–302 (2016).
Xie, Y. et al. Inhibition of aurora kinase A induces necroptosis in pancreatic carcinoma. Gastroenterology 153, 1429–1443 e1425 (2017).
Chen, W. et al. Ppm1b negatively regulates necroptosis through dephosphorylating Rip3. Nat. Cell Biol. 17, 434–444 (2015).
Onizawa, M. et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat. Immunol. 16, 618–627 (2015).
Huang, Z. et al. RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host. Microbe. 17, 229–242 (2015).
Thapa, R. J. et al. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc. Natl Acad. Sci. USA 110, E3109–E3118 (2013).
Robinson, N. et al. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat. Immunol. 13, 954–962 (2012).
Wang, H. et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 54, 133–146 (2014).
Dondelinger, Y. et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep 7, 971–981 (2014).
Hildebrand, J. M. et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc. Natl Acad. Sci. USA 111, 15072–15077 (2014).
Murphy, J. M. et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–453 (2013).
Chen, X. et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 24, 105–121 (2014).
Zhao, X. M. et al. Hsp90 modulates the stability of MLKL and is required for TNF-induced necroptosis. Cell Death Dis. 7, e2089 (2016).
Li, D. et al. Natural product kongensin A is a non-canonical HSP90 inhibitor that blocks RIP3-dependent necroptosis. Cell Chem Biol 23, 257–266 (2016).
Jacobsen, A. V. et al. HSP90 activity is required for MLKL oligomerisation and membrane translocation and the induction of necroptotic cell death. Cell Death Dis. 7, e2051 (2016).
Bigenzahn, J. W. et al. An inducible retroviral expression system for tandem affinity purification mass-spectrometry-based proteomics identifies mixed lineage kinase domain-like protein (MLKL) as an heat shock protein 90 (HSP90) client. Mol. Cell. Proteomics. 15, 1139–1150 (2016).
Dovey, C. M. et al. MLKL requires the inositol phosphate code to execute necroptosis. Mol. Cell 70, 936–948 e937 (2018).
Gong, Y. N. et al. ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell 169, 286–300 e216 (2017).
Yoon, S., Kovalenko, A., Bogdanov, K. & Wallach, D. MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity 47, 51–65 e57 (2017).
Vanden Berghe, T. et al. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 17, 922–930 (2010).
Wang, Z., Jiang, H., Chen, S., Du, F. & Wang, X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148, 228–243 (2012).
Zhang, Y. et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 8, 14329 (2017).
Yang, Z. et al. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat. Cell Biol. 20, 186–197 (2018).
Remijsen, Q. et al. Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. Cell Death Dis. 5, e1004 (2014).
Yoon, S., Bogdanov, K., Kovalenko, A. & Wallach, D. Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ. 23, 253–260 (2016).
Weber, K., Roelandt, R., Bruggeman, I., Estornes, Y. & Vandenabeele, P. Nuclear RIPK3 and MLKL contribute to cytosolic necrosome formation and necroptosis. Commun Biol 1, 6 (2018).
Wang, X., Yousefi, S. & Simon, H. U. Necroptosis and neutrophil-associated disorders. Cell Death Dis. 9, 111 (2018).
Newton, K. et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343, 1357–1360 (2014).
Lawlor, K. E. et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 6, 6282 (2015).
Shan, B., Pan, H., Najafov, A. & Yuan, J. Necroptosis in development and diseases. Genes Dev. 32, 327–340 (2018).
Dillon, C. P., Tummers, B., Baran, K. & Green, D. R. Developmental checkpoints guarded by regulated necrosis. Cell. Mol. Life Sci. 73, 2125–2136 (2016).
Galluzzi, L., Kepp, O., Chan, F. K. & Kroemer, G. Necroptosis: mechanisms and relevance to disease. Annu. Rev. Pathol. 12, 103–130 (2017).
Vanden Berghe, T. et al. Passenger mutations confound interpretation of all genetically modified congenic mice. Immunity 43, 200–209 (2015).
Fink, S. L. & Cookson, B. T. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 73, 1907–1916 (2005).
Brennan, M. A. & Cookson, B. T. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol. Microbiol. 38, 31–40 (2000).
Hersh, D. et al. The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc. Natl Acad. Sci. USA 96, 2396–2401 (1999).
Broz, P. & Dixit, V. M. Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 16, 407–420 (2016).
Chen, X. et al. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 26, 1007–1020 (2016).
Rathkey, J. K. et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci. Immunol. https://doi.org/10.1126/sciimmunol.aat2738 (2018).
He, Y., Zeng, M. Y., Yang, D., Motro, B. & Nunez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530, 354–357 (2016).
Fernandes-Alnemri, T. et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 11, 385–393 (2010).
Rathinam, V. A. et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 11, 395–402 (2010).
Kayagaki, N. et al. Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 (2011).
Shi, J. et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192 (2014).
Hagar, J. A., Powell, D. A., Aachoui, Y., Ernst, R. K. & Miao, E. A. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341, 1250–1253 (2013).
Kayagaki, N. et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249 (2013).
Vanaja, S. K. et al. Bacterial outer membrane vesicles mediate cytosolic localization of LPS and caspase-11 activation. Cell 165, 1106–1119 (2016).
Deng, M. et al. The endotoxin delivery protein HMGB1 mediates caspase-11-dependent lethality in sepsis. Immunity 49, 740–753 e747 (2018).
Rathinam, V. A. et al. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150, 606–619 (2012).
Man, S. M. et al. IRGB10 Liberates Bacterial Ligands For Sensing by the AIM2 and Caspase-11-NLRP3 Inflammasomes. Cell 167, 382–396 e317 (2016).
Lu, B. et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 488, 670–674 (2012).
Xie, M. et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat. Commun. 7, 13280 (2016).
Yang, L. et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat. Commun. 5, 4436 (2014).
Moon, J. S. et al. mTORC1-Induced HK1-dependent glycolysis regulates NLRP3 inflammasome activation. Cell Rep 12, 102–115 (2015).
Ding, J. et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116 (2016).
Liu, X. et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158 (2016).
Kayagaki, N. et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015).
Shi, J. et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015).
He, W. T. et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 25, 1285–1298 (2015).
Lee, B. L. et al. Caspase-11 auto-proteolysis is crucial for noncanonical inflammasome activation. J. Exp. Med. 215, 2279–2288 (2018).
Kang, R. et al. Lipid peroxidation drives gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host. Microbe. 24, 97–108 e104 (2018).
Chen, R. et al. cAMP metabolism controls caspase-11 inflammasome activation and pyroptosis in sepsis. Sci. Adv. https://doi.org/10.1126/sciadv.1601167 (2019).
Ruhl, S. et al. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362, 956–960 (2018).
Orning, P. et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362, 1064–1069 (2018).
Sarhan, J. et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl Acad. Sci. USA 115, E10888–E10897 (2018).
Wang, Y. et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103 (2017).
Kambara, H. et al. Gasdermin D exerts anti-inflammatory effects by promoting neutrophil death. Cell Rep 22, 2924–2936 (2018).
Sollberger, G. et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. https://doi.org/10.1126/sciimmunol.aar6689 (2018).
Chen, K. W. et al. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci. Immunol. https://doi.org/10.1126/sciimmunol.aar6676 (2018).
Evavold, C. L. et al. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity 48, 35–44 e36 (2018).
de Vasconcelos, N. M., Van Opdenbosch, N., Van Gorp, H., Parthoens, E. & Lamkanfi, M. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ. https://doi.org/10.1038/s41418-018-0106-7 (2018).
Dolma, S., Lessnick, S. L., Hahn, W. C. & Stockwell, B. R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 3, 285–296 (2003).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Friedmann Angeli, J. P. et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180–1191 (2014).
Neitemeier, S. et al. BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. 12, 558–570 (2017).
Hong, S. H. et al. Molecular crosstalk between ferroptosis and apoptosis: emerging role of ER stress-induced p53-independent PUMA expression. Oncotarget 8, 115164–115178 (2017).
Yang, W. S. et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl Acad. Sci. USA 113, E4966–E4975 (2016).
Feng, H. & Stockwell, B. R. Unsolved mysteries: how does lipid peroxidation cause ferroptosis? PLoS Biol. 16, e2006203 (2018).
Hayano, M., Yang, W. S., Corn, C. K., Pagano, N. C. & Stockwell, B. R. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 23, 270–278 (2016).
Yang, W. S. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 (2014).
Woo, J. H. et al. Elucidating compound mechanism of action by network perturbation analysis. Cell 162, 441–451 (2015).
Weiwer, M. et al. Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg. Med. Chem. Lett. 22, 1822–1826 (2012).
Cao, J. Y. & Dixon, S. J. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 73, 2195–2209 (2016).
Gaschler, M. M. et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 14, 507–515 (2018).
Hassannia, B. et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Invest. 128, 3341–3355 (2018).
Li, Q. et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2, e90777 (2017).
Yuan, H., Li, X., Zhang, X., Kang, R. & Tang, D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem. Biophys. Res. Commun. 478, 838–844 (2016).
Yagoda, N. et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868 (2007).
Gao, M. et al. Role of mitochondria in ferroptosis. Mol. Cell 73, 354–363 e353 (2019).
Xie, Y. et al. Ferroptosis: process and function. Cell Death Differ. 23, 369–379 (2016).
Seiler, A. et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 8, 237–248 (2008).
Ran, Q. et al. Embryonic fibroblasts from Gpx4+/- mice: a novel model for studying the role of membrane peroxidation in biological processes. Free Radic. Biol. Med. 35, 1101–1109 (2003).
Canli, O. et al. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood 127, 139–148 (2016).
Yang, W. S. & Stockwell, B. R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15, 234–245 (2008).
Doll, S. et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98 (2017).
Kagan, V. E. et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90 (2017).
Yuan, H., Li, X., Zhang, X., Kang, R. & Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 478, 1338–1343 (2016).
Wenzel, S. E. et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell 171, 628–641 e626 (2017).
Sun, X. et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63, 173–184 (2016).
Sun, X. et al. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 34, 5617–5625 (2015).
Zhu, S. et al. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. 77, 2064–2077 (2017).
Gao, M., Monian, P., Quadri, N., Ramasamy, R. & Jiang, X. Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 59, 298–308 (2015).
Sun, X. et al. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology 64, 488–500 (2016).
Chen, D. et al. NRF2 is a major target of ARF in p53-independent tumor suppression. Mol. Cell 68, 224–232 e224 (2017).
Chang, L. C. et al. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 416, 124–137 (2018).
Kwon, M. Y., Park, E., Lee, S. J. & Chung, S. W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 6, 24393–24403 (2015).
Jiang, L. et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62 (2015).
Zhang, Y. et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat. Cell Biol. https://doi.org/10.1038/s41556-018-0178-0 (2018).
Xie, Y. et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep 20, 1692–1704 (2017).
Tarangelo, A. et al. p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep 22, 569–575 (2018).
Kang, R., Kroemer, G. & Tang, D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic. Biol. Med. https://doi.org/10.1016/j.freeradbiomed.2018.05.074 (2018).
Jennis, M. et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. 30, 918–930 (2016).
Wu, Z. et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc. Natl Acad. Sci. USA 116, 2996–3005 (2019).
Gao, M. et al. Ferroptosis is an autophagic cell death process. Cell Res. 26, 1021–1032 (2016).
Hou, W. et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 12, 1425–1428 (2016).
Song, X. et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc(-) activity. Curr. Biol. 28, 2388–2399 e2385 (2018).
Zhou, B. et al. Ferroptosis is a type of autophagy-dependent cell death. Sem. Cancer Biol. pii: S1044-579X(19)30006-9. https://doi.org/10.1016/j.semcancer.2019.03.002 (2019).
Bai, Y. et al. Lipid storage and lipophagy regulates ferroptosis. Biochem. Biophys. Res. Commun. 508, 997–1003 (2019).
Kang, R. & Tang, D. Autophagy and ferroptosis - what’s the connection? Curr Pathobiol Rep 5, 153–159 (2017).
Stockwell, B. R. et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285 (2017).
Tan, S., Schubert, D. & Maher, P. Oxytosis: a novel form of programmed cell death. Curr. Top. Med. Chem. 1, 497–506 (2001).
Murphy, T. H., Malouf, A. T., Sastre, A., Schnaar, R. L. & Coyle, J. T. Calcium-dependent glutamate cytotoxicity in a neuronal cell line. Brain Res. 444, 325–332 (1988).
Lewerenz, J., Ates, G., Methner, A., Conrad, M. & Maher, P. Oxytosis/ferroptosis-(Re-) emerging roles for oxidative stress-dependent non-apoptotic cell death in diseases of the central nervous system. Front. Neurosci. 12, 214 (2018).
David, K. K., Andrabi, S. A., Dawson, T. M. & Dawson, V. L. Parthanatos, a messenger of death. Front. Biosci. (Landmark Ed) 14, 1116–1128 (2009).
Delettre, C. et al. AIFsh, a novel apoptosis-inducing factor (AIF) pro-apoptotic isoform with potential pathological relevance in human cancer. J. Biol. Chem. 281, 6413–6427 (2006).
Wang, H. et al. Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J. Neurosci. 24, 10963–10973 (2004).
Nicholson, D. W. et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 376, 37–43 (1995).
Tewari, M. et al. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell 81, 801–809 (1995).
Wang, R. et al. OGG1-initiated base excision repair exacerbates oxidative stress-induced parthanatos. Cell Death Dis. 9, 628 (2018).
Andrabi, S. A., Dawson, T. M. & Dawson, V. L. Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann. N. Y. Acad. Sci. 1147, 233–241 (2008).
Susin, S. A. et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397, 441–446 (1999).
Yu, S. W. et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297, 259–263 (2002).
Wang, Y. et al. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci. Signal. 4, ra20 (2011).
Mashimo, M., Kato, J. & Moss, J. ADP-ribosyl-acceptor hydrolase 3 regulates poly (ADP-ribose) degradation and cell death during oxidative stress. Proc. Natl Acad. Sci. USA 110, 18964–18969 (2013).
Andrabi, S. A. et al. Iduna protects the brain from glutamate excitotoxicity and stroke by interfering with poly(ADP-ribose) polymer-induced cell death. Nat. Med. 17, 692–699 (2011).
Wang, Y. et al. A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science https://doi.org/10.1126/science.aad6872 (2016).
Jang, K. H. et al. AIF-independent parthanatos in the pathogenesis of dry age-related macular degeneration. Cell Death Dis. 8, e2526 (2017).
Rodriguez-Vargas, J. M. et al. ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res. 22, 1181–1198 (2012).
Xu, X. et al. The role of PARP activation in glutamate-induced necroptosis in HT-22 cells. Brain Res. 1343, 206–212 (2010).
Overholtzer, M. et al. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell 131, 966–979 (2007).
Hamann, J. C. et al. Entosis Is Induced by Glucose Starvation. Cell Rep 20, 201–210 (2017).
Durgan, J. et al. Mitosis can drive cell cannibalism through entosis. Elife https://doi.org/10.7554/eLife.27134 (2017).
Brouwer, M., de Ley, L., Feltkamp, C. A., Elema, J. & Jongsma, A. P. Serum-dependent “cannibalism” and autodestruction in cultures of human small cell carcinoma of the lung. Cancer Res. 44, 2947–2951 (1984).
Krajcovic, M. et al. A non-genetic route to aneuploidy in human cancers. Nat. Cell Biol. 13, 324–330 (2011).
Durgan, J. & Florey, O. Cancer cell cannibalism: multiple triggers emerge for entosis. Biochim. Biophys. Acta Mol. Cell Res. 1865, 831–841 (2018).
Martins, I. et al. Entosis: the emerging face of non-cell-autonomous type IV programmed death. Biomed J 40, 133–140 (2017).
Wang, M. et al. Impaired formation of homotypic cell-in-cell structures in human tumor cells lacking alpha-catenin expression. Sci. Rep. 5, 12223 (2015).
Sun, Q., Cibas, E. S., Huang, H., Hodgson, L. & Overholtzer, M. Induction of entosis by epithelial cadherin expression. Cell Res. 24, 1288–1298 (2014).
Sottile, F., Aulicino, F., Theka, I. & Cosma, M. P. Mesenchymal stem cells generate distinct functional hybrids in vitro via cell fusion or entosis. Sci. Rep. 6, 36863 (2016).
Wen, S., Shang, Z., Zhu, S., Chang, C. & Niu, Y. Androgen receptor enhances entosis, a non-apoptotic cell death, through modulation of Rho/ROCK pathway in prostate cancer cells. Prostate 73, 1306–1315 (2013).
Sun, Q. et al. Competition between human cells by entosis. Cell Res. 24, 1299–1310 (2014).
Wan, Q. et al. Regulation of myosin activation during cell-cell contact formation by Par3-Lgl antagonism: entosis without matrix detachment. Mol. Biol. Cell. 23, 2076–2091 (2012).
Xia, P. et al. Aurora A orchestrates entosis by regulating a dynamic MCAK-TIP150 interaction. J. Mol. Cell Biol. 6, 240–254 (2014).
Hinojosa, L. S., Holst, M., Baarlink, C. & Grosse, R. MRTF transcription and Ezrin-dependent plasma membrane blebbing are required for entotic invasion. J. Cell. Biol. 216, 3087–3095 (2017).
Li, Y., Sun, X. & Dey, S. K. Entosis allows timely elimination of the luminal epithelial barrier for embryo implantation. Cell Rep 11, 358–365 (2015).
Heckmann, B. L., Boada-Romero, E., Cunha, L. D., Magne, J. & Green, D. R. LC3-associated phagocytosis and inflammation. J. Mol. Biol. 429, 3561–3576 (2017).
Brinkmann, V. et al. Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535 (2004).
Arazna, M., Pruchniak, M. P. & Demkow, U. Reactive Oxygen Species, Granulocytes, and NETosis. Adv. Exp. Med. Biol. 836, 1–7 (2015).
Kazzaz, N. M., Sule, G. & Knight, J. S. Intercellular interactions as regulators of NETosis. Front. Immunol. 7, 453 (2016).
Remijsen, Q. et al. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. 18, 581–588 (2011).
Branzk, N. & Papayannopoulos, V. Molecular mechanisms regulating NETosis in infection and disease. Semin. Immunopathol. 35, 513–530 (2013).
Albrengues, J. et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science https://doi.org/10.1126/science.aao4227 (2018).
Skendros, P., Mitroulis, I. & Ritis, K. Autophagy in neutrophils: from granulopoiesis to neutrophil extracellular traps. Front. Cell. Dev. Biol. 6, 109 (2018).
Remijsen, Q. et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 21, 290–304 (2011).
Yipp, B. G. et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 18, 1386–1393 (2012).
Li, P. et al. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 207, 1853–1862 (2010).
Hemmers, S., Teijaro, J. R., Arandjelovic, S. & Mowen, K. A. PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS ONE 6, e22043 (2011).
Mitroulis, I. et al. Neutrophil extracellular trap formation is associated with IL-1beta and autophagy-related signaling in gout. PLoS ONE 6, e29318 (2011).
Neubert, E. et al. Chromatin swelling drives neutrophil extracellular trap release. Nat. Commun. 9, 3767 (2018).
Okubo, K. et al. Lactoferrin suppresses neutrophil extracellular traps release in inflammation. EBioMedicine 10, 204–215 (2016).
Aits, S. & Jaattela, M. Lysosomal cell death at a glance. J. Cell. Sci. 126, 1905–1912 (2013).
Franko, J., Pomfy, M. & Prosbova, T. Apoptosis and cell death (mechanisms, pharmacology and promise for the future). Acta Medica (Hradec. Kralove) 43, 63–68 (2000).
Kroemer, G. & Jaattela, M. Lysosomes and autophagy in cell death control. Nat. Rev. Cancer 5, 886–897 (2005).
Gao, H. et al. Ferroptosis is a lysosomal cell death process. Biochem. Biophys. Res. Commun. 503, 1550–1556 (2018).
Repnik, U., Stoka, V., Turk, V. & Turk, B. Lysosomes and lysosomal cathepsins in cell death. Biochim. Biophys. Acta 1824, 22–33 (2012).
Kreuzaler, P. A. et al. Stat3 controls lysosomal-mediated cell death in vivo. Nat. Cell Biol. 13, 303–309 (2011).
Wu, G. S., Saftig, P., Peters, C. & El-Deiry, W. S. Potential role for cathepsin D in p53-dependent tumor suppression and chemosensitivity. Oncogene 16, 2177–2183 (1998).
Liu, N. et al. NF-kappaB protects from the lysosomal pathway of cell death. EMBO J. 22, 5313–5322 (2003).
Colletti, G. A. et al. Loss of lysosomal ion channel transient receptor potential channel mucolipin-1 (TRPML1) leads to cathepsin B-dependent apoptosis. J. Biol. Chem. 287, 8082–8091 (2012).
Terman, A. & Kurz, T. Lysosomal iron, iron chelation, and cell death. Antioxid. Redox. Signal. 18, 888–898 (2013).
Torii, S. et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem. J. 473, 769–777 (2016).
Platt, F. M., Boland, B. & van der Spoel, A. C. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J. Cell. Biol. 199, 723–734 (2012).
Gomez-Sintes, R., Ledesma, M. D. & Boya, P. Lysosomal cell death mechanisms in aging. Ageing Res. Rev. 32, 150–168 (2016).
Klionsky, D. J. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 8, 931–937 (2007).
Levine, B. & Kroemer, G. Biological functions of autophagy genes: a disease perspective. Cell 176, 11–42 (2019).
Dikic, I. & Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 19, 349–364 (2018).
Liu, Y. & Levine, B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 22, 367–376 (2015).
Bialik, S., Dasari, S. K. & Kimchi, A. Autophagy-dependent cell death—where, how and why a cell eats itself to death. J. Cell. Sci. https://doi.org/10.1242/jcs.215152 (2018).
Denton, D. & Kumar, S. Autophagy-dependent cell death. Cell Death Differ. https://doi.org/10.1038/s41418-018-0252-y (2018).
Kriel, J. & Loos, B. The good, the bad and the autophagosome: exploring unanswered questions of autophagy-dependent cell death. Cell Death Differ. https://doi.org/10.1038/s41418-018-0267-4 (2019).
Gump, J. M. et al. Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nat. Cell Biol. 16, 47–54 (2014).
He, W. et al. A JNK-mediated autophagy pathway that triggers c-IAP degradation and necroptosis for anticancer chemotherapy. Oncogene 33, 3004–3013 (2014).
Sousa, L. et al. Effects of iron overload on the activity of Na,K-ATPase and lipid profile of the human erythrocyte membrane. PLoS ONE 10, e0132852 (2015).
Song, X. et al. JTC801 induces pH-dependent death specifically in cancer cells and slows growth of tumors in mice. Gastroenterology 154, 1480–1493 (2018).
Zheng, C. J., Yang, L. L., Liu, J. & Zhong, L. JTC-801 exerts anti-proliferative effects in human osteosarcoma cells by inducing apoptosis. J. Recept. Signal. Transduct. Res. 38, 133–140 (2018).
Pochet, J. M., Laterre, P. F., Jadoul, M. & Devuyst, O. Metabolic alkalosis in the intensive care unit. Acta Clin. Belg. 56, 2–9 (2001).
Holze, C. et al. Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat. Immunol. 19, 130–140 (2018).
Saito, Y. et al. Turning point in apoptosis/necrosis induced by hydrogen peroxide. Free. Radic. Res. 40, 619–630 (2006).
Ingold, I. et al. Selenium Utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 172, 409–422 e421 (2018).
Casares, N. et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 202, 1691–1701 (2005).
Green, D. R., Ferguson, T., Zitvogel, L. & Kroemer, G. Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 9, 353–363 (2009).
Obeid, M. et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 13, 54–61 (2007).
Galluzzi, L., Buque, A., Kepp, O., Zitvogel, L. & Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 17, 97–111 (2017).
Tang, D., Kang, R., Coyne, C. B., Zeh, H. J. & Lotze, M. T. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol. Rev. 249, 158–175 (2012).
Hou, W. et al. Strange attractors: DAMPs and autophagy link tumor cell death and immunity. Cell Death Dis. 4, e966 (2013).
Yatim, N. et al. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science 350, 328–334 (2015).
Ahn, J., Xia, T., Rabasa Capote, A., Betancourt, D. & Barber, G. N. Extrinsic Phagocyte-dependent STING signaling dictates the immunogenicity of dying cells. Cancer Cell. 33, 862–873 e865 (2018).
Ma, Y. et al. Contribution of IL-17-producing gamma delta T cells to the efficacy of anticancer chemotherapy. J. Exp. Med. 208, 491–503 (2011).
Ren, J. et al. The RIP3-RIP1-NF-kappaB signaling axis is dispensable for necroptotic cells to elicit cross-priming of CD8(+) T cells. Cell. Mol. Immunol. 14, 639–642 (2017).
Michaud, M. et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 334, 1573–1577 (2011).
Vacchelli, E. et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science 350, 972–978 (2015).
Apetoh, L. et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 13, 1050–1059 (2007).
Yang, M. et al. TFAM is a novel mediator of immunogenic cancer cell death. Oncoimmunology 7, e1431086 (2018).
Kang, R. et al. HMGB1 in health and disease. Mol. Aspects. Med. 40, 1–116 (2014).
Kazama, H. et al. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 29, 21–32 (2008).
Li, C. et al. PINK1 and PARK2 suppress pancreatic tumorigenesis through control of mitochondrial iron-mediated immunometabolism. Dev. Cell. 46, 441–455 e448 (2018).
Ito, T. et al. Proteolytic cleavage of high mobility group Box 1 protein by thrombin-thrombomodulin complexes. Arterioscler. Thromb. Vasc. Biol. 28, 1825–1830 (2008).
Yu, H. et al. Role of high-mobility group Box 1 protein and poly(ADP-ribose) polymerase 1 degradation in Chlamydia trachomatis-induced cytopathicity. Infect. Immun. 78, 3288–3297 (2010).
Yu, Y., Tang, D. & Kang, R. Oxidative stress-mediated HMGB1 biology. Front. Physiol. 6, 93 (2015).
Fridman, J. S. & Lowe, S. W. Control of apoptosis by p53. Oncogene 22, 9030–9040 (2003).
Fujiki, K., Inamura, H., Sugaya, T. & Matsuoka, M. Blockade of ALK4/5 signaling suppresses cadmium- and erastin-induced cell death in renal proximal tubular epithelial cells via distinct signaling mechanisms. Cell Death Differ. https://doi.org/10.1038/s41418-019-0307-8 (2019).
Song, X. et al. FANCD2 protects against bone marrow injury from ferroptosis. Biochem. Biophys. Res. Commun. 480, 443–449 (2016).
Alvarez, S. W. et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 551, 639–643 (2017).
Brown, C. W., Amante, J. J., Goel, H. L. & Mercurio, A. M. The alpha6beta4 integrin promotes resistance to ferroptosis. J. Cell. Biol. 216, 4287–4297 (2017).
Liu, T., Jiang, L., Tavana, O. & Gu, W. The deubiquitylase OTUB1 mediates ferroptosis via stabilization of SLC7A11. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-18-3037 (2019).
Yoneda, T. et al. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 276, 13935–13940 (2001).
Kalai, M. et al. Regulation of the expression and processing of caspase-12. J. Cell. Biol. 162, 457–467 (2003).
Haberzettl, P. & Hill, B. G. Oxidized lipids activate autophagy in a JNK-dependent manner by stimulating the endoplasmic reticulum stress response. Redox Biol. 1, 56–64 (2013).
Barany, T. et al. Oxidative stress-related parthanatos of circulating mononuclear leukocytes in heart failure. Oxid. Med. Cell Longev. 2017, 1249614 (2017).
Palladino, E. N. D., Katunga, L. A., Kolar, G. R. & Ford, D. A. 2-Chlorofatty acids: lipid mediators of neutrophil extracellular trap formation. J. Lipid Res. 59, 1424–1432 (2018).
Zhou, R., Yazdi, A. S., Menu, P. & Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 (2011).
Silke, J., Rickard, J. A. & Gerlic, M. The diverse role of RIP kinases in necroptosis and inflammation. Nat. Immunol. 16, 689–697 (2015).
Polykratis, A. et al. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J. Immunol. 193, 1539–1543 (2014).
Newton, K. et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 23, 1565–1576 (2016).
Berger, S. B. et al. Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J. Immunol. 192, 5476–5480 (2014).
Kondylis, V., Kumari, S., Vlantis, K. & Pasparakis, M. The interplay of IKK, NF-kappaB and RIPK1 signaling in the regulation of cell death, tissue homeostasis and inflammation. Immunol. Rev. 277, 113–127 (2017).
Dondelinger, Y., Darding, M., Bertrand, M. J. & Walczak, H. Poly-ubiquitination in TNFR1-mediated necroptosis. Cell. Mol. Life Sci. 73, 2165–2176 (2016).
Dondelinger, Y. et al. NF-kappaB-independent role of IKKalpha/IKKbeta in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol. Cell 60, 63–76 (2015).
Menon, M. B. et al. p38(MAPK)/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat. Cell Biol. 19, 1248–1259 (2017).
Dondelinger, Y. et al. MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat. Cell Biol. 19, 1237–1247 (2017).
Jaco, I. et al. MK2 phosphorylates RIPK1 to prevent TNF-induced cell death. Mol. Cell 66, 698–710 e695 (2017).
Geng, J. et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat. Commun. 8, 359 (2017).
Xu, D. et al. TBK1 suppresses RIPK1-driven apoptosis and inflammation during development and in aging. Cell 174, 1477–1491 e1419 (2018).
Wegner, K. W., Saleh, D. & Degterev, A. Complex pathologic roles of RIPK1 and RIPK3: moving beyond necroptosis. Trends Pharmacol. Sci. 38, 202–225 (2017).
Choi, J. J., Reich, C. F. 3rd & Pisetsky, D. S. Release of DNA from dead and dying lymphocyte and monocyte cell lines in vitro. Scand. J. Immunol. 60, 159–166 (2004).
Chen, Q., Sun, L. & Chen, Z. J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 17, 1142–1149 (2016).
Holm, C. K. et al. Influenza A virus targets a cGAS-independent STING pathway that controls enveloped RNA viruses. Nat. Commun. 7, 10680 (2016).
Costa Franco, M. M. et al. Brucella abortus triggers a cGAS-independent STING pathway to induce host protection that involves guanylate-binding proteins and inflammasome activation. J. Immunol. 200, 607–622 (2018).
DeFilippis, V. R., Alvarado, D., Sali, T., Rothenburg, S. & Fruh, K. Human cytomegalovirus induces the interferon response via the DNA sensor ZBP1. J. Virol. 84, 585–598 (2010).
Zhang, Z. et al. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol. 12, 959–965 (2011).
Kondo, T. et al. DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc. Natl Acad. Sci. USA 110, 2969–2974 (2013).
Unterholzner, L. et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 11, 997–1004 (2010).
Zeng, L. et al. ALK is a therapeutic target for lethal sepsis. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.aan5689 (2017).
Barber, G. N. STING: infection, inflammation and cancer. Nat. Rev. Immunol. 15, 760–770 (2015).
Ahn, J., Son, S., Oliveira, S. C. & Barber, G. N. STING-dependent signaling underlies IL-10 controlled inflammatory colitis. Cell Rep 21, 3873–3884 (2017).
Ahn, J., Gutman, D., Saijo, S. & Barber, G. N. STING manifests self DNA-dependent inflammatory disease. Proc. Natl Acad. Sci. USA 109, 19386–19391 (2012).
Bakhoum, S. F. et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472 (2018).
Sliter, D. A. et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 561, 258–262 (2018).
Larkin, B. et al. Cutting edge: activation of STING in T cells induces type I IFN responses and cell death. J. Immunol. 199, 397–402 (2017).
Gulen, M. F. et al. Signalling strength determines proapoptotic functions of STING. Nat. Commun. 8, 427 (2017).
Gaidt, M. M. et al. TheDNA inflammasome in human myeloid cells is initiated by a STING-cell death program upstream of NLRP3. Cell 171, 1110–1124 e1118 (2017).
Cunha, L. D. et al. LC3-associated phagocytosis in myeloid cells promotes tumor immune tolerance. Cell https://doi.org/10.1016/j.cell.2018.08.061 (2018).
Man, S. M., Karki, R. & Kanneganti, T. D. AIM2 inflammasome in infection, cancer, and autoimmunity: role in DNA sensing, inflammation, and innate immunity. Eur. J. Immunol. 46, 269–280 (2016).
Wilson, J. E. et al. Inflammasome-independent role of AIM2 in suppressing colon tumorigenesis via DNA-PK and Akt. Nat. Med. 21, 906–913 (2015).
Kuriakose, T. & Kanneganti, T. D. ZBP1: innate sensor regulating cell death and inflammation. Trends Immunol. 39, 123–134 (2018).
Acknowledgements
We thank Christine Burr (Department of Surgery, University of Pittsburgh) and Dave Primm (Department of Surgery, University of Texas Southwestern Medical Center) for their critical reading of the manuscript. We apologize to all researchers whose great work could not be cited in this review due to space limitations. G.K. is supported by the Ligue contre le Cancer Comité de Charente-Maritime (équipe labelisée); Agence National de la Recherche (ANR) – Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Chancelerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); the European Commission (ArtForce); the European Research Council (ERC); Fondation Carrefour; Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology; the RHU Torino Lumière, the Searave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI).
Author information
Authors and Affiliations
Contributions
D.T. and G.K. took the lead in writing the manuscript. R.K., T.V.B., and P.V. discussed the contents and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tang, D., Kang, R., Berghe, T.V. et al. The molecular machinery of regulated cell death. Cell Res 29, 347–364 (2019). https://doi.org/10.1038/s41422-019-0164-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41422-019-0164-5
This article is cited by
-
Dysregulated dendritic cells in sepsis: functional impairment and regulated cell death
Cellular & Molecular Biology Letters (2024)
-
Ferroptotic therapy in cancer: benefits, side effects, and risks
Molecular Cancer (2024)
-
Calcium signaling from sarcoplasmic reticulum and mitochondria contact sites in acute myocardial infarction
Journal of Translational Medicine (2024)
-
Exploration of programmed cell death-associated characteristics and immune infiltration in neonatal sepsis: new insights from bioinformatics analysis and machine learning
BMC Pediatrics (2024)
-
Autophagy-mediated ferroptosis is involved in development of severe acute pancreatitis
BMC Gastroenterology (2024)
