
Abstract
Autism spectrum disorder (ASD) is a complex neurodevelopmental condition that affects social interaction and behavior. Mutations in the gene encoding chromodomain helicase DNA-binding protein 8 (CHD8) lead to autism symptoms and macrocephaly by a haploinsufficiency mechanism. However, studies of small animal models showed inconsistent findings about the mechanisms for CHD8 deficiency-mediated autism symptoms and macrocephaly. Using the nonhuman primate as a model system, we found that CRISPR/Cas9-mediated CHD8 mutations in the embryos of cynomolgus monkeys led to increased gliogenesis to cause macrocephaly in cynomolgus monkeys. Disrupting CHD8 in the fetal monkey brain prior to gliogenesis increased the number of glial cells in newborn monkeys. Moreover, knocking down CHD8 via CRISPR/Cas9 in organotypic monkey brain slices from newborn monkeys also enhanced the proliferation of glial cells. Our findings suggest that gliogenesis is critical for brain size in primates and that abnormal gliogenesis may contribute to ASD.
Similar content being viewed by others


Introduction
Autism spectrum disorder (ASD) comprises a group of neurodevelopmental conditions that affect social interaction and communication and display repetitive behavior. The gene encoding chromodomain helicase DNA-binding protein 8 (CHD8), an ATP-dependent chromatin remodeling factor that regulates gene transcription, has been described as an autism susceptibility/intellectual disability gene, as mutations in CHD8 were found to be strongly associated with ASD1,2,3,4,5,6. Individuals with heterozygous CHD8 mutations display various clinical features consisting of autistic behaviors, macrocephaly, and facial dysmorphisms6,7,8,9,10.
Because heterozygous CHD8 mutations were identified in ASD patients, the haploinsufficiency of the CHD8 gene is thought to account for the pathogenesis of CHD8-related ASD. In support of this theory, mice carrying heterozygous Chd8 mutations show autistic-like phenotypes and macrocephaly11,12,13 whereas homozygous Chd8 mutations cause embryonic lethality14. However, the behavioral phenotypes in mouse models are heterogenous and vary to a great extent. For example, social deficits were found in some Chd8 mutant mice11,12,13 but not others15,16 whereas repetitive behavior could occur16 or was absent12,13,15 in Chd8 deficient mice. In addition, the mechanism underlying CHD8 deficiency-mediated macrocephaly remains controversial. Although increased neurogenesis was reported to be the potential mechanism underlying increased brain volume15,17,18, neurogenesis was also found to be decreased11 or not altered13 in mice or mouse neural progenitor cells with Chd8 deficiency. Furthermore, increases in brain volume were observed in female but not male mice carrying a CHD8 mutation16. A potential role for CHD8 in gliogenesis has so far remained unexplored. Because of the lack of pathological studies of the brains of CHD8 mutant patients, there is no histological evidence for a correlation of abnormal neuronal development with macrocephaly and clinical phenotypes in patients carrying CHD8 mutations.
Our recent studies of nonhuman primate models of brain diseases indicate that there are substantial differences in brain pathological changes between rodent and primate models19,20,21,22,23. For example, loss of the SHANK3 or PINK1 gene can cause striking neuronal loss in the monkey brain, a phenomenon that has not been found in the Shank3 or Pink1 knockout mice19,23. Rodents exhibit many key features of the development and formation of the mammalian brains, which are characterized by the initial neurogenesis followed by the sequential production of glia. However, the timing and sources of proliferative cells for cerebral expansion are considerably different between rodents and primates24,25,26,27. A noticeable difference is that rodent brains lack gyrification or the folding of the cortical surface, a unique structure seen in large mammals and primate brains28. Gliogenesis was found to correlate with enlargement and gyrification of the primate cerebrum29, but this interesting finding remains to be verified with genetic evidence. Thus, it is important to establish a nonhuman primate model with CHD8 mutations to investigate how a CHD8 mutation affects the development of the primate brain.
In this study, we used CRISPR/Cas9 to target the CHD8 gene in the embryos of cynomolgus monkeys (Macaca fascicularis). As expected, CHD8 mutations affected early development and led to a low yield of live monkeys. Despite this, analysis of the brain tissues of aborted and stillborn monkeys and the behaviors of the live monkey revealed that CHD8 mutation abnormally increased gliogenesis to enlarge brain size and to alter social interaction and communication. To avoid embryonic lethality, we disrupted the CHD8 gene in fetal monkey brains prior to gliogenesis and in brain slices from newborn monkeys, resulting in an increased number of glial cells. These findings together show that in the primates, CHD8 mutations may primarily increase gliogenesis to cause macrocephaly and autism symptoms, providing a new insight into ASD pathogenesis.
Results
Generation and validation of CHD8 mutant monkeys
We used the embryonic targeting strategy to delete the CHD8 gene in fertilized monkey eggs (Fig. 1a). We designed gRNA to target exon 19 of CHD8, as exon 19 is shared by two CHD8 isoforms (NM_001170629.1 and NM_020920.3) (Fig. 1b and Supplementary Fig. S1a). Thus, a frameshift mutation or truncation in this exon is sufficient to disrupt CHD8 expression. After validating that gRNA was able to reduce CHD8 in cultured HEK 293 cells (Supplementary Fig. S1b, c), we injected CHD8 gRNA with Cas9 to one-cell stage embryos of cynomolgus monkeys using the same methods described in our previous studies19,21,23. It is known that only heterozygous mutations of CHD8 were seen in humans and that homozygous Chd8 mutations caused embryonic lethality in mice14. In our initial studies, we used different CHD8 gRNAs that might have higher targeting efficiency and failed to obtain any pregnancy (0/41 surrogates). We then used the gRNA described in Fig. 1a and obtained four (4/29) pregnant monkeys after transplanting 112 embryos into 29 surrogate mothers (Fig. 1c). It is likely that the potential lethality of homozygous or extensive mutations yielded a low rate of pregnancies. The four pregnancies we had obtained included a triplet pregnancy that was terminated to yield three aborted fetuses (T1, T2, and T3) at 53 days after embryo transfer. The other three pregnant monkeys produced one aborted fetus (M1) on day 125, one stillborn monkey (M2) that had developed full-term (158 days), and one live offspring (M3) that had been living for 54 months (Fig. 1d). The low birth rate of CHD8 mutant monkeys supports the idea that CHD8 is essential for early development.

a One-cell stage embryos of cynomolgus monkeys were injected with CHD8 gRNA and Cas9 mRNA. Embryos at 4–8-cell stage were transferred to surrogates to produce newborn monkeys with CHD8 mutations. b Schematic diagram of gRNA targeting exon 19 of CHD8. Part of the CHD8 gene structure was shown, Protospacer-adjacent motif (PAM) sequence is highlighted in blue. gRNA targeting site is highlighted in red. c The numbers of transferred embryos, surrogates, and pregnancies for generating monkeys with CHD8 mutations. d Summary of generated monkeys carrying CHD8 mutations. e Examination of CHD8 mutations in monkey offspring by PCR and T7E1 cleavage assay. M DNA marker; WT wild type. Brain tissues of M1, M2, and WT control, embryos from T1, T2, and T3 and umbilical cord from M3 were analyzed. The upper panel shows PCR products and the low panel shows DNA products after T7E1 digestion. Stars indicate DNA mutations. f Sequences of mutated CHD8 alleles in monkey offspring. PAM sequence is highlighted in blue; gRNA targeting site in red. Mutation sequences are marked in green. Insertions (+) or deletions (–) are indicated within parentheses. The numbers of DNA clones showing mutations of the total number of examined clones are also included in parentheses. g Identified CHD8 mutations result in truncation of the CHD8 protein. Amino acid sequences in yellow are present in wild-type CHD8 whereas sequences in green are mutated.
All the offspring were genotyped for mutations in exon 19 of CHD8 (Fig. 1e, f). The gender of triplets (T1, T2, T3) was determined by the presence of the sex-determining region Y (SRY) gene on the Y chromosome30. We used deep sequencing to analyze aborted triplets (T1, T2, T3), which showed low rates (5.1%–27.5%) of CHD8 mutations (Supplementary Fig. S2a). Thus, their fetal loss is unlikely due to CHD8 mutations because triplet fetuses in macaque monkeys are often miscarried. For M1, M2, and M3, their targeted DNAs were isolated by PCR and cloned for sequencing. Various mutations (–4/+4, –4/+2, +16, and –1) in CHD8 exon 19 of mutant offspring were identified by DNA sequencing (Fig. 1f). M1, which was miscarried at 125 days of pregnancy, carried bi-allelic mutations, in which one allele had 4 bp deletion and insertion (–4/+4) and the other had –4/+2 bp mutation in its brain (Fig. 1f). Such mutations resulted in two amino acid alternations (M1285N and S1286V) and a frameshift mutation to generate a truncated protein with 1298 amino acids (p.S1286fsX1299) (Fig. 1g). M2 had heterozygous +16 bp mutant sequences in the brain, leading to the truncation at 1289 amino acid position (Fig. 1f, g). The live M3 (male) has heterozygous –1 bp mutation in its umbilical cord and blood sample, which can lead to truncation of CHD8 protein with 1308 amino acids (p.S1286fsX1307), though the nature of CHD8 mutation in the brain remains to be determined (Fig. 1f, g).
To understand the mosaic nature of CHD8 mutant monkeys, we used DNA cloning and sequencing to analyze mutation rates in PCR products from various tissues. M1 monkey tissues showed more extensive mutations (>72.7%) than M2 monkey tissues (31.3%–73.7%) (Supplementary Fig. S2b). The more severe mutations in M1 may explain why M1 was terminated earlier than M2. To analyze the potential off-targets, we performed whole-genome sequencing of the blood sample of the live monkey (M3), and the results did not reveal obvious off-target events (Supplementary Fig. S3a). Whole-genome sequencing of the brain cortex DNAs from M1 and M2 did not detect any off-target events in the potential targeting sequences containing no more than five mismatched nucleotides, while M3 had one potential off-target (Supplementary Fig. S3b).
Increased gliogenesis in CHD8 mutant monkeys
The aborted M1 monkey provided us with tissues for immunocytochemical studies, and the stillborn M2 monkey offered more tissues for both immunocytochemical and western blotting analysis. We also obtained wild-type monkey (control-1) at E130 and a newborn wild-type (control-2) monkey for comparing with M1 and M2, respectively. Immunohistochemistry validated that CHD8 mutations could reduce the immunostaining of CHD8 at the protein level in the brains of M1 and M2 (Fig. 2a). We then used western blotting to analyze the expression of neuronal and glial proteins in control-2 and M2 monkey brain tissues. The results revealed striking increases in glial fibrillary acidic protein (GFAP) and myelin basic protein (MBP), which are marker proteins for astrocytes and oligodendrocytes, respectively, in the M2 monkey brain. However, examination of several neuronal proteins (NeuN, parvalbumin (PV), Homer (Hom), and doublecortin (Dcx)), did not show any significant difference between control-2 and M2 monkeys (Fig. 2b). Quantification of the ratios of examined proteins to the loading control tubulin on multiple western blots verified that CHD8 mutations selectively increased glial proteins but not neuronal proteins (Fig. 2c). Glia are found in both gray and white matter, but there are more oligodendrocytes in white matter31. We, therefore, used immunocytochemical staining of the white matter to identify oligodendrocytes with an antibody to olig2, a protein that is specifically expressed in oligodendrocytes. This examination also showed that there were more oligodendrocytes in the white matter of M1 and M2 (Fig. 2d, e).

a Immunostaining of the prefrontal cortex gray matter (GM) and white matter (WM) of control and CHD8 mutant monkeys (M1, M2) with anti-CHD8 antibody. CHD8 shows a higher expression in E125–E130 than newborn (E163 ± 5). Scale bar: 50 μm. b Western blotting of the prefrontal cortex of the newborn monkeys (control-2 and M2). The blots were probed with antibodies to neuronal proteins (NeuN, DcX, PV, and Hom) and glial proteins (GFAP, MBP). Tubulin served as a loading control. c Quantification of the ratios of neuronal and glial proteins to the loading control on western blots. The data were obtained from four western blotting experiments and are presented as means ± SEM. *P < 0.05; **P < 0.01. d Immunostaining of the white matter of control and CHD8 mutant monkeys with an antibody to olig2, a marker protein for oligodendrocytes. The nucleus was stained with hematoxylin. Scale bar: 50 μm. e Density of oligodendrocytes in the white matter of control and CHD8 mutant monkeys. The data were obtained by counting 12 images and presented as means ± SEM per mm2. ***P < 0.001.
The brains of newborn monkeys have been well developed and allowed us to compare the brain sizes of newborn control-2 and M2. This comparison revealed that the M2 brain was 28% larger in weight with enlarged white matter labeled by immunostaining of GFAP (Fig. 3a, b), which is selectively expressed in astrocytes, another major type of glial cells that are mainly distributed in the white matter. The gray matter thickness appears normal (Fig. 3b, c). Immunostaining of the entire monkey cortex layers clearly demonstrated more astrocytic staining in the cortex of CHD8 mutant monkeys (M1 and M2) than the control monkey cortex (Fig. 3c). Quantification of the numbers of astrocytes and GFAP-immunostaining intensity in the white matter verified that CHD8 mutations increased GFAP labeling (Fig. 3d, e). However, immunostaining of Iba1, a marker protein for microglial cells, did not show different staining between the control and CHD8 mutant monkeys (Supplementary Fig. S4a, b). In addition, the control and CHD8 mutant monkey brains showed no difference in immunostaining of neurons with an antibody to the neuronal marker NeuN (Supplementary Fig. S5a–c). In other brain regions such as the cerebellum, neuronal staining by antibodies to neuronal proteins (NeuN and calbindin) did not show any obvious difference between control-2 and M2 (Supplementary Fig. S5d). Thus, CHD8 mutations appear to selectively increase astrocytic and oligodendrocytic protein expression.

a Photographs of control-2 and M2 monkey brains. The brains were sliced at the same position (red lines) for immunostaining. Quantification of brain weights of M2 and six age-matched controls is shown on the right panel. Scale bar: 1 cm. b GFAP immunostaining of the white matter in control and CHD8 mutant monkeys. The thickness of gray matter, which is GFAP-negative, in the cortical regions of M2 and age-matched control-2 is shown on the right panel (n = 8 measures in each animal). Scale bar: 1 mm. c GFAP immunostaining of the entire cortical layers of control and CHD8 mutant monkeys. GM gray matter; WM white matter. Scale bar: 50 μm. d Large magnification micrographs of GFAP immunostaining in the white matter of control and CHD8 mutant monkeys. Scale bar: 20 μm. e Quantification of the density of GFAP-positive cells from GFAP-immunostaining results. The data were obtained by counting 4–11 images in each group and presented as means ± SEM per mm2. *P < 0.05; **P < 0.01, ***P < 0.001.
Since astrocytes and oligodendrocytes are derived from the same progenitor cells (radial glial cells) that also give rise to neurons before gliogenesis, the lack of NeuN alteration in M1 and M2 suggests that CHD8 mutations selectively increased the proliferation of astrocytes and oligodendrocytes. M1 monkey allowed us to explore this issue, as it was terminated at E125, a time after neurogenesis (E45–E102) and during gliogenesis (E90–E145) in the macaque monkey brain29,32 (Fig. 4a). Subventricular zone (SVZ) is critical for neurogenesis and gliogenesis in the mammalian brains and is divided into the inner (iSVZ) and outer (oSVZ) layer in the primate brains. oSVZ, which is absent in the rodent brain, is responsible for the expansion of the cerebral cortex in the primates25,33. Thus, we used brain slices containing iSVZ and oSVZ in the control-1 (E130) and M1 (E125) monkeys to assess the distribution of marker proteins for neurogenesis and gliogenesis (Fig. 4b, c). Examination of the progenitor markers such as Pax6 and EGFR revealed that Pax6-positive cells were mainly localized in iSVZ whereas EGFR-positive cells were distributed in all layers, which are consistent with results previously reported29. Importantly, more Pax6- and EGFR-positive cells were present in M1 than control-1 (Fig. 4c). These results suggest that more progenitor cells are in M1 for developing to neuronal or glial cells. However, anti-NeuN immunostaining showed weak or absent staining in the region of iSVZ and oSVZ in both control-1 and M1 (Supplementary Fig. S6a), suggesting that neurogenesis had been largely completed. In contrast, staining with antibodies to glial cells (olig2 and GFAP) revealed a number of positive cells in iSVZ and oSVZ. M1 displayed notably more olig2 and GFAP-positive cells than control-1 (Fig. 4d). This important difference was verified by quantitation of the density of progenitor cells and glial cells in the brain slices (Fig. 4e). We also examined the cerebellum but did not find any obvious difference for NeuN and GFAP staining between control-1 and M1 (Supplementary Fig. S6b). Postnatal gliogenesis occurs in the mammalian brains34,35. Examining the cortex near the lateral ventricle (LV) region revealed more glial cells and enlarged white matter in M2 than control-2 (Supplementary Fig. S7). Taken together, gliogenesis in the cerebral cortex is obviously enhanced in CHD8 mutant monkeys when compared with the control monkeys.

a The timeline for embryonic neurogenesis and gliogenesis in the macaque monkey. b The coronal section of the control-1 monkey (E130) showing the cortex, caudate, corpus callosum and LV. Rectangle indicates the location of brain slices that were used to examine the staining and numbers of neuronal and glial cells. Scale bar: 1 mm. c The brain slice used for examination consists of different lays including the inner subventricular zone (iSVZ), outer subventricular zone (oSVZ), intermediate zone (IZ), and subplate (SP). d Immunostaining of progenitor cells with antibodies to EGFR and Pax6. The nucleus was stained with hematoxylin. Scale bar: 100 μm. e Immunostaining of the white matter near the LV with antibodies to olig2 and GFAP, respectively. Scale bar: 100 μm. f Quantification of the relative numbers of EGFR- and Pax6-positive cells in iSVZ and the numbers of oligodendrocytes and astrocytes in the brain slices. The data were obtained by counting 4–7 brain slices per group and presented as means ± SEM. **P < 0.01; ***P < 0.001.
M2 monkey brain tissues also allowed us to perform RNA-seq to compare gene expression profiling with control-2, which could provide complementary information regarding the expression of neuronal and glial genes. We examined the expression of 18,224 genes in our data analysis by comparing RNA-seq results from the prefrontal cortex of control-2 and M2 and found 1298 up- and 423 downregulated genes above the threshold (Fig. 5a, b). In Gene Ontology analysis, upregulated genes were enriched in myelin-related processes, astrocyte and oligodendrocyte development/differentiation, and Wnt-signal pathway (Fig. 5c). Because of differential expression of neuronal and glial proteins in CHD8 mutant monkey brains, we focused on marker genes and transcription factors of neuronal and glial cells based on the published information for these genes36. Transcription factors specific for oligodendrocytes were upregulated to a greater extent than those for astrocytes whereas neuronal transcription factors were expressed at similar levels in control-2 and M2, consistent with greater increases in oligodendrocytic gene expression (Fig. 5d, e). Major oligodendrocyte transcription factors, including Olig2, Olig1, MYRF, Sox10, Sox8, Sox3, Nkx2-2, Nkx6-2, and Carhsp1, were significantly upregulated with fold change ranging from 1.7 to 74 in M2, indicating enhanced oligodendrogenesis (Fig. 5d, e). Since we previously investigated Myrf, an oligodendrocyte-specific transcription factor, in Huntington disease mice37, we examined MYRF expression via western blotting using a commercially available antibody. The results confirmed that both full-length and N-terminal MYRF were increased in the brain cortex of M2 monkey (Fig. 5f). Thus, RNA-seq results support the findings that the brain cortex in M2 has increased expression of glial proteins, particularly oligodendrocytic proteins, which were also revealed by western blotting and immunohistochemistry (Figs. 2 and 3).

a Summary of differentially expressed (DE) genes in M2 PFC compared to control-2 PFC. Significance cutoffs correspond to log2 (fold change) > 1.5 and adjusted P value (Padj) < 0.05. b Reduced CHD8 expression in M2 brain cortex in RNA-seq analysis. **P < 0.01. c Representative significant gene ontology (GO) terms for DE genes of up- and downregulation. Odds ratio > 2 and Padj < 0.05. d Expression levels of enriched transcription factors for neurons, astrocytes and oligodendrocytes demonstrate upregulation in gliogenesis, but not neurogenesis. The data were obtained by averaging three samples and presented as means ± SEM. e Heatmap analysis of neuron, astrocyte, and oligodendrocyte-specific genes revealing upregulation in oligodendrocyte-enriched genes but not in neuron-enriched genes. Fold change was plotted on a log2 color scale, with blue representing lower expression and red representing higher expression in M2 than control-2. f Western blotting showing the increased expression of full-length (fMYRF), N-terminal (nMYRF) MYRF and Sox9 in the brain cortex of M2 when compared with control-2. The ratios (means ± SEM) of MYRF and Sox9 to the loading control actin were obtained by quantifying the intensity of MYRF, Sox9, and actin on three independent western blots.
However, RNA-seq analysis of bulk tissues would be difficult to define gene alterations in specific types of cells or in specific regions. The proliferation of glial cells in SVZ in the primate brain is essential for the expansion and gyrification of the primate cerebrum29. We used immunostaining to examine the distribution of cells expressing Ki67, a proliferation marker, in the CHD8 mutant monkey brains. We found there was a significant increase in Ki67-positive cells in SVZ, but not in white matter, in M1 and M2 monkeys as compared with controls (Supplementary Fig. S8). This result suggests that increased gliogenesis in CHD8 mutant monkeys is more likely due to the enhanced proliferation of glial progenitor cells in SVZ rather than the proliferation of differentiated glial cells in the white matter.
More glial cells by knocking down CHD8 in the brains of fetal and newborn monkeys
The facts that homozygous CHD8 deletion is embryonic lethal in mice and that only heterozygous CHD8 mutations are seen in patients explain the low rate of survived CHD8 mutant monkeys in the CHD8 gene-targeting at the one-cell embryo stage. Transcription profiling studies indicate that in the monkey brain, neurogenesis peaks around gestation day 55 (G55) and continues until G80, which overlaps the beginning of gliogenesis38,39. To more rigorously investigate whether CHD8 expression is important for gliogenesis, we generated lentiviral vectors to express CHD8 gRNA and Cas9 (Supplementary Fig. S9a) and performed in utero delivery of lentiviral CHD8 gRNA/Cas9 into the fetal monkey brain at ~G55 (Fig. 6a). For the control, we used lentiviral control RNA/Cas9 that did not target any specific gene. The pregnancy was assessed by ultrasound examination, and injection of lentiviruses (50 μL lentiviral control or CHD8 gRNA mixed with 100 μL lentiviral Cas9, viral titer 1 × 109 vg/mL) into the LV of the fetal monkey brain was performed under the guidance of continuous ultrasound imaging (Fig. 6b and Supplementary Fig. S9b). We injected lentiviral control gRNA/Cas9 into one fetus, and lentiviral CHD8 gRNA/Cas9 into two fetuses, all at ~G55. All these three fetuses developed to newborn monkeys that were naturally delivered around days 161–164. The brains of these newborn monkeys were isolated at postnatal day 7–8 and revealed that lentiviral transduction was widespread in the cortical layers, as lentiviral vectors also expressed GFP (Fig. 6c). Lentiviral transduction led to the expression of transgene, reflected by GFP expression, in neuronal cells with normal morphology (Fig. 6d). The white matter, which was clearly labeled by anti-GFAP, appeared to be larger in CHD8-targeted monkey brain than in the control monkey brain (Fig. 6e and Supplementary Fig. S9c). Importantly, western blotting analysis of lentiviral-infected brain regions showed that CHD8 was decreased, accompanied by elevated levels of Olig2 and SOX9, which are marker proteins for oligodendrocytes and astrocytes (Fig. 6f).

a In utero delivery of lentiviral CHD8 gRNA/Cas9 into the LV of fetal monkey brain at ~G55. b Ultrasound image of injection of lentivirus into the LV of the fetal monkey brain at G55. Arrows indicate a needle. c Widespread expression of GFP, which reflects lentiviral CHD8 gRNA/Cas9 transduction in the brain of newborn monkey. Scale bar: 1 mm. d Neuronal expression of GFP in the newborn monkey brain cortex. Scale bar: 10 μm. e GFAP labeling of the white matter of the brain cortical region in control and CHD8-targeted newborn monkeys. Scale bar: 1 mm. f Western blotting of the lentiviral-infected brain regions showing that CHD8 gRNA/Cas9 expression reduced CHD8 expression and increased levels of glial proteins (Sox9 and Olig2). g Double immunostaining of lentiviral-infected brain regions showing that CHD8 gRNA/Cas9 expression increased Sox9-labeled astrocytes and Olig2-labeled oligodendrocytes when compared with control gRNA/Cas9 expression. Scale bar: 20 μm. h Quantitation of the relative number of oligodendrocytes and astrocytes. The data were obtained from one newborn monkey targeted by control gRNA/Cas9 and two newborn monkeys targeted by CHD8 gRNA/Cas9. Brain sections (1.3 mm apart from each other) from the brain cortical region in the control gRNA/Cas9 (six sections) and CHD8 gRNA/Cas9 (eight sections) targeted monkeys were used for quantitative analysis. At least four images (×20) from each brain section were taken to obtain the average numbers of glial cells per mm2. The data are presented as means ± SEM (n = 24 images for control gRNA/Cas9 and n = 32 images for CHD8 gRNA/Cas9). ***P < 0.001.
It is important to examine whether the density of glial cells was increased in the white matter of CHD8-targted monkey brain. Thus, we performed double immunofluorescent staining to verify that lentiviral CHD8 gRNA/Cas9 was expressed in both neuronal and glial cells (Supplementary Fig. S10a) and was able to reduce the level of CHD8 (Supplementary Fig. S10b). While CHD8 targeting did not influence neuronal processes, morphology, and density (Fig. 6d and Supplementary Fig. S11a), astrocytic proliferation, which was revealed by anti-GFAP labeling, appeared to be increased (Supplementary Fig. S11b). However, GFAP labeling could not clearly distinguish individual cells. We therefore used anti-SOX9 to label the nuclei of astrocytes and anti-olig2 to identify oligodendrocytes. Virus-mediated transgene (GFP) expression can decline overtime during brain development but disrupting the CHD8 gene by CRISPR/Cas9 in the brain cells including neuronal and glial progenitor cells is permanent. Thus, to assess the effect of targeting CHD8 gene at G55 on the glial cells in the newborn monkeys, we identified the brain regions expressing GFP in the newborn monkeys and counted glial cell numbers in the viral infected brain regions. The results demonstrated an increase in the density of astrocytes and oligodendrocytes in CHD8-targeted brain region as compared with control lentiviral infection (Fig. 6g and Supplementary Fig. S12). Quantitation of the relative numbers of glial cells in the examined areas also confirmed that targeting CHD8 by CRISPR/Cas9 at G55 could increase the numbers of astrocytes and oligodendrocytes in the newborn monkey brains (Fig. 6h).
Transcriptional profiling studies also showed that gliogenesis peaks after birth and continues through childhood in the primates38,39. To obtain additional evidence to support the idea that CHD8 plays an important role in gliogenesis in the primate brain, we used brain slices from newborn monkeys to examine the effect of CHD8 deficiency on glial cells after birth (Fig. 7a). The brain region containing the LV was infected by lentiviral CHD8 sgRNA/Cas9 (Supplementary Fig. S13a), as these regions contain glial cells that can proliferate and migrate to the cortical layers, a process important for enlarging brain size in the primates29. Under ex vivo culturing conditions, glial cells were preferentially infected by lentiviral vectors, and most of GFP-positive cells were negative to NeuN labeling despite use of the EF1-α promoter to express GFP (Fig. 7b, c), allowing us to examine the effect of targeting CHD8 on the proliferation of glial cells. Double immunofluorescent staining revealed that lentiviral infection apparently led to the expression of CHD8 gRNA in oligodendrocytes and astrocytes (Supplementary Fig. S13b) and decreased CHD8 expression in GFP-positive cells (Fig. 7d). Immunostaining of the brain region near the LV demonstrated the increased numbers of oligodendrocytes and astrocytes, which could be readily identified by the nuclear labeling of olig2 and SOX9, respectively (Fig. 7e).

a Cortical brain region near the LV and SVZ in the newborn monkeys was isolated for brain slice culture. OB olfactory bulb, Hippo hippocampus, Cere cerebellum. b After infection with lentivirus for 10 days, the brain slices showed strong GFP signaling, reflecting lentiviral transduction. Scale bar: 100 μm. c Immunostaining of the infected brain slices showing that GFP-positive cells contain NeuN positive and negative cells. Scale bar: 25 μm. d Double immunostaining showing that lentiviral CHD8 gRNA/Cas9 expression, reflected by GFP expression, reduced CHD8 expression and occurred in astrocytes and oligodendrocytes. Scale bar: 10 μm. e Double immunostaining of the brain slice region near the LV showing that the density of olig2-labeled oligodendrocytes and SOX9-labeled astrocytes was increased in the lentiviral CHD8 gRNA/Cas9 infected region as compared with lentiviral control gRNA/Cas9 infection. Scale bar: 25 μm. f Quantitation of the relative numbers of oligodendrocytes and astrocytes. Nine brain slices from three newborn monkeys were used for statistical analysis. The data are presented as means ± SEM. **P < 0.01; ***P < 0.001.
Enlarged white matter and macrocephaly in the live CHD8 mutant monkey
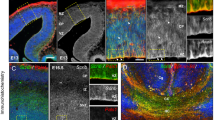
To examine whether CHD8 mutations caused macrocephaly, we carefully examined the brain size of the CHD8 mutant monkeys (M2 and M3). We noticed a greater weight of the isolated M2 brain (57.8 g) than controls (45 ± 2.8 g, n = 6), supporting 28% larger brain size in M2 at birth (Fig. 3a). This finding, along with increased gliogenesis in M1 and more glial cells in M2, led us to carefully examine whether the live M3 monkey displayed any abnormal size of its head. By comparing with 15 age- and body weight-matched male monkeys at 24 months of age, we found the size of M3’s head is apparently larger than the age- and sex-matched control monkeys. Figure 8a shows the photos of M3’s head and the control head of an age- and weight-matched male monkey at 54 months of age. Skeletal imaging of heads and measurements of the head circumference (n = 15 for control) verified that M3 monkey had a larger head (25.1 cm) than the control monkeys (23.26 ± 0.28 cm, n = 15) (Fig. 8b, c). MRI was then performed to examine the brain structure, especially the gray matter and white matter. Total brain volume of M3 monkey (115,916 mm3) was larger than that (101,622 ± 3626 mm3) of the age-matched control monkeys (n = 7) (Fig. 8c). The white matter volume of M3 monkey (36-m-old: 30,690 mm3; 42-m-old: 31,052 mm3) had a larger volume than that of the control monkeys (36-m-old: 26,413 ± 557 mm3; 42-m-old: 36,517 ± 501 mm3, n = 7). To rule out the influence of individual brain sizes on the relative areas of the white matter, we measured the ratios of white matter to gray matter (WM/GM) on the same brain regions in both controls and M3, which were determined by their imaging positions. This quantification demonstrated that the ratio of WM/GM of M3 monkey (36-m-old: 0.89; 42-m-old: 0.91) was higher than that of the control monkey (36-m-old:0.75 ± 0.02; 42-m-old: 0.78 ± 0.03, n = 7) (Fig. 8f). Thus, this imaging analysis revealed that the areas of the white matter were larger in the M3 monkey than the control monkeys (Fig. 8d, e). Considering the increased glial cells in M1 and M2 monkeys, these results suggest that CHD8 mutations can increase gliogenesis to enlarge white matter, resulting in macrocephaly.

a Head and face photos of M3 and an age-matched control monkey at 54 months. b Skeleton head images of M3 and control monkey. The sizes (height and width) of the head are indicated by red lines. Scale bar: 1 cm. c Head circumference (cm) and total brain volume (mm3) of M3 and 15 age-matched male control monkeys at 36 months. d Sagittal MRI images of M3 and a control monkey. The red lines indicate the same location and orientation of the slices that were used to obtain coronal MRI images containing the cerebral cortex. e Horizontal MRI images of M3 and control monkey showing white matter that appears whiter and gray matter that is darker. Scale bar: 5 cm. The coronal images were obtained from the respective brain slices shown in (d). The enlarged images in the dashed square are shown beneath each coronal image. M3 brain shows enlarged white matter in several lobes shown. f Quantitation of the volume of white matter and the ratio of white matter to gray matter. Controls consist of three age-matched monkeys. The data are presented as means ± SEM.
Discussion
The formation and maturation of the brain are largely determined by neurogenesis and gliogenesis during embryonic and postnatal development. Using the nonhuman primate models to investigate the effect of CHD8 on brain development, we found that CHD8 expression is important for normal gliogenesis because its deficiency can enlarge the brain size via abnormally increased gliogenesis. This finding highlights the specific function of CHD8 and unique pathogenesis of CHD8 deficiency in primates.
In the embryonic brain, both neurons and astrocytes/oligodendrocytes are derived from the common multipotent neuroepithelial cells (NECs) but neurogenesis precedes gliogenesis. However, early brain development in primates has distinct features. Unlike other animals, the primates contain white matter in a greater proportion40, and glial cell proportion increases faster than the gray matter in the primate brains41,42. Proliferation of progenitor cells occurs in the SVZ, which is small in rodents but much larger and complex in the primates25,43,44. Also, the expansion of progenitor cells in the oSVZ leads to cerebral gyrification, a process that is absent in rodents25,33. Oligodendrocytes are one of the major types of glial cells that are generated in late embryonic stage. During postnatal development, gliogenesis and myelination continue in the primate brains38,39,45.
Our findings provide several lines of evidence to support that CHD8 mutations enhance gliogenesis in the primate brain. First, western blotting and immunohistological studies of the brains of M1 and M2 monkeys show the elevated expression of glial proteins for astrocytes and oligodendrocytes before birth. Quantification of the numbers of astrocytes and oligodendrocytes in these two mutant monkey brains verified a greater number of glial cells in their brains than the age-matched control monkey brains. However, examining neuronal proteins and numbers did not show any significant differences between CHD8 mutant and control monkeys, suggesting that neurogenesis is not significantly altered by CHD8 mutations. Second, in the brain region around LV that was undergoing gliogenesis, we observed much more glial cells and their progenitor cells in M1 than the control monkey. Also, in the brain cortex of newborn monkeys, we observed more glial cells and enlarged white matter in M2 when compared with control monkey. The increased gliogenesis in M1 and M2 and macrocephaly in the live CHD8 mutant monkey provide critical evidence to support the idea that gliogenesis in the oSVZ, which is absent in rodents, might be responsible for the expansion and folding of the cortical surface in the primates29.
In support of the results from embryonic CHD8-targeted monkeys, we also conducted experiments to delete the CHD8 gene in the fetal monkey brains before gliogenesis occurs and in the brain slices from newborn monkeys. The results consistently showed that CHD8 deficiency could increase the numbers of glial cells without influencing neuronal density and differentiation. The more important finding is that targeting CHD8 prior to gliogenesis in monkey fetuses could also increase the number of astrocytes and oligodendrocytes, strongly supporting the idea that CHD8 regulates gliogenesis during early brain development. In addition, transcriptional profiling results from the M2 monkey are in line with the dysfunction of CHD8-mediated gene expression in the CHD8 mutant monkey brain. The effects of CHD8 mutations on the proliferation and number of glial cells in the macaque monkey are particularly relevant to the macrocephaly phenotype seen in the human brain6,8,9,10. Indeed, the live monkey with CHD8 mutations showed enlarged white matter as compared with the age-matched control monkeys. Glial proliferation and myelination in the primate brains continue after birth and last until adulthood38,39. This unique feature makes nonhuman primates an ideal model to investigate gliogenesis when compared with rodents in which glial proliferation and myelination are largely completed after birth46,47. Using brain slices from newborn monkeys, we could examine the proliferation of glial cells and found that CHD8 knockdown also increased the numbers of astrocytes and oligodendrocytes. This in vitro evidence strongly supports the in vivo finding that CHD8 mutations in monkey embryos led to the increased gliogenesis in the developing monkey brains.
CHD8 is one of the ATP-dependent chromatin remodeling factors that regulate the transcription of a large number of genes48,49. Although it remains unknown how CHD8 can differentially regulate gene expression in various types of cells at different developmental stages, it appears that the complex regulation of transcriptional networks by CHD8 may be cell type- and species-dependent. In the rodent progenitor cells, CHD8 can repress the activities of multiple transcription factors for neuronal differentiation such that CHD8 deficiency was thought to increase neurogenesis15,17. CHD8 deficiency was further found to affect axonal elongation and neuronal survival50. The effects of CHD8 deficiency may be species-dependent, as complete elimination of CHD8 in oligodendrocytes was found to inhibit the differentiation and maturation of oligodendrocytes in the rodent brain51,52. However, CHD8 deficiency-mediated suppression of oligodendrogenesis found in mouse models is hardly accountable for enlarged brain size that occurs in the majority of patients with CHD8 mutations. Instead, our studies of the nonhuman primate revealed an increase in gliogenesis caused by CHD8 deficiency. This increased gliogenesis was caused by CHD8 haploinsufficiency rather than complete ablation, because CRISPR/Cas9 targeting leads to knockdown or incomplete depletion effect due to the nature of mosaic mutations, which can mimic heterozygous inactivation of CHD8 in patients, at least to some extent. Since complete elimination of CHD8 can result in embryonic14 and postnatal51,52 lethality in mice, the extent to which CHD8 is suppressed may have differential effects on neuronal and glial cells as well as their progenitor cells. Indeed, CHD8’s effects as a transcriptional regulator are dosage-dependent17. Thus, it is possible that CHD8 mutation or haploinsufficiency preferentially affects transcription networks or/and signaling pathways for the differentiation of progenitor cells to glial cells and their proliferation in the primate brain.
Due to the complex and sophisticated regulation of gene transcription by CHD8 in different types of cells and at different developmental stages, understanding ASD pathogenesis related to CHD8 mutations faces considerable challenges. A large number of studies have employed transcription analysis of CHD8 mutant cellular and animal models but have not reached a conclusion regarding how CHD8 mutations cause macrocephaly53,54. Our analysis of gene expression in CHD8 mutant monkey brains suggests that knocking down CHD8 can upregulate a number of genes that are important for the proliferation of glial cells and increase the proliferation of Ki67-positive progenitor cells in SVZ. The more important evidence would be the increased number of glial cells in the CHD8 mutant monkey brains, which reflects the outcome of deficiency in the CHD8-mediated complex regulation of gene expression. To validate this important finding, we utilized in utero injection of the fetal monkey brain and brain slices from newborn monkeys to knockdown CHD8, and the results support those from the monkey model generated by embryonic CHD8 targeting.
Another important evidence for increased gliogenesis in the CHD8 mutant monkey brains is the macrocephaly of the live CHD8 mutant monkey, which mirrors the enlargement of head size seen in patients with CHD8 mutations7,8,9,55. The white matter of the cerebral cortex contains all axons that support long-range cortical connectivity and are essential to cognitive function40. Germline heterozygous mutations in PTEN have been identified in children with both ASD and macrocephaly, and are associated with marked abnormalities in the brain white matter and reduced cognitive ability56. Thus, any alterations in the development of either neuronal or glial cells would adversely affect neuronal activities, leading to altered behaviors. Indeed, MRI examination has revealed abnormal activity or morphology of myelin and white matter in the brains of ASD patients57,58,59,60. Although our findings demonstrated for the first time that CHD8 deficiency enhances gliogenesis in the primate brain and suggest that abnormal gliogenesis may play an important role in CHD8 mutation-mediated pathogenesis, further mechanistic studies are required to understand how CHD8 deficiency selectively alters gliogenesis. Despite this, the findings suggest that increased gliogenesis could account for the impaired white matter connectivity or function, at least in CHD8 mutant patients. The findings that CHD8 mutant monkeys show increased gliogenesis and macrocephaly underscore the importance in investigating the role of abnormal gliogenesis in ASD. These findings also have therapeutic implications for the correction of abnormal gliogenesis to achieve more effective treatments for ASD.
Materials and methods
Reagents
Reagent or resource source identifier | ||||
|---|---|---|---|---|
Antibodies | Source | Cat. # | IHC/WB dilution | Company |
CHD8 | Rabbit | A301-224A | IHC 1:1600 | Bethyl |
CHD8 | Rabbit | A301-225A | IHC 1:800 WB 1:1000 | Bethyl |
Doublecortin (DCX) | Rabbit | 4604 | WB 1:2000 IHC 1:500 | Cell Signaling |
GFAP | Mouse | KIT-0031 | IHC 1:300 | MAIXIN |
GFAP | Rabbit | Z0334 | WB 1:10000 | Dako |
NeuN | Mouse | MAB-0578 | IHC 1:100 | MAIXIN |
NeuN | Mouse | Ab104224 | WB 1:5000 | Abcam |
Olig2 | Rabbit | ZA-0561 | IHC 1:100 | ZSGB |
Olig2 | Rabbit AB9610 | WB 1:1000 Merck Millipore | ||
Homer | Mouse | Sc-17842 | WB 1:2000 | Santa Cruz |
MBP | Rat | ab7349 | WB 1:500 | Abcam |
Pax6 | Rabbit | 901301 | IHC 1:500 | Bio legend |
TBR1 | Rabbit | Ab31940 | IHC 1:500 | Abcam |
PV | Rabbit | Ab11427 | WB 1:1000 | Abcam |
Calbindin1 | Rabbit | AB1778 | IHC 1:500 | Millipore |
Tubulin | Mouse | T5168 | WB 1:20000 | Sigma |
Iba1 | Rabbit | 019-19741 | IHC 1:500 | Wako |
MYRF | Rabbit | ABN45 | WB 1:1000 | Merck Millipore |
Sox9 | Mouse | MABC277 | WB 1:2000 | Merck Millipore |
Sox9 | Rabbit | AB5535 | IHC 1:500 | Merck Millipore |
Actin | Mouse | MAB1501 | WB 1:5000 | Merck Millipore |
Vinculin | Rabbit | Ab129002 | WB 1:10,000 | Abcam |
Experimental models: cell lines | ||
|---|---|---|
HEK 293 cell | ATCC (cat# CRL-1573) | N/A |
Experimental models: organisms/strains | ||
|---|---|---|
CHD8 mutant monkeys | This paper | |
Gene | sequence | length | experiment |
|---|---|---|---|
SRY | TGCTTCTGCCATGTTAAGCGT CGGAAGCAAACTGCAACTGTTTTG | 427 bp | Gender detection in T1, T2, T3 |
CHD8 | TCAAGTTGGGATTGGATAAAG CACAATGACTCACTACAAACTTAT | 110 bp | Deep amplicon sequencing |
TGTCCACGCAGACTTTAGTG GGCACAGACAGAACATGCCTT AGTGTGCTGACACCTGGTCTAGATA CTTGGAGCCTTCATCATCTTCCT | Nested PCR 641 bp | Genotyping in CHD8 exon 19 |
Recombinant plasmid | ||
|---|---|---|
Plasmid: Cas9-MLM3613 T7 promoter | Cas9 with CMV | Addgene Plasmid#4225 |
Plasmid: pSpCas9(BB)-2A-GFP used in cultured cells | Cas9 together with sgRNA | Addgene Plasmid#48138 |
Plasmid: sgRNA with T7 promoter | sgRNA with T7 promoter | Liangxue Lai’s lab |
Software and algorithms | ||
|---|---|---|
GraphPad Prism 8 | Graphpad Software | |
Stereo Investigator 5.4.3 | Micro Bright Field Bioscience | |
Image J | National Institutes of Health | |
Aperio ImageScope | Leica | https://www.leicabiosystems.com/digital-pathology/manage/aperioimagescope/ |
MatLab (R2019b) | MathWorks | |
SPM 12 | WDCN | |
RadiAnt DICOM View | Medixant | |
Actical 3.1.1 | Respironics | https://bmedical.com.au/productcategory-/activity-heatresearch/actical/ |
MicroView 2.5.0 | Parallax Innovations | |
Other | ||
|---|---|---|
Axio Imager A2 | Zeiss | Carl Zeiss, Germany |
Zeiss LSM 800 Confocal Laser Scanning Microscope | Zeiss | Carl Zeiss, Germany |
Leica Aperio GT45 | Leica | Leica Biosystems, Germany |
GE Locus SP Micro CT Scanner | GE | GE, USA |
GE Discovery MR750 3.0 T Scanner | GE | GE, USA |
Actical Physical Monitors | N/A | Respironics, USA |
SONY FDR-AX45 Video Camera | SONY | SONY, Japan |
JVC GC-P100BAC Video Camera | JVC | JVC, Japan |
Infrared Video Camera C6p | HIKVISION | HIKVISION, China |
Actical Physical Monitors | Respironics | Respironics, USA |
Monkey
CHD8 mutant monkeys and age-, gender-matched control monkeys (M. fascicularis) were raised at Guangdong Landau Biotechnology Co. Ltd., which is an Association for Assessment and Accreditation of Laboratory Animal Care-accredited facility. Mutant and control monkeys used in this study were listed below. The commercial monkey diet (Ke-Ao, #HFZ-15kg, Beijing) was offered twice daily. Monkey health was monitored by veterinarians. All animal-related protocols were approved in advance by the Animal Care and Use Committee of Guangdong Landau Biotechnology Co. Ltd. This study was carried out in strict compliance with the “Guide for the Care and Use of Laboratory Animals of the Institute of Laboratory Animal Science (est. 2006)” and “the use of nonhuman primates in research of the Institute of Laboratory Animal Science (est. 2006)” to ensure the safety of personnel and animal welfare.
Animal information
Name | Gender | Genotype | Note | ||
|---|---|---|---|---|---|
M1 | M | CHD8 mutant | Aborted (E125) | ||
M2 | M | CHD8 mutant | Stillborn (E158) | ||
M3 | M | CHD8 mutant | Alive (54 M) | ||
T1, T2, T3 | F, M, M | CHD8 mutants | Aborted (E53) | ||
Control 1 | F | Wild type | Cesarean (E130) | ||
Control 2 | M | Wild type | Newborn (E160) | ||
Control 3 | M | Wild type | Alive (54 M) | ||
Control 4 | M | Wild type | Alive (54 M) | ||
Control 5 | M | Wild type | Alive (54 M) | ||
Newborn-1 | M | Wild type | Brain slice culture | ||
Newborn-2 | M | Wild type | Brain slice culture | ||
Newborn-3 | M | Wild type | Brain slice culture | ||
Newborn-4 | M | Wild type | Brain slice culture | ||
Newborn-5 | F | CHD8 gRNA | Brain development analysis | ||
injection at G55 | |||||
Newborn-6 | M | CHD8 gRNA injection at G55 | Brain development analysis | ||
Newborn-7 | M | Control gRNA injection at G55 | Brain development analysis | ||
Genotype analysis of targeted site
Genomic DNA was isolated from tissue lysate using the Quick-gDNATM MiniPrep (Zymo Research) according to the manufacturer’s protocol. The target sites were PCR amplified with primers listed above. The PCR products were purified with Qiaquick PCR Purification Kit (Qiagen, 28106). For T7E1 cleavage assay, purified PCR products were denatured and reannealed in NEBuffer 2 (NEB) using a thermocycler. Reannealed PCR products were digested with T7 endonuclease 1 (NEB, M0302L) and separated by 2.5% agarose gel. The PCR products were sub-cloned into the pEASY-blunt simple cloning vector (Transgene) and transformed to competent DH5α cells. After overnight culture at 37 °C, emerged colonies were picked up randomly and sequenced. Deep amplicon sequencing was performed on various tissues to analyze mosaic in mutant monkeys.
RNA preparation and sequencing
Total RNA was extracted from the prefrontal cortex (PFC) of CHD8 mutant M2 and age-matched control-2 (three samples each). Only samples with RNA integrity number (RIN) over 6.8 were used for cDNA library construction. Sequencing was performed on a single lane of an Illumina HiSeq 4000 to produce 150 bp paired-end reads. We performed three independent replicates from adjacent areas for each animal. The clean reads were aligned with Bowtie261 to the cynomolgus monkey genome (version 5.0). Duplicates were removed using Sambamba (version 0.6.8). Unique mapped reads were assigned to each gene according to Ensembl genome annotation. Differential expression analysis was performed by DEseq2.
Off-target analysis by whole-genome sequencing
Whole-genome sequencing data was used to analyze off-targets in CRISPR/Cas9 editing. Extracted DNA was sequenced as paired-end reads on the Illumina HiSeq2000 platform. Possible off-target sites of sgRNA with no more than five mismatched sites were identified using the bio-information-based search tool, Cas-OFFinder. A total of 1326 possible off-target regions with NAG or NGG PAM sequences were identified. Relative sequencing depth for most likely off-target loci by sgRNA in M3 was calculated by normalizing the number of mapped reads in those loci to the genome-wide average of mapped reads. We found no off-target mutations in mutant monkeys M1 and M2. A mutation site with 5 bp mismatches from CHD8 gRNA was found in M3, which is likely an inherited or de novo mutation.
Organotypic monkey brain slice culture and lentiviral infection
The brain cortex of newborn male monkeys at postnatal day 1–3 was carefully dissected on ice, directly transferred into ice-cold artificial cerebrospinal fluid (ACSF) (SL6630, Coolaber, Beijing, China) equilibrated with carbogen (95% O2, 5% CO2). The tissues were cut to generate brain slices at 300 μm thickness using a Leica VT1200S vibrating blade microtome (Leica, Wetzlar, Germany). The slices were then transferred onto 30 mm Millicell membrane inserts (0.4 μm; Millipore, Bedford, MA, USA) and kept in six-well cell culture plate containing 1.5 mL media (Neurobasal/DMEM 1:1, 5% fetal bovine serum, 5% horse serum, 1% N2, 2% B27, 2 mM lGlutamax, 5 ng/mL GDNF, 100 U/mL Pen-Strep; all from Gibco). The plates were incubated at 37 °C in a 5% CO2 humidified incubator. The medium was replaced with fresh medium three times each week. After 6 h of culture, virus infection was carried out according to experimental needs. The viral vectors used were lentiviral-CHD8 gRNA/CMV-GFP and lentiviral-EFs Cas9. These viral vectors had been packaged by Guangzhou IGE Biotechnology LTD to generate purified viruses. The genomic titer of purified viruses was ~109 vg/mL determined by PCR method.
We generated lentiviral vectors to express CHD8 gRNA or control gRNA that did not target any gene62 (Supplementary Fig. S9a). To examine the effect of knocking down CHD8 on glial proliferation, cultured monkey brain slices were infected with lentiviral viruses (each brain slice infected with 1 µL lentiviral gRNA and lentiviral Cas9). These viruses were diluted in culture medium and then added to the brain cortical slices containing the LV area. After 10–14 days of viral infection, immunofluorescent staining was performed to examine glial cell numbers and morphology. We used four newborn monkeys for organotypic brain slice culture and found 3 of them provided brain slices with effective lentiviral infection.
In utero delivery of lentiviruses to the fetal monkey brain
The pregnancy after timed mating was assessed periodically by ultrasound examination, and pregnancy was confirmed with ultrasound evidence of fetal heart activity. Pregnant monkeys having fetal monkeys at embryonic day 55 (E55) or gestational day 55 (G55) were selected for in utero injection. After induction of anesthesia of the pregnant monkey with intramuscular injection of Zoletil®50 at a dose of 5 mg/kg, a 25-G Quincke needle (BectonDickenson) was used to target the LV of the fetal monkey brain closest to the anterior maternal abdomen under continuous ultrasound imaging. Lentiviruses (50 μL lentivirus CHD8 or control gRNA mixed with 100 μL lentiviral Cas9 were injected as a slow bolus to deliver a total dose of 1.0 × 108 vg (lentiviral gRNA) and 2.0 × 108 vg (lentiviral Cas9) in each fetus. The correct delivery would result in ventricular swelling, and the needle was removed immediately. The injected fetus was monitored for another 15 min. We obtained two male and one female newborn monkeys, which were delivered naturally. These newborn monkeys were euthanized with intramuscular injection of Barbiturates with the dose of 100 mg/kg at postnatal day 7 or 8 to isolate their brains for analysis.
Western blotting analysis and immunohistography
For western blotting analysis, PFC or other brain region was homogenized in RIPA buffer (Hua Xing Bo Chuang, with 1× protease inhibitor cocktail) on ice. Protein lysates were separated by SDS-PAGE and transferred onto PVDF membranes (Millipore). The primary antibodies used are listed in Materials and methods. Specific bands were quantified by Image J and normalized to α-tubulin or other reference protein expression.
For immunohistography, the brains of controls and mutants were dissected out and fixed for 48 h in 4% paraformaldehyde. Different brain regions including PFC and cerebellum were paraffin-embedded. Paraffin-embedded tissues were sliced into 4-μm-thick sections. HRP-conjugated secondary antibodies (anti-mouse or anti-rabbit, 1:1000, Dako) were used. DAB (3, 3'-diaminobenzidine) staining was used for chemiluminescent detection. Hematoxylin staining was performed to label the nucleus. Images were acquired with a Leica Aperio GT450 slide scanner. For cell density analysis, cells within the certain area were counted manually. LFB staining (RY-0013, Zhong Ke Wan Bang) was performed to analyze myelin changes in mutant monkeys.
MRI image acquisition
Animals were anesthetized by ketamine (10 mg/kg, i.m.) and placed into the scanner in the prone position for T1-weighted 3-dimensional MR scan. The scans were collected on a 3.0 T MR machine (GE 750) using a custom-designed 8-channel radio-frequency surface head coil at Jinan University. We performed T1 sequence, with the following parameters: TR = 9.5 ms, TE = 4.0 ms, slice thickness = 0.5 mm, slice per slab = 196, matrix size = 256 × 256 mm, FOV = 150 × 150 mm, and voxel size = 0.5 × 0.5 × 0.5 mm.
Regional volumetric computation
The statistical parameter mapping software (SPM 12, Wellcome Department of Cognitive Neurology, London, UK) with in-house MatLab scripts was employed to perform voxel-based morphometry (VBM)63 for calculating the volume of brain regions of each animal. A study-specific anatomical template was built and tissue classification was performed using the Diffeomorphic Anatomical Registration Through Exponentiated Lie algebra (DARTEL) pipeline64. First, the T1-weighted images were registered to the rhesus monkey brain template64, resulting in individual T1-weighted images in the standard space with an isotropic spatial resolution of 0.3 mm. The unified segmentation method65 was then used to segment these images into gray matter (GM), white matter (WM) and cerebrospinal fluid (CSF). The tissue segmentations were used to generate the final study-specific template, GM, WM, and CSF probability maps using the DARTEL pipeline. The individual GM, WM, and CSF segmentation were then spatially normalized to the study-specific template. Modulated tissue maps were generated by using the Jacobian determinant of the deformation field obtained in the normalization process to encode the expansion or contraction of voxels. Therefore, the tissue volume in each voxel was preserved in the modulated images, and the tissue volume of a given region of interest (ROI) could be estimated as the summation of the modulated voxel intensities multiplied by the voxel size. The modulated tissue maps were spatially smoothed for noise reduction using a Gaussian kernel with full width at half maximum of 0.9 mm. The brain atlas66 was affine registered to the study-specific template generated with the DARTEL pipeline, and the ROIs defined by the regional boundaries in the atlas were used for brain region volume calculation. The volumes of gray matter and white matter in each ROI were calculated separately.
Quantification and statistical analysis
Analysis of RNA-seq data
Differentially expressed genes were identified using the following thresholds: log2-fold change > 1.5 and Benjamini–Hochberg adjusted P value < 0.05. For GO enrichment analysis, enriched terms were defined using fisher’s exact test with the following filters: odds ratio > 2 and Benjamini–Hochberg adjusted P value < 0.05.
Analysis of immunohistochemical data
For quantification of Olig2-positive cells in immunostaining of the brains of CHD8 mutant monkeys, rectangles of equal size (such as 0.04 mm2) per picture (×20) were quantified for positively stained cells. Positive nuclei in the assigned areas were counted manually and normalized for the area surface or total cell number. For quantification of GFAP staining intensities in control vs. mutant, the total intensity was measured using Image J software. Brightness and contrast were adjusted where needed.
For quantification of glial cells in newborn monkeys that were injected with lentiviral CHD8 gRNA and Cas9 at E55, we selected GFP-positive brain regions for quantification because lentiviral gRNA also expressed GFP so that GFP-positive areas reflect the expression of lentiviral gRNA and Cas9 when both were mixed for injection. Each brain was sectioned serially using a sliding microtome at 40 μm. Sections were collected into tissue collection solution and stored at −20 °C until processing. Brain sections were first examined under a fluorescent microscope to examine GFP signaling in order to ensure that the brain region had been transduced by lentiviral vectors. This process is necessary to obtain the viral transduced brain region, as lentiviral transduction in the fetal brain was restricted to some extent. We used anti-Sox9 to identify astrocytes and anti-olig2 to identify oligodendrocytes. Because these antibodies label glial nuclei so that glial cells could be unequivocally assessed for their numbers and density. In addition, DAPI staining was used to confirm the presence of nuclei of glial cells. To ensure antibody specificity, staining was also performed without the primary antibody but secondary antibody. Images containing Sox9 or olig2-positive cells at ×200 magnification or 0.31 mm2 were acquired by Zeiss (Carl Zeiss, Germany) and processed with Zeiss software (Carl Zeiss, Germany). Every 32nd section (or 1.3 mm apart) was used for quantitative analysis of immunostaining, and six and eight sections from the brain cortical region in control gRNA/Cas9 and CHD8 gRNA/Cas9, respectively, targeted monkeys were used to quantify the numbers of cells. At least four images (20×) from each brain section were taken to obtain the average numbers of glial cells per mm2. The staining results were scored by two investigators blinded to the genotypes (control and CHD8 targeted) of the monkey brains.
A similar method was also used to quantify glial cells in the monkey brain slices that were infected by lentiviral CHD8 or control gRNA with lentiviral Cas9. Because each monkey brain can provide multiple brain slices (400 µm each slice), at least three slices from each animal were infected by lentiviral CHD8 or control gRNA with lentiviral Cas9. After infection for 14 days, the infected brain slices were fixed and sectioned at 40 µm. Because each brain slice was infected by lentivirus and cultured independently, the number of brain slices represents biological replicates. We used a total nine brain slices from three newborn monkeys (three slices per animal) for quantitative analysis. At least three brain sections (40 µm) containing GFP-positive areas, which was identified under a fluorescent microscope, in each brain slice were used for quantification of astrocytes labeled by anti-Sox9 and oligodendrocytes labeled by anti-olig2. The infected brain regions next to the LV were selected for quantification. At least four images (×20) in each brain section were taken for counting the number of glial cells to provide the average number of glial cells in each brain slice that was used for quantification. The average numbers of glial cells per mm2 were used for comparison between control and CHD8-targeted monkey brain slices.
Statistical analysis
Data analysis was conducted using GraphPad Prism version 6 or 8. The normality of the data was analyzed first. Data were analyzed using two-tailed Student’s t test or Mann–Whitney U test for comparing two groups and univariate or two-way analysis of variance (ANOVA) and subsequent post hoc t tests using a Dunnett. All data were presented as means ± SE. The alpha level was set at P = 0.05 (NA, not significant, *P < 0.05; **P < 0.01; ***P < 0.001).
Data availability
All data generated or analyzed during this study are included in this article and its supplementary information files. The sequence data of RNA-seq and whole-genome sequencing of monkeys reported in this project have been deposited to Genome Sequence Archive (GSA, https://ngdc.cncb.ac.cn/gsa/) with accession number CRA003937.
References
O’Roak, B. J. et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250 (2012).
Talkowski, M. E. et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 149, 525–537 (2012).
Neale, B. M. et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245 (2012).
Krumm, N., O’Roak, B. J., Shendure, J. & Eichler, E. E. A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 37, 95–105 (2014).
Wang, T. et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat. Commun. 7, 13316 (2016).
An, Y. et al. De novo variants in the Helicase-C domain of CHD8 are associated with severe phenotypes including autism, language disability and overgrowth. Hum. Genet. 139, 499–512 (2020).
Bernier, R. et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158, 263–276 (2014).
Prontera, P. et al. Recurrent ∼100 Kb microdeletion in the chromosomal region 14q11.2, involving CHD8 gene, is associated with autism and macrocephaly. Am. J. Med. Genet. A 164a, 3137–3141 (2014).
Merner, N. et al. A de novo frameshift mutation in chromodomain helicase DNA-binding domain 8 (CHD8): a case report and literature review. Am. J. Med. Genet. A 170a, 1225–1235 (2016).
Douzgou, S. et al. The clinical presentation caused by truncating CHD8 variants. Clin. Genet. 96, 72–84 (2019).
Durak, O. et al. Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat. Neurosci. 19, 1477–1488 (2016).
Katayama, Y. et al. CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature 537, 675–679 (2016).
Platt, R. J. et al. Chd8 mutation leads to autistic-like behaviors and impaired striatal circuits. Cell Rep. 19, 335–350 (2017).
Nishiyama, M. et al. Early embryonic death in mice lacking the beta-catenin-binding protein Duplin. Mol. Cell. Biol. 24, 8386–8394 (2004).
Gompers, A. L. et al. Germline Chd8 haploinsufficiency alters brain development in mouse. Nat. Neurosci. 20, 1062–1073 (2017).
Jung, H. et al. Sexually dimorphic behavior, neuronal activity, and gene expression in Chd8-mutant mice. Nat. Neurosci. 21, 1218–1228 (2018).
Sood, S. et al. CHD8 dosage regulates transcription in pluripotency and early murine neural differentiation. Proc. Natl. Acad. Sci. USA 117, 22331–22340 (2020).
Sugathan, A. et al. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc. Natl. Acad. Sci. USA 111, E4468–E4477 (2014).
Zhao, H. et al. Altered neurogenesis and disrupted expression of synaptic proteins in prefrontal cortex of SHANK3-deficient non-human primate. Cell Res. 27, 1293–1297 (2017).
Tu, Z. et al. CRISPR/Cas9-mediated disruption of SHANK3 in monkey leads to drug-treatable autism-like symptoms. Hum. Mol. Genet. 28, 561–571 (2019).
Yang, W. et al. CRISPR/Cas9-mediated PINK1 deletion leads to neurodegeneration in rhesus monkeys. Cell Res. 29, 334–336 (2019).
Yin, P. et al. Caspase-4 mediates cytoplasmic accumulation of TDP-43 in the primate brains. Acta Neuropathol. 137, 919–937 (2019).
Yang, W. et al. PINK1 kinase dysfunction triggers neurodegeneration in the primate brain without impacting mitochondrial homeostasis. Protein Cell 13, 26–46 (2022).
Azevedo, F. A. et al. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J. Comp. Neurol. 513, 532–541 (2009).
Lui, J. H., Hansen, D. V. & Kriegstein, A. R. Development and evolution of the human neocortex. Cell 146, 18–36 (2011).
Workman, A. D. et al. Modeling transformations of neurodevelopmental sequences across mammalian species. J. Neurosci. 33, 7368–7383 (2013).
Otani, T. et al. 2D and 3D stem cell models of primate cortical development identify species-specific differences in progenitor behavior contributing to brain size. Cell Stem Cell 18, 467–480 (2016).
Ventura-Antunes, L., Mota, B. & Herculano-Houzel, S. Different scaling of white matter volume, cortical connectivity, and gyrification across rodent and primate brains. Front. Neuroanat. 7, 3 (2013).
Rash, B. G. et al. Gliogenesis in the outer subventricular zone promotes enlargement and gyrification of the primate cerebrum. Proc. Natl. Acad. Sci. USA 116, 7089–7094 (2019).
Graves, J. A. Weird mammals provide insights into the evolution of mammalian sex chromosomes and dosage compensation. J. Genet. 94, 567–574 (2015).
Dimou, L. et al. Progeny of Olig2-expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. J. Neurosci. 28, 10434–10442 (2008).
Rakic, P. Neurons in rhesus monkey visual cortex: systematic relation between time of origin and eventual disposition. Science 183, 425–427 (1974).
Hansen, D. V., Lui, J. H., Parker, P. R. & Kriegstein, A. R. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 464, 554–561 (2010).
Petrik, D. et al. Early postnatal in vivo gliogenesis from nestin-lineage progenitors requires cdk5. PLoS One 8, e72819 (2013).
Mony, T. J. et al. Valproic acid exposure during early postnatal gliogenesis leads to autistic-like behaviors in rats. Clin. Psychopharmacol. Neurosci. 14, 338–344 (2016).
Zhang, Y. et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947 (2014).
Huang, B. et al. Mutant huntingtin downregulates myelin regulatory factor-mediated myelin gene expression and affects mature oligodendrocytes. Neuron 85, 1212–1226 (2015).
Bakken, T. E. et al. A comprehensive transcriptional map of primate brain development. Nature 535, 367–375 (2016).
Zhu, Y. et al. Spatiotemporal transcriptomic divergence across human and macaque brain development. Science 362, eaat8077 (2018).
Fields, R. D. White matter in learning, cognition and psychiatric disorders. Trends Neurosci. 31, 361–370 (2008).
Herculano-Houzel, S., Mota, B., Wong, P. & Kaas, J. H. Connectivity-driven white matter scaling and folding in primate cerebral cortex. Proc. Natl. Acad. Sci. USA 107, 19008–19013 (2010).
Mota, B. et al. White matter volume and white/gray matter ratio in mammalian species as a consequence of the universal scaling of cortical folding. Proc. Natl. Acad. Sci. USA 116, 15253–15261 (2019).
Cheung, A. F. et al. The subventricular zone is the developmental milestone of a 6-layered neocortex: comparisons in metatherian and eutherian mammals. Cereb. Cortex 20, 1071–1081 (2010).
Molnár, Z. et al. Evolution and development of the mammalian cerebral cortex. Brain Behav. Evol. 83, 126–139 (2014).
Jakovcevski, I. et al. Oligodendrocyte development and the onset of myelination in the human fetal brain. Front. Neuroanat. 3, 5 (2009).
Privat, A. Postnatal gliogenesis in the mammalian brain. Int. Rev. Cytol. 40, 281–323 (1975).
Semple, B. D. et al. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 106-107, 1–16 (2013).
Marie, C. et al. Oligodendrocyte precursor survival and differentiation requires chromatin remodeling by Chd7 and Chd8. Proc. Natl. Acad. Sci. USA 115, E8246–E8255 (2018).
Goodman, J. V. & Bonni, A. Regulation of neuronal connectivity in the mammalian brain by chromatin remodeling. Curr. Opin. Neurobiol. 59, 59–68 (2019).
Xu, Q. et al. Autism-associated CHD8 deficiency impairs axon development and migration of cortical neurons. Mol. Autism 9, 65 (2018).
Zhao, C. et al. Dual requirement of CHD8 for chromatin landscape establishment and histone methyltransferase recruitment to promote CNS myelination and repair. Dev. Cell 45, 753–768 (2018).
Kawamura, A. et al. Oligodendrocyte dysfunction due to Chd8 mutation gives rise to behavioral deficits in mice. Hum. Mol. Genet. 29, 1274–1291 (2020).
Hoffmann, A. & Spengler, D. Chromatin remodeler CHD8 in autism and brain development. J. Clin. Med. 10, 366 (2021).
Weissberg, O. & Elliott, E. The mechanisms of CHD8 in neurodevelopment and autism spectrum disorders. Genes 12, 1133 (2021).
Yasin, H. et al. A distinct neurodevelopmental syndrome with intellectual disability, autism spectrum disorder, characteristic facies, and macrocephaly is caused by defects in CHD8. J. Hum. Genet. 64, 271–280 (2019).
Frazier, T. W. et al. Molecular and phenotypic abnormalities in individuals with germline heterozygous PTEN mutations and autism. Mol. Psychiatry 20, 1132–1138 (2015).
Boddaert, N. et al. MRI findings in 77 children with non-syndromic autistic disorder. PLoS ONE 4, e4415 (2009).
Deoni, S. C. et al. White-matter relaxation time and myelin water fraction differences in young adults with autism. Psychol. Med. 45, 795–805 (2015).
Hardan, A. Y. et al. A proton spectroscopy study of white matter in children with autism. Prog. Neuropsychopharmacol. Biol. Psychiatry 66, 48–53 (2016).
Thompson, A. et al. Age-related differences in white matter diffusion measures in autism spectrum condition. Mol. Autism 11, 36 (2020).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Yang, S. et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J. Clin. Invest. 127, 2719–2724 (2017).
Ashburner, J. & Friston, K. J. Voxel-based morphometry—the methods. Neuroimage 11, 805–821 (2000).
Ashburner, J. A fast diffeomorphic image registration algorithm. Neuroimage 38, 95–113 (2007).
Liu, C. et al. Rhesus monkey brain development during late infancy and the effect of phencyclidine: a longitudinal MRI and DTI study. Neuroimage 107, 65–75 (2015).
Ashburner, J. & Friston, K. J. Unified segmentation. Neuroimage 26, 839–851 (2005).
Acknowledgements
This work was supported by Guangzhou Key Research Program on Brain Science (202007030008, X.-J.L.); Department of Science and Technology of Guangdong Province (2021ZT09Y007; 2020B121201006, 2018B030337001, X.-J.L.), the Strategic Priority Research Program B of the Chinese Academy of Sciences (XDBS1020100 to Y.Q.Z.), the National Key Research and Development Program (2019YFA0707100 and 2021ZD0203901 to Y.Q.Z.), the National Science Foundation of China to Y.Q.Z. (31830036 and 31921002), H.Z. (32100783) and X.J.L. (81830032, 31872779), the Guangdong-Hong Kong-Macao Greater Bay Area Center for Brain Science and Brain-Inspired Intelligence Fund (2019018, B.L.), the Postdoctoral Science Foundation of China (2019M653275, B.L.), and the Fundamental Research Funds for the Central Universities (21619104, L.W.). We thank Drs. Heng Lu and Xiaoqun Wang for advice, and Qi Wang, Fei Zhang, Xudong Liu, An Yin, Qi Shi, Dazhang Bai, and Laiqiang Chen for technical assistance.
Author information
Authors and Affiliations
Contributions
Y.Z. and X.-J.L. conceptualized the project, supervised data collection and analysis. B.L. and H.Z. designed and performed the majority of experiments. Z.T., X.G., and S.Y. performed monkey embryo injection and transfer experiments. B.L., W.Y., R.H., X.P.L., M.P., X.C., J.Z., and P.Y. performed in utero injection, organoid brain slice cultures, and histological studies; H.J.X., J.L., Y, L., and Y.L. assisted animal experiments. L.W., Z.Y., B.Z., Z.T., H.X., and T.J. performed and analyzed MRI. Z.Z. analyzed RNA-seq data. X.-J.L. wrote the manuscript and Y.Z. edited the manuscript. S.L. and Y.J. provided advice.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, B., Zhao, H., Tu, Z. et al. CHD8 mutations increase gliogenesis to enlarge brain size in the nonhuman primate. Cell Discov 9, 27 (2023). https://doi.org/10.1038/s41421-023-00525-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41421-023-00525-3
This article is cited by
-
Tauopathy promotes spinal cord-dependent production of toxic amyloid-beta in transgenic monkeys
Signal Transduction and Targeted Therapy (2023)
