
Abstract
Ischemia-reperfusion injury (IRI) is a common cause of acute kidney injury (AKI). The kidney is susceptible to IRI under several clinical conditions, including hypotension, sepsis, and surgical procedures, such as partial nephrectomy and kidney transplantation. Extensive research has been conducted on the mechanism and intervention strategies of renal IRI in past decades; however, the complex pathophysiology of IRI-induced AKI (IRI-AKI) is not fully understood, and there remains a lack of effective treatments for AKI. Renal IRI involves several processes, including reactive oxygen species (ROS) production, inflammation, and apoptosis. Mitochondria, the centers of energy metabolism, are increasingly recognized as substantial contributors to the early phases of IRI. Multiple mitochondrial lesions have been observed in the renal tubular epithelial cells (TECs) of IRI-AKI mice, and damaged or dysfunctional mitochondria are toxic to the cells because they produce ROS and release cell death factors, resulting in TEC apoptosis. In this review, we summarize the recent advances in the mitochondrial pathology in ischemic AKI and highlight promising therapeutic approaches targeting mitochondrial dysfunction to prevent or treat human ischemic AKI.
Similar content being viewed by others

Facts
-
1.
The hallmarks of IRI-AKI are elevated levels of ROS, mitochondrial dysfunction, and inflammation.
-
2.
Mitochondrial dysfunction is a potential mechanism underlying energy metabolism disorder.
-
3.
Loss of transcription factor A (TFAM) caused by mtROS led to mitochondrial dysfunction in TECs.
-
4.
Mitophagy has a protective effect on TECs during AKI.
-
5.
Screening for effective interventions to regulate mitochondrial metabolism should be the focus of future research.
Open questions
-
1.
What is the specific protective mechanism of Sirt5 deficiency underlying renal tubule injury?
-
2.
Can UMI-77 targeting mitophagy improve AKI? What is the specific mechanism?
-
3.
Can drugs targeted to regulate mitochondrial metabolism treat CKD?
Introduction
Acute kidney injury (AKI) is a common clinical condition comprising complex pathophysiological processes. AKI is associated with high morbidity and mortality and lacks clear and effective treatments. A meta-analysis of the global incidence of AKI showed that the combined incidence of AKI was 23.2% when the KDIGO-equivalent AKI definition was used across 154 studies (130 adult and 24 child studies). The all-cause mortality associated with AKI was 23.0% in 110 studies (99 adult and 11 child studies) that used KDIGO-equivalent AKI to define and assess mortality. AKI affects up to 20% of hospitalized patients [1]. A British study showed that the total annual medical expenses related to inpatients with AKI are as high as 1.02 billion pounds, indicating that AKI exerts a great burden on the medical and social economy [2].
The kidneys of ischemia-reperfusion injury (IRI)-AKI mice exhibit severe mitochondrial damage, as evidenced by increased mitochondrial ROS levels and decreased ATP levels in the proximal tubule cells [3, 4]. Mitochondrial lesions, such as mitochondrial swelling and fragmentation, disruption of membrane integrity, and broken or absent cristae, have been observed in renal tubules of IRI-AKI mice [4]. Healthy mitochondria generate ATP, buffer cytosolic calcium, and produce appropriate ROS [5]. Furthermore, healthy mitochondria mediate energy utilization, signal transduction, and apoptosis in cells [6]. Dysfunctional mitochondria disrupt intracellular homeostasis and induce lethal injury. The accumulation of damaged mitochondria causes serious energy deficiency, impaired calcium buffering, increased ROS production, apoptosis due to pro-apoptotic factors released into the cytoplasm, and necrosis due to the opening of the mitochondrial permeability transition pore [7]. Thus, the maintenance of the number, morphology, and function of mitochondria, which act as energy powerhouses, is essential for cell health. Studies on mitochondria may provide greater insight into the mechanism underlying IRI-AKI. Furthermore, the restoration of healthy mitochondrial function and mass is critical for the recovery of kidney function.
Recent advancements regarding the role of mitochondria in IRI-AKI has led to their being used in an array of potential applications as biomarkers of kidney injury and targets for novel therapeutic strategies. In this review, we describe mitochondrial metabolic dysregulation in ischemic AKI and discuss how this knowledge may guide the development of novel therapies for ischemic AKI.
Abnormal morphology and function of mitochondria during ischemia-induced AKI
Mitochondria constitute the main energy producers in cells, and the kidneys are second only to the heart in terms of mitochondrial number and oxygen consumption. The primary cause of acute kidney disease is thought to be related to mitochondrial damage and impaired function [8,9,10]. In the physiological state, mitochondria exhibit normal tubular morphology [11]. Functionally, mitochondria produce ATP via oxidative phosphorylation during cellular energy metabolism [12]. In IRI-AKI, mitochondria become smaller and more rounded in atrophied tubules [13]. However, alterations in energy metabolism in TECs of IRI-AKI mice can lead to changes in the balance between mitochondrial and peroxisomal fatty acid oxidation (FAO), thereby aggravating AKI [14]. Additionally, mitochondrial damage increases ROS production and induces mitophagy. The loss of PINK1, PARK2, BINP3, and other proteins can block mitophagy and aggravate ROS accumulation, thereby causing mitochondrial accumulation and TEC damage, ultimately aggravating AKI [15, 16].
Mitochondrial pathology in atrophic tubules following IRI
In human kidneys, mitochondria in TECs of patients with AKI are swollen and fragmented [17]. In a Sprague-Dawley rat model subjected to bilateral renal ischemia for 45 min, endothelial cells, podocytes, and TECs showed significant mitochondrial damage after nine months of ischemia [18]. After the death of proximal tubule cells due to AKI, the surviving epithelial cells dedifferentiate, migrate, and proliferate [19]. Recovery of normal structure and function occurs through redifferentiation of the reconstituted epithelium. However, to varying degrees, proximal tubule cells proliferating after AKI fail to re-differentiate, undergo premature growth arrest, become atrophic, and exhibit a flat simplified cytoplasm. In rat kidneys, tubules undergoing atrophy following IRI and abnormal recovery show greatly reduced mitochondrial numbers, as well as smaller sizes, with round profiles. Moreover, atrophic tubules exhibit large complex autophagolysosomes containing degenerate mitochondria [13].
Mitochondrial energy metabolism changes in ischemia-induced AKI
Under normal conditions, TECs primarily depend on FAO as their energy source [20]. Following IRI in a Sprague-Dawley rat model, tubules that failed to re-differentiate continued to exhibit increased glycolysis and higher glycolytic enzyme expression, an alteration accompanied by increased expression of hypoxia markers. In atrophic tubules of IRI rat kidneys, the levels of carbonic anhydrase 9 (CA9), a hypoxic marker directly induced by HIF-1α, significantly increased. In addition, hypoxia can trigger TGF-β, which is involved in the pathogenesis of ischemic tubular atrophy. TGF-β signal in the rat kidney of IRI continuously increased, resulting in PTEN defects in renal tubules, which interfered with the differentiation of damaged tubules, thereby inducing tubule atrophy. When tubule atrophy occurs, mitochondrial mass markedly decreases and autophagy becomes abnormal (Fig. 1) [13, 21, 22]. Thus, renal tubule atrophy leads to increased CA9 expression, which further aggravates tubule atrophy, leading to abnormal mitochondrial metabolism. Potter et al. found that mitochondrial division and autophagy are enhanced after CA9 inhibition in malignant mesothelioma cells [23]. Therefore, the targeted regulation of CA9 expression may regulate mitochondrial energy metabolism, thereby improving IRI-AKI. In healthy mitochondria, electron transport along the electron transport chain is coupled with oxidative phosphorylation to produce ATP. In the ischemic state of the mitochondria, the activity of various complexes in the electron transport chain is reduced, leading to electron leakage and reducing oxygen formation of superoxide radicals when oxygen is available during reperfusion, ultimately reducing the amount of ATP available [24]. Nicotinamide adenine dinucleotide (NAD) is required for glycolysis, FAO, and the tricarboxylic acid cycle, leading to ATP generation via the electron transport chain in mitochondria. In healthy renal TECs, ATP provides the energy required for key functions, such as solute transport and the maintenance of membrane integrity. NAD in the mitochondria is the rate-limiting step in mitochondrial energy production [25]. Rats subjected to renal IRI show accelerated NAD consumption. Therefore, NAD cannot interact well with Sirtuins and PPARγ co-activator 1α (PGC1α) to alleviate renal tubular injury. Sirtuin 1 (SIRT1) activates PGC1α through NAD+-dependent deacetylation, and PGC1α promotes the biosynthesis of NAD+ via the de novo pathway by synergistically upregulating the expression of genes encoding the NAD+ pathway. Increased NAD+ levels can enhance FAO and active a broad cellular regulatory signaling network to protect against oxidative metabolism and mitochondrial damage, consequently protecting against renal tubular injury. NAD consumption may also reduce phosphorylation of NAD to NADP, which may weaken the defense of TECs against oxidative stress induced by IRI (Fig. 1) [26, 27].

Hypoxia can trigger TGF-β, and the increase in TGF-β leads to PTEN defects in renal tubules, inducing tubule atrophy, reducing mitochondrial mass, and causing abnormal autophagy. In healthy tubular epithelial cells (TECs), NAD+ produces ATP through the mitochondria, supplying solute transport and maintaining membrane integrity. In normal TECs, SIRT1 activates PGC1α through the de novo pathway, and promotes the production of NAD+, thereby enhancing fatty acid oxidation (FAO) and mitochondrial protection. In damaged TECs, the consumption of NAD+ inhibits PGC1α, reduces the production of NAD+, decreases FAO, and weakens the protective effect on mitochondria. The depletion of NAD+ inhibits the phosphorylation of NADP, and decreases the defense ability of TECs against ischemia-reperfusion injury-induced oxidative stress.
Additionally, Sirt5 regulates the balance between mitochondrial and peroxisomal FAO in proximal TECs to protect against AKI. Following ischemia-induced AKI, Sirt5 deficiency regulates proximal TECs to increase peroxisomal FAO when compared with mitochondrial FAO, thereby reducing oxygen demand, alleviating oxidative stress, significantly improving renal function and less tissue damage [14]. Mou, Luohe et al., found that compound 58(IC50 = 0.31 μM), an inhibitor of SIRT-5, regulates protein succinylation and proinflammatory cytokines in the kidneys of mice with septic AKI, thereby showing renal protection in vivo [28]. However, the specific protective mechanism of Sirt5 deficiency against IR-induced renal tubular injury remains unclear and requires further investigation.
During ischemia-induced AKI, the mitochondrial energy metabolic balance in TECs is altered. Thus, the inhibition of mitochondrial mass loss due to tubule atrophy, mitochondrial energy deficit due to NAD depletion, and mitochondrial FAO levels can improve renal function, thereby rendering mitochondria potential therapeutic targets.
Mitophagy in ischemia-induced AKI
Mitophagy is a type of selective autophagy that tags damaged or dysfunctional mitochondria for autophagic recognition and degradation. As a critical component of mitochondrial quality control, mitophagy can prevent excessive ROS production and release of mitochondrial pro-apoptotic factors, and inhibit pathological inflammatory responses [29, 30]. Two mitophagy priming mechanisms have been proposed that rely on the PTEN-induced kinase 1 (PINK1)-parkin RBR E3 ubiquitin-protein ligase (PARK2/PRKN) pathway [15], and mitophagy receptors, BCL2-interacting protein 3 (BNIP3L/NIX), BCL2-interacting protein 3 (BNIP3), or FUN14 domain-containing 1 [16, 31].
Renal IRI induces PINK1-PARK2-mediated mitophagy in TECs in mice. Mice deficient in Pink1 or Park2 alone or in combination show aggravated mitochondrial damage and enhanced inflammatory cell infiltration during renal IRI [15]. In damaged mitochondria, PINK1 accumulates in the mitochondrial outer membrane, which binds to the translocase of the outer mitochondrial membrane, and is activated by autophosphorylation. Activated PINK1 then phosphorylates ubiquitin, triggering PRKN recruitment to the mitochondria and activating their E3 ligase activity, ultimately ubiquitinating the mitochondrial substrate and initiating mitophagy [32]. BNIP3 deficiency reduces renal IRI-induced mitophagy in mice, resulting in the accumulation of damaged mitochondria and TECs, and exacerbation of renal injury (Fig. 2) [16]. However, BNIP3 overexpression improves kidney damage and increases mitophagy in tubular HIF-1α knockout mice with IRI (Fig. 2) [33]. Li et al. found that the inhibition of dynamin-related protein 1 (Drp1) phosphorylation by mitochondrial division inhibitor-1 (Mdivi-1) significantly inhibits renal IRI-induced mitophagy in rats without affecting general autophagy, and confirmed that the downregulation of mitophagy significantly aggravated cell apoptosis and IRI-induced renal dysfunction [34].

Mitochondrial damage increases reactive oxygen species (ROS) production, triggers mitochondrial DNA (mtDNA) damage, damages cells, and eventually leads to cell necrosis and apoptosis. BNIP3 deficiency reduces mitophagy, causes the accumulation of damaged mitochondria, and aggravates kidney injury. Overexpression of BNIP3 can increase mitophagy, reduce damaged mitochondria, and improve kidney injury.
These studies suggest that downregulation of mitophagy can lead to the accumulation of damaged and dysfunctional mitochondria, aggravating TEC injury, whereas upregulation of mitophagy can alleviate IR-induced AKI.
ROS metabolism in ischemia-induced AKI
The destructive role played by ROS in IRI is widely recognized. As a result of the disruption of the electron transport chain caused by ischemia, ROS are produced by several sources, including NADPH oxidase, xanthine oxidase-hypoxanthine, nitric oxide synthase, and parenchymal mitochondria [35]. Renal mitochondrial injury following ischemia is associated with cellular damage resulting from ROS generation. Mitochondrial DNA (mtDNA) damage is triggered by the loss of mitochondrial membrane potential and instability of the electron transport chain, which directly damages cellular components, resulting in necrosis and apoptosis (Fig. 2) [4]. Mitochondria constitute the main source of intracellular ROS and contain numerous enzymes that convert molecular oxygen into superoxides or their derivatives, such as hydrogen peroxide [36]. In an in vitro model of IRI-AKI, increased ROS production caused direct oxidative damage to mitochondrial proteins and lipids, thereby impairing mitochondrial bioenergetics by disrupting electron transport chain function and increasing mitochondrial membrane permeability. In addition, mtROS can induce kidney damage by activating pro-inflammatory signals, such as toll-like receptors and the NLRP3 inflammasome. Elevated ROS levels are a characteristic of IRI-induced AKI [4].
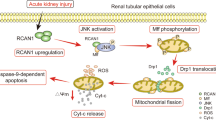
Lon, a quality control protease, has a unique function in the mitochondrial genome to regulate mtDNA metabolism and reshape nucleoid composition. Mitochondrial TFAM is a nuclear-encoded protein synthesized in the cytoplasm, imported into the mitochondria, and removed by Lon-mediated degradation [37]. In IRI-AKI mice, mitochondrial ROS induces mtDNA instability and cytokine release by suppressing TFAM transcription and promoting Lon-mediated TFAM degradation in TECs (Fig. 3). TFAM deficiency and mtDNA damage have also been observed in the kidneys of patients with AKI [4]. In healthy mitochondria, PINK1, a serine/threonine kinase targeting mitochondria, inserts into the mitochondrial inner membrane and intermembrane space by combining with the translocator of the outer mitochondrial membrane complex, and is rapidly degraded by the stromal processing peptidase. When mitochondria are injured, PINK1 accumulates in the mitochondrial outer membrane, and phosphorylates ubiquitin and the E3 ubiquitin ligase Parkin [38]. Parkin is recruited to depolarize mitochondria and ubiquitinates several mitochondrial proteins [39]. In an in vivo model of renal IRI, Pink1 or Park2 deficiency leads to the accumulation of damaged mitochondria in renal tubular cells, decreases mitochondrial protein ubiquitination, and increases ROS production (Fig. 3) [15]. BNIP3 is a BCL-2 family protein, primarily located in the outer membrane of mitochondria. BNIP3 has been identified as a pro-apoptotic protein that sensitizes BAX (BCL2-associated X, apoptosis regulator) and BAK (BCL2 antagonist/killer 1) insertion and activation in the mitochondria, and releases pro-apoptotic factors to initiate apoptotic cascades. Alternatively, BNIP3 is also a type of mitophagy receptor, which plays a role in promoting survival under some pathological conditions. Notably, BNIP3 deficiency increases mitochondrial fragmentation, swelling, and loss of mitochondrial matrix vacuoles and cristae. Tang et al. demonstrated that renal IRI in Bnip3-KO mice caused more mitochondrial damage in renal proximal tubular cells than in wild-type mice, and that ROS levels were also significantly elevated [16].

In damaged mitochondria, ROS induces mitochondrial DNA (mtDNA) instability and cytokine release by inhibiting mitochondrial transcription factor A (TFAM) transcription, while promoting Lon-mediated TFAM degradation. In damaged mitochondria, PINK1 aggregates in the mitochondrial outer membrane and phosphorylates ubiquitin and PARK2. After PARK2 aggregates in depolarized mitochondria, it ubiquitinates mitochondrial proteins. When Pink1 or Park2 is deficient, mitochondrial protein ubiquitination is inhibited and ROS production is increased.
These studies suggest that elevated ROS levels in kidneys following IRI aggravate damaged and dysfunctional mitochondria, thereby exacerbating AKI. This indicates that AKI can be ameliorated by inhibiting the increase in ROS levels in TECs following IRI.
Disruption of mitochondrial fusion and fission in AKI
Mitochondria are highly dynamic organelles that undergo fission and fusion, which are closely related to their functions [40]. Disruption of mitochondrial dynamics has been confirmed in AKI and may ultimately lead to cell damage and death [41].
Drp1, a key protein involved in mitochondrial fission, is activated during mitochondrial dysfunction during tubule cell injury and mediates mitochondrial breakage. During mitochondrial division, cytoplasmic localized Drp1 is recruited to the division site of the mitochondrial outer membrane, where it oligomerizes, leading to assembly into helical structures to mediate membrane splice [42]. Drp1 expression increases in the renal IRI model [43]. The key mediators of mitochondrial fusion include the outer membrane fusion proteins, Mitofusin-1(Mfn1), Mitofusin-2 (Mfn2), and the inner membrane fusion protein optic atrophy 1 (Opa1) [44, 45]. In the kidneys of IR mice, Opa1 expression is significantly decreased, which leads to the obstruction of mitochondrial intima fusion and secondary mitochondrial dysfunction, and finally aggravates proximal renal tubule injury [42, 46, 47]. Notably, the kidneys of IRI mice exhibit an increase in Mfn1 expression, and a significant decrease in Mfn2 expression when compared to those of the sham group [42, 48, 49]. These results indicate that mitochondrial fusion is partly suppressed following IRI.
Therapeutic targeting of mitochondria in ischemia-induced AKI
Targeting mitochondrial energy metabolism
Mitochondrial energy metabolism includes pyruvate oxidation, FAO, citric acid cycle (TCA), and five oxidative phosphorylation complexes [50]. The above-mentioned imbalance in mitochondrial FAO could be used as a potential target for mitochondria-targeted therapy. FAO levels decline following renal IRI. Portilla et al. found that upregulation of the peroxisome proliferator-activated receptor-α (PPARα)-regulated FAO gene exerts an important protective effect on renal tubular cells during IRI. Mitochondrial carnitine palmitoacyltransferase 1 (CPT1) activity inhibitors can be used as direct ligands of PPARα, and this ligand-mediated activation upregulates the activities of peroxisome acyl-coA oxidase and the microsomal somatic pigment P-450, thereby increasing FAO levels and ameliorating renal tubular injury following IR [50, 51]. Hypoxic preconditioning and extracellular MSC vesicles inhibit renal fibrosis by restoring CPT A-mediated mitochondrial FAO, which can be achieved by regulating mitochondrial homeostasis [52] (Table 1).
The pyruvate generated by glycolysis in the TCA cycle is converted into acetyl-CoA through the action of pyruvate dehydrogenase (PDH). This acetyl-CoA is then metabolized to carbon dioxide by enzymes such as citrate synthase (CS) and alpha-ketoglutarate dehydrogenase (AKGDH) [53]. During the TCA cycle, electrons generated undergo oxidative phosphorylation to generate ATP [53]. The mitochondrial respiratory chain complex regulates mitochondrial oxidative phosphorylation, which is a crucial component of mitochondrial energy metabolism [54]. Liao et al. have shown that in renal IRI models, fluorofenidone retains the expression of PDH, CS, AKGDH, complex I and complex II in the IRI model, alleviating the damage of TCA circulation and mitochondrial respiratory chain in IRI, thereby improving mitochondrial energy metabolism [55]. This suggests that by targeting key enzymes in the TCA cycle or oxidizing related complexes in the respiratory chain, we can enhance mitochondrial energy metabolism and protect against renal IRI.
In addition, mitochondria are a major site of glutamine metabolism in various cell types [56]. Katharina et al. demonstrated that glutamine treatment significantly upregulates proteins associated with oxidative phosphorylation in mitochondria, particularly a critical subunit of NADH: ubiquinone oxidoreductase (Complex I). It has been shown that NAD+ synthesis damage enhances the development of AKI [57]. Notably, in a Phase I clinical trial, enhanced biosynthesis of NAD+ reduced the incidence of postoperative AKI in patients undergoing cardiac surgery [57]. Glutamine is involved in the regulation of oxidative stress because it is a precursor for many antioxidant molecules, such as glutathione and NAD+ [58,59,60,61]. Therefore, targeted upregulation of glutamine in mitochondria can increase the expression of Complex I, elevate the NAD+/NADH ratio, enhance the oxidative phosphorylation process, and ultimately improve mitochondrial function and mitigate kidney injury.
Nutrient-sensing pathways can directly affect mitochondrial energetics in response to external stimuli such as hypoxia, oxidative stress, and energy depletion. In particular, two signaling pathways have been explored extensively in the kidney: the mechanistic target of rapamycin [62] and AMP-activated protein kinase (AMPK) signaling pathways [63]. The AMPK activator 5-aminoimidazole-4-carboxamide-1-β-D-riboside (AICAR) prevents glomerulopathy and tubulointerstitial fibrosis in mice by stimulating FAO. AICAR also has a therapeutic effect on mouse renal IRI [41, 64].
Targeting mitophagy
HIF-1α-BNIP3-mediated mitophagy in tubular cells protects against renal IRI [33]. Mechanistically, HIF-1α directly increases the transcription and expression of BNIP3 in IRI-AKI mice [33]. BNIP3 can bind to LC3 protein, and then connect autophagy vesicles to targeted mitochondria, directly induce mitophagy, while reducing the production of reactive oxygen species and improving renal IRI (Fig. 4) [32, 65]. Additionally, the PINK1-PRKN/PARK2 mitophagy pathway can be activated to protect against renal IRI [15]. This is due to ischemia-induced mitochondrial depolarization, during which Pink1 accumulates on the mitochondrial outer membrane and recruits Parkin from the cytoplasm. This activation of Parkin’s ubiquitin ligase activity triggers mitophagy, leading to the elimination of damaged mitochondria (Fig. 4) [66, 67]. After renal IRI, Drp1 is activated by S616 phosphorylation and translocated onto the mitochondrial membrane [34]. Subsequently, the Drp1-dependent mitophagy pathway can be activated, and mitophagic clearance of damaged mitochondria protects cells from IRI-induced apoptosis, thereby protecting the kidney (Fig. 4) [34]. Furthermore, ischemic preconditioning driven FUN14 domain protein 1 (Fundc1)-dependent mitophagy is activated by post-transcriptional phosphorylation of Ser17, which subsequently regulates mitochondrial fission to resist AKI, thereby ameliorating kidney injury [68]. Notably, Jin et al. found that UMI-77, a complex that induces mitophagy, attenuates renal TEC damage by enhancing mitophagy, downregulating the TGF-β/Smad signaling pathway, and subsequently inhibiting the epithelial-to-stromal transition phenotype, ultimately preventing renal fibrosis [69]. These findings suggest that targeting mitophagy may ameliorate kidney injury.

In the IRI-AKI state, HIF-1α increases the expression of BNIP3, which localizes to the mitochondrial outer membrane and binds to LC3 on the autophagosome membrane. This interaction ultimately triggers mitophagy. In the ischemic state, PINK1 accumulates on the depolarized mitochondrial outer membrane and recruits activated Parkin, leading to mitophagy activation. Additionally, in the IRI state, S616 phosphorylation activates Drp1, which is translocated to the mitochondrial membrane and triggers mitophagy.
Reducing ROS levels in renal tissue in ischemic AKI
Mitochondria are both the source and target of ROS in cells. Oxidative damage to mitochondria during reperfusion can further trigger or aggravate the induction of the cell death pathway, autophagy, and activation of the inflammasome, thus damaging renal function. Han et al. reported that mitochondrial NADP+-dependent isocitrate dehydrogenase (IDH2) deficiency exacerbates mitochondrial and cell damage following kidney IRI. Therefore, IDH2 is an important mitochondrial antioxidant enzyme that provides NADPH to promote ROS clearance and protects cells from IRI [70]. Bax inhibitor-1 (BI1), an evolutionarily conserved inhibitor of apoptosis, mediates the activation of extracellular signal-regulated kinase 1/2, leading to the inhibition of mitochondria-mediated ROS production. Wang et al. indicated that BI1 levels in the urine and plasma decrease in patients with AKI, and its expression inversely correlates with renal function. Mechanistically, BI1 interacts with anti-proliferative protein 2 (PHB2) to facilitate the translocation of cytoplasmic PHB2 into mitochondria with the assistance of TIM23. Once localized to mitochondria, PHB2 maintains mitochondrial morphology and function, while also inhibiting mitochondrial apoptosis. In addition, BI1 reconstitution in a murine AKI model reduced mitochondrial oxidative stress, inhibited excessive mitochondrial fission, improved mitophagy, and suppressed mitochondrial apoptosis [71]. Therefore, ROS production in the mitochondria can be inhibited by upregulating BI1 to improve kidney injury.
Mitoquinone (MitoQ), a mitochondria-targeted antioxidant that inhibits mitochondrial ROS production, accumulates in cells driven by the positive charge of the plasma membrane and further accumulates within mitochondria driven by the positive charge of the mitochondrial membrane. In the mitochondrial matrix, MitoQ is reduced to its active antioxidant form ubiquinol by the respiratory chain, thereby preventing oxidative damage such as lipid peroxidation. Antioxidant activity generates the ubiquinone form, which is subsequently recycled to ubiquinol by the respiratory chain, continuing its antioxidant effect. Dare et al. also found that MitoQ decreases mitochondrial oxidative damage, thus protecting against renal IRI in a mouse model [72].
Thus, treatment strategies that promote ROS clearance, inhibit ROS production, and protect mitochondria from oxidative damage may ameliorate renal IRI. BI1, the mitochondrial antioxidant enzyme IDH, and the mitochondria-targeted antioxidant MitoQ have been shown to reduce mitochondrial ROS levels and protect mitochondria from oxidative damage, thereby improving renal IRI.
Targeting disrupted mitochondrial fusion and fission
Proximal tubule-specific deletion of Drp1 prevented renal IRI-induced kidney injury, inflammation, and programmed cell death observed in wild-type mice, and promoted epithelial recovery [73]. Wang et al. found that Mdivi-1 inhibition of Drp1-mediated mitochondrial fission alleviates renal interstitial fibrosis induced by unilateral ureteral obstruction [74]. Qi et al. designed a new selective peptide inhibitor P110 that inhibits Drp 1/Fis 1 interactions, thereby inhibiting mitochondrial breakage and ROS production [75, 76]. The above-mentioned inhibitors of genes Mdivi-1 and P110 can inhibit mitochondrial division to improve the function of tissues and organs such as the kidney.
Mfn2 protects against renal tubular apoptosis during ischemic AKI [77]. Thymoquinone, a strong free radical and superoxide scavenger, mitigates diclofenac-induced mitochondrial viability and apoptosis by upregulating Mfn2 expression, thereby improving renal function [78, 79].
Therefore, the regulation of key proteins involved in mitochondrial fusion and fission can inhibit apoptosis and ameliorate kidney injury.
Other therapeutic approaches targeting mitochondria in AKI
The innate immune system has been implicated in AKI and CKD. Damaged mitochondria release noxious molecules, such as ROS, mtDNA, and cardiolipin, which can cause NLRP3 inflammasome activation, and the upregulation of IL-18 and IL-1b. Mitochondrial damage persists long after ischemia and sustains chronic inflammasome activation. However, using a mitoprotective agent SS-31 following ischemia preserves mitochondrial integrity, ameliorates expression levels of all inflammatory markers, restores glomerular capillaries and podocyte structure, and arrests glomerulosclerosis and interstitial fibrosis with the normalization of IL-18 and IL-1b expression. In the rat model of IRI, SS-31 treatment protected mitochondrial structure and respiration during early reperfusion, accelerated ATP recovery, reduced renal tubular cell necrosis, and reduced IR-mediated oxidative stress [80, 81].
Tan et al. suggested that fibroblast growth factor (FGF2) substantially ameliorates renal IRI by mitigating several mitochondrial damage parameters, including the pro-apoptotic alteration of Bcl2/Bax expression, caspase-3 activation, loss of mitochondrial membrane potential, and mitochondrial ATP-dependent potassium channel integrity. IRI alone results in mild activation of FGFR, whereas FGF2 treatment results in a more robust receptor activation. Notably, post-IRI administration of FGF2 also provides robust protection against IRI by reducing cell apoptosis, inhibiting the release of damage-associated molecular pattern molecule HMBG1, and activation of its downstream inflammatory cytokines, such as IL-1a, IL-6, and TNFα [82].
These findings suggest that mitochondrial protection is a novel therapeutic approach to arrest the progression of AKI to CKD. In addition, agents that open the mitochondrial ATP-dependent potassium channel are effective in preventing renal injury via the inhibition of mitochondrial DNA damage [83].
The replacement of damaged mitochondria also restores mitochondrial homeostasis. Jabbari et al. injected normal mitochondria isolated from healthy muscle cells into injured kidney cells via renal arteries in an animal model of AKI. Mitochondrial transplantation can prevent renal tubule cell death, restore renal function, improve renal injury, enhance renal tubule regeneration potential, and reduce IRI-induced apoptosis [84]. Although further research, including clinical trials, is urgently needed, mitochondrial transplantation offers a novel therapeutic strategy for AKI.
Mitochondrial-related biomarkers for ischemia-induced AKI
Previous studies have shown a time-dependent loss of renal cortical mitochondrial proteins including ATP synthase subunit b (ATPSb) following IRI-AKI, indicating renal mitochondrial dysfunction [49, 85, 86]. Moreover, urinary full-length ATPSb has been identified as a specific biomarker for renal mitochondrial dysfunction [87]. In patients who have undergone surgery and developed postoperative AKI, urinary ATPSb levels have been found to be elevated. Similarly, in the IRI-AKI mouse model, urinary full-length ATPSb levels increased with moderate and severe kidney injury. This increase preceded the elevation of other markers of kidney injury [87]. These findings suggest that urinary full-length ATPSb may serve as a sensitive and specific translational biomarker for renal mitochondrial dysfunction in IRI-AKI.
Elevated mtDNA levels may serve as surrogate biomarkers of mitochondrial dysfunction, kidney damage, and the progression and prognosis of kidney diseases [88]. Following ischemic AKI, considerable mitochondrial damage occurs in renal tubular cells and podocytes, resulting in the release of mtDNA into the extracellular space, and subsequently, in systemic circulation. Thereafter, free mtDNA is detected in the plasma and used as a biomarker of kidney injury. Simultaneously, damaged mtDNA from renal parenchymal cells leaks into urine because of kidney injury, and elevated urinary mitochondrial DNA levels can be used as a surrogate biomarker for kidney injury [89]. Therefore, the detection of elevated mtDNA levels in plasma and urine is significant to advance our understanding of IRI pathogenesis in AKI and the role of targeting mtDNA in IRI. The search for mitochondrial biomarkers of ischaemic-induced AKI is a new field and direction, and only a few studies have been conducted so far, which deserves our attention and further study.
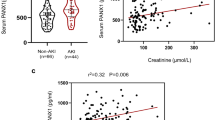
Pannexin 1 (PANX1) is a regulator of ATP release [90, 91]. In an oxidative environment, PANX1 activates autophagy signaling by increasing extracellular ATP efflux and engaging purinergic P2X and P2Y signaling pathways [92]. Furthermore, P2Y receptors promote the rapamycin target protein signaling pathway and regulate mitochondrial activity [93]. Therefore, PANX1 activation leads to ATP release, which stimulates P2Y receptors, resulting in activation of mTOR signaling and ultimately inhibiting mitophagy [94]. Research by Su et al. has shown that PANX1 levels are elevated in the serum of patients who develop AKI following cardiac surgery [94]. These findings suggest that PANX1 may serve as a potential mitochondria-related biomarker for the diagnosis of IRI-AKI.
There is a pressing need for early, specific, and non-invasive biomarkers of mitochondrial dysfunction in IRI-AKI. The identification and validation of such markers could provide valuable insights into the underlying mechanisms of renal mitochondrial dysfunction in AKI and lead to the development of novel therapeutic targets.
Conclusion and perspectives
Severe tubular injury is a characteristic of AKI. Extensive necrosis and apoptosis of TECs is observed, especially in the proximal tubules. Mitochondria in the proximal tubular epithelium play a key role in the pathophysiology of IRI-AKI. The morphology and function of mitochondria change considerably in AKI. The most obvious morphological changes are mitochondrial swelling and fragmentation, disruption of membrane integrity, and mitochondrial ridge fractures or deletions. Subsequent mitochondrial dysfunction manifested as reduced ATP production, elevated ROS levels, and the release of pro-apoptotic factors, further damages the kidneys.
In this paper, several targets of mitochondrial-targeted therapy are discussed. In targeted mitochondrial energy metabolism therapy, renal IRI can be improved by modulating CPT1 to increase PPARα-regulated FAO levels [51]. However, in the mouse kidney fibrosis model, it can still maintain mitochondrial homeostasis by affecting CPT1A, thereby improving kidney injury [52]. Previous studies have shown that AICAR has a protective effect against ischemia-reperfusion injury in the heart and liver, which further supports the great potential of AICAR in the treatment of IRI-AKI [64]. In addition, regulating key enzymes (such as PDH, CS, AKGDH, etc.) in the process of TCA and related complexes (such as complex I, complex II, etc.) in the process of oxidative phosphorylation can preserve or restore mitochondrial energy metabolism in IRI-AKI, thereby improving kidney injury [53,54,55]. Therapeutic measures targeting mitochondrial energy metabolism have been more studied and confirmed in other organ or disease models, which has a very positive significance in guiding the treatment of IRI-AKI.
In targeted mitophagy therapy, HIF-1α-BNIP3 and PINK-PRKN/PARK2 eliminate damaged mitochondria and reduce ROS levels by directly inducing or activating mitophagy, thereby reducing mitochondrial damage and achieving the purpose of protecting renal function [15, 33]. Although Drp1 and Fundc1 have also been confirmed in IRI-AKI to activate mitophagy and play a protective role in kidney injury, the specific molecular mechanisms are not fully understood [34, 68]. The protective effect of UMI-77 on the kidney by activating mitophagy in the renal fibrosis model also enables us to make a reasonable hypothesis that UMI-77 can activate mitophagy to improve kidney injury in IRI-AKI.
ROS is a key factor affecting mitochondrial damage in AKI, and targeting ROS is the main direction of AKI treatment. In addition, there are many effective antioxidant drugs (such as n-acetylcysteine, MitoQ, etc.) that have been shown to play a beneficial role in the ischemia/reperfusion process of the liver, heart, and kidney [95,96,97,98,99,100].
For IRI-AKI, several key targets of IDH2, BI1 and MitoQ are discussed in this paper. In particular, the therapeutic role of MitoQ in IRI-AKI has been well established, and studies have shown that MitoQ can be used as a potential drug for post-resuscitation myocardial infarction [101]. These studies fully demonstrate that MitoQ plays a positive therapeutic role in IRI-AKI. This also suggests that the development of more mitochondria-targeting antioxidants will be of great significance for the future treatment of IRI-AKI. In targeting mitochondrial fusion and fission, we focused on the therapeutic effect of Mdivi-1 on IRI-AKI. Mdivi-1 also plays a protective role in ischemic reperfusion injury in the brain and heart muscle [102, 103]. Other targets for mitochondrial therapy (such as SS-31 and FGF2) are also very meaningful and worth exploring. We believe that these are the hot spots and new directions of IRI-AKI therapy in the future.
Studies have confirmed multiple therapeutic targets for AKI; however, no targeted drugs are currently approved for the treatment of AKI in clinical practice. SIRT5, MitoQ, and BNIP3 have shown promising potential for treating mitochondrial damage and kidney disease in clinical trials concerning certain kidney injuries and diseases. In the future, we can regulate the levels of peroxisome and mitochondrial fatty acid oxidation in proximal tubule epithelial cells by inhibiting SIRT5, thereby improving AKI. Moreover, the development of BNIP3 activators to reduce ROS production will make it possible to alleviate AKI symptoms. In addition, we can also develop reducing agents for MitoQ to activate it to prevent oxidative damage and improve AKI. Therefore, targeted AKI therapy remains an active area of research. With the progress in human clinical trials on the efficacy and safety of mitochondria-targeted therapy approaches, we hope that mitochondrial therapeutic targets become a strategy to improve kidney injury and disease in the future.
Data availability
All data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Susantitaphong P, Cruz DN, Cerda J, Abulfaraj M, Alqahtani F, Koulouridis I, et al. World incidence of AKI: a meta-analysis. Clin J Am Soc Nephrology: Cjasn 2013;8:1482–93.
Hoste EAJ, Kellum JA, Selby NM, Zarbock A, Palevsky PM, Bagshaw SM, et al. Global epidemiology and outcomes of acute kidney injury. Nat Rev Nephrol 2018;14:607–25.
Emma F, Montini G, Parikh SM, Salviati L. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat Rev Nephrol 2016;12:267–80.
Zhao M, Wang Y, Li L, Liu S, Wang C, Yuan Y, et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 2021;11:1845–63.
Graier WF, Frieden M, Malli R. Mitochondria and Ca(2+) signaling: old guests, new functions. Pflug Arch: Eur J Physiol 2007;455:375–96.
Tao M, You CP, Zhao RR, Liu SJ, Zhang ZH, Zhang C, et al. Animal mitochondria: evolution, function, and disease. Curr Mol Med 2014;14:115–24.
Srinivasan S, Guha M, Kashina A, Avadhani NG. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochimica et Biophys Acta Bioenerg 2017;1858:602–14.
Panconesi R, Widmer J, Carvalho MF, Eden J, Dondossola D, Dutkowski P, et al. Mitochondria and ischemia reperfusion injury. Curr Opin Organ Transplant 2022;27:434–45.
Duann P, Lin PH. Mitochondria damage and kidney disease. Adv Exp Med Biol 2017;982:529–51.
Ratliff BB, Abdulmahdi W, Pawar R, Wolin MS. Oxidant mechanisms in renal injury and disease. Antioxid redox Signal 2016;25:119–46.
Rafelski SM. Mitochondrial network morphology: building an integrative, geometrical view. BMC Biol 2013;11:71.
Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell 2012;148:1145–59.
Lan R, Geng H, Singha PK, Saikumar P, Bottinger EP, Weinberg JM, et al. Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J Am Soc Nephrology: Jasn 2016;27:3356–67.
Chiba T, Peasley KD, Cargill KR, Maringer KV, Bharathi SS, Mukherjee E, et al. Sirtuin 5 regulates proximal tubule fatty acid oxidation to protect against AKI. J Am Soc Nephrology: Jasn 2019;30:2384–98.
Tang C, Han H, Yan M, Zhu S, Liu J, Liu Z, et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy 2018;14:880–97.
Tang C, Han H, Liu Z, Liu Y, Yin L, Cai J, et al. Activation of BNIP3-mediated mitophagy protects against renal ischemia-reperfusion injury. Cell Death Dis 2019;10:677.
Parekh DJ, Weinberg JM, Ercole B, Torkko KC, Hilton W, Bennett M, et al. Tolerance of the human kidney to isolated controlled ischemia. J Am Soc Nephrology: Jasn 2013;24:506–17.
Szeto HH, Liu S, Soong Y, Seshan SV, Cohen-Gould L, Manichev V, et al. Mitochondria protection after acute ischemia prevents prolonged upregulation of IL-1β and IL-18 and arrests CKD. J Am Soc Nephrology: Jasn 2017;28:1437–49.
Tang C, Cai J, Yin XM, Weinberg JM, Venkatachalam MA, Dong Z. Mitochondrial quality control in kidney injury and repair. Nat Rev Nephrol 2021;17:299–318.
Houten SM, Violante S, Ventura FV, Wanders RJ. The biochemistry and physiology of mitochondrial fatty acid β-oxidation and its genetic disorders. Annu Rev Physiol 2016;78:23–44.
Lan R, Geng H, Polichnowski AJ, Singha PK, Saikumar P, McEwen DG, et al. PTEN loss defines a TGF-β-induced tubule phenotype of failed differentiation and JNK signaling during renal fibrosis. Am J Physiol Ren Physiol 2012;302:F1210–23.
Potter C, Harris AL. Hypoxia inducible carbonic anhydrase IX, marker of tumour hypoxia, survival pathway and therapy target. Cell Cycle 2004;3:164–7.
Li Z, Jiang L, Chew SH, Hirayama T, Sekido Y, Toyokuni S. Carbonic anhydrase 9 confers resistance to ferroptosis/apoptosis in malignant mesothelioma under hypoxia. Redox Biol 2019;26:101297.
Pabla N, Bajwa A. Role of mitochondrial therapy for ischemic-reperfusion injury and acute kidney injury. Nephron 2022;146:253–8.
Akie TE, Liu L, Nam M, Lei S, Cooper MP. OXPHOS-mediated induction of NAD+ promotes complete oxidation of fatty acids and interdicts non-alcoholic fatty liver disease. PloS one 2015;10:e0125617.
Ralto KM, Rhee EP, Parikh SM. NAD(+) homeostasis in renal health and disease. Nat Rev Nephrol 2020;16:99–111.
Fernández ML, Marson ME, Ramirez JC, Mastrantonio G, Schijman AG, Altcheh J, et al. Pharmacokinetic and pharmacodynamic responses in adult patients with Chagas disease treated with a new formulation of benznidazole. Mem do Inst Oswaldo Cruz 2016;111:218–21.
Mou L, Yang L, Hou S, Wang B, Wang X, Hu L, et al. Structure-activity relationship studies of 2,4,5-Trisubstituted Pyrimidine derivatives leading to the identification of a novel and potent sirtuin 5 inhibitor against sepsis-associated acute kidney injury. J Med Chem 2023;66:11517–35.
Parikh SM, Yang Y, He L, Tang C, Zhan M, Dong Z. Mitochondrial function and disturbances in the septic kidney. Semin Nephrol 2015;35:108–19.
Tang C, He L, Liu J, Dong Z. Mitophagy: basic mechanism and potential role in kidney diseases. Kidney Dis 2015;1:71–9.
Lin C, Chen W, Han Y, Sun Y, Zhao X, Yue Y, et al. PTEN-induced kinase 1 enhances the reparative effects of bone marrow mesenchymal stromal cells on mice with renal ischaemia/reperfusion-induced acute kidney injury. Hum cell 2022;35:1650–70.
Su L, Zhang J, Gomez H, Kellum JA, Peng Z. Mitochondria ROS and mitophagy in acute kidney injury. Autophagy 2023;19:401–14.
Fu ZJ, Wang ZY, Xu L, Chen XH, Li XX, Liao WT, et al. HIF-1α-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol 2020;36:101671.
Li N, Wang H, Jiang C, Zhang M. Renal ischemia/reperfusion-induced mitophagy protects against renal dysfunction via Drp1-dependent-pathway. Exp Cell Res 2018;369:27–33.
Granger DN, Kvietys PR. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol 2015;6:524–51.
Kalogeris T, Bao Y, Korthuis RJ. Mitochondrial reactive oxygen species: a double-edged sword in ischemia/reperfusion vs preconditioning. Redox Biol 2014;2:702–14.
Lu B, Lee J, Nie X, Li M, Morozov YI, Venkatesh S, et al. Phosphorylation of human TFAM in mitochondria impairs DNA binding and promotes degradation by the AAA+ Lon protease. Mol Cell 2013;49:121–32.
Lin Q, Li S, Jiang N, Shao X, Zhang M, Jin H, et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol 2019;26:101254.
Ivankovic D, Chau KY, Schapira AH, Gegg ME. Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin-mediated mitophagy. J Neurochem 2016;136:388–402.
Al Ojaimi M, Salah A, El-Hattab AW. Mitochondrial fission and fusion: molecular mechanisms, biological functions, and related disorders. Membranes 2022;12:893.
Morigi M, Perico L, Rota C, Longaretti L, Conti S, Rottoli D, et al. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J Clin Investig 2015;125:715–26.
Long RT, Peng JB, Huang LL, Jiang GP, Liao YJ, Sun H, et al. Augmenter of liver regeneration alleviates renal hypoxia-reoxygenation injury by regulating mitochondrial dynamics in renal tubular epithelial cells. Mol Cells 2019;42:893–905.
Liu Z, Li H, Su J, Xu S, Zhu F, Ai J, et al. Numb depletion promotes Drp1-mediated mitochondrial fission and exacerbates mitochondrial fragmentation and dysfunction in acute kidney injury. Antioxid Redox Signal 2019;30:1797–816.
Hall AR, Burke N, Dongworth RK, Kalkhoran SB, Dyson A, Vicencio JM, et al. Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell Death Dis 2016;7:e2238.
Guan L, Che Z, Meng X, Yu Y, Li M, Yu Z, et al. MCU Up-regulation contributes to myocardial ischemia-reperfusion Injury through calpain/OPA-1-mediated mitochondrial fusion/mitophagy Inhibition. J Cell Mol Med 2019;23:7830–43.
Wu L, Li Q, Liu S, An X, Huang Z, Zhang B, et al. Protective effect of hyperoside against renal ischemia-reperfusion injury via modulating mitochondrial fission, oxidative stress, and apoptosis. Free Radic Res 2019;53:727–36.
Xiao X, Hu Y, Quirós PM, Wei Q, López-Otín C, Dong Z. OMA1 mediates OPA1 proteolysis and mitochondrial fragmentation in experimental models of ischemic kidney injury. Am J Physiol Ren Physiol 2014;306:F1318–26.
Neres-Santos RS, Junho CVC, Panico K, Caio-Silva W, Pieretti JC, Tamashiro JA, et al. Mitochondrial dysfunction in Cardiorenal Syndrome 3: Renocardiac effect of vitamin C. Cells 2021;10:3029.
Funk JA, Schnellmann RG. Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am J Physiol Ren Physiol 2012;302:F853–64.
Vidali S, Aminzadeh S, Lambert B, Rutherford T, Sperl W, Kofler B, et al. Mitochondria: The ketogenic diet–A metabolism-based therapy. Int J Biochem Cell Biol 2015;63:55–9.
Portilla D, Dai G, Peters JM, Gonzalez FJ, Crew MD, Proia AD. Etomoxir-induced PPARalpha-modulated enzymes protect during acute renal failure. Am J Physiol Ren Physiol 2000;278:F667–75.
Gao Z, Zhang C, Peng F, Chen Q, Zhao Y, Chen L, et al. Hypoxic mesenchymal stem cell-derived extracellular vesicles ameliorate renal fibrosis after ischemia-reperfusion injure by restoring CPT1A mediated fatty acid oxidation. Stem cell Res Ther 2022;13:191.
Martínez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun 2020;11:102.
Klopstock T, Priglinger C, Yilmaz A, Kornblum C, Distelmaier F, Prokisch H. Mitochondrial disorders. Dtsch Arzteblatt Int 2021;118:741–8.
Liao X, Lv X, Zhang Y, Han Y, Li J, Zeng J, et al. Fluorofenidone inhibits UUO/IRI-induced renal fibrosis by reducing mitochondrial damage. Oxid Med Cell Longev 2022;2022:2453617.
Matés JM, Segura JA, Campos-Sandoval JA, Lobo C, Alonso L, Alonso FJ, et al. Glutamine homeostasis and mitochondrial dynamics. Int J Biochem Cell Biol 2009;41:2051–61.
Poyan Mehr A, Tran MT, Ralto KM, Leaf DE, Washco V, Messmer J, et al. De novo NAD(+) biosynthetic impairment in acute kidney injury in humans. Nat Med 2018;24:1351–9.
Amores-Sánchez MI, Medina MA. Glutamine, as a precursor of glutathione, and oxidative stress. Mol Genet Metab 1999;67:100–5.
Matés JM, Pérez-Gómez C, Núñez de Castro I, Asenjo M, Márquez J. Glutamine and its relationship with intracellular redox status, oxidative stress and cell proliferation/death. Int J Biochem cell Biol 2002;34:439–58.
Cruzat VF, Bittencourt A, Scomazzon SP, Leite JS, de Bittencourt PI Jr., Tirapegui J. Oral free and dipeptide forms of glutamine supplementation attenuate oxidative stress and inflammation induced by endotoxemia. Nutrition 2014;30:602–11.
Massudi H, Grant R, Guillemin GJ, Braidy NNAD. metabolism and oxidative stress: the golden nucleotide on a crown of thorns. Redox Rep: Commun Free Radic Res 2012;17:28–46.
Fantus D, Rogers NM, Grahammer F, Huber TB, Thomson AW. Roles of mTOR complexes in the kidney: implications for renal disease and transplantation. Nat Rev Nephrol 2016;12:587–609.
Kim Y, Park CW. Adenosine monophosphate-activated protein kinase in diabetic nephropathy. Kidney Res Clin Pract 2016;35:69–77.
Lempiäinen J, Finckenberg P, Levijoki J, Mervaala E. AMPK activator AICAR ameliorates ischaemia reperfusion injury in the rat kidney. Br J Pharmacol 2012;166:1905–15.
Liu L, Sakakibara K, Chen Q, Okamoto K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res 2014;24:787–95.
Dhanabalan K, Mzezewa S, Huisamen B, Lochner A. Mitochondrial oxidative phosphorylation function and mitophagy in ischaemic/reperfused hearts from control and high-fat diet rats: effects of long-term melatonin treatment. Cardiovasc Drugs Ther 2020;34:799–811.
Narendra D, Walker JE, Youle R. Mitochondrial quality control mediated by PINK1 and Parkin: links to Parkinsonism. Cold Spring Harb Perspect Biol 2012;4:a011338.
Wang J, Zhu P, Li R, Ren J, Zhou H. Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biol 2020;30:101415.
Jin L, Yu B, Liu G, Nie W, Wang J, Chen J, et al. Mitophagy induced by UMI-77 preserves mitochondrial fitness in renal tubular epithelial cells and alleviates renal fibrosis. FASEB J: Off Publ Fed Am Soc Exp Biol 2022;36:e22342.
Han SJ, Jang HS, Noh MR, Kim J, Kong MJ, Kim JI, et al. Mitochondrial NADP(+)-dependent isocitrate dehydrogenase deficiency exacerbates mitochondrial and cell damage after kidney ischemia-reperfusion injury. J Am Soc Nephrol: Jasn 2017;28:1200–15.
Wang J, Zhu P, Li R, Ren J, Zhang Y, Zhou H. Bax inhibitor 1 preserves mitochondrial homeostasis in acute kidney injury through promoting mitochondrial retention of PHB2. Theranostics 2020;10:384–97.
Dare AJ, Bolton EA, Pettigrew GJ, Bradley JA, Saeb-Parsy K, Murphy MP. Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox Biol 2015;5:163–8.
Perry HM, Huang L, Wilson RJ, Bajwa A, Sesaki H, Yan Z, et al. Dynamin-related protein 1 deficiency promotes recovery from AKI. J Am Soc Nephrology: Jasn 2018;29:194–206.
Wang Y, Lu M, Xiong L, Fan J, Zhou Y, Li H, et al. Drp1-mediated mitochondrial fission promotes renal fibroblast activation and fibrogenesis. Cell Death Dis 2020;11:29.
Haileselassie B, Mukherjee R, Joshi AU, Napier BA, Massis LM, Ostberg NP, et al. Drp1/Fis1 interaction mediates mitochondrial dysfunction in septic cardiomyopathy. J Mol Cell Cardiol 2019;130:160–9.
Qi X, Qvit N, Su YC, Mochly-Rosen DA. novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J cell Sci 2013;126:789–802.
Yan Y, Ma Z, Zhu J, Zeng M, Liu H, Dong Z. miR-214 represses mitofusin-2 to promote renal tubular apoptosis in ischemic acute kidney injury. Am J Physiol Ren Physiol 2020;318:F878–f87.
Awad AS, Kamel R, Sherief MA. Effect of thymoquinone on hepatorenal dysfunction and alteration of CYP3A1 and spermidine/spermine N-1-acetyl-transferase gene expression induced by renal ischaemia-reperfusion in rats. J Pharm Pharmacol 2011;63:1037–42.
Hashem KS, Abdelazem AZ, Mohammed MA, Nagi AM, Aboulhoda BE, Mohammed ET, et al. Thymoquinone alleviates mitochondrial viability and apoptosis in diclofenac-induced acute kidney injury (AKI) via regulating Mfn2 and miR-34a mRNA expressions. Environ Sci Pollut Res Int 2021;28:10100–13.
Zhu Y, Luo M, Bai X, Li J, Nie P, Li B, et al. SS-31, a mitochondria-targeting peptide, ameliorates kidney disease. Oxid Med Cell Longev 2022;2022:1295509.
Szeto HH, Liu S, Soong Y, Wu D, Darrah SF, Cheng FY, et al. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J Am Soc Nephrology: Jasn 2011;22:1041–52.
Tan XH, Zheng XM, Yu LX, He J, Zhu HM, Ge XP, et al. Fibroblast growth factor 2 protects against renal ischaemia/reperfusion injury by attenuating mitochondrial damage and proinflammatory signalling. J Cell Mol Med 2017;21:2909–25.
Sun Z, Zhang X, Ito K, Li Y, Montgomery RA, Tachibana S, et al. Amelioration of oxidative mitochondrial DNA damage and deletion after renal ischemic injury by the KATP channel opener diazoxide. Am J Physiol Ren Physiol 2008;294:F491–8.
Jabbari H, Roushandeh AM, Rostami MK, Razavi-Toosi MT, Shokrgozar MA, Jahanian-Najafabadi A, et al. Mitochondrial transplantation ameliorates ischemia/reperfusion-induced kidney injury in rat. Biochim et Biophys acta Mol Basis Dis 2020;1866:165809.
Jesinkey SR, Funk JA, Stallons LJ, Wills LP, Megyesi JK, Beeson CC, et al. Formoterol restores mitochondrial and renal function after ischemia-reperfusion injury. J Am Soc Nephrol 2014;25:1157–62.
Stallons LJ, Funk JA, Schnellmann RG Mitochondrial Homeostasis in Acute Organ Failure. Current pathobiology reports. 2013;1.
Whitaker RM, Korrapati MC, Stallons LJ, Jesinkey SR, Arthur JM, Beeson CC, et al. Urinary ATP Synthase Subunit β is a novel biomarker of renal mitochondrial dysfunction in acute kidney injury. Toxicolog Sci 2015;145:108–17.
Feng J, Chen Z, Liang W, Wei Z, Ding G. Roles of mitochondrial DNA damage in kidney diseases: a new biomarker. Int J Mol Sci 2022;23:15166.
Zhang M, Zhang Y, Wu M, Li Z, Li X, Liu Z, et al. Importance of urinary mitochondrial DNA in diagnosis and prognosis of kidney diseases. Mitochondrion 2021;61:174–8.
Linden J, Koch-Nolte F, Dahl G. Purine release, metabolism, and signaling in the inflammatory response. Annu Rev Immunol 2019;37:325–47.
Sun M, Hao T, Li X, Qu A, Xu L, Hao C, et al. Direct observation of selective autophagy induction in cells and tissues by self-assembled chiral nanodevice. Nat Commun 2018;9:4494.
Poudel N, Okusa MD. Pannexins in acute kidney injury. Nephron 2019;143:158–61.
Bao Y, Ledderose C, Graf AF, Brix B, Birsak T, Lee A, et al. mTOR and differential activation of mitochondria orchestrate neutrophil chemotaxis. J Cell Biol 2015;210:1153–64.
Su L, Zhang J, Wang J, Wang X, Cao E, Yang C, et al. Pannexin 1 targets mitophagy to mediate renal ischemia/reperfusion injury. Commun Biol 2023;6:889.
Rushworth GF, Megson IL. Existing and potential therapeutic uses for N-acetylcysteine: the need for conversion to intracellular glutathione for antioxidant benefits. Pharmacol Ther 2014;141:150–9.
Maestro C, Leache L, Gutiérrez-Valencia M, Saiz LC, Gómez H, Bacaicoa MC, et al. Efficacy and safety of N-acetylcysteine for preventing post-intravenous contrast acute kidney injury in patients with kidney impairment: a systematic review and meta-analysis. Eur Radiol 2023;33:6569–81.
Azarkish F, Nematbakhsh M, Fazilati M, Talebi A, Pilehvarian AA, Pezeshki Z, et al. N-acetylcysteine prevents kidney and lung disturbances in renal ischemia/reperfusion injury in rat. Int J Prev Med 2013;4:1139–46.
Aki T, Tanaka H, Funakoshi T, Unuma K, Uemura K. Excessive N-acetylcysteine exaggerates glutathione redox homeostasis and apoptosis during acetaminophen exposure in Huh-7 human hepatoma cells. Biochem Biophys Res Commun 2023;676:66–72.
Zhao J, Li M, Tan C. Efficacy of N-acetylcysteine in preventing acute kidney injury and major adverse cardiac events after cardiac surgery: a meta-analysis and trial sequential analysis. Front Med 2022;9:795839.
Baker WL, Anglade MW, Baker EL, White CM, Kluger J, Coleman CI. Use of N-acetylcysteine to reduce post-cardiothoracic surgery complications: a meta-analysis. Eur J Cardio-Thorac Surg 2009;35:521–7.
Radajewska A, Szyller J, Krzywonos-Zawadzka A, Olejnik A, Sawicki G, Bil-Lula I. Mitoquinone alleviates donation after cardiac death kidney injury during hypothermic machine perfusion in rat model. Int J Mol Sci 2023;24:14772.
Li Y, Wang M, Wang S. Effect of inhibiting mitochondrial fission on energy metabolism in rat hippocampal neurons during ischemia/reperfusion injury. Neurol Res 2016;38:1027–34.
Yang Y, Ma J, Lin D. [Effect of dynamin-related protein 1 in rats with myocardial ischemia/reperfusion injury]. Zhonghua wei Zhong Bing Ji Jiu Yi Xue 2017;29:902–6.
Acknowledgements
We thank Editage for their linguistic assistance during the preparation of this manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (82000647 and 81974095), Zhanjiang Science and Technology Plan Project (2022A01158 and 2022A01160), Guangdong Provincial Key Laboratory of Autophagy and Major Chronic Non-Communicable Diseases (2022B1212030003), Discipline construction project of Guangdong Medical University(4SG21229G), the high-level talents scientific research start-up funds of the Affiliated Hospital of Guangdong Medical University (1007Z20220002 and 10403Z20190001), and National Clinical Key Specialty Construction Project (Institute of Nephrology, Affiliated Hospital of Guangdong Medical University).
Author information
Authors and Affiliations
Contributions
HL, JH and XL: design and conception; XL and HL: review and revision of the paper; YC, ZL and HZ: writing and revision of the paper; HC: technical and material support. All the authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics
Not applicable
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, Y., Li, Z., Zhang, H. et al. Mitochondrial metabolism and targeted treatment strategies in ischemic-induced acute kidney injury. Cell Death Discov. 10, 69 (2024). https://doi.org/10.1038/s41420-024-01843-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-024-01843-5
This article is cited by
-
FGF21 Inhibits Hypoxia/Reoxygenation-induced Renal Tubular Epithelial Cell Injury by Regulating the PPARγ/NF-κB Signaling Pathway
Cell Biochemistry and Biophysics (2024)
