
Abstract
Multiple myeloma (MM) remains an incurable hematological malignancy disease characterized by the progressive dysfunction of the patient’s immune system. In this context, immunotherapy for MM has emerged as a prominent area of research in recent years. Various targeted immunotherapy strategies, such as monoclonal antibodies, antibody-drug conjugates, bispecific antibodies, chimeric antigen receptor T cells/natural killer (NK) cells, and checkpoint inhibitors have been developed for MM. This review aims to discuss promising experimental and clinical evidence as well as the mechanisms of action underlying these immunotherapies. Specifically, we will explore the design of exosome-based bispecific monoclonal antibodies that offer cell-free immunotherapy options. The treatment landscape for myeloma continues to evolve with the development of numerous emerging immunotherapies. Given their significant advantages in modulating the MM immune environment through immune-targeted therapy, these approaches provide novel perspectives in selecting cutting-edge treatments for MM.
Similar content being viewed by others

Facts
-
The patients with Multiple myeloma present severely dysregulated immune systems.
-
The utilization of immunotherapy drugs offers a highly effective therapeutic option for relapsed/refractory multiple myeloma patients.
-
The application of immunotherapy demonstrates significant promise for newly diagnosed multiple myeloma patients.
Open questions
-
What are the potential applications for designing bispecific monoclonal antibodies based on exosomes?
-
what are the effects of immunomodulatory drugs on the multiple myeloma cellular microenvironment?
-
Can personalized combinations of immunotherapy drugs be provided to relapsed refractory patients to further enhance treatment outcomes?
Introduction
Multiple myeloma (MM) is the second most prevalent hematological malignancy, characterized by abnormal proliferation of clonal plasma cells, resulting in hypercalcemia, renal insufficiency, anemia, bone disease, and other manifestations [1]. Despite significant advancements in MM treatment, it remains incurable. In the new therapeutic era, induction chemotherapy regimens based on proteasome inhibitors and/or immunomodulators as three or four-drug combinations combined with autologous stem cell transplantation have significantly prolonged the median survival time of MM patients. However, there are still cases where remission cannot be achieved or relapse occurs after remission [2,3,4].
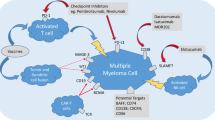
The pathogenesis of MM is complex, in which immune dysfunction plays a crucial role in disease recurrence and drug resistance [5]. It has been reported that myeloma cells escape immune detection, have insufficient antigen presentation cells, have T cell activation disorders, and other factors that can promote the survival of myeloma cells within the host [6]. With the continuous emergence of new drugs, the anti-tumor effect of the body’s autoimmune system can be stimulated by the action of plasma cell-specific antigens in patients with MM to achieve the purpose of targeting the killing of tumor cells. Therefore, the emergence of targeted immunotherapy has provided renewed hope for relapsed or refractory MM patients. Currently available, the targeted immunotherapies for MM mainly include chimeric antigen receptor T cell immunotherapy (CAR-T), CAR-NK cells, checkpoint-blocking antibodies, monoclonal antibodies, antibody-drug conjugates, and bispecific antibodies [7] (Fig. 1). This review aims to summarize the clinical application of these targeted immunotherapy strategies while introducing our novel exosome bispecific antibody-based immunotargeted therapy for MM. We hope this will inspire further advancements in targeted therapy.

CAR chimeric antigen receptor, PD-L1 programmed death-ligand 1, PD-1 programmed cell death protein 1, TCR T cell receptor, MHC major histocompatibility complex, NK cell natural killer cell.
Cellular immunotherapy
Chimeric antigen receptor T cells
Chimeric antigen receptors (CARs) are transmembrane proteins consisting of an extracellular domain for antigen recognition, a hinge region, a transmembrane domain, and an intracellular signaling domain. The antigen recognition domain typically comprises single-chain antibodies (scFVs) that specifically target tumor antigens. The intracellular signaling domain includes the T cell activation domain (CD3 ζ) and costimulatory molecules necessary for T cell activation, usually including the costimulatory domains 4-1BB or CD28 [8,9,10]. The process of CAR-T cell treatment begins with the collection of T cells from either patient or healthy donors through leukapheresis. Subsequently, these T cells are stimulated, virally transduced, and proliferated to generate CAR-T cells by employing lentiviral or γ-retroviral vectors to transfer the CAR-encoding gene into the genome of T cells. Before infusing CAR-T cells, patients are initially treated with lymphodepleting chemotherapy, commonly using a regimen combining fludarabine and cyclophosphamide (FC). During the infusion of CAR-T cells, close attention should be paid to observing drug infusion reactions such as cytokine release syndrome (CRS) and neurotoxicity [11] (Fig. 2).

FC fludarabine and cyclophosphamide.
The antigens targeted by CAR-T cells should exhibit specific expression exclusively on the surface of tumor cells. B cell maturation antigen (BCMA) is a member of the tumor necrosis factor receptor superfamily, expressed in MM cells, while normal plasma cells and mature B cells exhibit low or no expression, making it the primary target for CAR-T cell therapy in MM patients [12]. The American Cancer Institute first launched a phase 1 clinical trial of anti-BCMA-CAR-T cell therapy for relapsed/refractory MM (R/RMM) (NCT02215967) demonstrated that BCMA-CAR-T cells are effective against R/RMM, but further optimization is needed to mitigate adverse effects [13, 14]. In a subsequent phase 1 clinical trial (NCT03090659), LCAR-B38M, a CAR-T cell targeting two different BCMA epitopes (VH1 and VH2), was reported in 57 patients who had received an average of three treatments with an ORR of 88%, a complete response (CR) rate of 68%, and a median progression-free survival (PFS) of 15 months. CRS occurred in 90% of subjects, primarily in grades 1 or 2(83%) [15]. CT103A is another BCMA-targeted CAR-T cell containing a fully human single-chain antibody, CD8α hinge, and transmembrane region, 4-1BB costimulatory and CD3ζ activation domains. In a phase I clinical trial (ChiCTR1800018137) of R/RMM being studied in China, good efficacy (ORR 100%; CR or strict CR rate 72.2%) was shown in 18R/RMM patients treated with CT103A. A subsequent phase I/II study (ChiCTR2000033946) included 71R/RMM patients to evaluate the efficacy and safety of CT103A. Notably, among these patients, 18.3% had previously received murine BCMA CAR-T therapy. The results were similarly encouraging (ORR 94.4%, MRD negative rate 92.8%). CRS occurred in 93.0% patients, of which only 2.8% were grade 3 [16, 17]. In addition to CT103A, BB2121 is another second-generation CAR incorporating an anti-BCMA single-chain variable fragment, a CD137 (4-1BB) costimulatory motif, and aCD3-zeta signaling domain. Both phase I (NCT02658929) [18] and phase II (NCT03361748) [19] clinical trials have shown favorable results in R/RMM, which were safe and manageable. The results of the phase II study showed that the ORR of 128R/RMM patients was 73% (≥CR rate 33%, MRD negative rate 26%), and the median PFS was 8.8 months. Based on these promising results, BB2121 has been approved by the FDA in 2020.
Other targets of CRA-T cell therapy mainly include CD19, CD38, and other antigens. A clinical trial (NCT02135406) reported that CAR-T targeting CD19 antigen combined with autologous hematopoietic stem cell transplantation in treating 10 patients with R/RMM, the ORR was 80% [20]. In addition, some studies have combined infusion of CD19 and BCMA-specific CAR T cells to treat R/RMM or high-risk MM with ORR ranging from 90 to 100% [21,22,23]. Besides, a study (ChiCTR1800018143) had reported that the results of a bispecific CAR-T cell therapy targeting BCMA and CD38 for R/RMM showed that after a median follow-up period of 9.0 months, 12 patients (52%) achieved a stringent complete response, and extramedullary plasmacytoma completely resolved in 56% of the patients [24]. Currently, there are some CAR-T cell therapy studies targeting other targets (such as CD138, SLAMF7/CS1, kappa light chain, etc.), but few studies have reported [25]. Table 1 summarizes the significant clinical trials of CAR-T therapy for MM.
CAR-T therapy improves the survival and prognosis of patients with MM. However, current research on CAR-T therapy primarily focuses on R/RMM patients who exhibit resistance to multiple mechanistic drugs, necessitating further exploration of its therapeutic potential in other MM subgroups. Moreover, producing autologous CAR-T cells is a time-consuming process that requires strict control over transportation, storage, and management [26]. Additionally, optimizing and exploring off-target effects in CAR-T therapy, as well as addressing the severe side effects caused by CRS, remains crucial [27]. To enhance the safety of CAR-T cell therapy, studies have suggested incorporating a suicide switch into the CAR structure to trigger it when adverse reactions occur; however, only approximately 10% of clinical trials have successfully implemented this safety feature [28].
Chimeric antigen receptor NK cells
Car-modified NK cells use genetic engineering to add a chimeric antibody to NK cells that can recognize tumor cells and simultaneously activate NK cells to kill tumor cells. It is safer than CAR-T cell therapy because NK cells do not secrete inflammatory factors such as IL-1 and IL-6. Since NK cells do not need to be pre-sensitized and do not cause graft-versus-host disease, Car-modified NK cells have attracted much attention recently [29].
Allogeneic NK cells come from various sources, including peripheral blood, NK cell lines, umbilical cord blood, induced pluripotent stem cells, etc [30]. Currently, CAR-NK cell therapy products for the research and treatment of MM mostly use the NK92 cell line. The cell surface glycoprotein CD2 subset 1 (CS1) is a highly expressed protein on the surface of MM cells and is primarily involved in the adhesion and growth of MM cells [31]. CD138 is highly expressed in MM cells and is the primary diagnostic marker for MM [32]. In vitro studies have shown that CS1-CAR-NK92 cells and CD138-IFNα-CAR-NK92 cells designed with CS1 and CD138 targets, respectively, successfully inhibited the growth of MM cells and prolonged the survival of myeloma mice [33, 34]. CAR-NK cell therapy also has some limitations: although NK92 cell lines can be easily amplified, their activation and proliferation depend on IL-2 or IL-15. Systematic IL-2 administration in clinical application has serious side effects and can activate immunosuppressive signals. In addition, improvement of CAR structure and transfection methods needs to be further optimized [30].
Checkpoint inhibitors
Immune checkpoints refer to receptors and ligands involved in programmed cell death. The dysfunction of immune surveillance cells plays a crucial role in the development and progression of MM. Inhibiting the interaction between immune checkpoint receptors and their ligands can enhance the aggressiveness of the host immune system against tumor cells. Studies have demonstrated that aberrant activation of the programmed cell death protein 1 (PD-1)/ programmed death-ligand 1 (PD-L1) pathway is significant in MM [35]. The PD-1/PD-L1 axis functions as a negative costimulatory pathway, with increased expression of PD-1 on T cells and PD-L1 on plasma cells observed in MM patients. The binding of PD-L1 to PD-1 inhibits T cell proliferation and activation, creating an immunosuppressive microenvironment that promotes migration and proliferation of myeloma cells. By blocking the combination of PD-1 and PD-L1, inhibitors restore T-cell killing function against myeloma cells [36]. Furthermore, studies have shown elevated expression of PD-1 on other immune cells, such as NK cells and macrophages. Blocking the PD-1/PD-Ll pathway enhances NK-cell-mediated effects against MM and macrophage phagocytosis, thereby reducing tumor growth [37, 38]. Additionally, a study has demonstrated that pDCs and MM cells exhibit high expression of PD-L1, suggesting a dual inhibition of immune function in PD-1-expressing T cells and NK cells. Moreover, using PD-L1 inhibitors can augment the cytolytic activity of T cells and NK cells against MM cells [39] (Fig. 3).

In patients with MM, the expression of PD-1 on various subsets of immune cells, including T cells, NK cells, and macrophages, is upregulated. The interaction between PD-1 on immune cells and PD-L1 on MM cells leads to the inhibition of immune cell proliferation and activation, resulting in the formation of an immunosuppressive microenvironment. Moreover, plasmacytoid dendritic cells (pDCs) exhibit high levels of PD-L1 expression, which further hampers the immune killing function when combined with PD-1 on T or NK cells. However, the administration of either PD-1 or PD-L1 inhibitors can disrupt the binding between PD-1 and PD-L1 molecules and restore the cytotoxicity of immune cells against MM.
Nivolumab and Pembrolizumab are PD-1 inhibitors. A Phase Ib trial evaluated Nivolumab monotherapy for R/RMM with an ORR of only 4% [40]. Another phase 1b study (KEYNOTE-013, NCT01953692) evaluated the efficacy of pembrolizumab monotherapy in the treatment of R/RMM and showed that the best response was disease stabilization in the 30 R/RMM patients enrolled (n = 17; 56.7%), and eventually 80% of patients stopped treatment due to disease progression/clinical progression (n = 24) [41]. All the above studies showed that PD-1 inhibitor monotherapy had poor efficacy in treating R/RMM. Consequently, researchers further investigated the synergistic potential of combining PD-1 inhibitors with other therapeutic agents in MM.
KEYNOTE-023 is a Phase I clinical study (NCT02036502) designed to evaluate the efficacy and safety of pembrolizumab in combination with lenalidomide and low-dose dexamethasone (Rd) in R/RMM. A total of 62 patients were enrolled, with an overall response rate of 44%, median PFS of 7.2 months, and 3.2% (2/62) of patients discontinued treatment due to TRAEs [42]. Although this study showed an acceptable safety and efficacy of pembrolizumab in combination with lenalidomide and low-dose dexamethasone for R/RMM, the results were not reproduced in the subsequent phase III trial. KEYNOTE-183 (NCT02576977) is a phase 3 clinical study that evaluated the efficacy and safety of pembrolizumab plus pomalidomide and dexamethasone (Pd) for R/RMM patients. A total of 249 patients were enrolled and randomly assigned to the pembrolizumab +Pd group or the Pd group. However, the results of the study showed that the pembrolizumab +Pd group had a shorter median PFS (5.6 months vs. 8.4 months), a decrease in the 6-month OS rate (82% vs. 90%), and a significantly higher proportion of serious adverse events (AEs) (63% vs. 46%) than the Pd group [43]. Similarly, the EYNOTE-185 (NCT02579863) phase 3 clinical study evaluated the efficacy and safety of lenalidomide and dexamethasone (Rd) with or without pembrolizumab in newly diagnosed MM (NDMM) patients who were not eligible for transplantation. Patients were randomly divided into the pembrolizumab + Rd group (n = 151) and Rd group (n = 150). The results of the study showed that compared with the Rd group, the 6-month PFS rate of the pembrolizumab combination group was reduced (82% vs. 85%), and the incidence of serious AEs was significantly higher (54% vs. 39%) [44]. Since the risks of the pembrolizumab combination outweighed the benefits, On 3 July 2017, the FDA halted the above study (The ASCO Post, 2017).
In addition, the majority of clinical trials investigating other PD-L1 inhibitors, such as durvalumab and atezolizumab, have been either completely or partially halted. The efficacy and safety of combining durvalumab with daratumumab in patients with daratumumab-refractory relapsed/refractory MM patients were evaluated in a prospective phase 2 clinical study (NCT03000452). The results revealed that the incidence of serious AEs was 38.9%, but none of the 18 enrolled patients achieved a PR or higher. Inhibition of the PD-1/PD-L1 signaling pathway during daratumumab resistance proved insufficient to reverse daratumumab resistance [45]. Table 2 provides an overview of the significant clinical trials involving checkpoint inhibitors in MM.
However, no specific cause or distinct AEs associated with a higher risk of death were observed in phase III clinical trials comparing PD-1 monoantibody combination therapy to the control group [46]. Other studies exploring pomalidomide and dexamethasone combined with pembrolizumab for treating relapsed/refractory MM demonstrated persistent immune effects even after discontinuation of pembrolizumab [47]. Additionally, a study investigating lenalidomide and dexamethasone combined with pembrolizumab for post-transplant consolidation therapy in high-risk myeloma patients suggested that this combination offers potential long-term disease control as a short-term consolidation regimen [48]. Therefore, PD-1 /PD-L1 inhibitor combination therapy for MM remains a therapeutic option.
Monoclonal antibodies
At present, the FDA has approved three monoclonal antibodies for the treatment of MM, including daratumumab (anti-CD38), elotuzumab (antibody targeting signaling lymphocytic activation molecule F7 (SLAMF7)), approved in 2015, and isatuximab (anti-CD38), approved in 2020. The mechanism of CD38 monoclonal antibodies (mAbs) treatment of MM is similar, such as complement-dependent cytotoxicity (CDC), antibody-dependent cell-mediated cytotoxicity (ADCC), and antibody-dependent cell-mediated phagocytosis (ADCP). In addition, CD38 mAbs can also induce programmed cell death, reduce mitochondrial transfer, inhibit adenosine production and adhesion molecule function, and regulate enzyme activity to cause MM cell death. Besides, CD38 mAbs can combine with immunomodulatory cells to eliminate the inhibitory effect of these cells on T effector cells, thereby activating T cells to kill MM cells [49, 50]. Elotuzumab works against MM mainly by directly activating NK cells and mediating ADCC through the CD16 pathway [51] (Fig. 4).

Monoclonal antibodies have a variety of modes of action, mainly including antibody-mediated cross-linking induces programmed cell death (PCD) in MM cells; complement-dependent cytotoxicity (CDC) is initiated by the binding of C1q to the Fc tail of the antibody initiates the complement cascade, resulting in formation of the membrane attack complex (MAC); antibody-dependent cell-mediated cytotoxicity (ADCC) is the release of cytoplasmic granzymes and perforin by NK cells after being activated by antibodies, leading to apoptosis of MM cells; antibody-dependent cell-mediated phagocytosis (ADCP) is mainly the phagocytic destruction of MM cells mediated by macrophages; and immunoregulatory actions such as decreasing myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), regulatory B cells (Bregs), and pDCs, which is crucial for the activation of T cells. In addition, monoclonal antibodies can reduce mitochondrial transfer between bone marrow stromal cells (BMSCs) and MM cells, inhibit the function of adhesion molecules, modulate enzymatic activity, and reduce the production of adenosine to inhibit MM cells.
Daratumumab
Daratumumab is the first CD38 monoclonal antibody for the treatment of MM. The results of clinical studies have confirmed that daratumumab alone or in combination can achieve good results in R/RMM. In Phase 1–2 clinical trials GEN501 (NCT00574288) and SIRIUS (NCT01985126), 16 mg/kg was determined to be the optimal dosage concentration for daratumumab [52, 53]. Subsequent phase III trials POLLUX [54,55,56,57] and CASTOR [58, 59] confirmed that daratumumab in combination with lenalidomide or bortezomib improved the remission rate of patients with R/RMM, including higher MRD negative rates and longer PFS and OS [60]. The recent open-label phase III trial, CANDOR (NCT03158688), enrolled 466 patients with R/RMM and randomly divided them into daratumumab + carfilzomib + dexamethasone (Dara-Kd) and Kd group. After 27.8 months of median follow-up, Dara-Kd prolonged PFS in the patients (median 28.6 vs 15.2 months; P < 0.0001). The incidence of AEs leading to discontinuation was similar in the D-Kd and Kd groups [61]. Overall, daratumumab-based combination therapy enables R/RMM patients to achieve deep remission and prolonged clinical outcomes in different subgroup analyses, including age, cytogenetics, etc [62, 63].
Currently, the utilization of daratumumab in combination with other chemotherapy regimens has witnessed a progressive rise in NDMM patients. For MM patients who are suitable for transplantation, the GRIFFIN (NCT02874742) phase II clinical trial included 207 patients, who were randomly divided into daratumumab, bortezomib, lenalidomide and dexamethasone (Dara-VRd) group and VRd group. The results confirmed that the complete remission rate, including deep remission, and the long-term survival prognosis of the Dara-VRd group were superior to that of the VRd group [64]. In a phase III clinical trial (CASSIOPEIA, NCT02541383) of 1085 patients with NDMM, the addition of daratumumab to a regimen of bortezomib, thalidomide, and dexamethasone (Dara-VTd) was associated with a higher rate of ≥CR rate and MRD negativity rate compared with standard triple therapy (VTd) after 100 days of transplantation. ≥CR rate was observed in 39% of patients in the Dara-treated group, with 64% achieving MRD negativity (10−5), while only 26% of patients with ≥CR, 44% being MRD negativity in the VTd group [65]. In addition, in terms of patient quality of life, Dara-VTd resulted in more significant pain reduction, less cognitive deterioration, and greater improvement in emotional function compared with the VTd group [66].
For newly diagnosed MM patients who are not suitable for transplantation, a series of phase III clinical studies have also been conducted; for example, the ALCYONE (NCT02195479) phase III clinical trial included 706 patients treated with nine cycles of daratumumab in combination with bortezomib, melphalan, and prednisone (Dara-VMP) or VMP. After chemotherapy, the remission rate and long-term survival were better in the Dara-VMP group than in the VMP group [67, 68]. In addition, the phase III clinical trial MAIA (NCT02252172) enrolled 737 NDMM patients and randomly divided them into daratumumab combined with lenalidomide and dexamethasone (Dara-Rd) group and Rd group. The ≥CR rates in the Dara-Rd and Rd groups were 47.6% and 24.9%, respectively (p < 0.001), and the MRD negative rates (10−5) were 24.2% and 7.3%, respectively (P < 0.001). At a median follow-up of 56.2 months, the median PFS was not reached in the daratumumab group versus 34.4 months in the control group (p < 0.0001) [69, 70]. The clinical benefit of Dara-Rd in transplant-ineligible NDMM patients enrolled in MAIA, regardless of frailty status [71]. At the same time, Dara-Rd also improves patients’ quality of life faster than Rd [72]. In terms of economic benefits, at the current price of daratumumab, first-line use of daratumumab is not cost-effective [73]. The 2022 V3 NCCN guidelines propose that for patients who are not suitable for hematopoietic stem cell transplantation, an appropriately reduced Dara-Rd regimen can be utilized, based on the aforementioned studies.
In general, for R/RMM patients and NDMM patients, daratumumab combined with standard therapy can be used to improve the curative effect, survival outcomes, and the patients’ quality of life. Furthermore, daratumumab-based combination therapy improved the depth of remission, that is, the rate of MRD-negative conversion and the duration of MRD-negative, which was also closely related to longer PFS [74, 75].
Isatuximab
Isatuximab (SAR650984) is another humanized IgG1 monoclonal antibody targeting CD38-specific epitopes. The mechanism of action is similar to that of daratumumab. Still isatuximab can induce the internalization of CD38 in MM cells without releasing CD38 from MM cells, which is different from daratumumab [76]. There are also differences in the dose and frequency of administration. In comparison, the dose and frequency of Isatuxima are better than those of daratumumab [77].
Phase 1b clinical studies evaluated the favorable safety and efficacy of isatuximab in combination with lenalidomide and dexamethasone or isatuximab in combination with pomalidomide and dexamethasone in the treatment of patients with R/RMM [78, 79]. The phase III ICARIA study (NCT02990338) compared pomalidomide and dexamethasone in combination with Isatuximab in patients with bortezomib and/or lenalidomide-resistant R/RMM. A total of 307 patients were enrolled in this study, who were randomly divided into the Isatuximab/pomalidomide/dexamethasone group (Isa-Pd) and the Pd group. After a median follow-up of 11.6 months, the median PFS of the Isa-Pd group was better than that of the Pd group (11.5 vs. 6.5 months; p = 0.001). After a median follow-up of 35.3 months, the median OS of the Isa-Pd group was better than that of the Pd group (24.6 vs. 17.7 months; p = 0.001), and there was no significant difference in safety between the two groups [80, 81]. In addition, Isa-Pd improved ORR in patients with either lenalidomide-refractory, bortezomib-refractory, or double-refractory patients (59% vs. 31.4%; 60.2% vs. 32.2%; 58.6% vs. 29.9%; respectively) [82]. Subsequent subgroup analyses showed that the addition of Isa to Pd improved subpopulations with poor prognostic factors, including those with high-risk cytogenetics, those with renal impairment, those who are elderly, and those who are refractory to prior lines of treatment [83,84,85]. In addition, the IKEMA Phase III trial (NCT03275285) evaluated the efficacy of isatuximab + carfilzomib + dexamethasone (Isa-Kd) in R/RMM patients. A total of 302 patients were randomly divided into Isa-Kd and Kd groups. The results showed: Isa-Kd improved patients’ depth of response and median PFS (MRD negative rate 10−5: 30% vs. 16%, P = 0.0004; median PFS: not reached vs. 19.15 months, p = 0.0007; respectively). Serious AEs occurred similarly in both groups (59% vs. 57%) [86]. Subgroup analysis results showed that complete renal responses occurred more frequently with Isa-Kd (52.0%) versus Kd (30.8%) and were durable in 32.0% versus 7.7% of patients, respectively [87]. Based on these findings, the FDA has approved isatuximab in combination with pomalidomide, dexamethasone, and isatuximab in combination with carfilzomib, dexamethasone, respectively, for the treatment of R/RMM patients.
In addition, for newly diagnosed MM patients, a series of clinical studies have also been carried out. The GMMG-CONCEPT phase II clinical trial (NCT03104842) investigated the efficacy of four regimens of isatuximab, carfilzomib, lenalidomide, and dexamethasone (Isa-KRd) in high-risk (HR) newly diagnosed MM patients. The findings demonstrate encouraging rates of rapid and deep remissions in HR MM patients with Isa-KRd induction, with an ORR of 100% for the best induction response and an MRD-negative rate of 62.5%. After a median follow-up of 24.9 months, the median PFS was not reached, and the median 12-month PFS was 79.6%; the median 24-month PFS was 75.5% [88]. In addition, several ongoing clinical studies are currently being conducted to evaluate the combination of isatuximab with lenalidomide, bortezomib, and dexamethasone (GMMG-HD7), as well as the combination of isatuximab with carfilzomib, lenalidomide, and dexamethasone (NCT04430894) in NDMM patients eligible for transplantation. Furthermore, there are clinical studies assessing the efficacy and safety of isatuximab in combination with lenalidomide, bortezomib, and dexamethasone (Isa-VRd) for treating NDMM patients who are ineligible for transplantation (NCT02513186). The results from these studies are highly anticipated.
Elotuzumab
Elotuzumab is a humanized IgG1 anti-SLAMF7 monoclonal antibody that elicits its effect via a dual mechanism of action: direct activation of NK cells and ADCC [51]. Although the efficacy of elotuzumab monotherapy in relapsed/refractory multiple myeloma (R/RMM) is not significant [89], its combination with other antineoplastic agents enhances patient response rates and improves clinical outcomes.
ELOQUENT-2 (NCT01239797) is a phase 3, randomized clinical trial that investigated the efficacy and safety of elotuzumab plus lenalidomide, dexamethasone (E-Rd) compared with Rd in R/RMM patients. The ORR in the elotuzumab group was 79% versus 66% in the control group (P < 0.001). Regarding security, infusion reactions occurred in 33 patients (10%) in the elotuzumab group and almost were grade 1 or 2 [90]. Subsequent follow-up data showed that, after a minimum follow-up of 70.6 months, the elotuzumab group also demonstrated a PFS advantage (median PFS: 19.4 versus 14.9 months; P < 0.001) and an OS advantage (median OS: 48.3 versus 39.6 months; P = 0.0408). The magnitude of OS benefit was most significant among patients with adverse prognostic factors, including older age, ISS stage III, IMWG high-risk disease, and 2–3 prior lines of therapy [91]. In addition, ELOQUENT-3 (NCT02654132) clinical studies have reported whether elotuzumab combined with pomalidomide + dexamethasone (EPd) is effective in treating R/RMM. The results showed that the ORR (53% vs. 26%) and PFS (median PFS: 10.3 versus 4.7 months; P = 0.008) of the elotuzumab group were better than those of the pomalidomide + dexamethasone (Pd) group, with no significant difference in adverse drug reactions [92]. Median OS was significantly improved with EPd (29.8 [22.9 to 45.7] months) versus Pd (17.4 [13.8 to 27.7] months), with a hazard ratio of 0.59 (95% CI, 0.37 to 0.93; P = 0.0217). OS benefit with EPd was observed in most patient subgroups [93]. Moreover, HRQoL was not impacted by the addition of elotuzumab to Pd [94]. The FDA has approved the use of elotuzumab in combination with lenalidomide/ dexamethasone or pomalidomide/dexamethasone for the treatment of R/RMM patients who have undergone 1–3 prior regimens, based on the studies mentioned above demonstrating the therapeutic benefits of elotuzumab-based drug combinations.
The SWOG-1211 (NCT01668719) study was conducted to further investigate the efficacy and safety of elotuzumab combined with lenalidomide, bortezomib, and dexamethasone (E-VRd) in newly diagnosed high-risk MM patients. However, the results showed no difference in median PFS (VRd 33.64 months vs. E-VRd 31.47 months, p = 0.45), and the addition of elotuzumab to VRd induction and maintenance did not improve patient outcomes [95].
Other monoclonal antibodies
Currently, there are ongoing clinical developments for novel anti-CD38 monoclonal antibodies. For instance, phase I/II clinical studies have evaluated the efficacy and safety of MOR202 in combination with dexamethasone and/or lenalidomide and permadomide in R/RMM patients [96]. Furthermore, research is also underway to develop other targeted antibodies against malignant plasma cell surface molecules such as CD138 monoclonal antibody (indatuximab ravtansine), CD56 monoclonal antibody (lorvotuzumab mertansine), CD74 monoclonal antibody (Milatuzumab), CD40 monoclonal antibody (Dacetuzumab) and others. However, further studies are required to confirm the efficacy of these drugs’ efficacy and their combined [97,98,99,100].
Additionally, within the MM microenvironment consisting of stromal cells, endothelial cells, and osteoclasts, various growth factors, including interleukin-6 (IL-6), insulin-like growth factor 1(IGF1), vascular endothelial growth factor(VEGF) are secreted. These factors play crucial roles in activating intracellular signaling pathways that mediate MM cell growth, survival, migration, and drug resistance [101]. Therefore, the development of neutralizing growth factors or inhibiting growth-promoting receptor-related antibodies is underway. For instance, several studies have been conducted to assess the safety and efficacy of Siltuximab (anti-IL-6) in combination with conventional drugs for treating MM. Despite its good tolerability, the effectiveness of Siltuximab remains controversial [102,103,104]. Descamps et al. [105] discovered that AVE1642, a humanized IGF1 receptor monoclonal antibody, inhibited tumor growth and significantly enhanced bortezomib-induced apoptosis in CD45-negative tumor cell lines. However, the results of phase I studies of AVE1642 as a single agent and in combination with bortezomib in treating relapsed MM were not satisfactory [106]. Additionally, bevacizumab, a VEGF monoclonal antibody, can inhibit the activation of signaling cascades in MM cells and reduce their proliferation along with stromal cells [107]. Nevertheless, a randomized phase 2 clinical study evaluating the efficacy of bevacizumab combined with bortezomib in the treatment of R/RMM showed that it did not significantly improve the efficacy [108].
Monoclonal antibody immunotherapy has gradually emerged over recent years; FDA-approved examples include Dara monoclonal antibody, Isatuximab, and Elotuzumab. However, it remains crucial to further optimize drug performance and application methods while identifying specific targets for myeloma cells. Table 3 provides an overview of significant clinical trials involving monoclonal antibody therapy for MM.
Antibody-drug conjugates
Targeted monoclonal antibodies can also be used as carriers to deliver drugs to target cells. Among them, belantamab mafodotin (Blenrep, GSK-2857916) is a novel fucosylated anti-BCMA monoclonal antibody conjugated with the microtubule-disrupting agent monomethyl auristatin F (MMAF) via a protease-resistant linker. Blenrep targets and binds to BCMA on the surface of MM cells through an anti-BCMA monoclonal antibody, which is internalized by MM cells and releases MMAF, specifically blocks cell growth via G2/M arrest and induces caspase three dependent apoptosis in MM cells. At the same time, Blenrep inhibits B cell activation factor (BAFF)/a proliferation-inducing ligand (APRIL)-BCMA axis mediated NF-kB signaling by competitively binding to BCMA. Besides, Blenrep can also bind to effector cells (NK cells, monocytes, and macrophages) to promote ADCC and ADCP to kill MM cells [109, 110] (Fig. 5).

Blenrep is an antibody-drug conjugate that specifically binds to BCMA on the MM cell surface, delivering the cytotoxic monomethyl auristatin F (MMAF) to kill MM cells. Blenrep could compete for the binding of B cell activation factor (BAFF) and/or a proliferation-inducing ligand (APRIL) to BCMA, thereby blocking the NF-κB signaling pathway, which is essential for MM cell growth. In addition, Blenrep could mediate the Fc-dependent effects consisting of CDC, ADCC and ADCP to kill MM cells. Bispecific antibodies (BsAbs), including bispecific T cell engagers (BiTE), could bind to both CD3 on T cells and MM cell surface antigens such as BCMA, GPRC5D, FCRL5 and CD38. The interaction leads to the activation and proliferation of T cells to release granzymes and perforins to kill MM cells. GPRC5D G-protein-coupled receptor class 5 member D, FcRL5 Fc receptor-like 5, scFvs single-chain variable fragments, Fab antigen-binding fragment, Fc crystallizable fragment.
In the phase I study of the DREAMM-1 Study (NCT02064387), single-agent GSK2857916 had an ORR of 60% in R/RMM patients previously treated with alkylators, PIs, and immunomodulatory agents and refractory to the last line of therapy. After a median follow-up of 12.5 months, the median duration of response (DoR) was 14.3 months, and the median PFS was 12 months [111, 112]. The phase II study of DREAMM -2 (NCT03525678) enrolled 196 previously overtreated R/R MM patients to further evaluate the efficacy and safety of single-agent GSK2857916 in patients refractory to immunomodulators, PIs and refractory and/or intolerant to CD38-targeted monoclonal antibodies. Patients were randomly assigned to receive GSK2857916 at a dose of 2.5 mg/kg or 3.4 mg/kg once every 3 weeks. The ORR of GSK2857916 was 31% at 2.5 mg/kg and 34% at 3.4 mg/kg [113]. Subsequent analysis of GSK2857916 (3.4 mg/kg every 3 weeks) showed that after a median follow-up of 11.2 months, the median DoR was 9.0 months, the median PFS was 5.7 months, and the median OS was not reached. The most common grade 3/4 AEs were keratopathy (microcyst-like corneal epithelial changes, a pathological finding seen on eye examination [75%]); periodic ophthalmic evaluation is therefore required [114]. Based on these results, the FDA has approved GSK2857916 as a monotherapy for treating adult R/RMM patients who have received at least four prior therapies, including anti-CD38 monoclonal antibodies, PIs, and immunomodulators. Table 4 summarizes the significant clinical trials of GSK2857916 therapy for MM.
In addition, other antibody-drug conjugates (ADCs) targeting BCMA are currently under development. The cytotoxic part of MEDI2228, for example, is tesirine, a DNA-binding pyrrole benzodiazepine (PBD) dimer that binds to antibodies via Linker, which can be cleaved by proteases [115]. The investigational drug AMG 224, currently undergoing phase I evaluation, also targets BCMA and is conjugated with the tubulin inhibitor mertansine (also known as DM1) [116].
Bispecific antibodies
Bispecific antibodies (BsAbs) are antibodies capable of simultaneously targeting two antigens, typically the CD3 molecule of T cells and the tumor cell antigen. Recently, numerous BsAbs have been developed for MM treatment, with ideal tumor antigen targets including BCMA, G protein-coupled receptor 5D (GPRC5D), Fc receptor-like 5 (FCRL5), and CD38. BsAbs facilitate an immune synapse by recognizing and binding to surface antigens on T cells and MM cells, inducing T cell activation, proliferation, and subsequent tumor cell killing. There are various types of bispecific antibodies, mainly including the bispecific T cell engager (BiTE) containing two different scFvs connected by a flexible linker, and the IgG-like BsAbs consisting of fragment antigen-binding (Fab) domains and fragment-crystallizable (Fc) region [117]. While the Fc domain can mediate immune effects (such as ADCC, CDC, and ADCP), studies have shown that T-cell-directed BsAbs are designed to have an effector-silenced Fc region to prevent excessive cytokine secretion [118] (Fig. 5). Additionally, Table 4 summarizes significant clinical trials involving bispecific antibodies in MM.
Anti-CD3/BCMA bispecific antibodies
As mentioned above, BCMA is expressed explicitly in MM cells and is an ideal target for treating MM. AMG 420 is a BiTE that targets BCMA on MM cells and CD3 on T cells. A dose-escalation trial was given to evaluate the efficacy and safety of AMG420 in R/RMM patients with up to 10 cycles of AMG 420 (4-week infusions/6-week cycles). The results showed that the maximum tolerated dose of AMG 420 was 400 μg/d, and the response rate was 70%, including 50% MRD-negative complete responses [119]. However, due to the short half-life of AMG420, continuous infusion is required to maintain blood concentration, which brings great inconvenience.
ABBV-383, a BCMA/CD3 T-cell engaging bispecific antibody with a silent human IgG4 Fc segment, effectively restricts nonspecific activation while exhibiting an extended half-life. ABBV-383 is in an ongoing phase I clinical trial in R/RMM patients who have received at least three prior line therapies (NCT03933735). Preliminary findings indicate that ABBV-383 demonstrated favorable tolerability and achieved an ORR of 68% at doses equal to or greater than 40 mg [120]. In addition, teclistamab, a bispecific IgG4 antibody, also demonstrated an extended half-life. A phase I study (NCT03145181) evaluating the efficacy and safety of teclistamab in R/RMM patients showed that teclistamab was well tolerated at a subcutaneous dose of 1500 ug/kg once per week, with no discontinuations due to treatment-emergent adverse events [121]. With a median follow-up of 14.1 months, the ORR was 63.0%, the MRD-negativity rate was 26.7%, and the median duration of PFS was 11.3 months (95% CI, 8.8–17.1). Based on these findings, an international, open-label phase 2 expansion study of teclistamab in patients with R/RMM is underway (NCT04557098) [122]. CC-93269 is an asymmetric 2-arm humanized IgG TCE that binds bivalently to BCMA and monovalently to CD3ε in a 2 + 1 format. Interim results of phase 1 multicenter clinical study (NCT03486067) of CC-93269 in patients with R/RMM: Among the 12 patients treated with ≥6 mg CC-93269 in cycle 1, the ORR was 83.3%, MRD-negativity rate was 75.0%. The most common adverse reactions are CRS (89.5%); however, the majority of these events were classified as grade 1-2 toxicity levels [123]. MagnetisMM-1 (NCT03269136) is a phase 1 study of elranatamab (PF-06863135), a humanized bispecific molecule that targets BCMA and CD3 for patients with R/RMM. The ORR was 70% (14/20) in 20 patients with an effective dose range of 215–1000 μg/kg once per week. CR/sCR rate was 30%, and negative MRD was induced in 100% of patients with evaluable MRD. At the highest dose of 1000 μg/kg, the ORR was 83% (n = 5/6). These results suggest that elranatamab, as a single agent, administered either q1w or q2w, had a manageable safety profile for patients with R/RMM. Other developments in combination with lenalidomide or pomadomide are still to be expected [124].
Several anti-BCMA /CD3 BsAbs are being evaluated in clinical trials. For example, animal experimental results showed that REGN5458 and AMG701 significantly inhibited the formation of MM tumors in murine xenogeneic models and showed potent combinatorial efficacy with programmed cell death protein one blockade [125, 126]. Notably, the combination of AMG 701 with lenalidomide induced sustained inhibition of MM cell growth in SCID mice reconstituted with human T cells. Therefore, clinical studies of AMG 701 as monotherapy in R/RMM patients and in combination with IMiDs to improve the outcome of MM patients are ongoing (NCT03287908) [127]. A phase 1 study of REGN5458 in heavily pretreated patients (NCT03761108) is ongoing. Although BCMA as an MM cell antigen has been used to develop various bispecific antibodies, BCMA antigen loss may occur during treatment, so the development of other antigens is still very important.
Anti-CD3/GPRC5D bispecific antibodies
G-protein-coupled receptor class 5 member D (GPRC5D) is a 7-pass transmembrane protein. Studies have shown that GPRC5D is significantly expressed in malignant plasma cells and has limited expression in normal human tissue, making it an attractive novel antigen for MM cells to target. This study synthesized a GPRC5DxCD3 bispecific antibody (Jnj-64407564) that can recruit CD3+ T cells to GPRC5D+ MM cells and induce killing of GPRC5D+ cells. In vivo experiments in a mouse myeloma model showed that JNJ-64407564 recruited T cells and induced tumor regression [128].
Based on the results of previous studies, the efficacy and safety of talquetamab (JNJ-64407564) is currently being evaluated in a phase I clinical trial (NCT03399799) in R/RMM patients, 30% of whom had previously been treated with BCMA. The results showed that in 30 response-evaluable patients treated with the recommended phase 2 doses (RP2Ds 405 µ g/kg SC weekly), the ORR was 70%, and the 6-month DOR rate was 67% (95% CI: 41–84). Responses were durable and deepened over time. The most common AEs at 405 g/kg weekly dose were CRS (73%; 1 patient had grade 3 CRS), neutropenia (67%), and dysgeusia (60%). Overall, talquetamab had a tolerable safety profile at the RP2D dose, with no dose-limiting toxicity (DLT) observed [129]. At median follow-ups of 11.7 months, the percentage of patients with a response was 70% (95% CI, 51 to 85), and the median duration of response was 10.2 months. Further studies of talquetamab monotherapy (phase II, NCT04634552) and in combination with other therapies to treat patients with R/RMM are underway [130].
Anti-CD3/FcRL5 bispecific antibodies
Fc receptor-like 5 (FcRL5/FcRH5/IRTA2/CD307) is a type I membrane protein with an Ig domain. Studies have shown that the expression rate of FcRH5 in myeloma cells is 100%, which is significantly higher than that in normal B cells. This study developed and preclinically validated an anti- FcRL5/CD3 TDB as an immunotherapy for MM. Anti-FcRL5/CD3 TDB (BFCR4350A) induced mild/moderate cytokine release immediately after administration in primates, effectively killing myeloma cells and marrow plasma cells, but the effect in mouse models was significantly limited [131].
GO39775 (NCT03275103) is an ongoing multicenter phase I clinical trial designed to evaluate the safety, activity, pharmacodynamics, and pharmacokinetics of BFCR4350A monotherapy in R/RMM patients. All enrolled patients were previously exposed to CAR-T cells, T-cell-engaging bispecific antibodies, and antibody-drug conjugates, including those targeting BCMA, for which no established therapy is available, appropriate, or tolerated. At the cut-off time, ORR was 51.7% (15/29), observed at dose levels of 3.6/20 mg and above. Notably, treatment responses were also observed in high-risk patients: those with HR cytogenetics (9/17), triple-class refractory disease (10/20), and prior exposure to anti-CD38 monoclonal antibodies (11/22), CAR-T cells (2/3), or antibody-drug conjugates (2/2). The most common treatment-related AE was CRS (74.5%), most of which were grade 1–2 (72.2%), and most occurred in the first course of treatment. These results indicate that BFCR4350A monotherapy shows promising efficacy with manageable toxicity in heavily pretreated R/R MM [132].
Anti-CD3/CD38 bispecific antibodies
CD38 is a membrane protein universally expressed at high levels in MM cells. AMG 424, a novel humanized T cell-recruiting bispecific anti-CD3/CD38 antibody, has been described recently. In vivo, AMG 424 could inhibit tumor growth in bone marrow invasive mouse cancer models and the depletion of peripheral B cells in cynomolgus monkeys without triggering excessive cytokine release. However, the activity of AMG 424 against normal immune cells expressing CD38 is also presented [133]. Another study developed a new CD3/CD38 BiTE (BI38-3) consisting of two single-stranded variable fragments from mouse hybridomas. This study has shown that BI38-3 triggers T cell-mediated lysis of CD38+ MM cells both in vitro and in vivo. Bi38-3 does not impair the surface expression of CD38 and only triggers T-cell-mediated killing of cells expressing high levels of CD38 while showing no or limited toxicity to cells expressing intermediate levels of CD38 (e.g., hematopoietic progenitors, B, T, or NK cells). Therefore, compared with AMG424, BI38-3 can trigger the killing of MM cells more effectively. In addition, because BI38-3 recognizes a specific epitope on CD38 and does not have an Fc region, it is expected to be effective in patients who relapsed after daratumumab treatment [134]. However, the clinical trials of these two CD3/CD38 bispecific antibodies have not yet been investigated, and further research progress is anticipated.
Our new findings on immunotherapy for multiple myeloma
Despite advances in bispecific antibodies, many patients do not respond to them or relapse afterward. Therefore, it is crucial to optimize the performance of bispecific antibodies continuously [135]. Exosomes are vesicle-like nanobodies secreted by many kinds of cells, which have the advantages of low immunogenicity, strong stability, and good biocompatibility [136]. Signals are transferred from exosomes to recipient cells by receptor-ligand interactions, direct membrane fusion, and endocytosis/ phagocytosis [137]. Exosomes are widely present in human blood, saliva, and other body fluids. They can stably deliver microRNAs, small interfering RNAs, and anticancer drugs to target cells, which has an extensive application prospect in drug delivery [138, 139]. Although natural exosomes are not targeted, they can be genetically engineered or chemically engineered to obtain targeting to recipient cells [140]. Numerous preclinical and clinical studies have shown that exosome-based therapeutics have good efficacy and safety in treating many human diseases [141, 142].
One study expresses anti-CD3 and anti-tumor cell associated epidermal growth factor receptor (EGFR) molecules on the surface of exosomes by genetic engineering, thereby generating a drug (aCD3-a EGFR SMART-Exos) that simultaneously targets T cells (CD3) and tumor cells (EGFR), activating cytotoxic T cells to attack tumor cells expressing EGFR. aCD3-a EGFR SMART-Exos exhibited highly potent and specific anti-tumor activity in vitro and in vivo [143]. Another study also synthesized SMART-Exos that could target T cells (CD3) and tumor cells (HER2). The results demonstrated that aCD3-aHER2 SMART-Exos exhibited enhanced binding affinity towards both CD3+ and HER2+ cell lines in vitro, mediating the interaction between T cells and HER2+ tumor cells, thereby activating cytotoxic T cells to target HER2-expressing cancer cells effectively. Mice treated with aCD3-aHER2 SMART-Exos displayed remarkable suppression of tumor growth without any observed liver or kidney damage. Furthermore, the study also compared aCD3-aHER2 SMART-Exos with bispecific aCD3-aHER2 antibodies, revealing that αCD3-αHER2 SMART-Exos exhibited superior potency in inducing specific cytotoxicity against HER2-positive cells and promoting T-cell activation [144].
Since we have done much essential research on modifying targeted antigens and drug delivery on exosomes [145,146,147], we attempted to achieve its dual targeting to T cells and myeloma cells through exosome surface protein modification. A flexible (GGGGS)3-linker spliced CD3 scFvs gene sequence and CD38 nano antibody (Nb) gene sequence were linked to the Lamp2b gene to construct a double-targeted fusion expression plasmid. Subsequently, we transfected aCD3-aCD38 Nb-Lamp2b plasmid into Expi293F cells to obtain exosomes that could bind CD3 + T cells and CD38 + MM cells simultaneously. We next performed in vitro cell interaction experiments using CD3+ T cell line (Jurkat), CD38+ myeloma cell line (NCI-H929), and CD38-cell line (K562). We co-incubated red fluorescently labeled CD38+ cells, green fluorescently labeled CD38- cells, and CD3+ cells with aCD3-aCD38 -EXOs to observe the cell-cell interactions by confocal microscopy. Results showed that CD3+ Jurkat cells were significantly crosslinked with CD38 + NCI-H929 cells, but no cross-linking of CD3+ Jurkat cells and CD38- K562 cells. These data supported aCD3-aCD38 -EXOs induced cell-cell interactions (Fig. 6).

A Construction process and action mode of bispecific antibodies modified exosomes (aCD3-aCD38-EXOs); B Compared with aCD38-EXOs, aCD3-aCD38-EXOs mediated the binding of T cells and MM cells to each other.
For the first time, we genetically engineered exosomes to target both CD3+ T cells and CD38+ MM cells, giving them the benefits of bispecific antibodies. In addition, taking advantage of the drug-loading advantages of exosomes, cytotoxic drugs can be loaded into bispecific antibody-targeted exosomes in vitro to exert both immune-killing and cytotoxic effects. This study design may provide a theoretical basis for developing a novel immunotherapy-coupled cytotoxic drug therapy.
Conclusion
In recent years, the application of proteasome inhibitors and immunomodulatory agents in MM has significantly prolonged the survival of patients. However, due to the heterogeneity of MM, the disease is prone to recurrence and drug resistance. New immunotarget therapy has brought great promise for the treatment of MM. In particular, CAR-T and monoclonal antibody therapy have shown promising efficacy in inducing deep and durable remissions in both NDMM and R/RMM. In addition, BiTEs and antibody-drug conjugate treatment have also demonstrated encouraging efficacy in treating R/RMM patients who have previously received multiple therapies, including CD-38 monoclonal antibody therapy. However, immunotherapy also presents certain limitations, including the challenges associated with cell drug preparation, off-target effects, and toxic side effects. Therefore, it is imperative to continuously explore new MM antigen targets and further optimize the performance of immunotherapy drugs to maximize their efficacy while minimizing toxicity. Compared to traditional nanoparticles, exosomes offer advantages such as enhanced biocompatibility, stability, and reduced immunogenicity. This may provide a novel opportunity for targeted modification and loading of cytotoxic drugs in treating MM. However, the therapeutic efficacy, toxicity, in vivo distribution, stability, and mechanism of action should be further investigated in future studies.
In addition, synergistic effects between different immunotherapies and traditional chemotherapy agents may exist. With the increasing number of novel immunotherapy drugs entering clinical trials, it is imperative to evaluate the synergistic impact of diverse immunotherapies and conventional chemotherapeutic agents on anti-tumor immune responses while minimizing therapeutic toxicity. Furthermore, the efficacy and safety profiles of immunotherapy should be assessed across various stages of MM treatment and distinct prognostic groups to achieve a more rationalized and standardized drug regimen. It would be intriguing to investigate whether effective combinations can be achieved among different subsets of immune-binding drugs. In conclusion, the continuous advancement in new immunotherapy drugs has instilled fresh hope for achieving a cure for MM.
Literature search
The PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) methodology was employed in this review. An electronic search was conducted using PubMed and the blood journal database up to 2023. The search terms included MM, cellular immunotherapy, checkpoint inhibitors, monoclonal antibodies, antibody-drug conjugates, bispecific antibodies, and exosomes. A total of 143 articles were retrieved from PubMed, along with five abstracts from the blood journal.
References
van de Donk N, Pawlyn C, Yong KL. Multiple myeloma. Lancet. 2021;397:410–27.
Xu L, Liu J, Huang B, Kuang L, Gu J, Chen M, et al. Comparison of efficacy, safety, patients’ quality of life, and doctors’ occupational stress between lenalidomide‐based and bortezomib‐based induction in patients with newly diagnosed multiple myeloma. Cancer Med. 2021;10:1656–67.
Attal M, Lauwers-Cances V, Hulin C, Leleu X, Caillot D, Escoffre M, et al. Lenalidomide, bortezomib, and dexamethasone with transplantation for myeloma. N. Engl J Med. 2017;376:1311–20.
Jimenez-Zepeda VH, Reece DE, Trudel S, Chen C, Tiedemann R, Kukreti V. Early relapse after single auto-SCT for multiple myeloma is a major predictor of survival in the era of novel agents. Bone Marrow Transpl. 2015;50:204–08.
Soekojo CY, Ooi M, de Mel S, Chng WJ. Immunotherapy in multiple myeloma. Cells. 2020;9:601.
Rapoport AP. Myeloma escape from immunity: an “inside” job. Blood. 2015;126:1401–03.
Shah UA, Mailankody S. Emerging immunotherapies in multiple myeloma. BMJ. 2020;370:m3176.
Guedan S, Calderon H, Posey AJ, Maus MV. Engineering and design of chimeric antigen receptors. Mol Ther Methods Clin Dev. 2019;12:145–56.
Sadelain M, Riviere I, Riddell S. Therapeutic T cell engineering. Nature. 2017;545:423–31.
Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388–98.
Mikkilineni L, Kochenderfer JN. CAR T cell therapies for patients with multiple myeloma. Nat Rev Clin Oncol. 2021;18:71–84.
Novak AJ, Darce JR, Arendt BK, Harder B, Henderson K, Kindsvogel W, et al. Expression of BCMA, TACI, and BAFF-R in multiple myeloma: a mechanism for growth and survival. Blood. 2004;103:689–94.
Brudno JN, Maric I, Hartman SD, Rose JJ, Wang M, Lam N, et al. T Cells genetically modified to express an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J Clin Oncol. 2018;36:2267–80.
Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood. 2016;128:1688–700.
Zhao WH, Liu J, Wang BY, Chen YX, Cao XM, Yang Y, et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J Hematol Oncol. 2018;11:141.
Wang D, Wang J, Hu G, Wang W, Xiao Y, Cai H, et al. A phase 1 study of a novel fully human BCMA-targeting CAR (CT103A) in patients with relapsed/refractory multiple myeloma. Blood. 2021;137:2890–901.
Li C, Wang D, Song Y, Li J, Huang H, Chen B, et al. A phase 1/2 study of a novel fully human B-cell maturation antigen-specific CAR T cells (CT103A) in patients with relapsed and/or refractory multiple myeloma. Blood. 2021;138:547.
Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N. Engl J Med. 2019;380:1726–37.
Munshi NC, Anderson LJ, Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N. Engl J Med. 2021;384:705–16.
Garfall AL, Stadtmauer EA, Hwang WT, Lacey SF, Melenhorst JJ, Krevvata M, et al. Anti-CD19 CAR T cells with high-dose melphalan and autologous stem cell transplantation for refractory multiple myeloma. JCI Insight. 2018;3:e120505.
Yan L, Qu S, Shang J, Shi X, Kang L, Xu N, et al. Sequential CD19 and BCMA-specific CAR T-cell treatment elicits sustained remission of relapsed and/or refractory myeloma. Cancer Med. 2021;10:563–74.
Yan Z, Cao J, Cheng H, Qiao J, Zhang H, Wang Y, et al. A combination of humanised anti-CD19 and anti-BCMA CAR T cells in patients with relapsed or refractory multiple myeloma: a single-arm, phase 2 trial. Lancet Haematol. 2019;6:e521–29.
Shi X, Yan L, Shang J, Qu S, Kang L, Zhou J, et al. Tandom autologous transplantation and combined infusion of CD19 and Bcma-specific chimeric antigen receptor T cells for high risk MM: initial safety and efficacy report from a clinical pilot study. Blood. 2018;132:1009. https://doi.org/10.1182/blood-2018-99-117964.
Mei H, Li C, Jiang H, Zhao X, Huang Z, Jin D, et al. A bispecific CAR-T cell therapy targeting BCMA and CD38 in relapsed or refractory multiple myeloma. J Hematol Oncol. 2021;14:161.
Cohen AD. CAR T cells and other cellular therapies for multiple myeloma: 2018 update. Am Soc Clin Oncol Educ Book. 2018;38:e6–15.
Rodriguez-Lobato LG, Ganzetti M, Fernandez DLC, Hudecek M, Einsele H, Danhof S. CAR T-cells in multiple myeloma: state of the art and future directions. Front Oncol. 2020;10:1243.
Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transpl. 2019;25:625–38.
MacKay M, Afshinnekoo E, Rub J, Hassan C, Khunte M, Baskaran N, et al. The therapeutic landscape for cells engineered with chimeric antigen receptors. Nat Biotechnol. 2020;38:233–44.
Wang L, Dou M, Ma Q, Yao R, Liu J. Chimeric antigen receptor (CAR)-modified NK cells against cancer: Opportunities and challenges. Int Immunopharmacol. 2019;74:105695.
Pfefferle A, Huntington ND. You have got a fast CAR: chimeric antigen receptor NK cells in cancer therapy. Cancers. 2020;12:706.
Tai YT, Soydan E, Song W, Fulciniti M, Kim K, Hong F, et al. CS1 promotes multiple myeloma cell adhesion, clonogenic growth, and tumorigenicity via c-maf-mediated interactions with bone marrow stromal cells. Blood. 2009;113:4309–18.
Lutz RJ, Whiteman KR. Antibody-maytansinoid conjugates for the treatment of myeloma. Mabs. 2009;1:548–51.
Jiang H, Zhang W, Shang P, Zhang H, Fu W, Ye F, et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol Oncol. 2014;8:297–310.
Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28:917–27.
Le Calvez B, Moreau P, Touzeau C. Immune checkpoint inhibitors for the treatment of myeloma: novel investigational options. Expert Opin Investig Drugs. 2021;30:965–73.
Jelinek T, Paiva B, Hajek R. Update on PD-1/PD-L1 inhibitors in multiple myeloma. Front Immunol. 2018;9:2431.
Sue-A-Quan R, Patel PG, Shakfa N, Nyi MN, Afriyie-Asante A, Kang EY, et al. Prognostic significance of T cells, PD-L1 immune checkpoint and tumour associated macrophages in clear cell carcinoma of the ovary. Gynecol Oncol. 2021;162:421–30.
Benson DJ, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood. 2010;116:2286–94.
Ray A, Das DS, Song Y, Richardson P, Munshi NC, Chauhan D, et al. Targeting PD1-PDL1 immune checkpoint in plasmacytoid dendritic cell interactions with T cells, natural killer cells and multiple myeloma cells. Leukemia. 2015;29:1441–44.
Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: preliminary results of a phase Ib study. J Clin Oncol. 2016;34:2698–704.
Ribrag V, Avigan DE, Green DJ, Wise-Draper T, Posada JG, Vij R, et al. Phase 1b trial of pembrolizumab monotherapy for relapsed/refractory multiple myeloma: KEYNOTE-013. Br J Haematol. 2019;186:e41–44.
Mateos MV, Orlowski RZ, Ocio EM, Rodriguez-Otero P, Reece D, Moreau P, et al. Pembrolizumab combined with lenalidomide and low-dose dexamethasone for relapsed or refractory multiple myeloma: phase I KEYNOTE-023 study. Br J Haematol. 2019;186:e117–21.
Mateos MV, Blacklock H, Schjesvold F, Oriol A, Simpson D, George A, et al. Pembrolizumab plus pomalidomide and dexamethasone for patients with relapsed or refractory multiple myeloma (KEYNOTE-183): a randomised, open-label, phase 3 trial. Lancet Haematol. 2019;6:e459–69.
Usmani SZ, Schjesvold F, Oriol A, Karlin L, Cavo M, Rifkin RM, et al. Pembrolizumab plus lenalidomide and dexamethasone for patients with treatment-naive multiple myeloma (KEYNOTE-185): a randomised, open-label, phase 3 trial. Lancet Haematol. 2019;6:e448–58.
Frerichs KA, Verkleij C, Dimopoulos MA, Marin SJ, Zweegman S, Young MH, et al. Efficacy and safety of durvalumab combined with daratumumab in daratumumab-refractory multiple myeloma patients. Cancers. 2021;13:2452.
Gormley NJ, Pazdur R. Immunotherapy combinations in multiple myeloma—known unknowns. N. Engl J Med. 2018;379:1791–95.
Badros AZ, Ma N, Rapoport AP, Lederer E, Lesokhin AM. Long-term remissions after stopping pembrolizumab for relapsed or refractory multiple myeloma. Blood Adv. 2019;3:1658–60.
Biran N, Gourna PE, Feinman R, Vesole DH, Zenreich J, Wang S, et al. Pembrolizumab, lenalidomide and dexamethasone post autologous transplant in patients with high-risk multiple myeloma. Am J Hematol. 2021;96:E430–33.
Wu HT, Zhao XY. Regulation of CD38 on multiple myeloma and NK cells by monoclonal antibodies. Int J Biol Sci. 2022;18:1974–88.
Franssen LE, Stege C, Zweegman S, van de Donk N, Nijhof IS. Resistance mechanisms towards CD38-directed antibody therapy in multiple myeloma. J Clin Med. 2020;9:1195.
Ritchie D, Colonna M. Mechanisms of action and clinical development of elotuzumab. Clin Transl Sci. 2018;11:261–66.
Usmani SZ, Nahi H, Plesner T, Weiss BM, Bahlis NJ, Belch A, et al. Daratumumab monotherapy in patients with heavily pretreated relapsed or refractory multiple myeloma: final results from the phase 2 GEN501 and SIRIUS trials. Lancet Haematol. 2020;7:e447–55.
Lonial S, Weiss BM, Usmani SZ, Singhal S, Chari A, Bahlis NJ, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet. 2016;387:1551–60.
Bahlis NJ, Dimopoulos MA, White DJ, Benboubker L, Cook G, Leiba M, et al. Daratumumab plus lenalidomide and dexamethasone in relapsed/refractory multiple myeloma: extended follow-up of POLLUX, a randomized, open-label, phase 3 study. Leukemia. 2020;34:1875–84.
Dimopoulos MA, Oriol A, Nahi H, San-Miguel J, Bahlis NJ, Usmani SZ, et al. Overall survival with daratumumab, lenalidomide, and dexamethasone in previously treated multiple myeloma (POLLUX): a randomized, open-label, phase III trial. J Clin Oncol. 2023;41:1590–99.
Plesner T, Dimopoulos MA, Oriol A, San-Miguel J, Bahlis NJ, Rabin N, et al. Health-related quality of life in patients with relapsed or refractory multiple myeloma: treatment with daratumumab, lenalidomide, and dexamethasone in the phase 3 POLLUX trial. Br J Haematol. 2021;194:132–39.
Kaufman JL, Dimopoulos MA, White D, Benboubker L, Cook G, Leiba M, et al. Daratumumab, lenalidomide, and dexamethasone in relapsed/refractory myeloma: a cytogenetic subgroup analysis of POLLUX. Blood Cancer J. 2020;10:111.
Spencer A, Lentzsch S, Weisel K, Avet-Loiseau H, Mark TM, Spicka I, et al. Daratumumab plus bortezomib and dexamethasone versus bortezomib and dexamethasone in relapsed or refractory multiple myeloma: updated analysis of CASTOR. Haematologica. 2018;103:2079–87.
Weisel K, Spencer A, Lentzsch S, Avet-Loiseau H, Mark TM, Spicka I, et al. Daratumumab, bortezomib, and dexamethasone in relapsed or refractory multiple myeloma: subgroup analysis of CASTOR based on cytogenetic risk. J Hematol Oncol. 2020;13:115.
Sonneveld P, Chanan-Khan A, Weisel K, Nooka AK, Masszi T, Beksac M, et al. Overall survival with daratumumab, bortezomib, and dexamethasone in previously treated multiple myeloma (CASTOR): a randomized, open-label, phase III trial. J Clin Oncol. 2023;41:1600–09.
Usmani SZ, Quach H, Mateos MV, Landgren O, Leleu X, Siegel D, et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): updated outcomes from a randomised, multicentre, open-label, phase 3 study. Lancet Oncol. 2022;23:65–76.
Avet-Loiseau H, San-Miguel J, Casneuf T, Iida S, Lonial S, Usmani SZ, et al. Evaluation of sustained minimal residual disease negativity with daratumumab-combination regimens in relapsed and/or refractory multiple myeloma: analysis of POLLUX and CASTOR. J Clin Oncol. 2021;39:1139–49.
Mateos MV, Spencer A, Nooka AK, Pour L, Weisel K, Cavo M, et al. Daratumumab-based regimens are highly effective and well tolerated in relapsed or refractory multiple myeloma regardless of patient age: subgroup analysis of the phase 3 CASTOR and POLLUX studies. Haematologica. 2020;105:468–77.
Voorhees PM, Kaufman JL, Laubach J, Sborov DW, Reeves B, Rodriguez C, et al. Daratumumab, lenalidomide, bortezomib, and dexamethasone for transplant-eligible newly diagnosed multiple myeloma: the GRIFFIN trial. Blood. 2020;136:936–45.
Moreau P, Attal M, Hulin C, Arnulf B, Belhadj K, Benboubker L, et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): a randomised, open-label, phase 3 study. Lancet. 2019;394:29–38.
Roussel M, Moreau P, Hebraud B, Laribi K, Jaccard A, Dib M, et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab for transplantation-eligible patients with newly diagnosed multiple myeloma (CASSIOPEIA): health-related quality of life outcomes of a randomised, open-label, phase 3 trial. Lancet Haematol. 2020;7:e874–83.
Mateos MV, Cavo M, Blade J, Dimopoulos MA, Suzuki K, Jakubowiak A, et al. Overall survival with daratumumab, bortezomib, melphalan, and prednisone in newly diagnosed multiple myeloma (ALCYONE): a randomised, open-label, phase 3 trial. Lancet. 2020;395:132–41.
Mateos MV, Dimopoulos MA, Cavo M, Suzuki K, Jakubowiak A, Knop S, et al. Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma. N. Engl J Med. 2018;378:518–28.
Facon T, Kumar SK, Plesner T, Orlowski RZ, Moreau P, Bahlis N, et al. Daratumumab, lenalidomide, and dexamethasone versus lenalidomide and dexamethasone alone in newly diagnosed multiple myeloma (MAIA): overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22:1582–96.
Facon T, Kumar S, Plesner T, Orlowski RZ, Moreau P, Bahlis N, et al. Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N. Engl J Med. 2019;380:2104–15.
Facon T, Cook G, Usmani SZ, Hulin C, Kumar S, Plesner T, et al. Daratumumab plus lenalidomide and dexamethasone in transplant-ineligible newly diagnosed multiple myeloma: frailty subgroup analysis of MAIA. Leukemia. 2022;36:1066–77.
Perrot A, Facon T, Plesner T, Usmani SZ, Kumar S, Bahlis NJ, et al. Health-related quality of life in transplant-ineligible patients with newly diagnosed multiple myeloma: findings from the phase III MAIA trial. J Clin Oncol. 2021;39:227–37.
Patel KK, Giri S, Parker TL, Bar N, Neparidze N, Huntington SF. Cost-effectiveness of first-line versus second-line use of daratumumab in older, transplant-ineligible patients with multiple myeloma. J Clin Oncol. 2021;39:1119–28.
Cavo M, San-Miguel J, Usmani SZ, Weisel K, Dimopoulos MA, Avet-Loiseau H, et al. Prognostic value of minimal residual disease negativity in myeloma: combined analysis of POLLUX, CASTOR, ALCYONE, and MAIA. Blood. 2022;139:835–44.
San-Miguel J, Avet-Loiseau H, Paiva B, Kumar S, Dimopoulos MA, Facon T, et al. Sustained minimal residual disease negativity in newly diagnosed multiple myeloma and the impact of daratumumab in MAIA and ALCYONE. Blood. 2022;139:492–501.
Moreno L, Perez C, Zabaleta A, Manrique I, Alignani D, Ajona D, et al. The mechanism of action of the anti-CD38 monoclonal antibody isatuximab in multiple myeloma. Clin Cancer Res. 2019;25:3176–87.
Richter J, Sanchez L, Thibaud S. Therapeutic potential of isatuximab in the treatment of multiple myeloma: evidence to date. Semin Oncol. 2020;47:155–64.
Martin T, Baz R, Benson DM, Lendvai N, Wolf J, Munster P, et al. A phase 1b study of isatuximab plus lenalidomide and dexamethasone for relapsed/refractory multiple myeloma. Blood. 2017;129:3294–303.
Mikhael J, Richardson P, Usmani SZ, Raje N, Bensinger W, Karanes C, et al. A phase 1b study of isatuximab plus pomalidomide/dexamethasone in relapsed/refractory multiple myeloma. Blood. 2019;134:123–33.
Attal M, Richardson PG, Rajkumar SV, San-Miguel J, Beksac M, Spicka I, et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): a randomised, multicentre, open-label, phase 3 study. Lancet. 2019;394:2096–107.
Richardson PG, Perrot A, San-Miguel J, Beksac M, Spicka I, Leleu X, et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): follow-up analysis of a randomised, phase 3 study. Lancet Oncol. 2022;23:416–27.
Bringhen S, Pour L, Vorobyev V, Vural F, Warzocha K, Benboubker L, et al. Isatuximab plus pomalidomide and dexamethasone in patients with relapsed/refractory multiple myeloma according to prior lines of treatment and refractory status: ICARIA-MM subgroup analysis. Leuk Res. 2021;104:106576.
Dimopoulos MA, Leleu X, Moreau P, Richardson PG, Liberati AM, Harrison SJ, et al. Isatuximab plus pomalidomide and dexamethasone in relapsed/refractory multiple myeloma patients with renal impairment: ICARIA-MM subgroup analysis. Leukemia. 2021;35:562–72.
Richardson PG, Harrison SJ, Bringhen S, Schjesvold F, Yong K, Campana F, et al. Isatuximab for relapsed/refractory multiple myeloma: review of key subgroup analyses from the Phase III ICARIA-MM study. Future Oncol. 2021;17:4797–812.
Schjesvold F, Bringhen S, Richardson PG, Perrot A, Leleu X, Moreau P, et al. Isatuximab plus pomalidomide and dexamethasone in frail patients with relapsed/refractory multiple myeloma: ICARIA-MM subgroup analysis. Am J Hematol. 2021;96:E423–27.
Moreau P, Dimopoulos MA, Mikhael J, Yong K, Capra M, Facon T, et al. Isatuximab, carfilzomib, and dexamethasone in relapsed multiple myeloma (IKEMA): a multicentre, open-label, randomised phase 3 trial. Lancet. 2021;397:2361–71.
Capra M, Martin T, Moreau P, Baker R, Pour L, Min CK, et al. Isatuximab plus carfilzomib and dexamethasone versus carfilzomib and dexamethasone in relapsed multiple myeloma patients with renal impairment: IKEMA subgroup analysis. Haematologica. 2022;107:1397–409.
Leypoldt LB, Besemer B, Asemissen AM, Hanel M, Blau IW, Gorner M, et al. Isatuximab, carfilzomib, lenalidomide, and dexamethasone (Isa-KRd) in front-line treatment of high-risk multiple myeloma: interim analysis of the GMMG-CONCEPT trial. Leukemia. 2022;36:885–88.
Zonder JA, Mohrbacher AF, Singhal S, van Rhee F, Bensinger WI, Ding H, et al. A phase 1, multicenter, open-label, dose escalation study of elotuzumab in patients with advanced multiple myeloma. Blood. 2012;120:552–59.
Lonial S, Dimopoulos M, Palumbo A, White D, Grosicki S, Spicka I, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N. Engl J Med. 2015;373:621–31.
Dimopoulos MA, Lonial S, White D, Moreau P, Weisel K, San-Miguel J, et al. Elotuzumab, lenalidomide, and dexamethasone in RRMM: final overall survival results from the phase 3 randomized ELOQUENT-2 study. Blood Cancer J. 2020;10:91.
Dimopoulos MA, Dytfeld D, Grosicki S, Moreau P, Takezako N, Hori M, et al. Elotuzumab plus pomalidomide and dexamethasone for multiple myeloma. N. Engl J Med. 2018;379:1811–22.
Dimopoulos MA, Dytfeld D, Grosicki S, Moreau P, Takezako N, Hori M, et al. Elotuzumab plus pomalidomide and dexamethasone for relapsed/refractory multiple myeloma: final overall survival analysis from the randomized phase II ELOQUENT-3 trial. J Clin Oncol. 2023;41:568–78.
Weisel K, Dimopoulos MA, San-Miguel J, Paner A, Engelhardt M, Taylor F, et al. Impact of elotuzumab plus pomalidomide/dexamethasone on health-related quality of life for patients with relapsed/refractory multiple myeloma: final data from the phase 2 ELOQUENT-3 trial. Hemasphere. 2023;7:e843.
Usmani SZ, Hoering A, Ailawadhi S, Sexton R, Lipe B, Hita SF, et al. Bortezomib, lenalidomide, and dexamethasone with or without elotuzumab in patients with untreated, high-risk multiple myeloma (SWOG-1211): primary analysis of a randomised, phase 2 trial. Lancet Haematol. 2021;8:e45–54.
Raab MS, Engelhardt M, Blank A, Goldschmidt H, Agis H, Blau IW, et al. MOR202, a novel anti-CD38 monoclonal antibody, in patients with relapsed or refractory multiple myeloma: a first-in-human, multicentre, phase 1-2a trial. Lancet Haematol. 2020;7:e381–94.
Kelly KR, Ailawadhi S, Siegel DS, Heffner LT, Somlo G, Jagannath S, et al. Indatuximab ravtansine plus dexamethasone with lenalidomide or pomalidomide in relapsed or refractory multiple myeloma: a multicentre, phase 1/2a study. Lancet Haematol. 2021;8:e794–807.
Ailawadhi S, Kelly KR, Vescio RA, Jagannath S, Wolf J, Gharibo M, et al. A phase I study to assess the safety and pharmacokinetics of single-agent lorvotuzumab mertansine (IMGN901) in patients with relapsed and/or refractory CD-56-positive multiple myeloma. Clin Lymphoma Myeloma Leuk. 2019;19:29–34.
Kaufman JL, Niesvizky R, Stadtmauer EA, Chanan-Khan A, Siegel D, Horne H, et al. Phase I, multicentre, dose-escalation trial of monotherapy with milatuzumab (humanized anti-CD74 monoclonal antibody) in relapsed or refractory multiple myeloma. Br J Haematol. 2013;163:478–86.
Hussein M, Berenson JR, Niesvizky R, Munshi N, Matous J, Sobecks R, et al. A phase I multidose study of dacetuzumab (SGN-40; humanized anti-CD40 monoclonal antibody) in patients with multiple myeloma. Haematologica. 2010;95:845–48.
Hideshima T, Anderson KC. Signaling pathway mediating myeloma cell growth and survival. Cancers. 2021;13:216.
Shah JJ, Feng L, Thomas SK, Berkova Z, Weber DM, Wang M, et al. Siltuximab (CNTO 328) with lenalidomide, bortezomib and dexamethasone in newly-diagnosed, previously untreated multiple myeloma: an open-label phase I trial. Blood Cancer J. 2016;6:e396.
Orlowski RZ, Gercheva L, Williams C, Sutherland H, Robak T, Masszi T, et al. A phase 2, randomized, double-blind, placebo-controlled study of siltuximab (anti-IL-6 mAb) and bortezomib versus bortezomib alone in patients with relapsed or refractory multiple myeloma. Am J Hematol. 2015;90:42–49.
San-Miguel J, Blade J, Shpilberg O, Grosicki S, Maloisel F, Min CK, et al. Phase 2 randomized study of bortezomib-melphalan-prednisone with or without siltuximab (anti-IL-6) in multiple myeloma. Blood. 2014;123:4136–42.
Descamps G, Gomez-Bougie P, Venot C, Moreau P, Bataille R, Amiot M. A humanised anti-IGF-1R monoclonal antibody (AVE1642) enhances Bortezomib-induced apoptosis in myeloma cells lacking CD45. Br J Cancer. 2009;100:366–69.
Moreau P, Cavallo F, Leleu X, Hulin C, Amiot M, Descamps G, et al. Phase I study of the anti insulin-like growth factor 1 receptor (IGF-1R) monoclonal antibody, AVE1642, as single agent and in combination with bortezomib in patients with relapsed multiple myeloma. Leukemia. 2011;25:872–74.
Attar-Schneider O, Drucker L, Zismanov V, Tartakover-Matalon S, Rashid G, Lishner M. Bevacizumab attenuates major signaling cascades and eIF4E translation initiation factor in multiple myeloma cells. Lab Invest. 2012;92:178–90.
White D, Kassim A, Bhaskar B, Yi J, Wamstad K, Paton VE. Results from AMBER, a randomized phase 2 study of bevacizumab and bortezomib versus bortezomib in relapsed or refractory multiple myeloma. Cancer-Am Cancer Soc. 2013;119:339–47.
Tai YT, Anderson KC. Targeting B-cell maturation antigen in multiple myeloma. Immunotherapy. 2015;7:1187–99.
Tai YT, Mayes PA, Acharya C, Zhong MY, Cea M, Cagnetta A, et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood. 2014;123:3128–38.
Trudel S, Lendvai N, Popat R, Voorhees PM, Reeves B, Libby EN, et al. Antibody-drug conjugate, GSK2857916, in relapsed/refractory multiple myeloma: an update on safety and efficacy from dose expansion phase I study. Blood Cancer J. 2019;9:37.
Trudel S, Lendvai N, Popat R, Voorhees PM, Reeves B, Libby EN, et al. Targeting B-cell maturation antigen with GSK2857916 antibody-drug conjugate in relapsed or refractory multiple myeloma (BMA117159): a dose escalation and expansion phase 1 trial. Lancet Oncol. 2018;19:1641–53.
Lonial S, Lee HC, Badros A, Trudel S, Nooka AK, Chari A, et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): a two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020;21:207–21.
Richardson PG, Lee HC, Abdallah AO, Cohen AD, Kapoor P, Voorhees PM, et al. Single-agent belantamab mafodotin for relapsed/refractory multiple myeloma: analysis of the lyophilised presentation cohort from the pivotal DREAMM-2 study. Blood Cancer J. 2020;10:106.
Xing L, Lin L, Yu T, Li Y, Cho SF, Liu J, et al. A novel BCMA PBD-ADC with ATM/ATR/WEE1 inhibitors or bortezomib induce synergistic lethality in multiple myeloma. Leukemia. 2020;34:2150–62.
Lee HC, Raje NS, Landgren O, Upreti VV, Wang J, Avilion AA, et al. Phase 1 study of the anti-BCMA antibody-drug conjugate AMG 224 in patients with relapsed/refractory multiple myeloma. Leukemia. 2021;35:255–58.
Hosny M, Verkleij C, van der Schans J, Frerichs KA, Mutis T, Zweegman S, et al. Current state of the art and prospects of T cell-redirecting bispecific antibodies in multiple myeloma. J Clin Med. 2021;10:4593.
Liu H, Saxena A, Sidhu SS, Wu D. Fc engineering for developing therapeutic bispecific antibodies and novel scaffolds. Front Immunol. 2017;8:38.
Topp MS, Duell J, Zugmaier G, Attal M, Moreau P, Langer C, et al. Anti-B-cell maturation antigen BiTE molecule AMG 420 induces responses in multiple myeloma. J Clin Oncol. 2020;38:775–83.
D’Souza A, Shah N, Rodriguez C, Voorhees PM, Weisel K, Bueno OF, et al. A phase I first-in-human study of ABBV-383, a B-cell maturation antigen x CD3 bispecific T-cell redirecting antibody, in patients with relapsed/refractory multiple myeloma. J Clin Oncol. 2022;40:3576–86.
Usmani SZ, Garfall AL, van de Donk N, Nahi H, San-Miguel JF, Oriol A, et al. Teclistamab, a B-cell maturation antigen x CD3 bispecific antibody, in patients with relapsed or refractory multiple myeloma (MajesTEC-1): a multicentre, open-label, single-arm, phase 1 study. Lancet. 2021;398:665–74.
Moreau P, Garfall AL, van de Donk N, Nahi H, San-Miguel JF, Oriol A, et al. Teclistamab in relapsed or refractory multiple myeloma. N. Engl J Med. 2022;387:495–505.
Costa LJ, Wong SW, Bermúdez A, de la Rubia J, Mateos MV, Ocio EM, et al.: First Clinical Study of the B-Cell Maturation Antigen (BCMA) 2+1 T Cell Engager (TCE) CC-93269 in Patients (Pts) with Relapsed/Refractory Multiple Myeloma (RRMM): Interim Results of a Phase 1 Multicenter Trial. Blood 2019; 134:143. https://doi.org/10.1182/blood-2019-122895.
Sebag M, Raje NS, Bahlis NJ, Costello C, Dholaria B, Solh M, et al.: Elranatamab (PF-06863135), a B-Cell Maturation Antigen (BCMA) Targeted CD3-Engaging Bispecific Molecule, for Patients with Relapsed or Refractory Multiple Myeloma: Results from Magnetismm-1. Blood 2021;138:895. https://doi.org/10.1182/blood-2021-150519.
Goldstein RL, Goyos A, Li CM, Deegen P, Bogner P, Sternjak A, et al. AMG 701 induces cytotoxicity of multiple myeloma cells and depletes plasma cells in cynomolgus monkeys. Blood Adv. 2020;4:4180–94.
DiLillo DJ, Olson K, Mohrs K, Meagher TC, Bray K, Sineshchekova O, et al. A BCMAxCD3 bispecific T cell-engaging antibody demonstrates robust antitumor efficacy similar to that of anti-BCMA CAR T cells. Blood Adv. 2021;5:1291–304.
Cho SF, Lin L, Xing L, Li Y, Wen K, Yu T, et al. The immunomodulatory drugs lenalidomide and pomalidomide enhance the potency of AMG 701 in multiple myeloma preclinical models. Blood Adv. 2020;4:4195–207. Blood Adv 2020, 4:5772
Pillarisetti K, Edavettal S, Mendonca M, Li Y, Tornetta M, Babich A, et al. A T-cell-redirecting bispecific G-protein-coupled receptor class 5 member D x CD3 antibody to treat multiple myeloma. Blood. 2020;135:1232–43.
Krishnan AY, Minnema MC, Berdeja JG, Oriol A, van de Donk NWCJ, Rodriguez-Otero P, et al.: Updated Phase 1 Results from MonumenTAL-1: First-in-Human Study of Talquetamab, a G Protein-Coupled Receptor Family C Group 5 Member D x CD3 Bispecific Antibody, in Patients with Relapsed/Refractory Multiple Myeloma. Blood 2021;138:158. https://doi.org/10.1182/blood-2021-146868.
Chari A, Minnema MC, Berdeja JG, Oriol A, van de Donk N, Rodriguez-Otero P, et al. Talquetamab, a T-cell-redirecting GPRC5D Bispecific Antibody for Multiple Myeloma. N. Engl J Med. 2022;387:2232–44.
Li J, Stagg NJ, Johnston J, Harris MJ, Menzies SA, DiCara D, et al. Membrane-proximal epitope facilitates efficient T cell synapse formation by anti-FcRH5/CD3 and is a requirement for myeloma cell killing. Cancer Cell. 2017;31:383–95.
Cohen AD, Harrison SJ, Krishnan A, Fonseca R, Forsberg PA, Spencer A, et al.: Initial Clinical Activity and Safety of BFCR4350A, a FcRH5/CD3 T-Cell-Engaging Bispecific Antibody, in Relapsed/Refractory Multiple Myeloma. Blood. 2020;136:42–43. https://doi.org/10.1182/blood-2020-136985.
Zuch DZC, Fajardo F, Zhong W, Bernett MJ, Muchhal US, Moore GL, et al. Targeting multiple myeloma with AMG 424, a novel anti-CD38/CD3 bispecific T-cell-recruiting antibody optimized for cytotoxicity and cytokine release. Clin Cancer Res. 2019;25:3921–33.
Fayon M, Martinez-Cingolani C, Abecassis A, Roders N, Nelson E, Choisy C, et al. Bi38-3 is a novel CD38/CD3 bispecific T-cell engager with low toxicity for the treatment of multiple myeloma. Haematologica. 2021;106:1193–97.
Einsele H, Borghaei H, Orlowski RZ, Subklewe M, Roboz GJ, Zugmaier G, et al. The BiTE (bispecific T-cell engager) platform: Development and future potential of a targeted immuno-oncology therapy across tumor types. Cancer. 2020;126:3192–201.
Antimisiaris SG, Mourtas S, Marazioti A. Exosomes and exosome-inspired vesicles for targeted drug delivery. Pharmaceutics. 2018;10:218.
He C, Zheng S, Luo Y, Wang B. Exosome theranostics: biology and translational medicine. Theranostics. 2018;8:237–55.
Xu X, Xu L, Wen C, Xia J, Zhang Y, Liang Y. Programming assembly of biomimetic exosomes: an emerging theranostic nanomedicine platform. Mater Today Bio. 2023;22:100760.
Xu L, Liang Y, Xu X, Xia J, Wen C, Zhang P, et al. Blood cell-derived extracellular vesicles: diagnostic biomarkers and smart delivery systems. Bioengineered. 2021;12:7929–40.
Liang Y, Duan L, Lu J, Xia J. Engineering exosomes for targeted drug delivery. Theranostics. 2021;11:3183–95.
Xu X, Xu L, Xia J, Wen C, Liang Y, Zhang Y. Harnessing knee joint resident mesenchymal stem cells in cartilage tissue engineering. Acta Biomater. 2023;168:372–87.
Wiklander O, Brennan MA, Lotvall J, Breakefield XO, El AS. Advances in therapeutic applications of extracellular vesicles. Sci Transl Med. 2019;11:eaav8521.
Cheng Q, Shi X, Han M, Smbatyan G, Lenz HJ, Zhang Y. Reprogramming exosomes as nanoscale controllers of cellular immunity. J Am Chem Soc. 2018;140:16413–17.
Shi X, Cheng Q, Hou T, Han M, Smbatyan G, Lang JE, et al. Genetically engineered cell-derived nanoparticles for targeted breast cancer immunotherapy. Mol Ther. 2020;28:536–47.
Xu L, Xu X, Liang Y, Wen C, Ouyang K, Huang J, et al. Osteoclast-targeted delivery of anti-miRNA oligonucleotides by red blood cell extracellular vesicles. J Control Release. 2023;358:259–72.
Liang Y, Xu X, Xu L, Iqbal Z, Ouyang K, Zhang H, et al. Chondrocyte-specific genomic editing enabled by hybrid exosomes for osteoarthritis treatment. Theranostics. 2022;12:4866–78.
Xu X, Liang Y, Li X, Ouyang K, Wang M, Cao T, et al. Exosome-mediated delivery of kartogenin for chondrogenesis of synovial fluid-derived mesenchymal stem cells and cartilage regeneration. Biomaterials. 2021;269:120539.
Acknowledgements
This work was supported by Shandong Provincial Natural Science Foundation (ZR2023QH148); PhD Research Foundation of Affiliated Hospital of Jining Medical University (2022-BS-03, 2022-BS-04).
Author information
Authors and Affiliations
Contributions
LMX, YJL, and XX designed the review; LMX, CNW, JX, and XX performed the discussion; LMX and XX gave valuable suggestions to this article; LMX, YJL, and XX contributed to the manuscript writing and editing; HZ and XX supervised the project. All authors have approved the final article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, L., Wen, C., Xia, J. et al. Targeted immunotherapy: harnessing the immune system to battle multiple myeloma. Cell Death Discov. 10, 55 (2024). https://doi.org/10.1038/s41420-024-01818-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-024-01818-6
