
Abstract
Lower back pain (LBP) is a common degenerative musculoskeletal disease that imposes a huge economic burden on both individuals and society. With the aggravation of social aging, the incidence of LBP has increased globally. Intervertebral disc degeneration (IDD) is the primary cause of LBP. Currently, IDD treatment strategies include physiotherapy, medication, and surgery; however, none can address the root cause by ending the degeneration of intervertebral discs (IVDs). However, in recent years, targeted therapy based on specific molecules has brought hope for treating IDD. The tumor suppressor gene p53 produces a transcription factor that regulates cell metabolism and survival. Recently, p53 was shown to play an important role in maintaining IVD microenvironment homeostasis by regulating IVD cell senescence, apoptosis, and metabolism by activating downstream target genes. This study reviews research progress regarding the potential role of p53 in IDD and discusses the challenges of targeting p53 in the treatment of IDD. This review will help to elucidate the pathogenesis of IDD and provide insights for the future development of precision treatments.
Similar content being viewed by others

Facts
-
IDD is considered to be an IVD cell-mediated degeneration process involving molecules, cells and tissues, which impairs the load-bearing capacity of IVD by affecting its tissue composition and biomechanical properties.
-
As a transcription factor, p53 is the central hub of the molecular network that controls cell metabolism and survival, and regulates protein expression in various cellular processes.
-
In IDD, p53 is activated by multiple stress signals and exhibits different dynamic characteristics, which can lead to different cell fates.
-
p53 is involved in various signal transduction pathways in IVD cells, and participates in the IDD process by influencing IVD cell aging, apoptosis, ECM metabolism, and oxidative stress.
Open questions
-
In IDD, what are the changes in the dynamic characteristics of p53 in the face of different stress signals?
-
What is the role of p53 in signal transduction and phenotypic changes of IVD cells?
-
What are the implications of p53 in the formulation of accurate treatment strategies for IDD?
Introduction
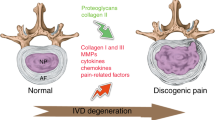
Lower back pain (LBP) is a major musculoskeletal disorder that leads to limited mobility and decreased quality of life in elderly individuals worldwide [1]. With the intensification of aging, the overall disability associated with LBP is on the rise globally [2, 3] and is most pronounced in low- and middle-income countries [4]. According to limited data, the one-year prevalence of LBP among adults in Africa and Latin America is 57% and 67%, respectively [5, 6]. The lifetime prevalence rate can be as high as 93% [7]. LBP seriously affects the quality of life of patients and creates a huge economic burden. In the United States, the total cost associated with LBP exceeds $100 billion annually [8], and the cost of spinal surgery in Brazil increased by 540% between 1995 and 2014 [9]. Intervertebral disc degeneration (IDD) is the primary cause of LBP [10]. However, the specific pathogenic mechanisms underlying IDD remain unclear. Currently, IDD is considered a degenerative process involving molecules, cells, and tissues mediated by intervertebral disc (IVD) cells that can lead to significant changes in IVD tissue composition and biomechanical properties, ultimately impairing the ability of IVDs to withstand loads [11] (Fig. 1). Currently, the treatment options for IDD include drugs and surgery, which relieve symptoms and reduce the incidence of disability; however, both treatment options have the disadvantages of multiple complications, high costs, and unknown efficacy [12] (Fig. 2). Neither approach resolves the underlying pathology by terminating the degenerative process of IVDs, and both are only applicable to end-stage disease. Therefore, it is important to further explore the pathogenic factors and related molecular mechanisms of IDD to guide treatment.

The normal function of an IVD relies on the structural integrity of its organization. During IVD degeneration, aberrant proteins, lipids, carbohydrates, and nucleic acids disrupt cellular homeostasis through apoptosis, senescence, and calcification processes. Consequently, these alterations in IVD composition adversely impact its organizational structure and ultimately compromise its mechanical functionality.

Conventional therapeutic approaches for IDD encompass pharmacotherapy, physical rehabilitation, minimally invasive procedures, and surgical interventions.
The IVD is a complex avascular connective tissue between the vertebrae, mainly composed of the nucleus pulposus (NP), annulus fibrosus (AF), and cartilage endplate (CEP). Additionally, it connects the spine, cushions spinal pressure, and increases spinal mobility [13,14,15]. As an age-related, multifactorial disease, the etiology of IDD remains unclear. Genetic susceptibility, age, obesity, smoking, occupational exposure, trauma, and abnormal nonphysiological mechanical loads contribute to its occurrence and progress [16,17,18,19,20,21] (Fig. 3). The proper mechanical function of the IVD depends on the quality and composition of the extracellular matrix (ECM) [22]. However, in the process of IVD degeneration, a series of internal, external, physical, or chemical factors promote the death and aging of IVD cells, leading to a decrease in the number of functional and viable cells and, thus, a decline in ECM synthesis [23, 24]. At the same time, dysfunctional IVD cells highly express matrix metalloproteinases (MMPs) and A disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS), which further promote ECM degradation [25,26,27]. The ECM components mainly include collagen (Col) networks and water-bound aggrecan (Agg), which have different proportions in the NP and AF [22]. The ECM in the NP mainly consists of Col II, elastin, and Agg, whereas the AF consists of alternating Col I [11]. The dysfunction of the NP and AF leads to the gradual loss of Col II and Agg, which causes the disappearance of the boundary between the NP and AF, reduces IVD height, and impairs the ability of the IVD to withstand mechanical loads [28]. Structural damage to the AF is conducive to infiltrating blood vessels and nerve endings, further promoting the deterioration of the IVD microenvironment and leading to IVD-derived LBP [29,30,31]. In addition, as a type of hyaline cartilage mainly composed of proteoglycans and collagen fibers, the CEP plays an important role in the exchange of nutrients and metabolic waste [32]. CEP degeneration can lead to nutritional disorders and metabolic waste accumulation in IVD cells, which helps accelerate IDD [33]. Therefore, various adverse factors lead to IVD cell death and dysfunction, which eventually leads to IDD by affecting ECM metabolism (Fig. 4).

Various factors have been associated with the development of IDD, including genetic predisposition, age, obesity, smoking, occupational exposure, trauma, and abnormal nonphysiologic mechanical loads.

Top: In a healthy IVD, an adequate number of IVD cells and normal cellular metabolic function are essential for maintaining the integrity of the ECM, which in turn preserves the structural and functional homeostasis of the IVD. Bottom: In degenerating IVDs, a reduction in the IVD cell population due to apoptosis, senescence, and calcification leads to the downregulation of ECM synthesis. Simultaneously, dysfunctional IVD cells exhibit high expression of pro-inflammatory cytokines, ROS, MMPs, and ADAMTS, further accelerating ECM degradation and ultimately deteriorating IVD tissue structure and biological function.
As a transcription factor, p53 is the central hub of the molecular network that controls cell metabolism and survival and is involved in the regulation of protein expression in cell cycle progression, aging, apoptosis, tumor metastasis, DNA repair, reactive oxygen species (ROS) metabolism, and glycolysis [34,35,36]. Recent studies have suggested that p53 activation and signal transduction are essential for the maintenance of IVD homeostasis. Zhang et al. [37] showed that p53 mRNA expression is significantly upregulated with increasing degrees of IVD degeneration. In addition, p53 knockdown in rats reduced the apoptotic phenotype of IVD cells by downregulating the expression of Bax and caspase 3, suggesting that the inhibition of p53 gene expression may be beneficial for delaying the progression of IDD [37]. In this review, we focus on the activation of p53 signaling in IVD, the signal transduction network, and its multiple biological functions in IVD cells, combined with existing evidence to comprehensively clarify the role of p53 in IDD.
The origin and structure of p53
In 1979, DeLeo et al. [38] first identified a protein with an apparent molecular weight of 53,000 in chemically induced sarcoma, leukemia, and spontaneously transformed fibroblasts and named it p53. Initially, researchers thought it was an oncogene because its mutant protein was expressed in many cancer tissues [39,40,41]. However, subsequent studies have demonstrated that wild-type p53 exhibits tumor suppression properties [42, 43]. Many studies have shown that p53 is an important tumor suppressor and plays an important role in the development of neurodegenerative diseases, cardiovascular diseases, diabetes, and osteoarthrosis [44,45,46,47].
The human p53 gene is located on the short arm of chromosome 17, which consists of 11 exons and 10 introns in a 2.2–2.5 Kb gene structure [48, 49]. Transcription of the p53 gene may start from five promoter regions: P0, P1, P1’, P2, and Pin, with P0, P1, and P1’ located in the first exon and the other two promoters located in introns [50]. From the P0, P1, and P1’ promoters, 2.5 Kb mRNA can be synthesized, encoding a full-length p53 protein containing 393 amino acids [50, 51]. The p53 protein consists of five main structural regions: the N-terminal transactivation domain, the proline-rich domain, the DNA-binding domain (DBD), the tetramerization domain, and the C-terminal domain [52] (Fig. 5). Detailed descriptions of the functions of major p53 structural regions have been documented previously [53,54,55,56,57].

Top: Chromosomal localization of the p53 gene. Middle: p53 mRNA. Bottom: p53 protein structure.
p53 and IDD
As a critical intracellular transcription factor, p53 is activated by cellular stress and promotes cell cycle arrest, DNA damage repair, and cell death through transcription-dependent and -independent pathways [58]. In these metabolic processes that determine the fate of cells, p53 often acts as a “double-edged sword,” complicating its role in specific diseases [59,60,61]. According to microarray and bioinformatics studies, p53 is a hub gene in a level-III-specific network, indicating that p53 may be involved in the progression of IDD [62, 63]. Analysis of degenerated IVD specimens from patients with lumbar disc herniation and rats showed that vascular endothelial growth factor and p53 expression increased in degenerated IVD tissues. The expression rate was significantly higher in tissues with vascular infiltration than those without, suggesting that they may be involved in late-stage vascularization and infiltration, accelerating IDD [64, 65]. Significantly increased p53 expression is involved in IVD cell apoptosis, aging, and ECM degradation by activating downstream target genes [37, 66,67,68,69,70,71]. These studies indicate that elevated p53 levels are important in promoting IDD progression. Xiong et al. [72] demonstrated that high p53 expression can protect nucleus pulposus cells (NPCs) from low cell viability and ECM degradation induced by low glucose concentrations, indicating that p53 can be used as a protective agent for NPCs under certain conditions. These studies suggest that p53 is essential for maintaining IVD homeostasis, and changes in its expression are directly involved in the progression of IDD.
p53 dynamics
General characteristics of p53 dynamics
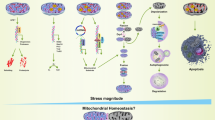
Traditionally, cell signaling dynamics refers to the temporal expression pattern of active signaling molecules, which is an important mechanism by which cells respond to complex stimuli [73, 74]. Dynamic regulation enables signaling molecules to respond to several stimuli and coordinate the activation of different and even conflicting downstream pathways to increase the specificity and function of signaling pathways [75]. As one of the most important tumor suppressors, p53 maintains genomic integrity by regulating the expression of many downstream target genes essential for relieving stress and regulating cell fate [76, 77]. The dynamics of p53 depend on the type, intensity, and duration of stimulation and show different patterns, including pulse, sustained platform, monotonic increase, and biphasic dynamics [73, 78]. The most intuitive dynamic change in p53 is a change in p53 protein abundance, which may be related to the selective activation of downstream target genes [79, 80]. Due to differential p53 thresholds for the activation of genes involved in cell cycle arrest and apoptosis, low levels of p53 primarily induce gene expression such as p21, leading to cell cycle arrest, while relatively higher levels of p53 tend to activate genes like Bax, triggering apoptosis [81]. In non-stressed cells, p53 remains low because of its rapid degradation. However, p53 can perform spontaneous transient pulses in non-stressed cells to cope with spontaneous DNA damage [82, 83]. Stressed cells can convert the p53 dynamic models induced by different stimuli into specific types of downstream responses [84] (Fig. 6). Ultraviolet (UV) light induces prolonged p53 pulses that increase in width and height with increasing UV doses, ultimately leading to apoptosis [85]. In contrast, ionizing radiation (IR) induces differential p53 kinetics. Low doses of IR induce low-frequency transient p53 pulses, which can lead to cell cycle arrest and DNA damage repair [86, 87]. However, high doses of IR can induce high-frequency p53 pulses and apoptosis [88]. In addition to stress types, there are significant differences in p53 kinetics among different cell types [89], reflecting that only cell-level p53 dynamics can accurately reflect the regulation of p53-mediated cellular signal transduction. Furthermore, p53 kinetics behave differently in different tissues. The small and large intestines are relatively insensitive to p53 levels. In contrast, the lymphoid organs are more sensitive to radiation, which consistently induces p53 expression [90]. The variability of p53 dynamics originates from the fact that p53 can activate the expression of genes that promote cell injury repair and death and induce the expression of genes that regulate their own gene expression and protein stability, leading to the formation of feedback loops and resulting in dynamic changes in p53 levels [91, 92]. Recent studies have shown that p53-MDM2 and p53-Wip1-ataxia telangiectasia-mutated (ATM) negative feedback loops are key factors in the synergistic control of p53 excitability, with the former being the topological basis of excitability in p53 kinetics and the latter regulating the sensitivity of p53 excitable responses [92,93,94,95,96]. However, it is difficult to explain the continuous p53 pulse that induces apoptosis through these two negative feedback loops. Therefore, a few scholars believe that a p53 positive feedback loop can induce a sustained p53 pulse to maintain high p53 levels [97]. For example, retinoid-related orphan receptor-α can stabilize p53 and activate p53 transcription in a HAUSP/Usp7-dependent manner, which has been shown to play a key role in persistent p53 pulses induced by DNA damage from extensive stimulation [97, 98]. Thus, positive and negative feedback regulation of p53 allows for the coordination of transient and continuous p53 pulses, constituting a flexible and effective regulatory network. In addition to regulating protein levels, p53 kinetics are characterized by changes in p53 protein activity, subcellular localization, post-translational modifications, and interactions among all p53 kinetic characteristics [96, 99, 100]. Therefore, p53 dynamics do not exhibit a simple linear relationship but a multifaceted dynamic network.

In the process of IVD degeneration, various intracellular and extracellular stress signals activate p53 by inducing DDR. Activation produces different dynamic changes in p53 by regulating p53-MDM2 and p53-Wip1-ATM negative feedback loops and potential positive feedback loops. These distinct alterations in p53 kinetics elicit diverse phenotypic modifications in IVD cells, thereby ultimately contributing to the progression of IDD.
Dynamics of p53 in IDD
p53 is involved in maintaining IVD microenvironment homeostasis [68, 101]. Compared with that in normal IVDs, the expression of p53 in degenerative IVDs is significantly increased, and the degree of increase is related to the grade of degeneration [37, 64,65,66, 71, 102]. In addition, multiple stimuli can lead to the upregulation of p53 levels, including oxidative stress, inflammatory responses, high glucose levels, and abnormal mechanical loading [101, 103,104,105]. However, the exact changes in p53 temporal dynamics under these stress conditions remain unclear. As a signaling molecule, p53 is continuously transported between the nucleus, cytoplasm, and mitochondria; this change in subcellular localization reflects the spatial dynamics of p53 [106, 107]. p53 is mainly located in the nucleus, which binds to the promoter or enhancer of target genes to regulate gene expression [58]. However, a small portion of the p53 protein is located in the mitochondria to perform its non-transcriptional functions [108]. Analysis of mitochondrial gene expression patterns showed that p53 was significantly upregulated in the mitochondria of AF cells with a higher grade of degeneration [102]. In addition, the expression of p53 in the mitochondria of NPCs increased under a compression load, which led to a significant upregulation of apoptosis in NPCs [109]. Therefore, the mitochondrial translocation of p53 may promote apoptotic activity. In addition, as one of the kinetic characteristics of p53, acetylation is an important modification required for p53-mediated cell senescence and apoptosis [110, 111]. Zhang et al. [112] showed that the acetylation level of p53 in NPCs was significantly increased under high glucose levels, accompanied by excessive senescence and NPC apoptosis. Although many studies have explored the role of p53 in IDD, our understanding of p53 dynamics is limited to changes in p53 protein levels. However, the biological significance of the differences in temporal dynamics, subcellular localization, and post-translational modification of p53 in response to different stimuli remains unclear. Different types of p53 kinetic changes and their interactions are essential for the regulation of specific cellular injury responses. Therefore, further exploration of the characteristics of p53 kinetics will help us understand its potential role in IDD.
Activation of p53
p53 is activated under various stress conditions, including DNA damage, inflammatory responses, hypoxia, high-glucose conditions, and oxidative stress [34, 113, 114]. p53 is extremely sensitive to DNA damage; therefore, it is called the “genome guardian” [113]. DNA damage response (DDR) consists of three key stages: damage perception, signal cascade, and damage repair [115]. Damage perception is accomplished by several kinases, including ATM, ataxia telangiectasia Rad3-related protein (ATR), and DNA-dependent protein kinase catalytic subunits (DNA-PKcs) [116, 117]. These kinases detect damage and signal to p53 through the Mre11-Rad50-NBS1 protein complex [96, 117]. ATM mainly responds to double-strand breaks, ATR mainly responds to single-strand breaks, and DNA-PKcs respond to double- and single-strand breaks [96, 118, 119]. In the resting state, ATM and ATR are inactive dimers recruited and activated by intermolecular autophosphorylation and homodimer dissociation following DNA damage [115, 116]. ATM, ATR, and DNA-PKcs can stabilize p53 by phosphorylating p53 and MDM2, and ATM-dependent MDM2 phosphorylation can accelerate MDM2 degradation, indirectly promoting p53 activation [96]. The occurrence and development of IDD are closely associated with DNA damage [120, 121]. In DNA repair-deficient Ercc1-/Δ mice, excessive DNA damage promoted IVD cell senescence and ECM degradation, accelerating IDD [122, 123]. Potential genotoxic stressors such as ionizing radiation and smoking can accelerate IDD in mice by inducing DNA damage [124, 125]. In addition, excessive ROS in degenerative IVDs is another source of DNA damage [126,127,128]. Recently, Han et al. [129] directly exposed human NPCs to the DNA-damaging agent cisplatin. In vitro, cisplatin-induced rapid phosphorylation of ATM in human NPCs led to ECM degradation and cell senescence; however, these effects were weakened after treatment with ATM-specific inhibitors. ATM knockdown reduced IVD cell senescence and ECM degradation in an accelerated senescent Ercc1-/Δ mouse model in vivo. This suggests that DNA damage affects the progression of IDD by activating ATM to promote the activation of p53. In addition, non-physiologically intense cyclic mechanical loading increases DNA damage in NPCs, activating the p53/p21/retinoblastoma protein (Rb) pathway and mediating premature NPC senescence [130]. Oxidative stress activates p53 through redox signaling and DDR induction, and oxidative post-translational modifications of p53 cysteine residues are thought to be associated with this process [131, 132]. In addition, redox signaling activates p53 through Jun N-terminal kinase (JNK) [133]. In IDD, treatment of human NPCs with H2O2 significantly increases intracellular ROS and p53 expression, in turn inducing cellular senescence by activating p21 [101]. In addition, a few inflammatory cytokines directly or indirectly promote p53 activation. IL-1β can upregulate the expression of p53 and its downstream genes, leading to cell cycle arrest and PUMA/Bax-mediated apoptosis of neural progenitor cells [134]. In ischemia/reperfusion-associated acute neuronal injury, IL-1β induces p53-dependent apoptosis by promoting NF-κB nuclear translocation [135]. Similarly, IL-1β can induce the degradation of NPC ECM through the NF-κB/p53 pathway, promoting the progress of IDD [136]. These studies suggest that multiple factors can directly or indirectly activate p53, initiating p53-related downstream responses that affect IDD progression.
Signal transduction of p53
As a transcription factor that responds to stress signals, p53 participates in complex signal transduction in cells [44]. Due to the central role of p53 in cellular stress and its involvement in various cellular processes such as DNA damage repair, cell cycle arrest, senescence, apoptosis, and metabolic regulation [137, 138], it can be speculated that the functional connections between p53 and other signal transduction molecules are diverse and regulate phenotypic changes in IVD cells by acting on dynamic signaling networks with complex feedback loops comprising different signaling pathways (Fig. 7). However, the exact molecular mechanisms of p53-mediated signaling remain elusive, and further studies are needed to elucidate the additional signaling pathways regulated by p53 and determine their mechanisms.

A Crosstalk between the NF-κB signaling pathway and p53. B Crosstalk between the MAPK/ERK signaling pathway and p53. C Crosstalk between the SIRT signaling pathway and p53. D Crosstalk between the PI3K-Akt signaling pathway and p53.
PI3K-Akt
Activation of the PI3K-Akt signaling pathway
The phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB/Akt) signaling pathway is a central pathway that regulates cell survival, proliferation, motility, and metabolism [139]. PI3Ks are a group of plasma membrane-associated lipid kinases with a common substrate, Akt [140]. Akt comprises a group of serine/threonine protein kinases with three isoforms, Akt1, Akt2, and Akt3 that are activated in response to upstream PI3Ks and regulate cellular metabolic processes by activating downstream target genes [141]. Various stimuli, including growth factors, cytokines, hormones, and stress, initiate PI3K/Akt signal transduction [142]. These molecules promote the activation of PI3K by activating the corresponding receptor tyrosine kinases, and activated PI3K converts phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-trisphosphate (PIP3) [143]. After generation, PIP3 acts as a second messenger to simultaneously recruit 3-phosphoinositide-dependent protein kinase 1 (PDK1) and Akt to the plasma membrane, such that PDK1 phosphorylates the threonine 308 site of the Akt protein, leading to its partial activation [144, 145]. Subsequently, mTOR complex 2 (mTORC2) phosphorylates the serine 473 site of Akt, resulting in its complete activation [146]. During this process, phosphatase and tensin homolog (PTEN) negatively regulate the PI3K/Akt signaling pathway by converting PIP3 to PIP2 [147].
Crosstalk between the PI3K-Akt signaling pathway and p53 in IDD
The PI3K/Akt signaling pathway functions through a variety of substrates, including forkhead box O (FOXO), glycogen synthase kinase 3 (GSK3) and mTORC1 [147, 148]. This pathway interacts with p53 through multiple mechanisms [149, 150]. When this pathway is activated, Akt leads to MDM2 translocation to the nucleus by phosphorylating serine 166 and 183 sites on MDM2. This promotes the ubiquitin-dependent proteasomal degradation of p53, thereby preventing ischemic injury-induced neuronal apoptosis and oxidative stress-induced cellular senescence in mice [151]. In addition, the p53-induced caspase cascade accelerates the degradation of MDM2 and Akt, thereby forming a negative feedback loop [152]. Regulation of p53 by the PI3K/Akt pathway is mediated by its downstream target, mTORC1. Under high-energy conditions, PI3K/Akt activates mTORC1, which in turn promotes MDM2 phosphorylation via the Akt/mTORC1/S6K1 signaling axis, leading to the inhibition of MDM2-mediated, ubiquitin-dependent p53 degradation and increasing the stability of p53 [153]. In addition, PTEN promotes the stability of p53 through the feedback inhibition of Akt activation and the downregulation of MDM2 transcription [154, 155]. In IDD, the PI3K/Akt pathway was significantly inhibited after NPCs were treated with IL-1β, which led to increased expression of p53 and induced cell senescence through the p53/p21 signal axis. In contrast, the expression of p53 was downregulated after pretreatment with a PI3K/Akt pathway activator [156]. Gong et al. [157] showed that bone morphogenetic protein-7 (BMP-7) attenuates replicative senescence in human NPCs by activating the PI3K/Akt pathway to downregulate the p53/p21 signaling axis, whereas treatment with the PI3K/Akt pathway inhibitor LY294002 partially inhibits the protective effect of BMP-7 on cellular senescence. These studies suggest that the activation of the PI3K/Akt signaling pathway inhibits p53 activation, which protects against IVD cell degeneration. Complex feedback regulation occurs between the PI3K/Akt signaling pathway and p53. Therefore, further exploration of the regulatory mechanism between the PI3K/Akt signaling pathway and p53 could help researchers elucidate the pathogenesis of IDD and develop new therapeutic approaches.
NF-κB
Activation of the NF-κB signaling pathway
NF-κBs are a group of transcription factors that bind to enhancer elements of the immunoglobulin κ light chain of activated B cells, which has two different activation mechanisms, classical and non-classical [158]. The activation of classical pathways plays an important role in tumors, neurodegenerative diseases, cardiovascular diseases, and musculoskeletal diseases by regulating various phenotypic changes, such as inflammation, immune response, cell proliferation, survival, and differentiation [11, 159,160,161]. Molecular cloning revealed that the human NF-κB transcription factor consists of five DBDs: NFKB1 (p50), NFKB2 (p52), REL (c-REL), RELA (p65), and RELB, the latter three of which possess transactivation domains [162]. In the classical signaling pathway, NF-κB is formed by heterologous DNA-binding dimers such as p65 and p50, which bind to their specific inhibitor IκB and are present as inactive complexes in the cytoplasm of most quiescent cells [163, 164]. IκB mainly consists of IKKα (IKK1), IKKβ (IKK2), and the regulatory subunit IKKγ, which forms the IKK complex with IKKα/IKKβ as a dimer [143, 165]. When cells are externally stimulated, IκB is phosphorylated and subjected to ubiquitin-dependent proteasomal degradation. This causes NF-κB to induce κB factor translocation to the nucleus and activate downstream target genes to regulate cell function [143, 164]. Meanwhile, NF-κB induces the expression of negative regulators such as IκBα, A20, and p105 to form a negative feedback regulatory mechanism [158, 166].
Crosstalk between the NF-κB signaling pathway and p53 in IDD
In the cellular signal transduction system, many intersections occur between the p53 and NF-κB pathways, indicating a wide range of mutual regulation between them. Regarding the ability to stimulate transcription, both p53 and NF-κB use the same histone acetyltransferase (p300/CBP) to promote the rapid transcription of target genes by acetylating related transcription factors and histones [167]. When both p53 and NF-κB respond to stress signals, the p65 subunit and p53 negatively regulate each other’s activities by competing for a limited protein pool of p300 and CBP coactivators [168,169,170]. The cross-transcriptional interference between p53 and NF-κB is caused by the direct interaction between these two transcription factors through the dimerization or tetramerization domain, leading to the inhibition of transcriptional activity independent of p300/CBP [171]. Another regulatory node in the p53 and NF-κB pathways is MDM2. MDM2 is a common downstream target gene of p53 and NF-κB signaling pathways, leading to MDM2 upregulation and p53 downregulation [172]. In addition, MDM2 acts as a cofactor for NF-κB, binding to the promoter binding site of its target gene and playing a role in activating NF-κB in sterile tissue inflammation [173]. As another regulatory node, an alternative reading frame (ARF) can bind and inhibit MDM2, thereby increasing p53 levels and activity [174, 175]. At the same time, ARF induces the formation of an ATR/checkpoint kinase 1 (CHK1) complex, which phosphorylates and inhibits NF-κB transcriptional capacity [176, 177]. In addition, the IKK protein family is associated with NF-κB activation and regulates p53 activity. Xia et al. [178] showed that IKKβ can phosphorylate the serine 362 and 366 sites of p53, leading to p53 recruitment by β-TrCP1 and ubiquitin-dependent degradation, thus reducing the level and activity of p53. Recent studies have reported that IKKα, another member of the IKK protein family, can induce the activation of CHK1 and promote the formation of a CHK1/p53 complex, which leads to the phosphorylation and activation of p53 [179]. In addition, IKKα triggers the formation of p300/p53 and CBP/p53 complexes, mediating UV-associated apoptosis by inducing p53 acetylation [180]. The above studies suggest that NF-κB activation usually inhibits p53 function and vice-versa; therefore, their relationship is antagonistic. p53 and NF-κB can synergistically regulate cellular processes under certain circumstances. In colon cancer cells containing wild-type p53, p53 activation induces nuclear translocation of the p65 subunit of NF-κB, leading to apoptosis, whereas a loss of NF-κB activity abolishes p53-induced apoptosis [181]. This suggests that NF-κB is essential for p53-mediated cell death in specific states. In addition, recent studies suggest that NF-κB and p53 may play a synergistic agonistic role in the regulation of pro-inflammatory responses. The coactivation of NF-κB and p53 leads to a significant elevation in the expression of pro-inflammatory genes in human macrophages, including pro-inflammatory cytokines and chemokines [182]. Zhang et al. [71] showed that the expression of p-p65 and p53 was significantly higher in degenerative NPCs than in normal human NPCs. In vitro, IL-1β significantly increased the expression of MMPs and ADAMTS by activating the NF-κB/p53 signal axis and decreased the expression of Agg and Col II, resulting in a decrease in ECM content [71]. In addition, both pro-inflammatory cytokines (IL-1β and TNF-α) and ROS can activate p53 by promoting NF-κB expression and nuclear transcription, inducing IVD cell senescence through the p53/p21 pathway, while the inhibition of NF-κB activity attenuates p53-induced cell senescence [68, 105, 183, 184]. This suggests that the coactivation of NF-κB and p53 may promote IDD progression. These studies suggest a complex network of mutual regulation between NF-κB and p53. Different stimuli and cell types may determine the outcome of regulation between NF-κB and p53. However, the crosstalk between NF-κB and p53 in IDD requires more studies to characterize further.
MAPK/ERK
Activation of the MAPK/ERK signaling pathway
The mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) signaling pathway plays an important role in the development of tumors, neurodegenerative diseases, infectious diseases, and musculoskeletal disorders by regulating cell survival, death, differentiation, proliferation, and metabolism [143, 185,186,187]. The complete MAPK cascade is divided into three modules: MAPK kinase (MAPKKK), MAPK kinase (MAPKK), and MAPK, which sense external stimuli and are activated by sequential phosphorylation [188]. Activated MAPKs phosphorylate many specific downstream substrates, leading to transcriptome and proteome reprogramming [189]. The existence of this sophisticated three-layer “core signal module” structure can associate different extracellular stimuli with a wide range of intracellular responses [190]. In mammals, multiple MAPK cascades have been identified, including ERK1/2/3/4/5/7/8, JNK1/2/3, p38 α/β/γ/δ, and nemo-like kinase [187, 188, 191, 192]. Among these, ERK is mainly activated by growth factors and insulin, whereas JNK and p38MAPK are mainly activated by environmental stress [193,194,195,196].
Crosstalk between the MAPK/ERK signaling pathway and p53 in IDD
Both MAPKs and p53 are activated by cellular stress, and their crosstalk controls the balance between cell survival and metabolism [197]. MAPKs control the activation of p53 by phosphorylation in response to the DDR [198, 199]. In ovarian cancer cells, cisplatin treatment mediates p53 serine 15 phosphorylation by activating ERK1/2 in response to DDR-induced apoptosis, whereas the selective MEK1 inhibitor PD98059 reduces p53 serine 15 phosphorylation and apoptosis [200]. Similarly, in human non-small-cell lung cancer (NSCLC) cells, the activation of JNK phosphorylates p53 at serine 15 and increases its stability by reducing the interaction between p53 and MDM2, thereby inducing G2/M arrest and cancer cell apoptosis [201]. In addition, activated JNK is directly involved in the phosphorylation of p53 at serine 20 and threonine 81, leading to an increase in p53-dependent transcriptional activity in response to DDR and stress inducers; this is inhibited in JNK knockout cells [202,203,204]. Many studies have shown that p38 can play an important role in activating p53 through phosphorylation. p38 can promote p53 activation and stabilization through the downregulation of MDM2 and phosphorylation of p53-serine 15 in response to apoptosis induced by hypoxia, carcinogenic toxicants, and UV exposure, which can be attenuated by the p38 inhibitors SB203580 and SB202190 [205,206,207]. In normal human keratinocytes, activated p38 phosphorylates p53 at serine 15, reducing p53 nuclear transcription and reducing UV-induced apoptosis [208]. This suggests that different stimuli and cell types affect the phosphorylation of p53 serine 15. In addition to serine 15, activated p38 phosphorylates p53 serine at sites 20, 33, 46, and 389, activating p53 [202, 209,210,211]. MAPKs can regulate p53 activity, and p53 can regulate MAPKs by activating downstream target genes. Wip1, a serine/threonine phosphatase, is induced by UV radiation in a p53-dependent manner in response to DNA damage [212]. In NSCLC, Wip1 promotes the expression of stemness-related proteins and cancer stem cell properties by inhibiting p38 activity [213]. Other studies have shown that Wip1 selectively reduces p38 activity by dephosphorylating conserved threonine residues [214]. In addition, other transcription products of p53, members of the bispecific protein phosphatase family, including mitogen-activated protein kinase phosphatase 1 (Mkp1), dual specific phosphatase 2 (DUSP2), and DUSP5, have been shown to inhibit the activities of ERK, JNK, and p38 [215, 216]. In IDD, both IL-1β and TNF-α can increase the phosphorylation of ERK, JNK, and p38 in NPCs, activating p53 and inducing cellular senescence through the p53/p21 pathway [68, 105]. Similarly, advanced oxidative protein products (AOPPs) can increase ERK, JNK, and p38 phosphorylation levels in AF cells and induce cellular senescence through the p53/p21 pathway, whereas intervention with pathway inhibitors ameliorates AOPP-induced senescence in AF cells [70]. Ge et al. [217] showed that syndecan-4 promotes IDD progression by affecting NPC function. Mechanistically, syndecan-4 overexpression significantly increases the expression of JNK, leading to p53 activation and promoting IDD progression by downregulating Col II and Agg expression and upregulating Col X expression. These phenomena were significantly improved when the cells were treated with the JNK inhibitor SP600125. This evidence suggests that complex regulatory mechanisms exist between MAPKs and p53 and that activating MAPK signaling pathways may accelerate IDD progression by promoting p53 activation and stabilization.
SIRT
Activation of the SIRT signaling pathway
The sirtuin (SIRT) protein family is an important gene-silencing complex composed of seven subtypes of type III histone deacetylase [218]. These protein families have highly conserved nicotinamide adenine dinucleotide+ (NAD+)-binding domains and catalytic functional domains, and each SIRT has different sequences and lengths of the N-terminal and C-terminal domains [219]. SIRT family proteins are functionally diverse, with SIRT1–SIRT3 having strong deacetylase activity and SIRT4–SIRT7 having weaker activity, which allows them to act specifically on different substrates depending on the biological processes they are involved in [143]. In addition, the SIRT protein family has different subcellular localizations: SIRT1, SIRT6, and SIRT7 are mainly located in the nucleus; SIRT3, SIRT4, and SIRT5 are distributed in the mitochondria; and SIRT2 is mainly present in the cytoplasm [219, 220]. SIRT regulates multiple cellular processes, including DNA repair, glucose metabolism, cell survival, inflammation, and senescence, through upstream and downstream signaling pathways and plays an important role in various diseases [221,222,223].
Crosstalk between the SIRT signaling pathway and p53 in IDD
The SIRT family of proteins regulates transcription factors, leading to complex regulatory interactions with p53 [224]. SIRT1, SIRT6, and SIRT7 are primarily located in the nucleus and interact with histones and transcription factors [225]. SIRT1 is the most widely studied member of the SIRT family [226,227,228]. SIRT1 negatively regulates the biological function of p53 by deacetylating the p53 gene, reducing p53-induced cell growth inhibition, cell cycle arrest, senescence, and apoptosis [229,230,231,232,233]. However, SIRT1 cannot inhibit all p53-mediated biological activities [234]. In contrast, SIRT1 acts synergistically with p53 to amplify the pharmacological effects of polydatin [235]. In addition, SIRT6 and SIRT7 can reduce p53-induced cellular phenotypic changes by deacetylating p53 [236, 237]. Unlike SIRT1, SIRT6, and SIRT7, SIRT2 is mainly located in the cytoplasm and deacetylates several cytoplasmic substrates, including tubulin [238]. SIRT2 is present in the nucleus and is associated with the deacetylation of histones and several transcription factors [239, 240]. SIRT2 induces p53 deacetylation at lysine 382, decreasing p53 transcriptional activity [241,242,243]. These studies indicate that SIRT2 activation regulates the activity of p53 and vice-versa [244]. SIRT2 expression significantly increases in senescent cells and decreases after p53 knockdown, suggesting that p53 may affect SIRT2 expression. Chromatin immunoprecipitation has revealed a p53 binding site in the SIRT2 promoter, indicating that it is regulated by p53 [244]. SIRT3, SIRT4, and SIRT5 are mainly localized in the mitochondria. In Alzheimer’s disease, SIRT3 attenuates p53-mediated neuronal apoptosis and mitochondrial dysfunction by inhibiting the acetylation of p53 lysine 320 [245]. In addition, SIRT5 interacts with p53 to inhibit its transcriptional activity. Mass spectrometric analysis showed that SIRT5 mediates the desuccinylation of p53 at lysine 120, resulting in the inhibition of p53 activity and p53-mediated apoptosis [246]. SIRT4 may play different regulatory roles in p53 activity. SIRT4 promotes p53 phosphorylation by inhibiting glutamine metabolism, which inhibits pancreatic ductal adenocarcinoma progression [247]. The modulation of p53 by SIRT is observed in IDD [248, 249]. In a high-glucose environment, SIRT1 activation inhibits p53 acetylation, which protects NPCs from high glucose-induced apoptosis and senescence [112]. In addition, CEP degeneration plays an important role in IDD associated with nutritional disorders [250]. Under sublethal oxidative stress, SIRT1 overexpression significantly reduced p53 acetylation and alleviated oxidative stress-induced senescence in human CEP cells by inhibiting the p53/p21 pathway [251]. In addition, SIRT2 mitigated oxidative stress and cellular senescence by inhibiting the IL-1β-induced p53/p21 pathway, thereby preventing IDD progression [252]. These studies suggest that SIRT1 and SIRT2 can reduce apoptosis and senescence in IVD cells and delay the progression of IDD by inhibiting the activity of p53. However, our understanding of the regulatory effects of other members of the SIRT protein family on p53 is limited, and further studies are needed.
The role of p53 in IDD
p53 and apoptosis
Apoptosis is the process by which cells stop growing and dividing, ultimately leading to cell death. Apoptosis is divided into endogenous and exogenous apoptosis; the former is known as mitochondrial apoptosis [253]. As key molecules in inducing endogenous apoptosis, Bcl-2 family proteins play a key role in mitochondrial apoptosis by regulating the permeability of the outer mitochondrial membrane and the release of cytochrome c [254]. Based on the modular structure of their homology domain, Bcl-2 family proteins can be divided into pro-apoptotic (Bak, Bax, and Bok) and anti-apoptotic proteins (Bcl-XL, Bcl-2, Bcl-w, and Mcl-1), the interaction between which is essential for maintaining the balance between cell death and survival [255, 256]. p53 induces transcription-dependent apoptosis through the transcriptional activation of various proapoptotic target genes, including Bak, Bax, and PUMA [257]. In addition to transcription-dependent apoptosis, p53 mediates transcription-independent apoptosis [258]. Nuclear magnetic resonance spectroscopy (NMR) shows that the H2 α-helix, L1, and L3 loops in the DBD of p53 bind directly to Bak, enhancing its oligomerization and leading to outer mitochondrial membrane permeability and cytochrome c release. In contrast, point mutations in these regions significantly weaken the ability of p53 oligomeric Bak to induce transcription-independent apoptosis [259, 260]. In addition, Bcl-XL inhibits Bak/Bax homo-oligomerization and apoptosis by forming an inhibitory complex with Bak and Bax [258]. The DBD of p53 can directly bind to Bcl-xL and release Bak and Bax, thereby enhancing the homo-oligomerization of Bak/Bax; this is applicable to Bcl-w, Mcl-1, and Bcl-2 [261,262,263,264,265]. In addition, PUMA is a direct target of p53-mediated mitochondrial apoptosis [266]. Using isothermal titration calorimetry, Han et al. [267] showed that PUMA binds to the DBD of p53 and thus induces apoptosis by a transcriptionally non-dependent mechanism. In IDD, basal p53 expression [37] and phosphorylated and acetylated p53 levels are significantly elevated in degenerative IVD cells [112, 268], leading to PUMA and Bax accumulation and mitochondrial dysfunction, which in turn leads to apoptosis [104, 269]. In addition, compression loading increases the expression of p53 in NPC mitochondria in a time-dependent manner, whereas the inhibition of p53 mitochondrial translocation by pifithrin (PFT) significantly reduces compression-induced NPC apoptosis, suggesting that p53 may promote apoptosis by interacting with Bcl-2 family proteins through mitochondrial translocation [109]. At the same time, compressive loading promotes p53-regulated apoptosis-inducing protein 1 (p53AIP1) expression and induces p53-dependent apoptosis by interacting with Bcl-2 family proteins [270]. In addition, elevated p53 levels in NPC stimulate apoptosis by upregulating the expression of NDRG2 and regulating the development and DDR 1 (REDD1)/thioredoxin-interacting protein (TXNIP) complex, which in turn aggravates IDD [37, 271]. These studies confirm that the expression of p53 increases in degenerative IVD, leading to excessive apoptosis of IVD cells; this inhibition of p53 expression and activity may delay IDD by reducing apoptosis in IVD cells.
p53 and cell senescence
As an age-related musculoskeletal disease, excessive cellular senescence in IVDs is an important feature of IDD. Cellular senescence can be divided into replicative and stress-induced senescence [272]. Replicative senescent cells accumulate during physiological or accelerated senescence processes and are aggravated by factors that promote the accumulation or inhibit the decomposition of senescent cells [273]. DNA damage, oxidative stress, and inflammation can significantly increase the burden of tissue cell senescence by inducing stress-related cell senescence [274,275,276]. In addition, senescent cells “infect” surrounding healthy cells through the paracrine effect of the senescence-associated secretory phenotype (SASP), which induces cellular senescence [277,278,279]. For instance, Moiseeva et al. [280] demonstrated that senescent cells can induce an inflammatory microenvironment that impedes the proliferation and regeneration of stem cells while alleviating the burden on senescent cells or neutralizing their inflammatory secretions to expedite regeneration in both juvenile and aged mice. High levels of SASP-associated MMPs have been shown to induce the shedding of autocrine ligands, making senescent cells less susceptible to immune surveillance and clearance [281]. These rapid accumulation, persistence, and amplification mechanisms lead to the accumulation of senescent cells in tissues, ultimately promoting the progression of IDD. p53-mediated cellular senescence is mainly derived from the DDR and can be induced by oxidative stress, inflammatory responses, telomere shortening, and oncogene activation [282, 283]. The DDR activates p53 through ATM and ATR, and the activated p53 subsequently undergoes phosphorylation and acetylation, which protects p53 from MDM2-mediated ubiquitin-dependent proteasome degradation [284]. After stabilization, p53 accumulates in the nucleus and activates the expression of effectors involved in the regulation of cellular senescence, among which p21 plays the most important role [285]. Once activated, p21 induces cell cycle arrest in the G1/S or G2/M phases by inhibiting apoptosis [286]. Histological analysis of human IVD specimens revealed that the proportion of senescence-associated β-galactosidase-positive cells was significantly higher in Pfirrmann grade IV/V than in grade I/II, and the expression of p53, p21, and Rb was high [66]. In addition, the expression of p53 mRNA and protein in rat degenerative IVD tissues was significantly increased [287]. In vitro, inflammation, oxidative stress, high glucose, and DNA damage can all induce senescence in IVD cells by activating the p53/p21/Rb signaling axis. In contrast, the downregulation of p53 expression can significantly improve cell senescence [68, 70, 101, 130, 183, 288, 289]. In addition, polo-like kinase 1 (PLK1), an evolutionarily conserved serine/threonine kinase, plays multiple roles in the cell cycle, including regulating cell proliferation and senescence [290]. Zhang et al. [67] showed significantly decreased expression of PLK1 in the NPCs of patients with IDD. Further functional experiments showed that inhibiting the activity of PLK1 in normal NPCs increased the activity of the p53/p21 signaling axis, inhibited cell proliferation, and induced cell senescence. In addition, N-acetylated proline-glycine-proline (N-Ac-PGP) is a collagen-derived tripeptide that plays an important role in inflammatory diseases by binding to CXC receptor 1/2 (CXCR1/2) [291]. N-Ac-PGP induces DNA damage and ROS accumulation in NPCs, which induces senescence by binding to CXCR1 and activating the p53/p21 signaling axis [128]. These results demonstrate that p53 is closely related to IVD cell senescence and that the inhibition of p53 expression and activation can effectively improve premature IVD cell senescence and delay IDD progression.
p53 and ECM metabolism
As a central component of the stress response mechanism, p53 controls the safeguard mechanism against the accumulation of abnormal cells and their transformation by regulating DNA repair, cell cycle progression, cellular senescence, and death [292]. Recently, various cellular processes regulated by p53 have been shown to control glucose metabolism, oxidative stress, and ECM remodeling [293]. In a mouse model of dimethylnitrosamine (DMN) liver injury, the effect of DMN led to a dose-dependent increase in p53, accompanied by an increase in the expression of MMP9, 10, and 12 [294]. In addition, p53 overexpression significantly reduced the ECM of mouse fibroblasts [295]. Similarly, p53 overexpression significantly increased the expression of MMP13 and ADAMTS5 and inhibited the expression of SOX9 in mice with osteoarthritis, which significantly improved after the inhibition of p53 [296]. These studies suggest that p53 induces ECM remodeling by promoting the expression of matrix-degrading enzymes and inhibiting the expression of matrix proteins. In IDD, IL-1β significantly increased the expression level of p53 in NPCs, which resulted in the upregulation of mRNA and protein levels of MMP-3, MMP-13, ADAMTS-4, and ADAMTS-5 and the downregulation of Col II and Agg [103]. In addition, several studies have shown that abnormal activation of the NF-κB and JNK signaling pathways in IVD is an important factor contributing to IDD progression [143]. Activated NF-κB and JNK signaling pathways upregulate p53 expression, accelerating ECM degradation by promoting the expression of MMPs and ADAMTS and inhibiting the expression of Col II and Agg [71, 217]. Recently, Zheng et al. [103] reported that p53 binds to the promoter regions of prolyl endopeptidase to promote their transcription. Overexpression of prolyl endopeptidases impairs the ECM quality of NPCs by inhibiting the expression of Col II and Agg and promoting the expression of MMP3. p53 may have opposing roles in specific environments. Xiong et al. [72] reported that p53 plays an important role in maintaining NPC activity and integrity. Mechanistic studies have shown that high levels of p53 can promote ATP production by inhibiting the G6PD and pentose phosphate pathways. In addition, the expression of SOX9 and Col II in p53-normal controls was significantly higher than that in p53-knockdown NPCs. These results suggest that the selective activation of p53 downstream target genes under different stress induction conditions may lead to the differential expression of metabolism-related phenotypes within the IVD.
p53 and oxidative stress
Oxidative stress can be located either upstream or downstream of p53. In normal IVD, NPCs exist in a hypoxic microenvironment, and high oxygen partial pressure enhances ROS production in NPC mitochondria [297]. At less than 20% oxygen partial pressure, the level of ROS in NPCs increases significantly, leading to p53 activation and cell senescence [298]. Furthermore, treatment of IVD cells with H2O2 resulted in a significant increase in intracellular ROS levels, which induced DDR and ATM activation, eventually leading to the activation and rapid accumulation of p53 [101, 299, 300]. These results suggest that increased ROS levels in IVD cells promote the expression and activation of p53. As a redox-sensitive transcription factor, p53 regulates various genes with antioxidant or pro-oxidative properties, affecting cellular redox homeostasis [301, 302]. The contradictory roles of p53 in oxidative stress regulation depend on various factors, including the type, intensity, and duration of stress [303]. Under physiological and mild stress conditions, p53 exerts its antioxidant activity by activating downstream antioxidant target genes, including TP53-inducible glycolysis and apoptosis regulator, sestrin 1/2 (SESN1/2), TP53-induced nuclear protein 1, and p21 [301]. These target genes reduce the overall level of intracellular oxidation by regulating cellular metabolism and maintaining mitochondrial function [304]. In addition, SESN1/2 and p21 interfere with Keap1–Nrf2 binding and enhance the stability and activity of Nrf2, thereby inhibiting intracellular oxidative stress [305, 306]. However, the phenomenon and related mechanism by which p53 inhibits oxidative stress in IDD remain unclear, and further research is needed. Under extensive and sustained stress, activated p53 exhibits pro-oxidative activity by turning on p53-inducible gene 3 and proline oxidase, leading to apoptosis by inducing the expression of Bax and PUMA [59, 304]. Additionally, TXNIP promotes intracellular oxidative stress by inhibiting thioredoxin reduction capacity [307]. In IDD, p53 expression increases with H2O2 treatment, and activated p53 further enhances oxidative stress [271]. These studies indicate a complex regulatory mechanism between p53 and oxidative stress. Further elucidation of the regulatory mechanisms between p53 and oxidative stress in IDD will be beneficial for developing potential therapeutic targets.
p53-related targeted therapy
The incidence and burden of IDD, an age-related disease, are increasing annually worldwide [3]. Currently, the treatment of IDD mainly includes conservative and surgical treatments, both aimed at relieving clinical symptoms. Therefore, the further exploration of the pathogenic mechanism of IDD and the adoption of targeted treatment strategies are current research hotspots. Recent studies have shown that p53 plays an important role in the occurrence and progression of IDD, and the inhibition of p53 overactivation in IVD may help delay the progression of IDD. However, extensive p53 inhibition may not be an applicable approach to alleviate IDD because the negative effects of this approach cannot be ignored. Therefore, p53-targeted functional inhibitors in combination with exosomes, biomaterials, and cellular therapies may yield greater benefits (Fig. 8).

Top: Establishment of a drug delivery system for inhibitors of p53 function. Middle: Amplification and purification of the drug delivery system. Bottom: Injection of the drug delivery system.
p53 inhibitor
In 1999, to reduce the serious side effects of p53-mediated chemotherapy and radiotherapy in cancer, Komarov et al. [308] isolated a small fraction of PFT to block the side effects caused by p53-dependent transcriptional activation. PFT protected mice from p53-related cell death induced by radiation and multiple cytotoxic drugs without promoting tumor formation [308]. Studies have confirmed that PFT can alleviate adriamycin-induced apoptosis in human umbilical vein endothelial cells by inhibiting basic and induced levels of the p53 protein [309]. In IDD, compression treatment induces NPC apoptosis by promoting the mitochondrial translocation of p53. In contrast, PFT significantly attenuates compression-induced NPC apoptosis and alleviates IDD progression by inhibiting p53 mitochondrial translocation [109]. These results suggest that PFT may delay the progression of IDD; however, further basic and clinical studies are needed to determine the optimal dose and route of administration.
p53 homologous sequence
In addition to the typical full-length p53, TP53 produces at least 12 truncated subtypes through the alternative initiation of translation, the use of alternative promoters, and alternative splicing, which can positively or negatively regulate the activity and function of full-length p53 [281, 310]. Among these p53 isoforms, Δ133p53α and p53β are thought to be endogenous regulators of cellular senescence [311]. Under stress conditions, p53β forms a complex with full-length p53, which in turn enhances the transcriptional activities of the Bax and p21 promoters, suggesting that p53β acts synergistically with full-length p53 to induce apoptosis and senescence [281, 311]. ∆133p53α acts in contrast to p53β. ∆133p53α was abundantly expressed in early-passage normal human fibroblasts and was significantly reduced in late-passage and senescent cells, mainly due to excessive autophagic degradation of ∆133p53α in senescent cells [311]. In addition, in Hutchinson–Gilford progeria syndrome (HGPS) fibroblasts, ∆133p53α inhibited cellular senescence by downregulating the p53 senescence-associated genes p21 and miR-34a. ∆133p53α overexpression restored the replicative capacity of HGPS fibroblasts [312]. As mentioned above, the selective, dominant-negative effect of ∆133p53α on full-length p53 suggests that ∆133p53α may be a potent target for the inhibition of cellular senescence. However, the expression and role of 133p53α in IVD cells are unclear, and more studies are needed.
Natural molecules
Numerous natural compounds have been investigated for their p53-inhibitory activities in the context of IDD. Naringin (Nar) and eupatilin (Eup), the primary flavonoids derived from Citrus and Artemisia, respectively, have been demonstrated to possess diverse biological effects [313, 314]. Both Nar and Eup regulate the expression of Col II, Agg, MMP-3, MMP-13, and ADAMTS-4 to maintain a high-quality ECM [68, 136]. Moreover, Eup suppresses TNF-α-induced senescence of NP cells through the downregulation of p21 and p53 expression [68]. Related mechanistic studies have shown that Nar and Eup can protect NPCs from damage by inhibiting the NF-κB/p53 signaling axis. In addition to Nar and Eup, quercetin (Que) and myricetin (Myr) belong to a family of natural flavonoids found in plants that exhibit anti-inflammatory, anti-aging, and antioxidant properties [315, 316]. Que and Myr can mitigate oxidative stress-induced IVD cell senescence by activating the SIRT1 signaling pathway, thereby inhibiting p53 expression [317, 318]. Morroniside (Mor), which belongs to the class of iridoid glycosides [319], has been reported to effectively attenuate H2O2-induced NP cell senescence by modulating the ROS-Hippo-p53 signaling pathway [101]. In vivo, Mor significantly ameliorated lumbar disc degeneration in rats after an 8-week treatment period. Furthermore, the inhibition of p53 by proanthocyanidins and resveratrol has been documented to effectively impede the progression of IDD [156, 320].
Conclusions and prospects
IDD is the initial step in the evolution and progression of a range of degenerative spinal disorders and is a major cause of LBP and disability, the prevalence of which increases with age [321]. Currently, the main treatment for IDD is to control the clinical symptoms through physiotherapy, oral drugs, and surgery; however, it is not to prevent the progression of IDD or reverse its degeneration by treating the causes of degenerative disease [322]. However, these methods lead to a high recurrence rate, treatment cost, and risk of adjacent IVD degeneration [323]. Therefore, the pathogenic mechanisms of IDD and the development of targeted therapeutic approaches are major clinical issues that must be addressed. Transcription factor p53 plays an important role in malignant tumors, cardiovascular diseases, neurodegenerative diseases, and osteoarticular diseases by regulating cell cycle progression, senescence, apoptosis, angiogenesis, DNA repair, and cell metabolism [44, 45, 138, 324]. Recent studies have shown that p53 activation and signal transduction maintain homeostasis in IVD. p53 reduces the number of normal IVD cells by activating intrinsic death mechanisms and senescence-related genes in IVD cells. In contrast, p53 alters the microenvironment where IVD cells survive by promoting the expression of a metabolic phenotype characterized by SASP, leading to an imbalance between anabolic and catabolic cellular metabolism and a decrease in IVD ECM content. The combined action of these two factors promotes abnormal changes in the organizational structure of the IVD, eventually leading to IDD. Current research suggests that inhibiting the activation of p53 and its downstream signaling pathways delays the progression of IDD. However, a few studies have shown that p53 plays an important role in maintaining the activity and functional integrity of NPCs under low-glucose conditions [72]. This suggests that p53 may play different roles at different stages of IDD. Further studies are required to elucidate the role of p53.
Studies have shown that p53 responds differently based on the severity and duration of stimulation. Mild and transient stimuli induce the repair of cell damage and transient growth arrest. In contrast, severe and sustained stimuli can lead to cell senescence and apoptosis [325]. Currently, studies on p53 in IDD mainly focus on the dynamic changes and role of p53 in late degeneration and severe stress. However, the dynamic changes and activation of related signaling pathways of p53 in early-stage IVD and sublethal stress have not yet been elucidated. Therefore, future studies should focus on the expression pattern of p53 in the early stages of IDD and its role in related phenotypic changes to explore targeted treatment strategies.
In addition, studies on the role of p53 in IDD are still in the exploratory stage, with most studies focusing on rodent models. However, there are still certain species-specific differences in the shape, size, and biochemical composition of the IVDs and the anatomical structure of the spine between rodents and large mammals [326], which makes many current studies inapplicable to human IDD, making it difficult to guide treatment. Therefore, selecting an appropriate animal model is significant for future basic research and clinical applications. Therefore, sheep may be more suitable than rodents for studying human IDD. Sheep IVDs have a shape and size similar to human IVDs and do not exhibit the persistence of notochord cells with age [323]. Additionally, current studies have focused on the NP, and future treatments should focus on the synergistic recovery of the NP, AF, and CEP. Simultaneously, because the progression of IDD is a chronic process, the long-term efficacy of related treatment strategies must be evaluated.
In conclusion, the molecular mechanism underlying the potential role of p53 in IDD has not yet been fully elucidated and requires further investigation. Future research should focus on the role of p53 in early IDD, which appears to be crucial for further elucidation of its pathogenesis and the development of targeted therapies.
References
Wong CK, Mak RY, Kwok TS, Tsang JS, Leung MY, Funabashi M, et al. Prevalence, incidence, and factors associated with non-specific chronic low back pain in community-dwelling older adults aged 60 years and older: a systematic review and meta-analysis. J Pain. 2022;23:509–34.
Buchbinder R, van Tulder M, Öberg B, Costa LM, Woolf A, Schoene M, et al. Low back pain: a call for action. Lancet. 2018;391:2384–8.
Hartvigsen J, Hancock MJ, Kongsted A, Louw Q, Ferreira ML, Genevay S, et al. What low back pain is and why we need to pay attention. Lancet. 2018;391:2356–67.
March L, Smith EUR, Hoy DG, Cross MJ, Sanchez-Riera L, Blyth F, et al. Burden of disability due to musculoskeletal (MSK) disorders. Best Pr Res Clin Rheumatol. 2014;28:353–66.
Morris LD, Daniels KJ, Ganguli B, Louw QA. An update on the prevalence of low back pain in Africa: a systematic review and meta-analyses. BMC Musculoskelet Disord. 2018;19:196.
Garcia JBS, Hernandez-Castro JJ, Nunez RG, Pazos MA, Aguirre JO, Jreige A, et al. Prevalence of low back pain in Latin America: a systematic literature review. Pain Physician. 2014;17:379–91.
Sharma S, Jensen MP, Pathak A, Sharma S, Pokharel M, Abbott JH. State of clinical pain research in Nepal: a systematic scoping review. Pain Rep. 2019;4:e788.
Katz JN. Lumbar disc disorders and low-back pain: socioeconomic factors and consequences. J Bone Jt Surg Am. 2006;88:21–24.
Teles AR, Righesso O, Gullo MCR, Ghogawala Z, Falavigna A. Perspective of value-based management of spinal disorders in Brazil. World Neurosurg. 2016;87:346–54.
Kamali A, Ziadlou R, Lang G, Pfannkuche J, Cui S, Li Z, et al. Small molecule-based treatment approaches for intervertebral disc degeneration: current options and future directions. Theranostics. 2021;11:27–47.
Zhang G-Z, Liu M-Q, Chen H-W, Wu Z-L, Gao Y-C, Ma Z-J, et al. NF-κB signalling pathways in nucleus pulposus cell function and intervertebral disc degeneration. Cell Prolif. 2021;54:e13057.
Kloppenburg M, Berenbaum F. Osteoarthritis year in review 2019: epidemiology and therapy. Osteoarthr Cartil. 2020;28:242–8.
Yang S, Zhang F, Ma J, Ding W. Intervertebral disc ageing and degeneration: the antiapoptotic effect of oestrogen. Ageing Res Rev. 2020;57:100978.
Zhu L, Yu C, Zhang X, Yu Z, Zhan F, Yu X, et al. The treatment of intervertebral disc degeneration using traditional Chinese Medicine. J Ethnopharmacol. 2020;263:113117.
Krut Z, Pelled G, Gazit D, Gazit Z. Stem cells and exosomes: new therapies for intervertebral disc degeneration. Cells. 2021;10:2241.
Dickinson PJ, Bannasch DL. Current understanding of the genetics of intervertebral disc degeneration. Front Vet Sci. 2020;7:431.
Theodore N, Ahmed AK, Fulton T, Mousses S, Yoo C, Goodwin CR, et al. Genetic predisposition to symptomatic lumbar disk herniation in pediatric and young adult patients. Spine. 2019;44:E640–E649.
Silva MJ, Holguin N. Aging aggravates intervertebral disc degeneration by regulating transcription factors toward chondrogenesis. FASEB J. 2020;34:1970–82.
Zehra U, Flower L, Robson-Brown K, Adams MA, Dolan P. Defects of the vertebral end plate: implications for disc degeneration depend on size. Spine J. 2017;17:727–37.
Chen Z, Li X, Pan F, Wu D, Li H. A retrospective study: does cigarette smoking induce cervical disc degeneration? Int J Surg. 2018;53:269–73.
Cannata F, Vadalà G, Ambrosio L, Fallucca S, Napoli N, Papalia R, et al. Intervertebral disc degeneration: a focus on obesity and type 2 diabetes. Diabetes Metab Res Rev. 2020;36:e3224.
Liang H, Luo R, Li G, Zhang W, Song Y, Yang C. The proteolysis of ECM in intervertebral disc degeneration. Int J Mol Sci. 2022;23:1715.
Chen S, Lei L, Li Z, Chen F, Huang Y, Jiang G, et al. Grem1 accelerates nucleus pulposus cell apoptosis and intervertebral disc degeneration by inhibiting TGF-β-mediated Smad2/3 phosphorylation. Exp Mol Med. 2022;54:518–30.
Novais EJ, Tran VA, Johnston SN, Darris KR, Roupas AJ, Sessions GA, et al. Long-term treatment with senolytic drugs Dasatinib and Quercetin ameliorates age-dependent intervertebral disc degeneration in mice. Nat Commun. 2021;12:5213.
Xing H, Zhang Z, Mao Q, Wang C, Zhou Y, Zhou X, et al. Injectable exosome-functionalized extracellular matrix hydrogel for metabolism balance and pyroptosis regulation in intervertebral disc degeneration. J Nanobiotechnol. 2021;19:264.
Ma H, Xie C, Chen Z, He G, Dai Z, Cai H, et al. MFG-E8 alleviates intervertebral disc degeneration by suppressing pyroptosis and extracellular matrix degradation in nucleus pulposus cells via Nrf2/TXNIP/NLRP3 axis. Cell Death Discov. 2022;8:209.
Wang W-J, Yu X-H, Wang C, Yang W, He W-S, Zhang S-J, et al. MMPs and ADAMTSs in intervertebral disc degeneration. Clin Chim Acta. 2015;448:238–46.
Vergroesen PPA, Kingma I, Emanuel KS, Hoogendoorn RJW, Welting TJ, van Royen BJ, et al. Mechanics and biology in intervertebral disc degeneration: a vicious circle. Osteoarthr Cartil. 2015;23:1057–70.
Shu CC, Smith MM, Smith SM, Dart AJ, Little CB, Melrose J. A Histopathological scheme for the quantitative scoring of intervertebral disc degeneration and the therapeutic utility of adult mesenchymal stem cells for intervertebral disc regeneration. Int J Mol Sci. 2017;18:1049.
Salo J, Kaigle Holm A, Indahl A, Mackiewicz Z, Sukura A, Holm S, et al. Expression of vascular endothelial growth factor receptors coincide with blood vessel in-growth and reactive bone remodelling in experimental intervertebral disc degeneration. Clin Exp Rheumatol. 2008;26:1018–26.
Sun K, Jiang J, Wang Y, Sun X, Zhu J, Xu X, et al. The role of nerve fibers and their neurotransmitters in regulating intervertebral disc degeneration. Ageing Res Rev. 2022;81:101733.
Chen H-W, Zhou J-W, Zhang G-Z, Luo Z-B, Li L, Kang X-W. Emerging role and therapeutic implication of mTOR signalling in intervertebral disc degeneration. Cell Prolif. 2023;56:e13338.
Jiang C, Guo Q, Jin Y, Xu J-J, Sun Z-M, Zhu D-C, et al. Inhibition of EZH2 ameliorates cartilage endplate degeneration and attenuates the progression of intervertebral disc degeneration via demethylation of Sox-9. EBioMedicine. 2019;48:619–29.
Liu Y, Gu W. p53 in ferroptosis regulation: the new weapon for the old guardian. Cell Death Differ. 2022;29:895–910.
Engeland K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 2022;29:946–60.
Liu Y, Gu W. The complexity of p53-mediated metabolic regulation in tumor suppression. Semin Cancer Biol. 2022;85:4–32.
Zhang K, Zhang Y, Zhang C, Zhu L. Upregulation of P53 promotes nucleus pulposus cell apoptosis in intervertebral disc degeneration through upregulating NDRG2. Cell Biol Int. 2021;45:1966–75.
DeLeo AB, Jay G, Appella E, Dubois GC, Law LW, Old LJ. Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proc Natl Acad Sci USA. 1979;76:2420–4.
Crawford LV, Pim DC, Bulbrook RD. Detection of antibodies against the cellular protein p53 in sera from patients with breast cancer. Int J Cancer. 1982;30:403–8.
Lübbert M, Miller CW, Crawford L, Koeffler HP. p53 in chronic myelogenous leukemia. Study of mechanisms of differential expression. J Exp Med. 1988;167:873–86.
Koeffler HP, Miller C, Nicolson MA, Ranyard J, Bosselman RA. Increased expression of p53 protein in human leukemia cells. Proc Natl Acad Sci USA. 1986;83:4035–9.
Nigro JM, Baker SJ, Preisinger AC, Jessup JM, Hostetter R, Cleary K, et al. Mutations in the p53 gene occur in diverse human tumour types. Nature. 1989;342:705–8.
Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–21.
Jazvinšćak Jembrek M, Oršolić N, Mandić L, Sadžak A, Šegota S. Anti-Oxidative, anti-inflammatory and anti-apoptotic effects of flavonols: targeting Nrf2, NF-κB and p53 pathways in neurodegeneration. Antioxidants. 2021;10:1628.
Men H, Cai H, Cheng Q, Zhou W, Wang X, Huang S, et al. The regulatory roles of p53 in cardiovascular health and disease. Cell Mol Life Sci. 2021;78:2001–18.
Kung C-P, Murphy ME. The role of the p53 tumor suppressor in metabolism and diabetes. J Endocrinol. 2016;231:R61–R75.
Jacques C, Tesfaye R, Lavaud M, Georges S, Baud’huin M, Lamoureux F, et al. Implication of the p53-Related miR-34c, -125b, and -203 in the osteoblastic differentiation and the malignant transformation of bone sarcomas. Cells. 2020;9:810.
Nataraj AJ, Trent JC, Ananthaswamy HN. p53 gene mutations and photocarcinogenesis. Photochem Photobio. 1995;62:218–30.
Smith ND, Rubenstein JN, Eggener SE, Kozlowski JM. The p53 tumor suppressor gene and nuclear protein: basic science review and relevance in the management of bladder cancer. J Urol. 2003;169:1219–28.
Swiatkowska A, Zydowicz P, Sroka J, Ciesiołka J. The role of the 5’ terminal region of p53 mRNA in the p53 gene expression. Acta Biochim Pol. 2016;63:645–51.
Courtois S, Verhaegh G, North S, Luciani M-G, Lassus P, Hibner U, et al. DeltaN-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene. 2002;21:6722–8.
Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53. Annu Rev Biochem. 2008;77:557–82.
Raj N, Attardi LD. The transactivation domains of the p53 protein. Cold Spring Harb Perspect Med. 2017;7:a026047.
Hoyos D, Greenbaum B, Levine AJ. The genotypes and phenotypes of missense mutations in the proline domain of the p53 protein. Cell Death Differ. 2022;29:938–45.
Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018;25:104–13.
Chène P. The role of tetramerization in p53 function. Oncogene. 2001;20:2611–7.
Kim H, Kim K, Choi J, Heo K, Baek HJ, Roeder RG, et al. p53 requires an intact C-terminal domain for DNA binding and transactivation. J Mol Biol. 2012;415:843–54.
Hassin O, Oren M. Drugging p53 in cancer: one protein, many targets. Nat Rev Drug Discov. 2023;22:127–44.
Beyfuss K, Hood DA. A systematic review of p53 regulation of oxidative stress in skeletal muscle. Redox Rep. 2018;23:100–17.
Ou H-L, Schumacher B. DNA damage responses and p53 in the aging process. Blood. 2018;131:488–95.
Hao Q, Chen J, Lu H, Zhou X. The ARTS of p53-dependent mitochondrial apoptosis. J Mol Cell Biol. 2023;14:mjac074.
He J, Xue R, Li S, Lv J, Zhang Y, Fan L, et al. Identification of the potential molecular targets for human intervertebral disc degeneration based on bioinformatic methods. Int J Mol Med. 2015;36:1593–1600.
Zhang Z, Wang Q, Li Y, Li B, Zheng L, He C. Hub genes and key pathways of intervertebral disc degeneration: bioinformatics analysis and validation. Biomed Res Int. 2021;2021:5340449.
Lu X-Y, Ding X-H, Zhong L-J, Xia H, Chen X-D, Huang H. Expression and significance of VEGF and p53 in degenerate intervertebral disc tissue. Asian Pac J Trop Med. 2013;6:79–81.
Liu X-W, Kang J, Fan X-D, Sun L-F. Expression and significance of VEGF and p53 in rat degenerated intervertebral disc tissues. Asian Pac J Trop Med. 2013;6:404–6.
Kim K-W, Chung H-N, Ha K-Y, Lee J-S, Kim Y-Y. Senescence mechanisms of nucleus pulposus chondrocytes in human intervertebral discs. Spine J. 2009;9:658–66.
Zhang Z, Huang Y, Xu N, Wang J, Yao T, Xu Y, et al. PLK1 mitigates intervertebral disc degeneration by delaying senescence of nucleus pulposus cells. Front Cell Dev Biol. 2022;10:819262.
Yang H, Yang X, Rong K, Liang J, Wang Z, Zhao J, et al. Eupatilin attenuates the senescence of nucleus pulposus cells and mitigates intervertebral disc degeneration via inhibition of the MAPK/NF-κB signaling pathway. Front Pharmacol. 2022;13:940475.
Shi S, Kang X-J, Zhou Z, He Z-M, Zheng S, He S-S. Excessive mechanical stress-induced intervertebral disc degeneration is related to Piezo1 overexpression triggering the imbalance of autophagy/apoptosis in human nucleus pulpous. Arthritis Res Ther. 2022;24:119.
Dai X, Chen Y, Yu Z, Liao C, Liu Z, Chen J, et al. Advanced oxidation protein products induce annulus fibrosus cell senescence through a NOX4-dependent, MAPK-mediated pathway and accelerate intervertebral disc degeneration. PeerJ. 2022;10:e13826.
Zhang L, Li X, Kong X, Jin H, Han Y, Xie Y. Effects of the NF‑κB/p53 signaling pathway on intervertebral disc nucleus pulposus degeneration. Mol Med Rep. 2020;22:1821–30.
Xiong X, Dai L, Liang W, Zhang J, Qin S, Cao W, et al. Protective effect of p53 on the viability of intervertebral disc nucleus pulposus cells under low glucose condition. Biochem Biophys Res Commun. 2017;490:1414–9.
Heltberg MS, Lucchetti A, Hsieh F-S, Minh Nguyen DP, Chen S-H, Jensen MH. Enhanced DNA repair through droplet formation and p53 oscillations. Cell. 2022;185:4394–4408.
Son M, Frank T, Holst-Hansen T, Wang AG, Junkin M, Kashaf SS, et al. Spatiotemporal NF-κB dynamics encodes the position, amplitude, and duration of local immune inputs. Sci Adv. 2022;8:eabn6240.
Aqdas M, Sung M-H. NF-κB dynamics in the language of immune cells. Trends Immunol. 2023;44:32–43.
Hafner A, Bulyk ML, Jambhekar A, Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Biol. 2019;20:199–210.
Steffens Reinhardt L, Groen K, Newton C, Avery-Kiejda KA. The role of truncated p53 isoforms in the DNA damage response. Biochim Biophys Acta Rev Cancer. 2023;1878:188882.
Wang P, Wang H-Y, Gao X-J, Zhu H-X, Zhang X-P, Liu F, et al. Encoding and decoding of p53 dynamics in cellular response to stresses. Cells. 2023;12:490.
Purvis JE, Karhohs KW, Mock C, Batchelor E, Loewer A, Lahav G. p53 dynamics control cell fate. Science. 2012;336:1440–4.
Paek AL, Liu JC, Loewer A, Forrester WC, Lahav G. Cell-to-Cell variation in p53 dynamics leads to fractional killing. Cell. 2016;165:631–42.
Kracikova M, Akiri G, George A, Sachidanandam R, Aaronson SA. A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell Death Differ. 2013;20:576–88.
Tsabar M, Mock CS, Venkatachalam V, Reyes J, Karhohs KW, Oliver TG, et al. A switch in p53 dynamics marks cells that escape from DSB-induced cell cycle arrest. Cell Rep. 2020;32:107995.
Kim JK, Jackson TL. Mechanisms that enhance sustainability of p53 pulses. PLoS One. 2013;8:e65242.
Jangid A, Malik MZ, Ramaswamy R, Singh RKB. Transition and identification of pathological states in p53 dynamics for therapeutic intervention. Sci Rep. 2021;11:2349.
Batchelor E, Loewer A, Mock C, Lahav G. Stimulus-dependent dynamics of p53 in single cells. Mol Syst Biol. 2011;7:488.
Mirzayans R, Andrais B, Scott A, Wang YW, Murray D. Ionizing radiation-induced responses in human cells with differing TP53 status. Int J Mol Sci. 2013;14:22409–35.
Stewart-Ornstein J, Lahav G. p53 dynamics in response to DNA damage vary across cell lines and are shaped by efficiency of DNA repair and activity of the kinase ATM. Sci Signal. 2017;10:eaah6671.
Liu B, Bhatt D, Oltvai ZN, Greenberger JS, Bahar I. Significance of p53 dynamics in regulating apoptosis in response to ionizing radiation, and polypharmacological strategies. Sci Rep. 2014;4:6245.
Xie J, Zhang L, Liu B, Liang X, Shi J. Single-cell analysis of p53 transitional dynamics unravels stimulus- and cell type-dependent signaling output motifs. BMC Biol. 2022;20:85.
Stewart-Ornstein J, Iwamoto Y, Miller MA, Prytyskach MA, Ferretti S, Holzer P, et al. p53 dynamics vary between tissues and are linked with radiation sensitivity. Nat Commun. 2021;12:898.
Zhu H, Gao H, Ji Y, Zhou Q, Du Z, Tian L, et al. Targeting p53-MDM2 interaction by small-molecule inhibitors: learning from MDM2 inhibitors in clinical trials. J Hematol Oncol. 2022;15:91.
Koo N, Sharma AK, Narayan S. Therapeutics targeting p53-MDM2 interaction to induce cancer cell death. Int J Mol Sci. 2022;23:5005.
Espadinha M, Lopes EA, Marques V, Amaral JD, Dos Santos DJVA, Mori M, et al. Discovery of MDM2-p53 and MDM4-p53 protein-protein interactions small molecule dual inhibitors. Eur J Med Chem. 2022;241:114637.
Pezone A, Olivieri F, Napoli MV, Procopio A, Avvedimento EV, Gabrielli A. Inflammation and DNA damage: cause, effect or both. Nat Rev Rheumatol. 2023;19:200–11.
Overstreet JM, Gifford CC, Tang J, Higgins PJ, Samarakoon R. Emerging role of tumor suppressor p53 in acute and chronic kidney diseases. Cell Mol Life Sci. 2022;79:474.
Luo Q, Beaver JM, Liu Y, Zhang Z. Dynamics of p53: a master decider of cell fate. Genes. 2017;8:66.
Kim H, Lee JM, Lee G, Bhin J, Oh SK, Kim K, et al. DNA damage-induced RORα is crucial for p53 stabilization and increased apoptosis. Mol Cell. 2011;44:797–810.
Kim H-S, An CH, Teller D, Moon S-J, Hwang GW, Song JW. The role of retinoid-related orphan receptor-α in cigarette smoke-induced autophagic response. Respir Res. 2022;23:110.
Ryu H, Nam K-Y, Kim JS, Hwang S-G, Song J-Y, Ahn J. The small molecule AU14022 promotes colorectal cancer cell death via p53-mediated G2/M-phase arrest and mitochondria-mediated apoptosis. J Cell Physiol. 2018;233:4666–76.
Sahin E, DePinho RA. Axis of ageing: telomeres, p53 and mitochondria. Nat Rev Mol Cell Biol. 2012;13:397–404.
Zhou C, Yao S, Fu F, Bian Y, Zhang Z, Zhang H, et al. Morroniside attenuates nucleus pulposus cell senescence to alleviate intervertebral disc degeneration via inhibiting ROS-Hippo-p53 pathway. Front Pharmacol. 2022;13:942435.
Gruber HE, Watts JA, Hoelscher GL, Bethea SF, Ingram JA, Zinchenko NS, et al. Mitochondrial gene expression in the human annulus: in vivo data from annulus cells and selectively harvested senescent annulus cells. Spine J. 2011;11:782–91.
Zheng H-L, Xu W-N, Chen P-B, Jiang L-S, Zheng X-F, Jiang S-D. Increased expression of Prolyl endopeptidase induced by oxidative stress in nucleus pulposus cells aggravates intervertebral disc degeneration. Oxid Med Cell Longev. 2022;2022:9731800.
Feng Y, Wang H, Chen Z, Chen B. High glucose mediates the ChREBP/p300 transcriptional complex to activate proapoptotic genes Puma and BAX and contributes to intervertebral disc degeneration. Bone. 2021;153:116164.
Chen ZB, Yu YB, Wa QB, Zhou JW, He M, Cen Y. The role of quinazoline in ameliorating intervertebral disc degeneration by inhibiting oxidative stress and anti-inflammation via NF-κB/MAPKs signaling pathway. Eur Rev Med Pharm Sci. 2020;24:2077–86.
Johnson RF, Perkins ND. Nuclear factor-κB, p53, and mitochondria: regulation of cellular metabolism and the Warburg effect. Trends Biochem Sci. 2012;37:317–24.
Yamada K, Yoshida K. Mechanical insights into the regulation of programmed cell death by p53 via mitochondria. Biochim Biophys Acta Mol Cell Res. 2019;1866:839–48.
Kim YY, Um J-H, Yoon J-H, Lee D-Y, Lee YJ, Kim DH, et al. p53 regulates mitochondrial dynamics by inhibiting Drp1 translocation into mitochondria during cellular senescence. FASEB J. 2020;34:2451–64.
Lin H, Zhao L, Ma X, Wang B-C, Deng X-Y, Cui M, et al. Drp1 mediates compression-induced programmed necrosis of rat nucleus pulposus cells by promoting mitochondrial translocation of p53 and nuclear translocation of AIF. Biochem Biophys Res Commun. 2017;487:181–8.
Nagasaka M, Miyajima C, Aoki H, Aoyama M, Morishita D, Inoue Y, et al. Insights into Regulators of p53 Acetylation. Cells. 2022; 11.
Sankunny M, Eng C. KLLN-mediated DNA damage-induced apoptosis is associated with regulation of p53 phosphorylation and acetylation in breast cancer cells. Cell Death Discov. 2018;4:31.
Zhang Z, Lin J, Nisar M, Chen T, Xu T, Zheng G, et al. The Sirt1/P53 axis in diabetic intervertebral disc degeneration pathogenesis and therapeutics. Oxid Med Cell Longev. 2019;2019:7959573.
Vaddavalli PL, Schumacher B. The p53 network: cellular and systemic DNA damage responses in cancer and aging. Trends Genet. 2022;38:598–612.
Shi D, Jiang P. A different facet of p53 function: regulation of immunity and inflammation during tumor development. Front Cell Dev Biol. 2021;9:762651.
Kciuk M, Gielecińska A, Mujwar S, Mojzych M, Kontek R. Cyclin-dependent kinases in DNA damage response. Biochim Biophys Acta Rev Cancer. 2022;1877:188716.
Deng S, Vlatkovic T, Li M, Zhan T, Veldwijk MR, Herskind C. Targeting the DNA damage response and DNA repair pathways to enhance radiosensitivity in colorectal cancer. Cancers. 2022;14:4874.
Matsumoto Y. Development and evolution of DNA-dependent protein kinase inhibitors toward cancer therapy. Int J Mol Sci. 2022;23:4264.
Schumacher B, Pothof J, Vijg J, Hoeijmakers JHJ. The central role of DNA damage in the ageing process. Nature. 2021;592:695–703.
Huang R, Zhou P-K. DNA damage repair: historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct Target Ther. 2021;6:254.
Feng C, Yang M, Lan M, Liu C, Zhang Y, Huang B, et al. ROS: crucial Intermediators in the pathogenesis of intervertebral disc degeneration. Oxid Med Cell Longev. 2017;2017:5601593.
Cao G, Yang S, Cao J, Tan Z, Wu L, Dong F, et al. The role of oxidative stress in intervertebral disc degeneration. Oxid Med Cell Longev. 2022;2022:2166817.
Nasto LA, Wang D, Robinson AR, Clauson CL, Ngo K, Dong Q, et al. Genotoxic stress accelerates age-associated degenerative changes in intervertebral discs. Mech Ageing Dev. 2013;134:35–42.
Vo N, Seo H-Y, Robinson A, Sowa G, Bentley D, Taylor L, et al. Accelerated aging of intervertebral discs in a mouse model of progeria. J Orthop Res. 2010;28:1600–7.
Zhong J, Chen J, Oyekan AA, Epperly MW, Greenberger JS, Lee JY, et al. Ionizing radiation induces disc annulus fibrosus senescence and matrix catabolism via MMP-mediated pathways. Int J Mol Sci. 2022;23:4014.
Nasto LA, Ngo K, Leme AS, Robinson AR, Dong Q, Roughley P, et al. Investigating the role of DNA damage in tobacco smoking-induced spine degeneration. Spine J. 2014;14:416–23.
Suzuki S, Fujita N, Hosogane N, Watanabe K, Ishii K, Toyama Y, et al. Excessive reactive oxygen species are therapeutic targets for intervertebral disc degeneration. Arthritis Res Ther. 2015;17:316.
Guo Q, Zhu D, Wang Y, Miao Z, Chen Z, Lin Z, et al. Targeting STING attenuates ROS induced intervertebral disc degeneration. Osteoarthr Cartil. 2021;29:1213–24.
Feng C, Zhang Y, Yang M, Lan M, Liu H, Wang J, et al. The matrikine N-acetylated proline-glycine-proline induces premature senescence of nucleus pulposus cells via CXCR1-dependent ROS accumulation and DNA damage and reinforces the destructive effect of these cells on homeostasis of intervertebral discs. Biochim Biophys Acta Mol Basis Dis. 2017;1863:220–30.
Han Y, Zhou C-M, Shen H, Tan J, Dong Q, Zhang L, et al. Attenuation of ataxia telangiectasia mutated signalling mitigates age-associated intervertebral disc degeneration. Aging Cell. 2020;19:e13162.
Feng C, Yang M, Zhang Y, Lan M, Huang B, Liu H, et al. Cyclic mechanical tension reinforces DNA damage and activates the p53-p21-Rb pathway to induce premature senescence of nucleus pulposus cells. Int J Mol Med. 2018;41:3316–26.
Shi T, Dansen TB. Reactive oxygen species induced p53 activation: DNA damage, redox signaling, or both? Antioxid Redox Signal. 2020;33:839–59.
Butturini E, Butera G, Pacchiana R, Carcereri de Prati A, Mariotto S, Donadelli M. Redox sensitive cysteine residues as crucial regulators of wild-type and mutant p53 isoforms. Cells. 2021;10:3149.
Li Y, Ding H, Liu L, Song Y, Du X, Feng S, et al. Non-esterified fatty acid induce dairy cow hepatocytes apoptosis via the mitochondria-mediated ROS-JNK/ERK signaling pathway. Front Cell Dev Biol. 2020;8:245.
Guadagno J, Swan P, Shaikh R, Cregan SP. Microglia-derived IL-1β triggers p53-mediated cell cycle arrest and apoptosis in neural precursor cells. Cell Death Dis. 2015;6:e1779.
Shan H, Bian Y, Shu Z, Zhang L, Zhu J, Ding J, et al. Fluoxetine protects against IL-1β-induced neuronal apoptosis via downregulation of p53. Neuropharmacology. 2016;107:68–78.
Gao G, Chang F, Zhang T, Huang X, Yu C, Hu Z, et al. Naringin protects against interleukin 1β (IL-1β)-induced human nucleus pulposus cells degeneration via downregulation nuclear factor kappa B (NF-κB) pathway and p53 expression. Med Sci Monit : Int Med J Exp Clin Res. 2019;25:9963–72.
Hu J, Cao J, Topatana W, Juengpanich S, Li S, Zhang B, et al. Targeting mutant p53 for cancer therapy: direct and indirect strategies. J Hematol Oncol. 2021;14:157.
Duffy MJ, Synnott NC, O’Grady S, Crown J. Targeting p53 for the treatment of cancer. Semin Cancer Biol. 2022;79:58–67.
Wang J, Hu K, Cai X, Yang B, He Q, Wang J, et al. Targeting PI3K/AKT signaling for treatment of idiopathic pulmonary fibrosis. Acta Pharm Sin B. 2022;12:18–32.
He Y, Sun MM, Zhang GG, Yang J, Chen KS, Xu WW, et al. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct Target Ther. 2021;6:425.
Hua H, Zhang H, Chen J, Wang J, Liu J, Jiang Y. Targeting Akt in cancer for precision therapy. J Hematol Oncol. 2021;14:128.
Long H-Z, Cheng Y, Zhou Z-W, Luo H-Y, Wen D-D, Gao L-C. PI3K/AKT signal pathway: a target of natural products in the prevention and treatment of Alzheimer’s disease and Parkinson’s disease. Front Pharm. 2021;12:648636.
Wang Y, Cheng H, Wang T, Zhang K, Zhang Y, Kang X. Oxidative stress in intervertebral disc degeneration: molecular mechanisms, pathogenesis and treatment. Cell Prolif. 2023:e13448.
Tewari D, Patni P, Bishayee A, Sah AN, Bishayee A. Natural products targeting the PI3K-Akt-mTOR signaling pathway in cancer: A novel therapeutic strategy. Semin Cancer Biol. 2022;80:1–17.
Chen K, Li Y, Zhang X, Ullah R, Tong J, Shen Y. The role of the PI3K/AKT signalling pathway in the corneal epithelium: recent updates. Cell Death Dis. 2022;13:513.
Hoxhaj G, Manning BD. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer. 2020;20:74–88.
Zhu K, Wu Y, He P, Fan Y, Zhong X, Zheng H, et al. PI3K/AKT/mTOR-targeted therapy for breast cancer. Cells. 2022;11:2508.
Vasan N, Cantley LC. At a crossroads: how to translate the roles of PI3K in oncogenic and metabolic signalling into improvements in cancer therapy. Nat Rev Clin Oncol. 2022;19:471–85.
Yarmohammadi F, Ebrahimian Z, Karimi G. MicroRNAs target the PI3K/Akt/p53 and the Sirt1/Nrf2 signaling pathways in doxorubicin-induced cardiotoxicity. J Biochem Mol Toxicol. 2023;37:e23261.
Zhu Z, McGray AJR, Jiang W, Lu B, Kalinski P, Guo ZS. Improving cancer immunotherapy by rationally combining oncolytic virus with modulators targeting key signaling pathways. Mol Cancer. 2022;21:196.
Chibaya L, Karim B, Zhang H, Jones SN. Mdm2 phosphorylation by Akt regulates the p53 response to oxidative stress to promote cell proliferation and tumorigenesis. Proc Natl Acad Sci USA. 2021;118:e2003193118.
Gottlieb TM, Leal JFM, Seger R, Taya Y, Oren M. Cross-talk between Akt, p53 and Mdm2: possible implications for the regulation of apoptosis. Oncogene. 2002;21:1299–303.
Ma Y, Vassetzky Y, Dokudovskaya S. mTORC1 pathway in DNA damage response. Biochim Biophys Acta Mol Cell Res. 2018;1865:1293–311.
Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, et al. Regulation of PTEN transcription by p53. Mol Cell. 2001;8:317–25.
Zhao K, Zhou Y, Qiao C, Ni T, Li Z, Wang X, et al. Oroxylin A promotes PTEN-mediated negative regulation of MDM2 transcription via SIRT3-mediated deacetylation to stabilize p53 and inhibit glycolysis in wt-p53 cancer cells. J Hematol Oncol. 2015;8:41.
Chen H-W, Liu M-Q, Zhang G-Z, Zhang C-Y, Wang Z-H, Lin A-X, et al. Proanthocyanidins inhibit the apoptosis and aging of nucleus pulposus cells through the PI3K/Akt pathway delaying intervertebral disc degeneration. Connect Tissue Res. 2022;63:650–62.
Gong C, Pan W, Hu W, Chen L. Bone morphogenetic protein-7 retards cell subculture-induced senescence of human nucleus pulposus cells through activating the PI3K/Akt pathway. Biosci Rep. 2019;39:BSR20182312.
Yu H, Lin L, Zhang Z, Zhang H, Hu H. Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Target Ther. 2020;5:209.
Capece D, Verzella D, Flati I, Arboretto P, Cornice J, Franzoso G. NF-κB: blending metabolism, immunity, and inflammation. Trends Immunol. 2022;43:757–75.
Li Y, Xia Y, Yin S, Wan F, Hu J, Kou L, et al. Targeting microglial α-Synuclein/TLRs/NF-kappaB/NLRP3 inflammasome axis in Parkinson’s disease. Front Immunol. 2021;12:719807.
Liu Y, Wang J, Zhang X. An update on the multifaceted role of NF-kappaB in endometriosis. Int J Biol Sci. 2022;18:4400–13.
Kaltschmidt C, Greiner JFW, Kaltschmidt B. The transcription factor NF-κB in stem cells and development. Cells. 2021;10:2042.
Di Francesco B, Verzella D, Capece D, Vecchiotti D, Di Vito Nolfi M, Flati I, et al. NF-κB: a druggable target in acute myeloid Leukemia. Cancers. 2022;14:3557.
Kaltschmidt B, Helweg LP, Greiner JFW, Kaltschmidt C. NF-κB in neurodegenerative diseases: recent evidence from human genetics. Front Mol Neurosci. 2022;15:954541.
Antonia RJ, Hagan RS, Baldwin AS. Expanding the View of IKK: new substrates and new biology. Trends Cell Biol. 2021;31:166–78.
Jimi E, Katagiri T. Critical roles of NF-κB signaling molecules in bone metabolism revealed by genetic mutations in osteopetrosis. Int J Mol Sci. 2022;23:7995.
Miyazato A, Sheleg S, Iha H, Li Y, Jeang K-T. Evidence for NF-kappaB- and CBP-independent repression of p53’s transcriptional activity by human T-cell leukemia virus type 1 Tax in mouse embryo and primary human fibroblasts. J Virol. 2005;79:9346–50.
Webster GA, Perkins ND. Transcriptional cross talk between NF-kappaB and p53. Mol Cell Biol. 1999;19:3485–95.
Pise-Masison CA, Mahieux R, Jiang H, Ashcroft M, Radonovich M, Duvall J, et al. Inactivation of p53 by human T-cell lymphotropic virus type 1 Tax requires activation of the NF-kappaB pathway and is dependent on p53 phosphorylation. Mol Cell Biol. 2000;20:3377–86.
Culmsee C, Siewe J, Junker V, Retiounskaia M, Schwarz S, Camandola S, et al. Reciprocal inhibition of p53 and nuclear factor-kappaB transcriptional activities determines cell survival or death in neurons. J Neurosci. 2003;23:8586–95.
Ikeda A, Sun X, Li Y, Zhang Y, Eckner R, Doi TS, et al. p300/CBP-dependent and -independent transcriptional interference between NF-kappaB RelA and p53. Biochem Biophys Res Commun. 2000;272:375–9.
Gudkov AV, Gurova KV, Komarova EA. Inflammation and p53: a tale of two stresses. Genes Cancer. 2011;2:503–16.
Thomasova D, Mulay SR, Bruns H, Anders H-J. p53-independent roles of MDM2 in NF-κB signaling: implications for cancer therapy, wound healing, and autoimmune diseases. Neoplasia. 2012;14:1097–101.
Timmerman DM, Remmers TL, Hillenius S, Looijenga LHJ. Mechanisms of TP53 pathway inactivation in embryonic and somatic cells-relevance for understanding (Germ Cell) Tumorigenesis. Int J Mol Sci. 2021;22:5377.
Kung C-P, Weber JD. It’s getting complicated-A fresh look at p53-MDM2-ARF triangle in tumorigenesis and cancer therapy. Front Cell Dev Biol. 2022;10:818744.
Rocha S, Garrett MD, Campbell KJ, Schumm K, Perkins ND. Regulation of NF-kappaB and p53 through activation of ATR and Chk1 by the ARF tumour suppressor. EMBO J. 2005;24:1157–69.
Hyder U, McCann JL, Wang J, Fung V, Bayo J, D’Orso I. The ARF tumor suppressor targets PPM1G/PP2Cγ to counteract NF-κB transcription tuning cell survival and the inflammatory response. Proc Natl Acad Sci USA. 2020;117:32594–605.
Xia Y, Padre RC, De Mendoza TH, Bottero V, Tergaonkar VB, Verma IM. Phosphorylation of p53 by IkappaB kinase 2 promotes its degradation by beta-TrCP. Proc Natl Acad Sci USA. 2009;106:2629–34.
Xu X, Zhang C, Xu H, Wu L, Hu M, Song L. Autophagic feedback-mediated degradation of IKKα requires CHK1- and p300/CBP-dependent acetylation of p53. J Cell Sci. 2020;133:jcs246868.
Wang H, Zhang M, Xu X, Hou S, Liu Z, Chen X, et al. IKKα mediates UVB-induced cell apoptosis by regulating p53 pathway activation. Ecotoxicol Environ Saf. 2021;227:112892.
Ryan KM, Ernst MK, Rice NR, Vousden KH. Role of NF-kappaB in p53-mediated programmed cell death. Nature. 2000;404:892–7.
Lowe JM, Menendez D, Bushel PR, Shatz M, Kirk EL, Troester MA, et al. p53 and NF-κB coregulate proinflammatory gene responses in human macrophages. Cancer Res. 2014;74:2182–92.
Li J, Li J, Cao C, Sun J, Wang S, Ruan Z, et al. Melatonin inhibits annulus fibrosus cell senescence through regulating the ROS/NF-κB pathway in an inflammatory environment. Biomed Res Int. 2021;2021:3456321.
Li P, Gan Y, Xu Y, Wang L, Ouyang B, Zhang C, et al. 17beta-estradiol Attenuates TNF-α-induced premature senescence of nucleus pulposus cells through regulating the ROS/NF-κB pathway. Int J Biol Sci. 2017;13:145–56.
Yuan J, Dong X, Yap J, Hu J. The MAPK and AMPK signalings: interplay and implication in targeted cancer therapy. J Hematol Oncol. 2020;13:113.
Kciuk M, Gielecińska A, Budzinska A, Mojzych M, Kontek R. Metastasis and MAPK pathways. Int J Mol Sci. 2022;23:3847.
Falcicchia C, Tozzi F, Arancio O, Watterson DM. Origlia N involvement of p38 MAPK in synaptic function and dysfunction. Int J Mol Sci. 2020;21:5624.
Cheng Y, Chen J, Shi Y, Fang X, Tang Z. MAPK signaling pathway in oral squamous cell carcinoma: biological function and targeted therapy. Cancers. 2022;14:4625.
Chen J, Wang L, Yuan M. Update on the roles of rice MAPK Cascades. Int J Mol Sci. 2021;22:1679.
Hepworth EMW, Hinton SD Pseudophosphatases as Regulators of MAPK Signaling. Int J Mol Sci. 2021;22:12595.
Yue J, López JM. Understanding MAPK signaling pathways in apoptosis. Int J Mol Sci. 2020;21:2346.
Lee S, Rauch J, Kolch W. Targeting MAPK signaling in cancer: mechanisms of drug resistance and sensitivity. Int J Mol Sci. 2020;21:1102.
Tan H, Chen J, Li Y, Li Y, Zhong Y, Li G, et al. Glabridin, a bioactive component of licorice, ameliorates diabetic nephropathy by regulating ferroptosis and the VEGF/Akt/ERK pathways. Mol Med. 2022;28:58.
Mubarak SA, Otaibi AA, Qarni AA, Bakillah A, Iqbal J. Reduction in insulin mediated ERK phosphorylation by palmitate in liver cells is independent of fatty acid induced ER stress. nutrients. 2022;14:3641.
Yuan D, Huang S, Berger E, Liu L, Gross N, Heinzmann F, et al. Kupffer cell-derived Tnf Triggers Cholangiocellular Tumorigenesis through JNK due to chronic mitochondrial dysfunction and ROS. Cancer Cell. 2017;31:771–789.
Yao Y, Cui L, Ye J, Yang G, Lu G, Fang X, et al. Dioscin facilitates ROS-induced apoptosis via the p38-MAPK/HSP27-mediated pathways in lung squamous cell carcinoma. Int J Biol Sci. 2020;16:2883–94.
Stramucci L, Pranteda A, Bossi G. Insights of crosstalk between p53 protein and the MKK3/MKK6/p38 MAPK signaling pathway in cancer. Cancers. 2018;10:131.
Wu GS. The functional interactions between the p53 and MAPK signaling pathways. Cancer Biol Ther. 2004;3:156–61.
Milne DM, Campbell DG, Caudwell FB, Meek DW. Phosphorylation of the tumor suppressor protein p53 by mitogen-activated protein kinases. J Biol Chem. 1994;269:9253–60.
Persons DL, Yazlovitskaya EM, Pelling JC. Effect of extracellular signal-regulated kinase on p53 accumulation in response to cisplatin. J Biol Chem. 2000;275:35778–85.
Hsu Y-L, Cho C-Y, Kuo P-L, Huang Y-T, Lin C-C. Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) induces apoptosis and cell cycle arrest in A549 cells through p53 accumulation via c-Jun NH2-terminal kinase-mediated phosphorylation at serine 15 in vitro and in vivo. J Pharm Exp Ther. 2006;318:484–94.
She Q-B, Ma W-Y, Dong Z. Role of MAP kinases in UVB-induced phosphorylation of p53 at serine 20. Oncogene. 2002;21:1580–9.
Buschmann T, Potapova O, Bar-Shira A, Ivanov VN, Fuchs SY, Henderson S, et al. Jun NH2-terminal kinase phosphorylation of p53 on Thr-81 is important for p53 stabilization and transcriptional activities in response to stress. Mol Cell Biol. 2001;21:2743–54.
Topisirovic I, Gutierrez GJ, Chen M, Appella E, Borden KLB, Ronai ZA. Control of p53 multimerization by Ubc13 is JNK-regulated. Proc Natl Acad Sci USA. 2009;106:12676–81.
She QB, Chen N, Dong Z. ERKs and p38 kinase phosphorylate p53 protein at serine 15 in response to UV radiation. J Biol Chem. 2000;275:20444–9.
Zhu Y, Mao XO, Sun Y, Xia Z, Greenberg DA. p38 Mitogen-activated protein kinase mediates hypoxic regulation of Mdm2 and p53 in neurons. J Biol Chem. 2002;277:22909–14.
Kwon Y-W, Ueda S, Ueno M, Yodoi J, Masutani H. Mechanism of p53-dependent apoptosis induced by 3-methylcholanthrene: involvement of p53 phosphorylation and p38 MAPK. J Biol Chem. 2002;277:1837–44.
Chouinard N, Valerie K, Rouabhia M, Huot J. UVB-mediated activation of p38 mitogen-activated protein kinase enhances resistance of normal human keratinocytes to apoptosis by stabilizing cytoplasmic p53. Biochem J. 2002;365:133–45.
Huang C, Ma WY, Maxiner A, Sun Y, Dong Z. p38 kinase mediates UV-induced phosphorylation of p53 protein at serine 389. J Biol Chem. 1999;274:12229–35.
ElKeeb AM, Collier MEW, Maraveyas A, Ettelaie C. Accumulation of tissue factor in endothelial cells induces cell apoptosis, mediated through p38 and p53 activation. Thromb Haemost. 2015;114:364–78.
Bulavin DV, Saito S, Hollander MC, Sakaguchi K, Anderson CW, Appella E, et al. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J. 1999;18:6845–54.
Song J-y, Han H-S, Sabapathy K, Lee B-M, Yu E, Choi J. Expression of a homeostatic regulator, Wip1 (wild-type p53-induced phosphatase), is temporally induced by c-Jun and p53 in response to UV irradiation. J Biol Chem. 2010;285:9067–76.
Deng K, Liu L, Tan X, Zhang Z, Li J, Ou Y, et al. WIP1 promotes cancer stem cell properties by inhibiting p38 MAPK in NSCLC. Signal Transduct Target Ther. 2020;5:36.
Takekawa M, Adachi M, Nakahata A, Nakayama I, Itoh F, Tsukuda H, et al. p53-inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J. 2000;19:6517–26.
Li D, Yang L, Wang W, Song C, Xiong R, Pan S, et al. Eriocitrin attenuates sepsis-induced acute lung injury in mice by regulating MKP1/MAPK pathway mediated-glycolysis. Int Immunopharmacol. 2023;118:110021.
Camps M, Nichols A, Arkinstall S. Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB J. 2000;14:6–16.
Ge J, Cheng X, Yuan C, Qian J, Wu C, Cao C, et al. Syndecan-4 is a novel therapeutic target for intervertebral disc degeneration via suppressing JNK/p53 pathway. Int J Biol Sci. 2020;16:766–76.
Zhang X-Y, Li W, Zhang J-R, Li C-Y, Zhang J, Lv X-J. Roles of sirtuin family members in chronic obstructive pulmonary disease. Respir Res. 2022;23:66.
Bian C, Ren H. Sirtuin family and diabetic kidney disease. Front Endocrinol. 2022;13:901066.
Fiorentino F, Mautone N, Menna M, D’Acunzo F, Mai A, Rotili D. Sirtuin modulators: past, present, and future perspectives. Future Med Chem. 2022;14:915–39.
Lee S-H, Lee J-H, Lee H-Y, Min K-J. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019;52:24–34.
Wu Q-J, Zhang T-N, Chen H-H, Yu X-F, Lv J-L, Liu Y-Y, et al. The sirtuin family in health and disease. Signal Transduct Target Ther. 2022;7:402.
Roh E, Kim M-S. Hypothalamic NAD+-Sirtuin axis: function and regulation. Biomolecules. 2020;10:396.
Kida Y, Goligorsky MS. Sirtuins, Cell Senescence, and Vascular Aging. Can J Cardiol. 2016;32:634–41.
Beegum F, Anuranjana PV, George KT, Divya KP, Begum F, Krishnadas N. et al. Sirtuins as therapeutic targets for improving delayed wound healing in diabetes. J Drug Target. 2022;30:911–26.
Wang L, Xu C, Johansen T, Berger SL, Dou Z. SIRT1 - a new mammalian substrate of nuclear autophagy. Autophagy. 2021;17:593–5.
Yang Y, Liu Y, Wang Y, Chao Y, Zhang J, Jia Y, et al. Regulation of SIRT1 and Its Roles in Inflammation. Front Immunol. 2022;13:831168.
Chen C, Zhou M, Ge Y, Wang X. SIRT1 and aging related signaling pathways. Mech Ageing Dev. 2020;187:111215.
Sarma P, Bag I, Ramaiah MJ, Kamal A, Bhadra U, Pal Bhadra M. Bisindole-PBD regulates breast cancer cell proliferation via SIRT-p53 axis. Cancer Biol Ther. 2015;16:1486–501.
Busch F, Mobasheri A, Shayan P, Stahlmann R, Shakibaei M. Sirt-1 is required for the inhibition of apoptosis and inflammatory responses in human tenocytes. J Biol Chem. 2012;287:25770–81.
Liu L, Xia G, Li P, Wang Y, Zhao Q. Sirt-1 regulates physiological process and exerts protective effects against oxidative stress. Biomed Res Int. 2021;2021:5542545.
Lee H, Jung T-Y, Lim SH, Choi EJ, Lee J, Min DS. Phospholipase D2 is a positive regulator of sirtuin 1 and modulates p53-mediated apoptosis via sirtuin 1. Exp Mol Med. 2021;53:1287–97.
Suzuki M, Ikeda A, Bartlett JD. Sirt1 overexpression suppresses fluoride-induced p53 acetylation to alleviate fluoride toxicity in ameloblasts responsible for enamel formation. Arch Toxicol. 2018;92:1283–93.
Kamel C, Abrol M, Jardine K, He X, McBurney MW. SirT1 fails to affect p53-mediated biological functions. Aging Cell. 2006;5:81–88.
Sun Z, Wang X, Xu Z. SIRT1 provides new pharmacological targets for polydatin through its role as a metabolic sensor. Biomed Pharmacother. 2021;139:111549.
Zhao J, Wozniak A, Adams A, Cox J, Vittal A, Voss J, et al. SIRT7 regulates hepatocellular carcinoma response to therapy by altering the p53-dependent cell death pathway. J Exp Clin Cancer Res. 2019;38:252.
Wood M, Rymarchyk S, Zheng S, Cen Y. Trichostatin A inhibits deacetylation of histone H3 and p53 by SIRT6. Arch Biochem Biophys. 2018;638:8–17.
Zeng Q-Z, Yang F, Li C-G, Xu L-H, He X-H, Mai F-Y, et al. Paclitaxel enhances the innate immunity by promoting NLRP3 inflammasome activation in macrophages. Front Immunol. 2019;10:72.
Zhang Y, Long X, Ruan X, Wei Q, Zhang L, Wo L, et al. SIRT2-mediated deacetylation and deubiquitination of C/EBPβ prevents ethanol-induced liver injury. Cell Discov. 2021;7:93.
Ren H, Hu F, Wang D, Kang X, Feng X, Zhang L, et al. Sirtuin 2 prevents liver steatosis and metabolic disorders by deacetylation of hepatocyte nuclear factor 4α. Hepatology. 2021;74:723–40.
Agborbesong E, Zhou JX, Li LX, Harris PC, Calvet JP, Li X. Prdx5 regulates DNA damage response through autophagy-dependent Sirt2-p53 axis. Hum Mol Genet. 2023;32:567–79.
Xu Y, Nasri M, Dannenmann B, Mir P, Zahabi A, Welte K, et al. NAMPT/SIRT2-mediated inhibition of the p53-p21 signaling pathway is indispensable for maintenance and hematopoietic differentiation of human iPS cells. Stem Cell Res Ther. 2021;12:112.
Katare PB, Nizami HL, Paramesha B, Dinda AK, Banerjee SK. Activation of toll like receptor 4 (TLR4) promotes cardiomyocyte apoptosis through SIRT2 dependent p53 deacetylation. Sci Rep. 2020;10:19232.
Anwar T, Khosla S, Ramakrishna G. Increased expression of SIRT2 is a novel marker of cellular senescence and is dependent on wild type p53 status. Cell Cycle. 2016;15:1883–97.
Lee J, Kim Y, Liu T, Hwang YJ, Hyeon SJ, Im H, et al. SIRT3 deregulation is linked to mitochondrial dysfunction in Alzheimer’s disease. Aging Cell. 2018;17:e12679.
Liu X, Rong F, Tang J, Zhu C, Chen X, Jia S, et al. Repression of p53 function by SIRT5-mediated desuccinylation at Lysine 120 in response to DNA damage. Cell Death Differ. 2022;29:722–36.
Li J, Zhan H, Ren Y, Feng M, Wang Q, Jiao Q, et al. Sirtuin 4 activates autophagy and inhibits tumorigenesis by upregulating the p53 signaling pathway. Cell Death Differ. 2023;30:313–26.
Yao C, Guo G, Huang R, Tang C, Zhu Q, Cheng Y, et al. Manual therapy regulates oxidative stress in aging rat lumbar intervertebral discs through the SIRT1/FOXO1 pathway. Aging. 2022;14:2400–17.
Zhang Z, Kakutani K, Maeno K, Takada T, Yurube T, Doita M, et al. Expression of silent mating type information regulator 2 homolog 1 and its role in human intervertebral disc cell homeostasis. Arthritis Res Ther. 2011;13:R200.
Habib M, Hussien S, Jeon O, Lotz JC, Wu PIK, Alsberg E, et al. Intradiscal treatment of the cartilage endplate for improving solute transport and disc nutrition. Front Bioeng Biotechnol. 2023;11:1111356.
Zhou N, Lin X, Dong W, Huang W, Jiang W, Lin L, et al. SIRT1 alleviates senescence of degenerative human intervertebral disc cartilage endo-plate cells via the p53/p21 pathway. Sci Rep. 2016;6:22628.
Yang M, Peng Y, Liu W, Zhou M, Meng Q, Yuan C. Sirtuin 2 expression suppresses oxidative stress and senescence of nucleus pulposus cells through inhibition of the p53/p21 pathway. Biochem Biophys Res Commun. 2019;513:616–22.
Green DR. The mitochondrial pathway of apoptosis Part II: the BCL-2 protein family. Cold Spring Harb Perspect Biol. 2022;14:a041046.
Warren CFA, Wong-Brown MW, Bowden NA. BCL-2 family isoforms in apoptosis and cancer. Cell Death Dis. 2019;10:177.
Siddiqui WA, Ahad A, Ahsan H. The mystery of BCL2 family: Bcl-2 proteins and apoptosis: an update. Arch Toxicol. 2015;89:289–317.
Banjara S, Suraweera CD, Hinds MG, Kvansakul M. The Bcl-2 family: ancient origins, conserved structures, and divergent mechanisms. Biomolecules. 2020;10:128.
Mellert HS, Stanek TJ, Sykes SM, Rauscher FJ, Schultz DC, McMahon SB. Deacetylation of the DNA-binding domain regulates p53-mediated apoptosis. J Biol Chem. 2011;286:4264–70.
Matissek KJ, Mossalam M, Okal A, Lim CS. The DNA binding domain of p53 is sufficient to trigger a potent apoptotic response at the mitochondria. Mol Pharm. 2013;10:3592–602.
Pietsch EC, Perchiniak E, Canutescu AA, Wang G, Dunbrack RL, Murphy ME. Oligomerization of BAK by p53 utilizes conserved residues of the p53 DNA binding domain. J Biol Chem. 2008;283:21294–304.
Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458:1127–30.
Lee D-H, Ha J-H, Kim Y, Jang M, Park SJ, Yoon HS, et al. A conserved mechanism for binding of p53 DNA-binding domain and anti-apoptotic Bcl-2 family proteins. Mol Cells. 2014;37:264–9.
Yao H, Mi S, Gong W, Lin J, Xu N, Perrett S, et al. Anti-apoptosis proteins Mcl-1 and Bcl-xL have different p53-binding profiles. Biochemistry. 2013;52:6324–34.
Tomita Y, Marchenko N, Erster S, Nemajerova A, Dehner A, Klein C, et al. WT p53, but not tumor-derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization. J Biol Chem. 2006;281:8600–6.
Hagn F, Klein C, Demmer O, Marchenko N, Vaseva A, Moll UM, et al. BclxL changes conformation upon binding to wild-type but not mutant p53 DNA binding domain. J Biol Chem. 2010;285:3439–50.
Petros AM, Gunasekera A, Xu N, Olejniczak ET, Fesik SW. Defining the p53 DNA-binding domain/Bcl-x(L)-binding interface using NMR. FEBS Lett. 2004;559:171–4.
Wang J, Thomas HR, Li Z, Yeo NCF, Scott HE, Dang N, et al. Puma, noxa, p53, and p63 differentially mediate stress pathway induced apoptosis. Cell Death Dis. 2021;12:659.
Han CW, Lee HN, Jeong MS, Park SY, Jang SB. Structural basis of the p53 DNA binding domain and PUMA complex. Biochem Biophys Res Commun. 2021;548:39–46.
Zhao R, Liu W, Wang M, Zhang Y, Pan L, Feng F, et al. Lysyl oxidase inhibits TNF-α induced rat nucleus pulposus cell apoptosis via regulating Fas/FasL pathway and the p53 pathways. Life Sci. 2020;260:118483.
Xie S, Zhao C, Chen W, Li G, Xiong Z, Tang X, et al. Recombinant human bone morphogenetic protein 2 and 7 inhibit the degeneration of intervertebral discs by blocking the Puma-dependent apoptotic signaling. Int J Biol Sci. 2021;17:2367–79.
Yurube T, Hirata H, Kakutani K, Maeno K, Takada T, Zhang Z, et al. Notochordal cell disappearance and modes of apoptotic cell death in a rat tail static compression-induced disc degeneration model. Arthritis Res Ther. 2014;16:R31.
Yin H, Wang K, Das A, Li G, Song Y, Luo R, et al. The REDD1/TXNIP complex accelerates oxidative stress-induced apoptosis of nucleus Pulposus cells through the mitochondrial pathway. Oxid Med Cell Longev. 2021;2021:7397516.
Di Micco R, Krizhanovsky V, Baker D, d’Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. 2021;22:75–95.
Wiley CD, Campisi J. The metabolic roots of senescence: mechanisms and opportunities for intervention. Nat Metab. 2021;3:1290–301.
Ebert T, Tran N, Schurgers L, Stenvinkel P, Shiels PG. Ageing - Oxidative stress, PTMs and disease. Mol Asp Med. 2022;86:101099.
Guerrero A, De Strooper B, Arancibia-Cárcamo IL. Cellular senescence at the crossroads of inflammation and Alzheimer’s disease. Trends Neurosci. 2021;44:714–27.
Shimizu I, Yoshida Y, Suda M, Minamino T. DNA damage response and metabolic disease. Cell Metab. 2014;20:967–77.
Birch J, Gil J. Senescence and the SASP: many therapeutic avenues. Genes Dev. 2020;34:1565–76.
Coryell PR, Diekman BO, Loeser RF. Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat Rev Rheumatol. 2021;17:47–57.
Ohtani N. The roles and mechanisms of senescence-associated secretory phenotype (SASP): can it be controlled by senolysis? Inflamm Regen. 2022;42:11.
Moiseeva V, Cisneros A, Sica V, Deryagin O, Lai Y, Jung S, et al. Senescence atlas reveals an aged-like inflamed niche that blunts muscle regeneration. Nature. 2023;613:169–78.
Beck J, Turnquist C, Horikawa I, Harris C. Targeting cellular senescence in cancer and aging: roles of p53 and its isoforms. Carcinogenesis. 2020;41:1017–29.
Wei W, Ji S. Cellular senescence: Molecular mechanisms and pathogenicity. J Cell Physiol. 2018;233:9121–35.
Shi T, van Soest DMK, Polderman PE, Burgering BMT, Dansen TB. DNA damage and oxidant stress activate p53 through differential upstream signaling pathways. Free Radic Biol Med. 2021;172:298–311.
Bourgeois B, Madl T. Regulation of cellular senescence via the FOXO4-p53 axis. FEBS Lett. 2018;592:2083–97.
Pawge G, Khatik GL. p53 regulated senescence mechanism and role of its modulators in age-related disorders. Biochem Pharm. 2021;190:114651.
Mijit M, Caracciolo V, Melillo A, Amicarelli F, Giordano A. Role of p53 in the regulation of cellular senescence. Biomolecules. 2020;10:420.
Li Y, Cao L, Li J, Sun Z, Liu C, Liang H, et al. Influence of microgravity-induced intervertebral disc degeneration of rats on expression levels of p53/p16 and proinflammatory factors. Exp Ther Med. 2019;17:1367–73.
Zhao L, Tian B, Xu Q, Zhang C, Zhang L, Fang H. Extensive mechanical tension promotes annulus fibrosus cell senescence through suppressing cellular autophagy. Biosci Rep. 2019;39:BSR20190163.
Feng C, Liu H, Yang M, Zhang Y, Huang B, Zhou Y. Disc cell senescence in intervertebral disc degeneration: causes and molecular pathways. Cell Cycle. 2016;15:1674–84.
Iliaki S, Beyaert R, Afonina IS. Polo-like kinase 1 (PLK1) signaling in cancer and beyond. Biochem Pharm. 2021;193:114747.
Feng C, Zhang Y, Yang M, Huang B, Zhou Y. Collagen-Derived N-Acetylated Proline-Glycine-Proline in Intervertebral Discs Modulates CXCR1/2 expression and activation in cartilage endplate stem cells to induce migration and differentiation toward a pro-inflammatory phenotype. Stem Cells. 2015;33:3558–68.
Lahalle A, Lacroix M, De Blasio C, Cissé MY, Linares LK, Le Cam L. The p53 pathway and metabolism: the tree that hides the forest. Cancers. 2021;13:133.
Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009;9:691–700.
Syed I, Rathod J, Parmar M, Corcoran GB, Ray SD. Matrix metalloproteinase-9, -10, and -12, MDM2 and p53 expression in mouse liver during dimethylnitrosamine-induced oxidative stress and genomic injury. Mol Cell Biochem. 2012;365:351–61.
Alexandrova A, Ivanov A, Chumakov P, Kopnin B, Vasiliev J. Changes in p53 expression in mouse fibroblasts can modify motility and extracellular matrix organization. Oncogene. 2000;19:5826–30.
Xu M, Feng M, Peng H, Qian Z, Zhao L, Wu S. Epigenetic regulation of chondrocyte hypertrophy and apoptosis through Sirt1/P53/P21 pathway in surgery-induced osteoarthritis. Biochem Biophys Res Commun. 2020;528:179–85.
Nasto LA, Robinson AR, Ngo K, Clauson CL, Dong Q, St Croix C, et al. Mitochondrial-derived reactive oxygen species (ROS) play a causal role in aging-related intervertebral disc degeneration. J Orthop Res. 2013;31:1150–7.
Feng C, Zhang Y, Yang M, Lan M, Liu H, Huang B, et al. Oxygen-sensing Nox4 generates genotoxic ROS to induce premature senescence of nucleus pulposus cells through MAPK and NF-κB Pathways. Oxid Med Cell Longev. 2017;2017:7426458.
Dimozi A, Mavrogonatou E, Sklirou A, Kletsas D. Oxidative stress inhibits the proliferation, induces premature senescence and promotes a catabolic phenotype in human nucleus pulposus intervertebral disc cells. Eur Cell Mater. 2015;30:89–102.
Sheng K, Li Y, Wang Z, Hang K, Ye Z. p-Coumaric acid suppresses reactive oxygen species-induced senescence in nucleus pulposus cells. Exp Ther Med. 2022;23:183.
Eriksson SE, Ceder S, Bykov VJN, Wiman KG. p53 as a hub in cellular redox regulation and therapeutic target in cancer. J Mol Cell Biol. 2019;11:330–41.
Kang R, Kroemer G, Tang D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic Biol Med. 2019;133:162–8.
Liu X, Fan L, Lu C, Yin S, Hu H. Functional role of p53 in the regulation of chemical-induced oxidative stress. Oxid Med Cell Longev. 2020;2020:6039769.
Liu D, Xu Y. p53, oxidative stress, and aging. Antioxid Redox Signal. 2011;15:1669–78.
Rhee SG, Bae SH. The antioxidant function of sestrins is mediated by promotion of autophagic degradation of Keap1 and Nrf2 activation and by inhibition of mTORC1. Free Radic Biol Med. 2015;88:205–11.
Chen W, Sun Z, Wang X-J, Jiang T, Huang Z, Fang D, et al. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol Cell. 2009;34:663–73.
Ji Cho M, Yoon S-J, Kim W, Park J, Lee J, Park J-G, et al. Oxidative stress-mediated TXNIP loss causes RPE dysfunction. Exp Mol Med. 2019;51:1–13.
Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285:1733–7.
Lorenzo E, Ruiz-Ruiz C, Quesada AJ, Hernández G, Rodríguez A, López-Rivas A, et al. Doxorubicin induces apoptosis and CD95 gene expression in human primary endothelial cells through a p53-dependent mechanism. J Biol Chem. 2002;277:10883–92.
Hafsi H, Hainaut P. Redox control and interplay between p53 isoforms: roles in the regulation of basal p53 levels, cell fate, and senescence. Antioxid Redox Signal. 2011;15:1655–67.
Fujita K. p53 isoforms in cellular senescence- and ageing-associated biological and physiological functions. Int J Mol Sci. 2019;20:6023.
von Muhlinen N, Horikawa I, Alam F, Isogaya K, Lissa D, Vojtesek B, et al. p53 isoforms regulate premature aging in human cells. Oncogene. 2018;37:2379–93.
Nan L-P, Wang F, Ran D, Zhou S-F, Liu Y, Zhang Z, et al. Naringin alleviates H2O2-induced apoptosis via the PI3K/Akt pathway in rat nucleus pulposus-derived mesenchymal stem cells. Connect Tissue Res. 2020;61:554–67.
Bai D, Sun T, Lu F, Shen Y, Zhang Y, Zhang B, et al. Eupatilin suppresses OVA-induced asthma by inhibiting NF-κB and MAPK and activating Nrf2 signaling pathways in mice. Int J Mol Sci. 2022;23:1582.
Chiang SCC, Owsley E, Panchal N, Chaturvedi V, Terrell CE, Jordan MB, et al. Quercetin ameliorates XIAP deficiency-associated hyperinflammation. Blood. 2022;140:706–15.
Chen G, Xu H, Wu Y, Han X, Xie L, Zhang G, et al. Myricetin suppresses the proliferation and migration of vascular smooth muscle cells and inhibits neointimal hyperplasia via suppressing TGFBR1 signaling pathways. Phytomedicine. 2021;92:153719.
Zhao W-J, Liu X, Hu M, Zhang Y, Shi P-Z, Wang J-W, et al. Quercetin ameliorates oxidative stress-induced senescence in rat nucleus pulposus-derived mesenchymal stem cells via the miR-34a-5p/SIRT1 axis. World J Stem Cells. 2023;15:842–65.
Xie T, Pan R, Huang W, Dong S, Wu S, Ye Y. Myricetin alleviates H2O2-induced senescence and apoptosis in rat nucleus pulposus-derived mesenchymal stem cells. Folia Histochem Cytobiol. 2023;61:98–108.
Yi X, Tao J, Qian Y, Feng F, Hu X, Xu T, et al. Morroniside ameliorates inflammatory skeletal muscle atrophy via inhibiting canonical and non-canonical NF-κB and regulating protein synthesis/degradation. Front Pharmacol. 2022;13:1056460.
Li X, Lin F, Wu Y, Liu N, Wang J, Chen R, et al. Resveratrol attenuates inflammation environment-induced nucleus pulposus cell senescence in vitro. Bioscience Reports. 2019;39:BSR20190126.
Xiang Q, Zhao Y, Lin J, Jiang S, Li W. The Nrf2 antioxidant defense system in intervertebral disc degeneration: Molecular insights. Exp Mol Med. 2022;54:1067–75.
Li L, He J, Zhang G, Chen H, Luo Z, Deng B, et al. Role of caspase family in intervertebral disc degeneration and its therapeutic prospects. Biomolecules. 2022;12:1074.
Liu W, Ma Z, Wang Y, Yang J. Multiple nano-drug delivery systems for intervertebral disc degeneration: current status and future perspectives. Bioact Mater. 2023;23:274–99.
Gong Z, Wang Y, Li L, Li X, Qiu B, Hu Y. Cardamonin alleviates chondrocytes inflammation and cartilage degradation of osteoarthritis by inhibiting ferroptosis via p53 pathway. Food Chem Toxicol. 2023;174:113644.
Spallarossa P, Altieri P, Aloi C, Garibaldi S, Barisione C, Ghigliotti G, et al. Doxorubicin induces senescence or apoptosis in rat neonatal cardiomyocytes by regulating the expression levels of the telomere binding factors 1 and 2. Am J Physiol Heart Circ Physiol. 2009;297:H2169–H2181.
Wang Y, Kang J, Guo X, Zhu D, Liu M, Yang L, et al. Intervertebral disc degeneration models for pathophysiology and regenerative therapy -benefits and limitations. J Invest Surg. 2022;35:935–52.
Acknowledgements
This work was supported by the Key Research and Development Program of Shaanxi Province (Program No. 2021SF-237), the Key Research and Development Program of Shaanxi Province (Program No. 2021SF-250) and the Cultivation Project of Xi’an Health and Construction Commission (Program No.2021ms10).
Author information
Authors and Affiliations
Contributions
YW, WZ and SH contributed equally to this work and is listed as a co-first author. All the authors contributed to the manuscript and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, Y., Hu, S., Zhang, W. et al. Emerging role and therapeutic implications of p53 in intervertebral disc degeneration. Cell Death Discov. 9, 433 (2023). https://doi.org/10.1038/s41420-023-01730-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-023-01730-5
