
Abstract
Lysophosphatidic acid (LPA) is an active phospholipid signaling molecule that binds to six specific G protein-coupled receptors (LPA1-6) on the cell surface and exerts a variety of biological functions, including cell migration and proliferation, morphological changes, and anti-apoptosis. The earliest study from our group demonstrated that LPA treatment could restore cochlear F-actin depolymerization induced by noise exposure, reduce hair cell death, and thus protect hearing. However, whether LPA could protect against cisplatin-induced ototoxicity and which receptors play the major role remain unclear. To this end, we integrated the HEI-OC1 mouse cochlear hair cell line and zebrafish model, and found that cisplatin exposure induced a large amount of reactive oxygen species accumulation in HEI-OC1 cells, accompanied by mitochondrial damage, leading to apoptosis and autophagy. LPA treatment significantly attenuated autophagy and apoptosis in HEI-OC1 cells after cisplatin exposure. Further investigation revealed that all LPA receptors except LPA3 were expressed in HEI-OC1 cells, and the mRNA expression level of LPA1 receptor was significantly higher than that of other receptors. When LPA1 receptor was silenced, the protective effect of LPA was reduced and the proportion of apoptosis cells was increased, indicating that LPA-LPA1 plays an important role in protecting HEI-OC1 cells from cisplatin-induced apoptosis. In addition, the behavioral trajectory and in vivo fluorescence imaging results showed that cisplatin exposure caused zebrafish to move more actively, and the movement speed and distance were higher than those of the control and LPA groups, while LPA treatment reduced the movement behavior. Cisplatin caused hair cell death and loss in zebrafish lateral line, and LPA treatment significantly protected against hair cell death and loss. LPA has a protective effect on hair cells in vitro and in vivo against the cytotoxicity of cisplatin, and its mechanism may be related to reducing apoptosis, excessive autophagy and ROS accumulation.

Similar content being viewed by others

Introduction
Hearing loss, a globally prevalent sensory disability, mainly occurs because of aging, medication use, or excessive noise exposure [1,2,3,4]. The platinum-based agent cisplatin is commonly recommended for treating solid tumors in humans, for example, melanoma, head and neck, lung, bladder, and ovarian tumors [5,6,7]. To date, ~2 billion dollars worth of platinum-based anticancer drugs have been marketed worldwide, and almost 50% of cancer patients are administered cisplatin for treatment [8]. However, Cisplatin is an ototoxic drug that can cause hearing loss [9]. A high incidence of severe ototoxicity is associated with cisplatin, which triggers hair cell death and subsequently results in loss of hearing [10, 11]. Cisplatin ototoxicity limits its therapeutic effects in patients with cancer, as approximately 80% of cisplatin-treated patients have at least 20 dB hearing loss [12]. Moreover, although several research studies are being conducted on cisplatin-induced ototoxicity, there are no available licensed drugs to prevent ototoxicity [13, 14]. Therefore, new agents or effective approaches are urgently required to prevent and treat cisplatin-induced ototoxicity.
Cisplatin induces apoptosis of cancer cells by inducing DNA damage, preventing DNA synthesis, and activating several transduction pathways [15, 16]. Ototoxicity induced by cisplatin remains to be fully characterized, and this effect may be mediated by several factors [17]. Cisplatin-induced ototoxicity promotes reactive oxygen species (ROS) accumulation, mitochondrial dysfunction, apoptosis, and autophagy [18, 19]. Hazlitt et al. reported that this ototoxicity might be due to the production of toxic ROS levels and cellular inflammatory responses toward apoptosis [20]. Inflammatory responses, combined with oxidative stress, lead to apoptosis and necrosis at the base and middle of the cochlea [21]. Moreover, ROS-induced cytotoxicity causes mitochondrial damage and reduces mitochondrial membrane potential, resulting in mitochondrial apoptosis and death [22]. Although autophagy activation is beneficial in some pathological states such as metabolic and neurodegenerative diseases, an excessive level of activation results in apoptotic cell death and promotes hepatic fibrosis development [4, 23]. Therefore, it remains controversial whether autophagy activation promotes or reduces cell survival.
The active phospholipid signaling molecule LPA (lysophosphatidic acid) exerts various biological effects such as cell migration and proliferation, morphological alterations, and antiapoptotic effects [24]. LPA binds to specific cell surface receptors (LPA1–6) to exert its influence. There are two subfamilies of LPA receptors: LPA1-3, which are G-protein-coupled (EDG) receptors; LPA4-6, which are non-EDG receptors [25]. LPA receptors are expressed in varying spatiotemporal patterns from embryonic development to adulthood, and LPA may activate different receptor isoforms to exert a variety of biological effects [26, 27]. LPA induces a motile and proinflammatory microglial phenotype via LPA5/protein kinase D-dependent pathways [28]. Selective LPA1 and LPA3 antagonists can inhibit the change in shape and aggregation of human platelets induced by LPA, thus indicating the regulatory role of LPA1/3 [29]. LPA’s effects on inner ear auditory cells are rarely studied, and the mechanisms underlying LPA’s effects on auditory cells remain unclear. At present, some compounds used to treat or prevent cisplatin-induced ototoxicity, including anti-inflammatory compounds (flunarizine and etanercept) and antioxidants (dexamethasone and sodium thiosulfate), are currently in clinical trials [30]. Based on our earlier findings on the efficacy of LPA in preventing noise-induced deafness [31], we speculate that LPA may prevent cisplatin ototoxicity. We also suggest that cochlea cell survival following cisplatin treatment may be influenced by LPA through binding to specific receptors (LPA1–6). In the present study, we validated these hypotheses by analyzing how LPA prevents the death of HEI-OC1 cells due to cisplatin exposure and the underlying mechanisms of action; we evaluated how cisplatin and LPA exposure influence the motor behavior in a zebrafish model by using a behavioral trajectory tracking system (EthoVision XT 17.0). We preliminarily investigated the influence of LPA1-6 expression and LPA1 silencing on the protective effect of LPA. These findings could enable to clarify the synergistic mechanism of LPA and its receptor.
Results
LPA protects HEI-OC1 cells against cytotoxicity of cisplatin
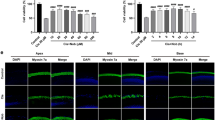
LPA pretreatment protects mice from noise-induced hearing loss (NIHL), thus suggesting that LPA is a potential drug to prevent hearing loss [31]. Here, we used HEI-OC1 cells to determine whether LPA shows preventive and protective effects on hearing loss due to cisplatin. First, we tested the viability of HEI-OC1 cells by using the CCK-8 kit (GlpBio Technology USA, USA, GK10001) to estimate LPA’s optimum concentration as well as pretreatment time. Various LPA concentrations (0, 1, 3, 5, 10, 15, and 20 µM, n = 6) were used for treating the cells for varying time points (i.e., 0, 4, 12, 24, and 48 h). Figure 1A shows that different concentrations (1–20 uM) of LPA-treated HEI-OC1 cells for 0–48 h. There was no significant difference in cell viability between groups, indicating that LPA is not toxic to cells and the length of incubation. Thus, the pretreatment time of LPA in our subsequent study was chosen to be 4 h.

A HEI-OC1 cells were treated with increasing concentrations of LPA: 0 μM (vehicle control), 1, 3, 5, 10, 15, and 20 µM, and incubated for 0, 4, 12, 24, and 48 h. Cell viability was assessed by cell counting kit-8 (n = 6, Data are represented as means ± SD, *p < 0.05). B HEI-OC1 cells were exposed to LPA (1, 3, 5, 10, 15, and 20 µM) for 4 h and treated with cisplatin (10 µM) for 24 h. (n = 6, Data are represented as means ± SD. *p < 0.05 versus cisplatin group; **p < 0.01; ***p < 0.001; ns, non-significance). C Flow cytometric analysis of apoptotic cells in various treatment groups. Viable cells are shown by the Annexin V-negative and propidium iodide-negative quadrant (Q3, lower left quadrant). The Annexin V-positive and PI-negative quadrant (Q4, lower right quadrant) shows early apoptotic cells. The Annexin V-positive and PI-positive quadrant (Q2, upper right quadrant) shows late apoptotic and necrotic cells. The total number of apoptotic cells was the sum of early (Q4) and late (Q2) apoptotic cells (i.e., Q4 + Q2). n = 3, Data are represented as means ± SD. *p < 0.05 versus control group or LPA group. D Data shown in C are quantified. ****p < 0.0001 and *p < 0.05.
To determine the effect and optimal concentration of LPA to protect cells from cisplatin damage. Cells were pretreated with different concentrations of LPA (1, 3, 5, 10, 15, and 20 μ M) for 4 h and then treated with 10 μM cisplatin for 24 h. Cell viability assays were performed. The CCK-8 assay demonstrated that the cell viability percentage was 100%, 27.56 ± 6.65%, 30.55% ± 8.05%, 41.63% ± 8.99%, 50.51% ± 6.74%, 61.82% ± 8.45%, and 57.44% ± 4.61% for the control, 1, 3, 5, 10, 15, and 20 µM LPA groups, respectively, and 24.59 ± 1.58% for cisplatin-treated cells (Fig. 1B). The results showed that pretreatment of HEI-OC1 cells with 3–20 μM of LPA significantly increased cell viability (*p < 0.05 versus cisplatin group) and the protective effect showed a dose-dependence. In all, 15 μM of LPA showed a relatively better protective effect, and therefore, subsequent immunofluorescence, Immunoblot detection, Therefore, 15 μM LPA was used to treat the cells in subsequent immunofluorescence, immunoblot detection, flow cytometry, and other experiments.
LPA alleviates apoptosis of HEI-OC1 cells caused by cisplatin
For validating and evaluating LPA’s effect on cell damage caused by cisplatin, TUNEL assay, and FCM were conducted to assess apoptotic cells. In FCM analysis, PI and Annexin V were used to mark dead cells as well as cells undergoing apoptosis, respectively. First, experimental samples were divided into the following four groups: PBS, 10 µM cisplatin, 15 µM LPA, and 10 µM cisplatin + 15 µM LPA. The apoptotic cell percentages in the cisplatin and LPA + cisplatin groups were 8.87% ± 0.55% and 6.93% ± 0.35% (Fig. 1C, D). Figure 1C, D data indicated that remarkable reduction in the apoptotic cell percentage by LPA pretreatment before cisplatin exposure (****p < 0.0001 and *p < 0.05, n = 3, means ± SD).
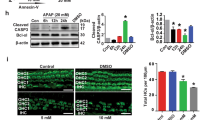
To further validate that LPA attenuated cisplatin-induced apoptosis in HEI-OC1 cells. We used the TUNEL assay to detect the number of apoptotic cells upon cisplatin exposure and LPA pretreatment. As shown in Fig. 2A, B, apoptotic cells were significantly increased in the cisplatin-exposed group, and LPA pretreatment significantly alleviated cisplatin-induced apoptosis. Immunoblot detection was conducted to evaluate expression of apoptosis-related proteins. As show in Fig. 2C–H, cleaved caspase-3 and Bax expression levels were significantly increased in HEI-OC1 cells after exposure to cisplatin when compared with those in the control and LPA groups (****p < 0.0001 and *p < 0.05, n = 3). Bax and cleaved caspase-3 were significantly downregulated after LPA (15 µM) treatment in comparison to those in the cisplatin group. The expression of Bcl-2 expression was significantly upregulated. The above results suggest that LPA treatment significantly reduces cisplatin-induced apoptosis in HEI-OC1 cells.

A Immunofluorescence staining with TUNEL and DAPI in the HEI-OC1 cells after different treatments, Scale bar = 100 um, n = 3. B Data shown in A were quantified (**p < 0.01). C Immunoblot detection of caspase 3/cleaved caspase 3 and β-actin expression in different treatment groups cells. D and E data shown in C were quantified (****p < 0.0001; ns, non-significance; n = 3). F Immunoblot detection of Bax and β-actin expression in different groups of HEI-OC1 cells. G Data shown in F were quantified. H WB assay of Bcl-2 and β-actin expression in different groups of HEI-OC1 cells. I Data shown in H were quantified (****p < 0.0001, **p < 0.01 and *p < 0.05, n = 3).
Cisplatin-induced ROS production and cell apoptosis are inhibited by pretreatment with LPA
Cisplatin treatment can increase ROS levels in cells [32,33,34,35,36], thereby leading to cell apoptosis and death. Intracellular ROS were detected using a DCFH-DA fluorescent probe [37] in HEI-OC1 cells of different groups (Fig. 3A, B). Thus, LPA pretreatment reduced ROS levels in HEI-OC1 cells, which were increased by cisplatin.

A DCFH-DA assay to detect intracellular ROS. HEI-OC1 cells were exposed to 0 µM cisplatin (vehicle control), 15 µM LPA, 15 µM LPA + 10 µM cisplatin, and 10 µM cisplatin. After 4-h LPA pretreatment, cisplatin was used to treat the cells for 24 h. B Data shown in A were quantified. ***p < 0.001, Scale bar = 100 μm.
Pretreatment with LPA inhibits cisplatin-induced ultrastructural changes in cells
F/G-actin is an important protein for maintaining the cytoskeleton structure. We previously reported that long-term exposure to noise led to hearing loss and caused a change in the F/G-actin ratio, which may be related to damage to the cytoskeleton structure [31]. Here, we studied alterations in F-actin caused by cisplatin and LPA treatment. The control and LPA group cells showed normal morphology, and F-actin was arranged in a proper order and loosely distributed within the cell body. In contrast, cells in the cisplatin-treated group became slender and spindle-shaped, and F-actin in the cells was decreased and tightly arranged (Fig. 4A). LPA pretreatment remarkably reduced the ultrastructural changes in cells caused by cisplatin. Thus, LPA reduced cytoskeletal rearrangement induced by cisplatin.

A Morphological and structural changes in the different treatment groups. Scale bar: 50 mm. B Transmission electron microscopy study for the presence of HEI-OC-1 cells that underwent apoptosis. Cells were exposed to 0 µM cisplatin (vehicle control), 15 µM LPA, 15 µM LPA + 10 µM cisplatin, and 10 µM cisplatin. After 4-h LPA pretreatment, cells were subjected to cisplatin treatment for 24 h. B′ is a locally enlarged picture of the site shown by the red dashed box in each treatment group of Fig. B, mark the mitochondria with red arrows.
We also examined the morphology of mitochondria and apoptotic bodies by TEM. We observed that exposure to cisplatin led to mitochondrial shrinkage and loss of mitochondrial crest (Fig. 4B). However, mitochondrial ultrastructure was maintained in the LPA-treated group. Thus, LPA attenuated the mitochondrial damage of HEI-OC1 cells due to cisplatin. Because cisplatin-treated cells showed several apoptotic bodies, cisplatin exposure may activate apoptotic pathways.
HEI-OC1 cell treatment with LPA inhibits autophagy due to cisplatin
We assessed how LPA is involved in regulating autophagy in HEI-OC1 cells. Expression and distribution of the autophagy-related protein LC3B (Fig. 5A–D) in treated cells were determined by immunofluorescence (Fig. 5A). LPA treatment markedly inhibited the upregulation of cisplatin-induced autophagy, thus showing that LPA inhibited autophagy induced by cisplatin.

A Immunofluorescence staining images of LC3B in HEI-OC1 cells exposed to cisplatin (10 µM) for 24 h and with or without 15 µM LPA. B Immunoblot detection of LC3B-II and LC3B-I expression in cisplatin- and/or LPA-exposed cells. C, D LC3B expression quantification in B ****p < 0.0001, *p < 0.05, Scale bar = 50 mm.
Pretreatment with LPA treatment alleviates cisplatin-induced ototoxicity in zebrafish
Zebrafish is an important model to test compounds that protect against toxicity caused by cisplatin in vivo; this is because their lateral line system has hair cells identical to those found in the inner ear of mammals. These cells can be readily reached through in vivo administration of drugs [38,39,40]. Therefore, we investigated the effects of cisplatin and LPA alone and in combination on Zebrafish embryonic development (10–96 hpf) and zebrafish larvae on day 5 following fertilization. The LPA and LPA + cisplatin groups were pretreated for 4 h with LPA (15 µM) and subjected to 24-h treatment with cisplatin (10 µM). A behavioral analysis system [41] was used to record and analyze the larvae’s locomotion parameters. Figure 6A–F shows that when compared with the other treatment groups, regardless of the tapping stimulation or light/dark cycle changes, cisplatin exposure caused zebrafish to move more actively, and their movement speed and distance were quite higher as compared to those of the control and LPA groups. Neuromast cell vitality was evaluated by Zebrafish [Et (krt4: EGFP)] labeled with lateral neuromast hair cells, cisplatin causes partial hair cell death and loss, and LPA treatment can significantly reduce hair cell death and loss, and has a significant protective effect (Fig. 7).

A, D Heatmap of the tracked path of tap and light/dark modes for each experimental group containing six zebrafish individuals. Blue marks show the area of the passage of zebrafish, and the color intensity represents the tracked path’s cumulative frequency [77]. B Quantification of the running distance of zebrafish in each treatment group. C Quantification of the movement velocity of zebrafish in each treatment group. E, F Running distance and movement velocity analysis of zebrafish in each treatment group under light/dark conditions, respectively (light and dark cycles were run for 10 min each). G Quantification of the movement velocity of zebrafish in each treatment group. H Quantification of the running distance of zebrafish in each treatment group. Six zebrafish were studied in each group. p < 0.0001, ***p < 0.001, **p < 0.01 and *p < 0.05, n = 6.

A Schematic diagram of the distribution of hair cells in the lateral line neuromast throughout the body of zebrafish and the observation of the dorsal and lateral view of hair cells. B Survival of neuromast hair cells in the lateral line of zebrafish in different treatment groups. C A partial enlarged view of the box in Figure B. Zebrafish strains: [Et(krt4: EGFP)]; control group, do not process; Cisplatin group, 10 μM Cisplatin ; LPA group, 15 μM LPA; LPA+Cis group, adding 15 μM LPA for 4 h, then adding 10 μM Cisplatin for 24 h; The above groups were treated for the same time.
Silencing of the LPA1 receptor blocks protective effects of LPA on HEI-OC1 cells from apoptosis due to cisplatin
We investigated the mRNA expression levels of the LPA1-6 receptor in cochlear cells by qRT-PCR. LPA1 receptor expression was remarkably greater as compared to that of the other receptors (Fig. 8A). To investigate the relationship between the LPA1 receptor and LPA’s protective effect on cisplatin-treated HEI-OC1 cells, LPA1-silenced cells were generated by transfecting cells with siRNA(LPA1), and siRNA(control)transfected cells served as controls. The siRNA(LPA1) mRNA expression level was remarkably decreased in LPA1-silenced HEI-OC1 cells in comparison to that in the control cells (Fig. 8B), thus confirming that the LPA1 receptor was efficiently silenced. We measured the apoptosis levels after transfection with siRNA(LPA1) by flow cytometry. Dead cells and apoptotic cells were marked with PI and Annexin V, respectively [22]. Compared to the siRNA(control) group, the siRNA(LPA1) group showed a significant increase in apoptotic cell percentage after LPA and cisplatin treatment (Fig. 8C, D). Thus, silencing of the LPA1 receptor blocked the protective effect of LPA.

A qRT-PCR for the relative expression of the LPA receptors 1–6 (n = 3). B Effective silencing of the LPA1 receptor using siRNA. siRNA(LPA1) expression was significantly decreased in HEI-OC1 cells as compared to that in cells transfected with siRNA(control) ****p < 0.0001, n = 3, as estimated by one-way ANOVA. C FCM of apoptotic cells after transfection with siRNA(LPA1). D Quantization of flow detection data in C ****p < 0.0001, n = 3, as determined by one-way ANOVA.
Discussion
Ototoxic drugs, including aminoglycosides and chemotherapeutic agents, are drugs that induce toxicity in the inner ear’s structure [42, 43]. The potent anticancer agent cisplatin is used to treat various tumors; however, it can also cause ototoxicity and neurotoxicity, which restricts its utility and clinical applications [21, 44]. LPA, which is a bioactive phospholipid that is present at nanomolar to micromolar concentrations in most tissues and biological fluids, participates in several cellular functions such as cell proliferation and migration, neurite retraction, smooth muscle cell contraction, and aggregation of platelets [45,46,47]. Here, we assessed whether LPA exhibits protective effects on ototoxicity induced by cisplatin. We revealed that a high ROS accumulation in HEI-OC1 cells exposed to cisplatin, accompanied by mitochondrial damage, led to apoptosis and autophagy. Treatment of HEI-OC1 cells with LPA significantly attenuated apoptosis and autophagy induced by cisplatin exposure. Alternatively, LPA1 receptor silencing blocked LPA’s protective effect on apoptosis induced by cisplatin.
Cisplatin has been shown to increase ROS levels [32,33,34,35,36]. Moreover, although cisplatin is highly cytotoxic to proliferating cells, it is also toxic to nonproliferative terminally differentiated cochlear hair cells. In the cochlea, cisplatin causes cytotoxicity mainly because of its interference of the cellular redox and inflammatory response to apoptosis [36, 48,49,50]. In the present study, cisplatin-treated HEI-OC1 cells showed high ROS accumulation, accompanied by mitochondrial damage, resulting in apoptosis, which is consistent with previous reports. Additionally, autophagy may also be involved in ototoxicity caused by cisplatin. At present, it remains unclear whether autophagy activation is destructive or protective [4, 22]. An excess amount of autophagy activation can cause cell death and pathological alterations [51,52,53,54]. The induction of autophagy after aminoglycoside-induced ototoxicity can suppress apoptosis and protect cochlear hair cells through the prevention of ROS accumulation [23, 34, 55, 56]. Here, exposure to cisplatin increased the autophagic marker LC3B expression and promoted apoptosis; however, these effects were alleviated by LPA pretreatment, thus indicating that LPA inhibited cisplatin-induced autophagy. These findings agree with the results of a previous study in which cisplatin combined with meclofenamic acid 2 (MA2) reduced cisplatin-induced ROS accumulation, apoptosis, and upregulation of autophagy in HEI-OC1 cells, indicating that MA2 suppresses autophagy induced by cisplatin [4]. Therefore, LPA might have a similar role to that of MA2. Additionally, in the present study, the motion behavior track experiment of zebrafish revealed that cisplatin exposure increased zebrafish activity and their motion speed and distance as compared to those of the control and LPA groups. These findings show that LPA pretreatment remarkably reduced cisplatin-induced motion behavior activity of zebrafish.
LPA and its receptors are involved in several pathophysiological and physiological processes [57,58,59]. Suckau [60] et al. described LPA1, LPA2, LPA4, and LPA6 receptor expression in mouse brain development. LPA4 and LPA6 show selective expression in HEV endothelial cells (ECs), and LPA4 has a critical role in the transmigration of lymphocytes across HEVs in mice [61]. Matsumoto et al. detected LPA1 and LPA4 expression, but not that of LPA2 and LPA3, in adult guinea pigs’ outer hair cells [62]. Signals mediated through LPA4 enable the formation of the vascular network, which restores normal vascular barrier function in subcutaneous tumor cells [63]. LPA also induces diverse adhesion molecule expression in human umbilical vein endothelial cells through LPA1 and LPA3 and several downstream effectors such as RhoA-ROCK2-NF-κB [64, 65]. LPA-induced ovarian cancer cell migration requires ezrin-radixin-moesin (ERM) protein activation through LPA1 and LPA2 [66]. Here, we first determined LPA receptor (LPA1-6) expression levels and found that all receptors were expressed in HEI-OC1 cells, except LPA3. The LPA1 receptor exhibited the highest expression. Moreover, silencing of LPA1 in HEI-OC1 cells by siRNA transfection promoted apoptosis by cisplatin despite LPA pretreatment, thus suggesting that the LPA1 receptor is critically involved in LPA’s protective effect on cisplatin-induced apoptosis in HEI-OC1 cells. LPA1 couples with G proteins (including Gi/o, Gq, and G12/13), and it induces various cellular responses through LPA1, including Akt, Rho, and MAPK pathway activation that induce physiological responses, which include cytoskeleton changes, cell migration and proliferation, and cell survival [67, 68]. Moreover, the LPA1 receptor is expressed in many tissues such as the brain, lungs, heart, spleen, stomach, and skeletal muscle [69, 70]. LPA1-deficient mice show defects in brain development and reduced neural precursor cell proliferation [71]. LPA controls cytoskeletal rearrangement and protects against lung injury by increasing the integrity and remodeling of the lung epithelial cell barrier [72, 73]. Our previous studies showed that LPA activates the Rho-ROCK2/p-ERM pathway to regulate the death of hair cells and NIHL by targeting the cytoskeleton [31, 74]. Here, we preliminarily found that LPA1 receptor silencing blocked LPA’s protective effect on apoptosis due to cisplatin. Further studies are required to understand the precise regulatory mechanism of the signaling pathway.
In summary, cisplatin treatment induces apoptosis in HEI-OC1 cells; cisplatin and LPA combination can decrease cisplatin-induced accumulation of ROS and HEI-OC1 cell apoptosis. In addition, LPA treatment remarkably suppressed cisplatin-induced autophagy, thereby protecting HEI-OC1 cells. In vivo fluorescence imaging results indicated that cisplatin caused hair cell death and loss, and LPA treatment significantly protected hair cell death and loss. Our findings provide novel evidence that the LPA1 receptor has a major involvement in LPA’s protective effect on HEI-OC1 cell apoptosis by cisplatin. This study reports that LPA-LPA1 acts as an important regulator of cisplatin ototoxicity, providing insights into the underlying mechanisms and potential therapeutic targets to prevent ototoxicity caused by cisplatin.
Materials and methods
Cell culture and drugs
Cisplatin from Hansoh Pharma (Jiangsu, China; #160203) was used. Caspase 3/cleaved caspase 3 (#WL02117), bax (#WL01637), bcl-2 (#WL01556), β-actin (# WL01845) were purchased from Wanleibio (Shenyang, China). LPA was supplied by Cayman Chemical (Ann Arbor, MI; #62215) and diluted in phosphate-buffered saline (PBS) to yield 10 mM stock concentration. The HEI-OC1 cell line was grown in a DMEM high-glucose medium, which was provided by Thermo Fisher Scientific (Waltham, MA); the medium included fetal bovine serum (10%, FBS) from Gibco (Gibco, USA). Culturing was performed at 33 °C under 5% CO2 condition. The cell line was used to study the mechanisms by which cisplatin induces ototoxicity at the cellular and molecular levels.
Flow cytometry-based analysis of apoptosis
The Annexin V kit (RUO; BD Biosciences, Franklin Lakes, NJ) was used to detect cell apoptosis. After collecting and washing the cells twice with PBS, they were resuspended in binding buffer (1×) at 1 × 106 cells/mL concentration. This was followed by incubation of the cell suspension at room temperature in dark with Annexin V and propidium iodide (PI). A BD Biosciences FACS Calibur system was used for data collection, and the collected data were analyzed by FlowJo 7.6 (FlowJo LLC., Oregon, USA).
ROS accumulation was measured by DCFH-DA assay (WLA131; Wanlei Bio, Shenyang, China). After collecting the standing cells, they were washed in PBS and diluted 1:1000 by using the diluent DCFH-DA (dichloro-dihydro-fluorescein diacetate). During incubation at 37 °C, the cells were mixed vigorously every 5 min. Subsequently, the cells were kept at 37 °C for 30 min and mixed rigorously every 5 min. The cells were then subjected to washing for three times with PBS to fully remove the excess amount of DCFH-DA. Finally, the cells in each treatment group were resuspended in 500 µL of PBS for flow cytometry (FCM) analysis.
Real-time polymerase chain reaction analysis
The RNAeasy Mini kit (#74004; Qiagen, Hilden, Germany) was employed for RNA extraction from HEI-OC1 cells. The RTreagent kit (#330401; Qiagen) was employed for generating cDNA. Next, RT-PCR was performed using Stratagene (M3003; New England Biolabs, Ipswich, MA). Subsequently, PCR was conducted in a 20-μL reaction volume with Luna universal qPCR Mix (10 μL), 1 μL of forward primer, 1 μL of reverse primer, cDNA template (1 μL), and deionized water. By using GAPDH as a standard, target gene levels were normalized, and gene expression was determined using the 2−ΔΔCt method. Primer sequences are shown in Table 1.
Transmission electron microscopy
First, trypsinization of HEI-OC1 cells was performed. The cells were then fixed for 1 h in a fixative solution of glutaraldehyde (3%, pH 7.4). The cells were again fixed for 1–2 h in osmic acid (1%) in a 0.1 M sodium cacodylate buffer at pH 7.2. After dehydration with acetone, araldite CY212 was used to embed the cells. The follow-up experimental sample treatment and process were performed by referring to a previous study [4].
ROS detection and immunofluorescence staining
Mitochondrial ROS levels were detected by DCFH-DA (#WLA131; Wanlei Bio, China). After 24-h exposure to cisplatin (10 μM) with or without 15 μM LPA, the cells were subjected to PBS washing and incubated with 1:1000 diluted DCFH-DA for 30 min. After fixation with 4% paraformaldehyde, a confocal microscope (Olympus, Tokyo, Japan) was used for cell visualization.
DMEM containing 10% FBS was used to culture HEI-OC1 cells. After the treatment, the cells from each group were aspirated with fluid, and the cells were fixed at −20 °C for 15 min with 4% paraformaldehyde. This was followed by washing with 1× PBS (5 min per wash, three times) and immunostaining. Blocking was performed in PBS with Triton X-100 (1%) and BSA (5%) for 1 h. HEI-OC1 cells were incubated at 4 °C overnight with rabbit anti-LC3B antibodies (dilution 1:200; #3868; Cell Signaling Technology, Danvers, MA); the cells were re-incubated for 12 h with secondary antibodies, counterstained for 20 min using DAPI (1:1000; Sigma-Aldrich), and visualized with a confocal microscope (Olympus).
siRNA transfection
To perform siRNA transfection, siRNA(control) and a target-specific siRNA(LPA1) were purchased from Thermo Fisher Scientific (siRNA ID: s66922; siRNA(LPA1): #4390771; siRNA(control): #4390843) to knockdown gene expression. After seeding HEI-OC1 cells onto 6-well plates (at 0.2 × 106 cells/well concentration), the plates were incubated overnight. Cell transfection was performed using 2 mL Opti-MEM (Gibco) with 9 μL of Lipofectamine RNAiMAX provided by Thermo Fisher Scientific and siRNA duplexes (200 nM), and the cells were transfected with siRNA(control) (Gibco, USA) by using the same method for 6 h. The cells were then removed, and the medium was then replaced by a complete culture medium. Next, the cells were subjected to 4-h LPA treatment; this was followed by 24-h cisplatin exposure. After cell harvesting, FCM and Western blotting (WB) assay were performed.
Protein extraction and Immunoblot detection
RIPA lysis buffer supplied by Beyotime Biotechnology (Shanghai, China) was used for total protein extraction from HEI-OC1 cells. In accordance with the instructions of the manufacturer, protein concentrations were estimated with a BCA protein quantification kit from Beyotime Biotechnology. Protein detection and estimation methods were similar to those described in a previous study. The process of protein detection and analysis is the same as that described in reference [4]. β-actin was taken as the loading control. β-Actin and caspase 3/cleaved caspase 3 rabbit monoclonal antibodies (1:1000 dilution) were used as primary antibodies. The membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (1:2000 dilution) for 1 h at room temperature.
Evaluation of cisplatin’s and LPA’s effects on zebrafish larvae behavior and In vivo imaging
For determining the protective effect of LPA, 5 d *AB WT (wild type) zebrafish larvae were selected as an animal model to investigate LPA’s and cisplatin’s effects in combination and when administered alone on motor behavior. The experimental protocols were approved by the Institutional Animal Care and Use Committee of the Air Force Medical University (Formerly known as the Fourth Military Medical University). Four treatment groups were set in a 24-well plate (round hole), and each treatment group was established with six replicates (one juvenile fish was placed in each hole). The LPA and LPA + cisplatin groups were subjected to pretreatment with LPA for 4 h. The groups were then treated with 10 µM cisplatin for 24 h. No treatment was administered to the control group. EthoVision XT 17.0, a behavioral analysis system, provided by Noldus (Wageningen, Netherlands) was used to record and analyze the motion trajectories of the larvae in light/dark exploration and tapping acoustic stimulation models. The light was switched on/off for 10 min in the light/dark cycle mode, and the procedure was repeated twice. Neuromast cell vitality was evaluated by Zebrafish [Et (krt4: EGFP)] labeled with lateral neuromast hair cells, Et (krt4: EGFP) [75] zebrafish that express green fluorescent protein in sensory hair cells. A large field fluorescence biological stereo microscope (Axio Zoom V16; Zeiss) was connected to a CCD camera (Axiocam506 mono; Zeiss) to record the fluorescence images Et (krt4: EGFP) Zebrafish.
Statistical analysis
GraphPad Prism 8.0 was employed for statistical analyses. We used Student’s t test to compare differences between two groups. Differences among more than two groups were compared by two-way ANOVA followed by Tukey’s multiple comparison test. P < 0.05 was assumed to be a significant value [76].
Data availability
The data supporting the study findings are available following reasonable request from the corresponding author.
References
Kayyali MN, Wooltorton JRA, Ramsey AJ, Lin M, Chao TN, Tsourkas A, et al. A novel nanoparticle delivery system for targeted therapy of noise-induced hearing loss. J Control Release. 2018;279:243–50.
An X, Wang R, Chen E, Yang Y, Fan B, Li Y, et al. A forskolin-loaded nanodelivery system prevents noise-induced hearing loss. J Control Release. 2022;348:148–57.
Naz S, Friedman TB. Growth factor and receptor malfunctions associated with human genetic deafness. Clin Genet. 2020;97:138–55.
Li H, Song Y, He Z, Chen X, Wu X, Li X, et al. Meclofenamic acid reduces reactive oxygen species accumulation and apoptosis, inhibits excessive autophagy, and protects hair cell-like HEI-OC1 cells from cisplatin-induced damage. Front Cell Neurosci. 2018;12:139.
Fritzsche S, Strauss C, Scheller C, Leisz S. Nimodipine treatment protects auditory hair cells from cisplatin-induced cell death accompanied by upregulation of LMO4. Int J Mol Sci. 2022;23:5780.
Krukowski K, Ma J, Golonzhka O, Laumet GO, Gutti T, van Duzer JH, et al. HDAC6 inhibition effectively reverses chemotherapy-induced peripheral neuropathy. PAIN. 2017;158:1126–37.
Ngo LP, Owiti NA, Swartz C, Winters J, Su Y, Ge J, et al. Sensitive CometChip assay for screening potentially carcinogenic DNA adducts by trapping DNA repair intermediates. Nucleic Acids Res. 2020;48:e13.
Ghosh S. Cisplatin: the first metal based anticancer drug. Bioorg Chem. 2019;88:102925.
Lieu JEC, Kenna M, Anne S, Davidson L. Hearing loss in children: a review. Jama. 2020;324:2195–205.
Cho SI, Jo ER, Song H. Mitophagy impairment aggravates cisplatin-induced ototoxicity. BioMed Res Int. 2021;2021:5590973.
Drögemöller BI, Wright GEB, Lo C, Le T, Brooks B, Bhavsar AP, et al. Pharmacogenomics of cisplatin-induced ototoxicity: successes, shortcomings, and future avenues of research. Clin Pharmacol Ther. 2019;106:350–9.
Tserga E, Moreno-Paublete R, Sarlus H, Björn E, Guimaraes E, Göritz C, et al. Circadian vulnerability of cisplatin-induced ototoxicity in the cochlea. FASEB J. 2020;34:13978–92.
Qi L, Luo Q, Zhang Y, Jia F, Zhao Y, Wang F. Advances in toxicological research of the anticancer drug cisplatin. Chem Res Toxicol. 2019;32:1469–86.
Rybak LP, Whitworth CA, Mukherjea D, Ramkumar V. Mechanisms of cisplatin-induced ototoxicity and prevention. Hearing Res. 2007;226:157–67.
Martinho N, Santos TCB, Florindo HF, Silva LC. Cisplatin-membrane interactions and their influence on platinum complexes activity and toxicity. Front Physiol. 2018;9:1898.
Achkar IW, Abdulrahman N, Al-Sulaiti H, Joseph JM, Uddin S, Mraiche F. Cisplatin based therapy: the role of the mitogen activated protein kinase signaling pathway. J Transl Med. 2018;16:96.
Yu D, Gu J, Chen Y, Kang W, Wang X, Wu H. Current strategies to combat cisplatin-induced ototoxicity. Front Pharmacol. 2020;11:999.
Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–79.
Rocha CRR, Silva MM, Quinet A, Cabral-Neto JB, Menck CFM. DNA repair pathways and cisplatin resistance: an intimate relationship. Clinics. 2018;73:e478s.
Hazlitt RA, Min J, Zuo J. Progress in the development of preventative drugs for cisplatin-induced hearing loss. J Med Chem. 2018;61:5512–24.
Kros CJ, Steyger PS. Aminoglycoside- and cisplatin-induced ototoxicity: mechanisms and otoprotective strategies. Cold Spring Harb Perspect Med. 2019;9:a033548.
Wang D, Shi S, Ren T, Zhang Y, Guo P, Wang J, et al. U0126 pretreatment inhibits cisplatin-induced apoptosis and autophagy in HEI-OC1 cells and cochlear hair cells. Toxicol Appl Pharmacol. 2021;415:115447.
He Z, Guo L, Shu Y, Fang Q, Zhou H, Liu Y, et al. Autophagy protects auditory hair cells against neomycin-induced damage. Autophagy. 2017;13:1884–904.
Meduri B, Pujar GV, Durai Ananda Kumar T, Akshatha HS, Sethu AK, Singh M, et al. Lysophosphatidic acid (LPA) receptor modulators: Structural features and recent development. Eur J Med Chem. 2021;222:113574.
Brown A, Hossain I, Perez LJ, Nzirorera C, Tozer K, D’Souza K, et al. Lysophosphatidic acid receptor mRNA levels in heart and white adipose tissue are associated with obesity in mice and humans. PLoS ONE. 2017;12:e0189402.
Park F, Miller DD. Role of lysophosphatidic acid and its receptors in the kidney. Physiol Genomics. 2017;49:659–66.
Yanagida K, Ishii S. Non-Edg family LPA receptors: the cutting edge of LPA research. J Biochem. 2011;150:223–32.
Plastira I, Bernhart E, Goeritzer M, DeVaney T, Reicher H, Hammer A, et al. Lysophosphatidic acid via LPA-receptor 5/protein kinase D-dependent pathways induces a motile and pro-inflammatory microglial phenotype. J Neuroinflammation. 2017;14:253.
Rother E, Brandl R, Baker DL, Goyal P, Gebhard H, Tigyi G, et al. Subtype-selective antagonists of lysophosphatidic Acid receptors inhibit platelet activation triggered by the lipid core of atherosclerotic plaques. Circulation. 2003;108:741–7.
Waissbluth S. Clinical trials evaluating transtympanic otoprotectants for cisplatin-induced ototoxicity: what do we know so far? Eur Arch Oto-Rhino-Laryngol. 2020;277:2413–22.
Han Y, Wang XR, Chen J, Sha SH. Noise-induced cochlear F-actin depolymerization is mediated via ROCK2/p-ERM signaling. J Neurochem. 2015;133:617–28.
Nan B, Zhao Z, Jiang K, Gu X, Li H, Huang X. Astaxanthine attenuates cisplatin ototoxicity in vitro and protects against cisplatin-induced hearing loss in vivo. Acta Pharm Sin B. 2022;12:167–81.
He Y, Zheng Z, Liu C, Li W, Zhao L, Nie G, et al. Inhibiting DNA methylation alleviates cisplatin-induced hearing loss by decreasing oxidative stress-induced mitochondria-dependent apoptosis via the LRP1-PI3K/AKT pathway. Acta Pharm Sin B. 2022;12:1305–21.
Liu W, Xu L, Wang X, Zhang D, Sun G, Wang M, et al. PRDX1 activates autophagy via the PTEN-AKT signaling pathway to protect against cisplatin-induced spiral ganglion neuron damage. Autophagy. 2021;17:4159–81.
He Y, Li W, Zheng Z, Zhao L, Li W, Wang Y, et al. Inhibition of Protein arginine methyltransferase 6 reduces reactive oxygen species production and attenuates aminoglycoside- and cisplatin-induced hair cell death. Theranostics. 2020;10:133–50.
Sheth S, Mukherjea D, Rybak LP, Ramkumar V. Mechanisms of cisplatin-induced ototoxicity and otoprotection. Front Cell Neurosci. 2017;11:338.
Liang L, Wang H, Yao J, Wei Q, Lu Y, Wang T, et al. NPC1 deficiency contributes to autophagy-dependent ferritinophagy in HEI-OC1 auditory cells. Front Mol Biosci. 2022;9:952608.
Vlasits AL, Simon JA, Raible DW, Rubel EW, Owens KN. Screen of FDA-approved drug library reveals compounds that protect hair cells from aminoglycosides and cisplatin. Hearing Res. 2012;294:153–65.
Teitz T, Fang J, Goktug AN, Bonga JD, Diao S, Hazlitt RA, et al. CDK2 inhibitors as candidate therapeutics for cisplatin- and noise-induced hearing loss. J Exp Med. 2018;215:1187–203.
Ingersoll MA, Malloy EA, Caster LE, Holland EM, Xu Z, Zallocchi M, et al. BRAF inhibition protects against hearing loss in mice. Sci Adv. 2020;6:eabd0561.
Zheng S, Huang W, Liu C, Xiao J, Wu R, Wang X, et al. Behavioral change and transcriptomics reveal the effects of 2, 2’, 4, 4’-tetrabromodiphenyl ether exposure on neurodevelopmental toxicity to zebrafish (Danio rerio) in early life stage. Sci Total Environ. 2021;752:141783.
Yin H, Sun G, Yang Q, Chen C, Qi Q, Wang H, et al. NLRX1 accelerates cisplatin-induced ototoxity in HEI-OC1 cells via promoting generation of ROS and activation of JNK signaling pathway. Sci Rep. 2017;7:44311.
Ho GY, Woodward N, Coward JI. Cisplatin versus carboplatin: comparative review of therapeutic management in solid malignancies. Crit Rev Oncol Hematol. 2016;102:37–46.
Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–78.
Aikawa S, Hashimoto T, Kano K, Aoki J. Lysophosphatidic acid as a lipid mediator with multiple biological actions. J Biochem. 2015;157:81–9.
Mutoh T, Rivera R, Chun J. Insights into the pharmacological relevance of lysophospholipid receptors. Br J Pharmacol. 2012;165:829–44.
Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003;3:582–91.
Ross CJ, Katzov-Eckert H, Dubé MP, Brooks B, Rassekh SR, Barhdadi A, et al. Genetic variants in TPMT and COMT are associated with hearing loss in children receiving cisplatin chemotherapy. Nat Genet. 2009;41:1345–9.
Ghosh S, Sheth S, Sheehan K, Mukherjea D, Dhukhwa A, Borse V, et al. The endocannabinoid/cannabinoid receptor 2 system protects against cisplatin-induced hearing loss. Front Cell Neurosci. 2018;12:271.
McKeage MJ. Comparative adverse effect profiles of platinum drugs. Drug Saf. 1995;13:228–44.
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–52.
Xue F, Cheng J, Liu Y, Cheng C, Zhang M, Sui W, et al. Cardiomyocyte-specific knockout of ADAM17 ameliorates left ventricular remodeling and function in diabetic cardiomyopathy of mice. Signal Transduct Target Ther. 2022;7:259.
Wang L, Li D, Su X, Zhao Y, Huang A, Li H, et al. AGO4 suppresses tumor growth by modulating autophagy and apoptosis via enhancing TRIM21-mediated ubiquitination of GRP78 in a p53-independent manner. Oncogene. 2023;42:62–77.
Sarkar J, Das M, Howlader MSI, Prateeksha P, Barthels D, Das H. Epigallocatechin-3-gallate inhibits osteoclastic differentiation by modulating mitophagy and mitochondrial functions. Cell Death Dis. 2022;13:908.
Zhou H, Qian X, Xu N, Zhang S, Zhu G, Zhang Y, et al. Disruption of Atg7-dependent autophagy causes electromotility disturbances, outer hair cell loss, and deafness in mice. Cell Death Dis. 2020;11:913.
Guo L, Cao W, Niu Y, He S, Chai R, Yang J. Autophagy regulates the survival of hair cells and spiral ganglion neurons in cases of noise, ototoxic drug, and age-induced sensorineural hearing loss. Front Cell Neurosci. 2021;15:760422.
Plastira I, Bernhart E, Joshi L, Koyani CN, Strohmaier H, Reicher H, et al. MAPK signaling determines lysophosphatidic acid (LPA)-induced inflammation in microglia. J Neuroinflammation. 2020;17:127.
Yung YC, Stoddard NC, Chun J. LPA receptor signaling: pharmacology, physiology, and pathophysiology. J Lipid Res. 2014;55:1192–214.
Kano K, Arima N, Ohgami M, Aoki J. LPA and its analogs-attractive tools for elucidation of LPA biology and drug development. Curr Med Chem. 2008;15:2122–31.
Suckau O, Gross I, Schrötter S, Yang F, Luo J, Wree A, et al. LPA(1), LPA(2), LPA(4), and LPA(6) receptor expression during mouse brain development. Dev Dyn. 2019;248:375–95.
Hata E, Sasaki N, Takeda A, Tohya K, Umemoto E, Akahoshi N, et al. Lysophosphatidic acid receptors LPA4 and LPA6 differentially promote lymphocyte transmigration across high endothelial venules in lymph nodes. Int Immunol. 2016;28:283–92.
Matsumoto N, Kitani R, Maricle A, Mueller M, Kalinec F. Pivotal role of actin depolymerization in the regulation of cochlear outer hair cell motility. Biophys J. 2010;99:2067–76.
Eino D, Tsukada Y, Naito H, Kanemura Y, Iba T, Wakabayashi T, et al. LPA4-mediated vascular network formation increases the efficacy of anti-PD-1 therapy against brain tumors. Cancer Res. 2018;78:6607–20.
Lee H, Lin CI, Liao JJ, Lee YW, Yang HY, Lee CY, et al. Lysophospholipids increase ICAM-1 expression in HUVEC through a Gi- and NF-kappaB-dependent mechanism. Am J Physiol Cell Physiol. 2004;287:C1657–1666.
Zhou Y, Little PJ, Ta HT, Xu S, Kamato D. Lysophosphatidic acid and its receptors: pharmacology and therapeutic potential in atherosclerosis and vascular disease. Pharm Ther. 2019;204:107404.
Park J, Jang JH, Oh S, Kim M, Shin C, Jeong M, et al. LPA-induced migration of ovarian cancer cells requires activation of ERM proteins via LPA(1) and LPA(2). Cell Signal. 2018;44:138–47.
Fukushima N, Ishii I, Contos JJ, Weiner JA, Chun J. Lysophospholipid receptors. Annu Rev Pharmacol Toxicol. 2001;41:507–34.
Ishii I, Fukushima N, Ye X, Chun J. Lysophospholipid receptors: signaling and biology. Annu Rev Biochem. 2004;73:321–54.
Matas-Rico E, García-Diaz B, Llebrez-Zayas P, López-Barroso D, Santín L, Pedraza C, et al. Deletion of lysophosphatidic acid receptor LPA1 reduces neurogenesis in the mouse dentate gyrus. Mol Cell Neurosci. 2008;39:342–55.
Shano S, Moriyama R, Chun J, Fukushima N. Lysophosphatidic acid stimulates astrocyte proliferation through LPA1. Neurochem Int. 2008;52:216–20.
Contos JJ, Ishii I, Fukushima N, Kingsbury MA, Ye X, Kawamura S, et al. Characterization of lpa(2) (Edg4) and lpa(1)/lpa(2) (Edg2/Edg4) lysophosphatidic acid receptor knockout mice: signaling deficits without obvious phenotypic abnormality attributable to lpa(2). Mol Cell Biol. 2002;22:6921–9.
Kim JH, Adelstein RS. LPA(1) -induced migration requires nonmuscle myosin II light chain phosphorylation in breast cancer cells. J Cell Physiol. 2011;226:2881–93.
Jeong KJ, Park SY, Cho KH, Sohn JS, Lee J, Kim YK, et al. The Rho/ROCK pathway for lysophosphatidic acid-induced proteolytic enzyme expression and ovarian cancer cell invasion. Oncogene. 2012;31:4279–89.
Chen FQ, Zheng HW, Hill K, Sha SH. Traumatic noise activates Rho-family GTPases through transient cellular energy depletion. J Neurosci. 2012;32:12421–30.
Zamora LY, Miguel KC, Lu Z. The alcohol-sensitive period during early octavolateral organ development in zebrafish (Danio rerio). J Neurosci Res. 2017;95:1194–203.
Wang X, Chen Y, Tao Y, Gao Y, Yu D, Wu H. A666-conjugated nanoparticles target prestin of outer hair cells preventing cisplatin-induced hearing loss. Int J Nanomed. 2018;13:7517–31.
Lv X, Zhang R, Xu L, Wang G, Yan C, Lin P. Tcap deficiency in zebrafish leads to ROS production and mitophagy, and idebenone improves its phenotypes. Front Cell Dev Biol. 2022;10:836464.
Acknowledgements
We would like to thank all the lab colleagues for their assistance, and thank TopEdit (www.topeditsci.com) for its linguistic assistance during the preparation of this manuscript. The present work was funded by grants from the National Natural Science Foundation of China (81870719), the Teaching Reform Research Project of Air Force Medical University (KJJYDXYB025), the Xijing Hospital Promotion Project (XJZT21CM10), the Foundation of Shaanxi Province(2023-YBSF-209), Sensorineural hearing loss basic and clinical research innovation team (Shaanxi Province, 2023-CX-TD-70).
Author information
Authors and Affiliations
Contributions
XA, CZ, and BH conceived the study. YH and DZ supervised the experimental design. XA, BH EC, and QZ conducted the experiments. YY, RL, and RY performed statistical analysis. CZ, DZ, and YH supervised data interpretation. XA, CZ, and BH wrote the manuscript. All authors have edited the manuscript and have approved it for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
During the experiment, raising and obtaining animal materials complied with the relevant rules and regulations of the Animal Management Committee of Air Force Medical University of Science and Technology and the International Association for Animal Research (IASP) on the management and protection of experimental animals.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
An, X., Zhong, C., Han, B. et al. Lysophosphatidic acid exerts protective effects on HEI-OC1 cells against cytotoxicity of cisplatin by decreasing apoptosis, excessive autophagy, and accumulation of ROS. Cell Death Discov. 9, 415 (2023). https://doi.org/10.1038/s41420-023-01706-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-023-01706-5
