
Abstract
Psoriasis is a chronic, systemic immune-mediated disease caused by abnormal proliferation, decreased apoptosis, and over-differentiation of keratinocytes. The psoriatic skin lesions due to abnormal keratinocytes are closely associated with ROS produced by inflammatory cells. Peroxiredoxin II (Prx II) is an efficient antioxidant enzyme, which were highly expressed in skin tissues of psoriasis patient. However, the detailed mechanical functions of Prx II on psoriatic skin remain to be elucidated. Present study showed that depletion of Prx II results in alleviation of symptoms of IMQ-induced psoriasis in mice, but no significant differences in the amounts of serum inflammatory factors. Prx II-knockdown HaCaT cells were susceptible to H2O2-induced apoptosis mediated by Ca2+ release from the endoplasmic reticulum through 1,4,5-triphosphate receptors (IP3Rs), the PI3K/AKT pathway and phosphorylated GSK3β (Ser9) were significant downregulated. Additionally, significantly reduced sensitivity of Prx II-knockdown HaCaT cells to apoptosis was evident post NAC, 2-APB, BAPTA-AM, SC79 and LiCl treated. These results suggest that Prx II regulated apoptosis of keratinocytes via the PI3K/AKT/GSK3β signaling axis. Furthermore, treatment with the Prx II inhibitor Conoidin A significantly alleviated psoriatic symptoms in IMQ model mice. These findings have important implications for developing therapeutic strategies through regulate apoptosis of keratinocytes in psoriasis, and Prx II inhibitors may be exploited as a therapeutic drug to alleviate psoriatic symptoms.
Similar content being viewed by others


Introduction
The skin disease psoriasis may occur as a consequence of several factors including infectious microorganisms, and environmental, immunological, and genetic factors [1]. The appearance of psoriatic skin lesions characterized by erythema as well as painful and itchy hardened scaly skin is the most common presentation [2,3,4]. The appearance of psoriatic lesions on normal skin drastically modifies visual appearance, and thereby results in deterioration of a patient’s quality of life. In recent years, psoriasis has witnessed the emergence of improved treatment strategies. Despite these advances, however, it remains a manageable but incurable disease. A better comprehension of the pathogenic mechanisms responsible for disease onset and progression, will therefore, help in developing effective therapeutic options.
Abnormal proliferation, over differentiation, and decreased apoptosis of keratinocytes are the main cellular events that lead to the occurrence and development of psoriasis [5,6,7]. Neutrophils at lesions sites have strong antibacterial action, including degranulation and production of reactive oxygen species (ROS) [8]. Excess production of ROS induces oxidative modification of macromolecules in epidermal keratinocytes, inhibits various protein functions, and ultimately affects cell viability [9]. However, the ubiquitous presence of antioxidant enzymes that reduce or counteract cellular peroxidation induced by ROS, including glutathione (GSH), superoxide dismutase (SOD), catalase (CAT) (337), glutathione peroxidase (GPX), and peroxiredoxin (Prxs) have been reported in a variety of cells [10,11,12]. The effects exerted by these enzymes reduce ROS-mediated cellular damage in epidermal keratinocytes, which is closely associated with abnormal proliferation and decreased apoptosis. Consequently, Prxs may play an important regulatory role in the development and severity of psoriatic skin lesions.
Comparative studies of the proteome of normal and psoriatic skin have revealed differences in proteins that maintain the redox balance between the two. The expression of Peroxiredoxin 2 (Prx II), a member of the Peroxiredoxin (Prxs) family, was upregulated in psoriatic skin tissue [13]. Prx II function as peroxide reductase, and is widely distributed among various cell types. This protein is responsible for the removal of low levels of ROS, and consequently play an important role in cellular proliferation and apoptosis [14,15,16]. We therefore speculated on the possible regulatory role of Prx II in psoriatic skin lesions. The present study examined this hypothesis by analyzing the effect of imiquimod (IMQ) that induces psoriatic skin lesions in Prx II-depleted skin tissue. The study also explored in vitro the regulatory mechanisms involving Prx II in these lesions.
Results
Amelioration of symptoms and reduction of epidermal thickness of IMQ induced psoriatic skin lesions in Prx II-knockout mice
The regulatory role of Prx II was studied in psoriasis models that were generated in Prx II+/+ and Prx II−/− mice by the application of IMQ on their backs. The degree of skin damage on the backs of Prx II−/− mice was found to be significantly lesser (Fig. 1A), with significantly thinner skin (Fig. 1B, C), than that on the backs of Prx II+/+ mice on the seventh day. Although microabscesses were observed, and neutrophils were seen to gather in the upper part of the spinous and granular layers, no significant differences were observed between the epidermis of the two mice (Fig. 1D). Further, no significant differences in the amounts of serum inflammatory factors, including IL-17 and IL-22, were observed between the two mice post application of IMQ (Fig. 1E, F). These results suggest that depletion of Prx II results in alleviation of skin lesions in IMQ-induced psoriasis in mice.

A Damage to skin on the back of mice post IMQ treatment in Prx II+/+ mice and Prx II−/− mice (n = 6 in each group). B Histological changes in mouse skin as seen by H&E staining. C Skin thickness is reduced in Prx II−/− mice compared to Prx II+/+ mice. The data are represented by the mean ± SD (n = 6). D Histological evaluation reveals the presence of microabscesses. E, F Levels of serum L-17 and IL-22 as measured by enzyme-linked immunosorbent assay (ELISA). The data are represented by the mean ± SEM of three separate samples. (**p < 0.01).
Sensitivity of Prx II-knockdown HaCaT cells to H2O2 cytotoxicity
Efficacious knockdown of the Prx II gene in HaCaT cells required successful intracellular vector transfer. The possible interference of the vector with Prx II gene expression was analyzed by comparing Prx II protein and mRNA levels by western blotting and real time PCR in both control (mock) and Prx II-knockdown (sh Prx II) cells (Fig. 2A, B), which revealed significantly lower levels of Prx II in the latter. A concomitant significant effect on cell growth and activity was not evident (Fig. 2C, D). Previous studies have demonstrated the importance of apoptosis in the treatment of psoriasis in HaCaT cells [6]. This was assessed by treating HaCaT cells with H2O2, IMQ, CaCl2, and inflammatory factors such as IL-22 and TNF-α, followed by the detection of apoptosis by the MTT assay. The results revealed that H2O2 significantly induced apoptosis in Prx II-knockdown HaCaT cells in a time- and concentration-dependent manner (Fig. 2E, F); however, no significant effect was evident in the other treatment groups (Fig. 2G–J).

A, B Expression levels of Prx II (Protein and mRNA) in mock and shprx II HaCaT cells as assessed by western blotting and real time PCR. C Measurement of mock and shprx II HaCaT cells viability by MTT. D Detection of apoptosis by assessing fluorescent intensity of Annexin V-PE by flow cytometry. E, F Time and H2O2 concentration-dependent cell viability of mock and shprx II HaCaT cells. G–J Cell viability of mock and shprx II HaCaT cells post IMQ, CaCl2, IL-22, and TNF-α treatment. The data are represented by the mean ± SEM of three separate samples. (*p < 0.05, **p < 0.01).
Induction of mitochondrial dependent apoptosis by H2O2 in Prx II-knockdown HaCaT cells
Intracellular ROS levels in HaCaT cells were estimated post treatment with H2O2 to shed light on underlying mechanisms involved in the sensitivity of Prx II-knockdown HaCaT cells to H2O2 cytotoxicity. The results revealed significant elevation in the accumulation of ROS in Prx II knockdown cells as compared to that in control cells after H2O2 treatment (Fig. 3A, B). Moreover, the accumulation of cytoplasmic Ca2+ significantly elevated in Prx II knockdown HaCaT cells after H2O2 treatment (Fig. 3C, D). As expected, the level of apoptosis was significantly higher in shPrx II HaCaT cells than in mock cells (Fig. 3E, F). Further, western blot analysis was used to detect the expression levels of apoptosis related proteins, which revealed a significant downregulation of Bcl-2, and upregulation of Bax and cleaved Caspase-3 in shPrx II HaCaT cells (Fig. 3G). The Bax/Bcl-2 and cle-Cas3/pro-Cas3 ratio, which were closely associated with accumulated cytoplasmic ROS and Ca2+ induced apoptosis were found to be significantly increased (Fig. 3H, I). These findings demonstrate H2O2 induced mitochondrial-dependent apoptosis in Prx II-knockdown HaCaT cells.

A, B Detection and quantitation of intracellular ROS by flow cytometry. C, D Detection and quantitation of intracellular Ca2+ by flow cytometry. E, F Detection and quantification of apoptosis in mock and shPrx II HaCaT cells post H2O2 treatment by flow cytometry. G–I Analysis and quantification of expression levels of Prx II and apoptosis-related proteins including Bax/Bcl-2 and cle-Cas3/Pro-Cas3 by western blotting post 6 h treatment with H2O2 in HaCaT cells. The data are represented by the mean ± SEM of three separate samples. (*p < 0.05, **p < 0.01, ***p < 0.001).
Reversal of H2O2 induced upregulation of intracellular ROS and Ca2+ levels and apoptosis in Prx II-knockdown HaCaT cells by NAC treatment
Treatment of NAC significantly reversed the increase in ROS and Ca2+ levels induced by H2O2 in HaCaT cells (Fig. 4A–D). Treatment with NAC also significantly inhibited the upregulation of Bax and cle-Caspase3, while concomitantly inducing the upregulation of Bcl-2 (Fig. 4E). The Bax/Bcl-2 and cle-Cas3/pro-Cas3 ratio were found to be significantly decreased (Fig. 4F, G). These findings suggest that the reduction of intracellular ROS attenuates apoptosis induced by H2O2 in Prx II-knockdown HaCaT cells by NAC treatment.

A, B Detection and quantitation of intracellular ROS by flow cytometry. C, D Detection and quantitation of intracellular Ca2+ by flow cytometry. E–G Analysis and quantification of expression levels of Bax/Bcl-2 and cle-Cas3/pro-Cas3 by western blotting post pre-treated with NAC for 30 min, followed by H2O2 treatment for 1 h. in HaCaT cells. The data are represented by the mean ± SEM of three separate samples. (*p < 0.05, **p < 0.01, ***p < 0.001).
Reversal of apoptosis induced by H2O2 in Prx II-knockdown HaCaT cells by BAPTA-AM and 2-APB treatment
Treatment with BAPTA-AM, a selective Ca2+ chelating agent significantly reversed apoptosis in Prx II-knockdown HaCaT cells induced by H2O2 treatment (Fig. 5A, B). The underlying mechanism responsible for the reversal of mitochondria-dependent apoptosis induced by H2O2, consequent to elimination of intracellular Ca2+ was explored by employing 2-aminoethyl diphenylborinate (2-APB), a commonly used inositol 1,4,5-trisphosphate receptor(IP3R) inhibitor [17]. Treatment with 2-APB significantly reversed apoptosis in Prx II-knockdown HaCaT cells induced by H2O2 treatment (Fig. 5C, D). Simultaneously, both BAPTA-AM and 2-APB treatment also significantly inhibited the upregulation of Bax and cle-Caspase3 induced by H2O2, and induced the upregulation of Bcl-2 in Prx II-knockdown HaCaT cells, the Bax/Bcl-2 and cle-Cas3/pro-Cas3 ratio were found to be significantly decreased (Fig. 5E–J). These results provide further evidence that the elimination of intracellular Ca2+ prevents apoptosis induced by H2O2 in Prx II-knockdown HaCaT cells.

A, B Detection of apoptosis in Prx II-knockdown HaCaT cells by flow cytometry. C, D Detection of apoptosis in Prx II-knockdown HaCaT cells by flow cytometry. E–G Analysis of expression levels of Bax/Bcl-2 and cle-Cas3/pro-Cas3 by western blotting pre-treated with BAPTA-AM for 30 min, followed by H2O2 treatment for 1 h. H–J Analysis of expression levels of Bax/Bcl-2 and cle-Cas3/pro-Cas3 by western blotting pre-treated with 2-APB for 30 min, followed by H2O2 treatment for 1 h. The data are represented by the mean ± SEM of three separate samples. (*p < 0.05, **p < 0.01, ***p < 0.001).
Prx II mediated H2O2-induced apoptosis in HaCaT cells via PI3K/AKT/GSK3β pathway
GSK3β plays a key role in regulating the activity of IP3R. phosphorylated GSK3β (Ser9) level was significantly downregulated in Prx II-knockdown HaCaT cells after H2O2 treatment, accompanied by a marked decrease in the phosphorylation levels of its upstream kinase PI3K and AKT (Fig. 6A–D). Treated with GSK3β inhibitor LiCl, the expression levels of the phosphorylated GSK3β (Ser9) was significantly upregulated, Bax and cle-Caspase3 protein were found to be significantly downregulated, and that of Bcl-2 was significantly upregulated in Prx II-knockout HaCaT cells after H2O2 treatment(Fig. 6E–H). Then treated with AKT activator SC79, the expression levels of the phosphorylated AKT, phosphorylated GSK3β (Ser9) were significantly upregulated, Bax and cle-Caspase3 protein were found to be significantly downregulated, and that of Bcl-2 was significantly upregulated in cells treated with SC79 as compared to that in control cells (Fig. 6I–M). Its shows that treatment with Licl and Sc79 can significantly reversed apoptosis signaling induced by H2O2 in Prx II-knockdown HaCaT cells. These results indicate that the Prx II mediated H2O2-induced apoptosis in HaCaT cells by regulating PI3K/AKT/GSK3β pathway.

A–D Analysis of expression levels of p-PI3K/PI3K, p-Akt/Akt and p-Gsk3β/Gsk3β by western blotting by H2O2 treatment for 1 h. E–H Analysis of expression levels of p-Gsk3β/Gsk3β, Bax/Bcl-2 and cle-Cas3/pro-Cas3 by western blotting pre-treated with Licl for 1 h, followed by H2O2 treatment for 1 h. I–M Analysis of expression levels of p-Akt/Akt, p-Gsk3β/Gsk3β, Bax/Bcl-2, and cle-Cas3/pro-Cas3 by western blotting pre-treated with SC79 for 30 min, followed by H2O2 treatment for 1 h. The data are represented by the mean ± SEM of three separate samples. (*p < 0.05, **p < 0.01).
Improvement of symptoms of psoriasis in mice by Conoidin A treatment
Since the depletion of Prx II significantly reduced psoriatic symptoms in mice, Conoidin A, an inhibitor of Prx II was employed to evaluate the possibility of exploiting Prx II as a potential therapeutic target for the disease. Skin on the back of Prx II+/+ mice was treated with IMQ alone for 4 days, followed by combined treatment with 5 mg/kg of Conoidin A and IMQ for an additional 3 days. Mice in the Conoidin A treatment group displayed a significantly lesser degree of skin surface erythema, scaling, and psoriatic lesions as compared to those in the IMQ alone treatment group (Fig. 7A). Histological examination and statistical analysis revealed significantly thinner skin in mice that additionally received Conoidin A as compared to those that only received IMQ (Fig. 7B, C). This indicates that Prx II may serve as a potential therapeutic target for the development of effective treatments for psoriasis.

A Conoidin A and IMQ were used in combination to treat the skin on the back of Prx II+/+ mice (n = 6). B Histological changes in the skin on the back of a mouse as assessed by H&E staining; the yellow line indicates the diameter of the skin. C Changes in skin thickness as compared to psoriatic mice post Conoidin A treatment. The data are represented by mean ± SEM (n = 6). D Volcano map manifestation of 3996 DEGs associated with the skin of psoriatic mice post Conoidin A treatment as determined by RNA sequencing (control vs. Conoidin A group). E GO terms enriched for expression in skin post Conoidin A treatment as compared to mice with psoriasis; the horizontal axis indicates number of DEGs and the vertical axis indicates enriched GO terms. F Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways analyses. G The 10 genes that were significantly altered and closely associated with mitochondrial damage and mitochondria-dependent apoptosis. The data are represented by the mean ± SEM of three separate samples. (**p < 0.01).
The molecular mechanisms underlying the therapeutic effects of Conoidin A were analyzed by RNA sequencing of psoriatic skin samples of mice before and after treatment with Conoidin A. The results revealed the presence of 3996 genes that were differentially expressed (Fig. 7D). GO enrichment analysis revealed significant enrichment of differentially expressed genes (DEGs) involved in cell apoptosis and mitochondrial dysfunction process (Fig. 7E). KEGG pathway analysis showed that the DEGs were mainly associated with Calcium signaling pathway (Fig. 7F). Moreover, genes associated with mitochondria-dependent apoptosis and mitochondrial damage were significantly upregulated. This suggests that Conoidin A may exert its therapeutic effects by inhibiting Prx II to regulate keratinocytes apoptosis, and may consequently serve as a key novel gene targets for the treatment of psoriatic skin lesion (Fig. 7G).
Discussion
Psoriasis is a chronic inflammatory skin disease characterized by the appearance of red plaques comprising an obvious boundary and white scales. Histological examination of these plaques has revealed abnormal proliferation of keratinocytes, as well as immunocyte and vascular proliferation and infiltration [18]. The present study demonstrates that Prx II knock- down HaCaT cells are sensitive to H2O2 stimulation. This effect exerted was found to be mediated by the subsequent activation of the PI3K/Akt/GSK3β signaling pathway, followed by the activation of IP3R in the endoplasmic reticulum that resulted in the elevation of intracytoplasmic Ca2+, which finally culminated in mitochondria-dependent apoptosis. Our results conclusively demonstrate that Prx II is a key player in ROS induced mitochondria-dependent apoptosis of keratinocytes, and may therefore serve as a therapeutic target for psoriasis at the genetic level.
A comparison of the proteome of psoriatic skin with that of normal skin, revealed a significant elevation in the expression of Prx II. The protein is known to be involved in the maintenance of the redox balance, and may thus play an important role in the pathogenesis of psoriasis by regulating ROS [13]. We have demonstrated the cytoplasmic localization of Prx II, as well as its efficaciousness in the elimination ROS in dermal mesenchymal stem cells, hippocampal neurons, and L02 hepatocytes [19,20,21,22]. Further, the protein inhibits the phosphorylation of GSK3β (ser9) in hippocampal neurons and consequently inhibits cell apoptosis [19]. However, the mechanisms employed by GSK3β to exert these effects remain unclear. Our results indicated that GSK3β may regulate mitochondria-dependent apoptosis by modulating intracellular Ca2+ levels. ROS have been shown to play a key role in the regulation of Ca2+ release from the endoplasmic reticulum [23,24,25]. We utilized CaCl2 and H2O2 to treat control, and Prx II knocked down HaCaT cells, to explore the role of Prx II in exogenous and endogenous Ca2+ induced apoptosis of HaCaT cells. Figure 3 shows that Ca2+ release from the endoplasmic reticulum, which is modulated in some degree by the activity of Prx II, induces HaCaT cell apoptosis. IP3Rs are known to contain the GSK3β consensus sequence for phosphorylation at Ser1756, which activates these channels and subsequently promotes Ca2+ outflow from ER [26]. Pretreatment of HaCaT cells with an IP3R inhibitor indicates that GSK3β may regulate the activation of IP3R, and thereby modulate the release of Ca2+ from the endoplasmic reticulum into the cytoplasm, thus indirectly regulating mitochondrial damage, and apoptosis.
We have previously demonstrated that Prx II inhibits GSK3β (Ser9) activity, regulates mitochondrial damage, and thereby results in the reversal of apoptosis in mouse hippocampal neuronal cells [19]. However, the underlying mechanisms behind the regulation of GSK3β (Ser9) phosphorylation by Prx II remain unclear. Our results indicated that Prx II regulates GSK3β (Ser9) phosphorylation through the PI3K/AKT pathway. Prx II is known to contain two Cys residues, and is mainly localized to the cytoplasm. An increase in the level of intracellular ROS, is accompanied by the gradual oxidation of the Cys residues into the sulfenic form (Cys-SOH), sulfinic form (Cys-SO2H) and Prx-sulfonic acid, which concomitantly results in the lowering of intracellular ROS levels [27, 28]. Many studies have shown that ROS mediated PI3K/Akt Pathway activation [29,30,31,32]. However, the exact mechanism has not yet been clarified and needs further study.
The regulatory role of Prx II in psoriasis was further examined by treating a skin mouse model of psoriasis with a crystalline, cell permeable solid compound referred to as Conoidin A. The compound is known to inhibit Prx II activity by covalently binding to its cysteine residues [33]. The epidermis of the psoriasis model mice was found to be significantly thinner if they were treated with Conoidin A. This indicates that Conoidin A may improve symptoms of psoriasis by possibly targeting Prx II, which is a key regulatory factor in the onset and development of psoriatic skin lesion. RNA sequencing of skin tissue treated with either IMQ, or IMQ in combination with Conoidin A revealed that the GO terms as well as specific genes involved in cell death, and apoptosis process were significantly modified in skin tissue post Conoidin A treatment. This implies that Conoidin A may alleviate the symptoms of psoriasis via regulating the apoptosis of epidermal cells.
Conclusion
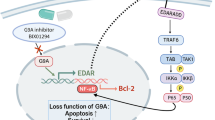
In conclusion, our findings demonstrate that ROS regulate GSK3β dephosphorylation via the PI3K/Akt pathway, which in turn activates IP3R on the ER membrane to induce the release Ca2+ into the cytoplasm. These further results in mitochondria-dependent apoptosis in HaCaT cells. Furthermore, the inhibition of Prx II was found to improve psoriatic symptoms in a mouse model of psoriasis. These findings suggest that Prx II regulated apoptosis of keratinocytes via the PI3K/AKT/GSK3β signaling pathway (Fig. 8), Prx II may therefore serve as a gene target for the development of therapeutic options that mediate apoptosis in order to treat psoriasis.

This effect exerted was found to be mediated by the subsequent activation of the PI3K/Akt/GSK3β signaling pathway, followed by the activation of IP3R in the endoplasmic reticulum that resulted in the elevation of intracytoplasmic Ca2+, which finally culminated in mitochondria-dependent apoptosis.
Materials and methods
Cell culture
Human immortalized keratinocytes (HaCaT) were procured from Bo Gu (Shanghai, China), and cultured in 1640 medium supplemented with 10% FBS and 100 μg/mL penicillin and streptomycin at 37 °C in air supplemented with 5% CO2. Cells were sub-cultured after treatment with 0.25% Trypsin–EDTA once they reached coverage of 90%.
Experimental animal
The mice were exposed to 12 h dark/light cycles, with free access to food and water at 20–22 °C ambient temperature and 50–60% humidity. All experiment treatments conformed to the guidelines laid down by the Animal Care and Use Committee. Thirty male 129/SVJ mice 4–8 weeks were used in the experiments. An area of 2 × 2 cm2 on the back of these mice was cleared of hair. The mice were then randomly divided into five groups, namely, the Prdx2+/+ (wild) control mice, Prdx2–/– (knockout) control mice, Prdx2+/+ mice treated with Imiquimod (IMQ), Prdx2–/– mice treated with IMQ, and Prdx2+/+ as well as Prdx2–/– mice treated with Conoidin A and IMQ. Vaseline and IMQ were topically applied for 7 days to the backs of control mice and those that received only IMQ, respectively. Mice that received combination therapy with Conoidin A and IMQ were topically administered IMQ for the first three days, followed by Conoidin A and IMQ for the next four days. After a total of 8 days post application of the drugs, blood samples were collected from the orbit, and the levels of cytokines. Mice were subsequently sacrificed, and skin samples from their backs were processed for histological evaluation.
Cell viability assay
A total of 8 × 103 HaCaT cells were seeded in 96-well plates. Cells were subsequently treated with varying concentrations of H2O2 (0–20 μM), CaCl2 (0–2 mM), TNF-α (0–100 ng/mL), and IL-22 (0–100 ng/mL). Cell viability was assessed by the MTT assay. Absorbance was measured at 490 nm after completion of the assay.
Flow cytometry analysis
A total of 4 × 105 HaCaT cells were plated per well in 6-well plates and cultured for 24 h and treated with varying concentrations of H2O2 20 μM. The cells were incubated respectively with dihydroethdium (DHE) dye, Fluo3-AM, and Annexin V-PE in cell binding buffer. The level of intracellular ROS, Ca2+ and the level of apoptosis were detected respectively by flow cytometry. Each experiment was performed in triplicates.
Western blot analysis
A total of 4 × 105 HaCaT cells were plated per well in 6-well plates and cultured for 24 h and treated with varying concentrations of H2O2 20 μM. Total protein HaCaT cell lysates were separated by SDS AGE using a 12% SDS polyacrylamide gel. The separated proteins were subsequently transferred to a nitrocellulose membrane, which was blocked with 5% skim milk prepared in TBST. The blocked membrane was incubated overnight at 4 °C with anti-Prx II (cat. no. LF-MA0144, AbFrontier), anti-Bax (cat. no. E-AB-13814, Elabscience Biotechnology), anti-Bcl2 (cat. no. E-AB-60788, Elabscience Biotechnology), anti-cleaved-Caspase 3/Caspase3 (cat. no. ab184787, abcam), anti-β-actin (cat. no. sc-47778, Santa Cruz Biotechnology), anti-pGSK3β(Ser9) (cat. no. AF5830, Beyotime Institute of Biotechnology), anti-GSK3β (cat. no. AF1543, Beyotime Institute of Biotechnology), and anti-pAKT (cat. no. sc-7985-R, Santa Cruz Biotechnology), anti-AKT(cat. no. sc‑8044, Santa Cruz Biotechnology), anti-pPI3K (cat. no. AF3242, Affinity Biosciences), and anti-PI3K (cat. no. 4257s, Cell Signaling Technology). The membrane was subsequently washed and incubated with mouse and rabbit IgG secondary antibodies (cat. no. D110087 and D110058; Shanghai Sangon Biotech Co.) at room temperature for 2 h. Post washing with TBST to remove excess antibodies, a chemiluminescence detection system was used to identify the presence of the probed proteins. Image J software was used to quantify the band intensity.
Statistical analysis
All data are presented as mean ± SEM values from at least three independent experiments. Repeated measures one-way ANOVA and Tukey’s honest significant difference tests were used to compare groups. A p value <0.05 was considered statistically significant. SPSS Statistics v. 25 software (IBM, Armonk, NY, USA) was used for statistical analysis.
Data availability
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
References
Baker BS, Owles AV, Fry L. A possible role for vaccination in the treatment of psoriasis? G Ital Dermatol Venereol. 2008;143:105–17.
Roszkiewicz M, Dopytalska K, Szymańska E, Jakimiuk A, Walecka I. Environmental risk factors and epigenetic alternations in psoriasis. Ann Agric Environ Med. 2020;27:335–42.
Armstrong AW, Read C. Pathophysiology, clinical presentation, and treatment of psoriasis: a review. JAMA. 2020;323:1945–60.
Griffiths CE, Barker JN. Pathogenesis and clinical features of psoriasis. Lancet. 2007;370:263–71.
Jiang BW, Zhang WJ, Wang Y, Tan LP, Bao YL, Song ZB, et al. Convallatoxin induces HaCaT cell necroptosis and ameliorates skin lesions in psoriasis-like mouse models. Biomed Pharmacother. 2020;121:109615.
Yang XG, Jiang BW, Jing QQ, Li WJ, Tan LP, Bao YL, et al. Nitidine chloride induces S phase cell cycle arrest and mitochondria-dependent apoptosis in HaCaT cells and ameliorates skin lesions in psoriasis-like mouse models. Eur J Pharmacol. 2019;863:172680.
Duan X, Liu X, Liu N, Huang Y, Jin Z, Zhang S, et al. Inhibition of keratinocyte necroptosis mediated by RIPK1/RIPK3/MLKL provides a protective effect against psoriatic inflammation. Cell Death Dis. 2020;11:1–14.
Chiang C-C, Cheng W-J, Korinek M, Lin C-Y, Hwang T-L. Neutrophils in psoriasis. Front Immunol. 2019;10:2376.
Wang WM, Jin HZ. Role of neutrophils in psoriasis. J Immunol Res. 2020;2020:3709749.
Sinha K, Das J, Pal PB, Sil PC. Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol. 2013;87:1157–80.
Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 2010;48:749–62.
Lei XG, Zhu J-H, Cheng W-H, Bao Y, Ho Y-S, Reddi AR, et al. Paradoxical roles of antioxidant enzymes: basic mechanisms and health implications. Physiol. Rev. 2016;96:307–64.
Ryu J, Park SG, Park BC, Choe M, Lee KS, Cho JW. Proteomic analysis of psoriatic skin tissue for identification of differentially expressed proteins: up-regulation of GSTP1, SFN and PRDX2 in psoriatic skin. Int J Mol Med. 2011;28:785–92.
Hampton MB, O'Connor KM. Peroxiredoxins and the regulation of cell death. Mol Cells. 2016;39:72–6.
Chandimali N, Huynh DL, Zhang JJ, Lee JC, Yu DY, Jeong DK, et al. MicroRNA-122 negatively associates with peroxiredoxin-II expression in human gefitinib-resistant lung cancer stem cells. Cancer Gene Ther. 2019;26:292–304.
Chandimali N, Jeong DK, Kwon T. Peroxiredoxin II regulates cancer stem cells and stemness-associated properties of cancers. Cancers. 2018;10:305.
Shibukawa Y, Suzuki T. Ca2+ signaling mediated by IP3-dependent Ca2+ releasing and store-operated Ca2+ channels in rat odontoblasts. J Bone Min Res. 2003;18:30–8.
Griffiths CEM, Armstrong AW, Gudjonsson JE, Barker J. Psoriasis. Lancet. 2021;397:1301–15.
Jin MH, Yu JB, Sun HN, Jin YH, Shen GN, Jin CH, et al. Peroxiredoxin II maintains the mitochondrial membrane potential against alcohol-induced apoptosis in HT22 cells. Antioxidants. 2019;9:1.
Jin MH, Yu NN, Jin YH, Mao YY, Feng L, Liu Y, et al. Peroxiredoxin II with dermal mesenchymal stem cells accelerates wound healing. Aging. 2021;13:13926–40.
Han YH, Jin MH, Jin YH, Yu NN, Liu J, Zhang YQ, et al. Deletion of peroxiredoxin II inhibits the growth of mouse primary mesenchymal stem cells through induction of the G(0)/G(1) cell-cycle arrest and activation of AKT/GSK3β/β-catenin signaling. In Vivo. 2020;34:133–41.
Han YH, Li WL, Jin MH, Jin YH, Zhang YQ, Kong LZ, et al. Peroxiredoxin II inhibits alcohol-induced apoptosis in L02 hepatocytes through AKT/β-catenin signaling pathway. Anticancer Res. 2020;40:4491–504.
Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012;19:1880–91.
Ly LD, Xu S, Choi S-K, Ha C-M, Thoudam T, Cha S-K, et al. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp Mol Med. 2017;49:e291.
Gleichmann M, Mattson MP. Neuronal calcium homeostasis and dysregulation. Antioxid Redox Signal. 2011;14:1261–73.
Gomez L, Thiebaut PA, Paillard M, Ducreux S, Abrial M, Crola Da Silva C, et al. The SR/ER-mitochondria calcium crosstalk is regulated by GSK3β during reperfusion injury. Cell Death Differ. 2016;23:313–22.
Ishii T, Warabi E, Yanagawa T. Novel roles of peroxiredoxins in inflammation, cancer and innate immunity. J Clin Biochem Nutr. 2012;50:91–105.
Kim Y, Jang HH. Role of cytosolic 2-Cys Prx1 and Prx2 in redox signaling. Antioxidants. 2019;8:169.
Deng S, Dai G, Chen S, Nie Z, Zhou J, Fang H, et al. Dexamethasone induces osteoblast apoptosis through ROS-PI3K/AKT/GSK3β signaling pathway. Biomed Pharmacother. 2019;110:602–8.
Xie C, Yi J, Lu J, Nie M, Huang M, Rong J, et al. N-acetylcysteine reduces ROS-mediated oxidative DNA damage and PI3K/Akt pathway activation induced by Helicobacter pylori infection. Oxid Med Cell Longev. 2018;2018:1874985.
Su X, Shen Z, Yang Q, Sui F, Pu J, Ma J, et al. Vitamin C kills thyroid cancer cells through ROS-dependent inhibition of MAPK/ERK and PI3K/AKT pathways via distinct mechanisms. Theranostics. 2019;9:4461.
Li F, Li J, Wang P-H, Yang N, Huang J, Ou J, et al. SARS-CoV-2 spike promotes inflammation and apoptosis through autophagy by ROS-suppressed PI3K/AKT/mTOR signaling. Biochim Biophys Acta Mol Basis Dis. 2021;1867:166260.
Haraldsen JD, Liu G, Botting CH, Walton JG, Storm J, Phalen TJ, et al. Identification of conoidin A as a covalent inhibitor of peroxiredoxin II. Org Biomol Chem. 2009;7:3040–8.
Acknowledgements
This research was a part of the project titled [Development and advancement of mass production process for Ishige okamurae, a functional material for improving sensitive skin condition], funded by the Ministry of Oceans and Fisheries, Korea.
Funding
This work was supported by Heilongjiang Provincial Natural Science Foundation of China (LH2021C061). This research was supported by the Korea Research Institute of Bioscience and Biotechnology (KRIBB) Research Initiative Program (KGM5162322, KGM5242322).
Author information
Authors and Affiliations
Contributions
Y-HH, LF, S-JL, S-IP, H-NS, and TK contributed to the conception of the study. S-IP, A-GW, LF, and Y-QZ contributed to the execution of the experiment, specially to the acquisition of skin samples and data collection and analysis. H-NS, M-HJ, and TK performed the analysis and the quality assessment of the study. Y-HH, LF, S-JL, H-NS, and TK were responsible for the study design, writing the manuscript, and performing the literature search. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All mouse procedures in this study were approved by the Institutional Animal Care and Use Committee protocol approved by the Committee for Care and Use of Laboratory Animals at Dalian Medical University (AEE21038, A-GW).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Han, YH., Feng, L., Lee, SJ. et al. Depletion of peroxiredoxin II promotes keratinocyte apoptosis and alleviates psoriatic skin lesions via the PI3K/AKT/GSK3β signaling axis. Cell Death Discov. 9, 263 (2023). https://doi.org/10.1038/s41420-023-01566-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-023-01566-z
