
Abstract
Acute myeloid leukemia (AML) is the most common hematopoietic malignancies, and chemotherapy resistance is one of the main causes of relapse. Because of lower survival rate for patients with relapse, it is pivotal to identify etiological factors responsible for chemo-resistance. In this work, direct MeRIP-seq analysis of sequential samples at stage of complete remission (CR) and relapse identifies that dysregulated N6-methyladenosine (m6A) methylation is involved in this progression, and hypomethylated RNAs are related to cell differentiation. m6A demethylase FTO is overexpressed in relapse samples, which enhances the drug resistance of AML cells in vivo and in vitro. In addition, FTO knockdown cells exhibit stronger capacity of differentiation towards granules and myeloid lineages after cytosine arabinoside (Ara-C) treatment. Mechanistically, FOXO3 is identified as a downstream target of FTO, the hypomethylation of FOXO3 mRNA affects its RNA degradation and further reduces its own expression, which ultimately result in attenuated cell differentiation. Collectively, these results demonstrate that FTO-m6A-FOXO3 is the main regulatory axis to affect the chemotherapy resistance of AML cells and FTO is a potential therapeutic target of chemotherapy resistance in AML.
Similar content being viewed by others


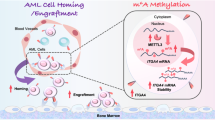
Introduction
Acute myeloid leukemia (AML) is one of the most common hematopoietic malignancies, characterized with high heterogeneity, abnormal clonal expansion and differentiation block of progenitor cells [1], which is a serious risk to human health.
Currently, the main treatment for AML is still chemotherapy. Although most of patients can achieve complete remission (CR) undergoing conventional chemotherapy [2], 5-year survival rates for younger (aged <60 years) and older patients (aged >60 years) reached only 40% and <20% respectively, indicates most of them are diagnosed refractory disease or relapse [3,4,5,6,7,8]. Because of limited treatment options, relapse is the leading cause of death in AML [9,10,11,12]. The main factor of relapse is chemo-resistance, so it is pivotal to identify etiological factors responsible for chemo-resistance, which relies on comprehensive understanding of the molecular mechanism underlying drug response. In the prevailing view, gene mutation, abnormal regulation of RNA and protein may lead to drug resistance. For example, some gene mutations occurred in autophagy signal pathway and drug resistance related protein, abnormal expression of mRNAs and microRNAs will lead to drug resistance in AML patients [3, 13,14,15].
Epigenetics has been proved to play an important role in physiological and pathological processes. RNA N6-methyladenosine (m6A) is the most abundant internal modification in mRNAs, including the hematopoietic system development and the maintenance of hematopoietic stem cell function [16,17,18,19,20,21]. Under the regulation of writers, erasers and readers, m6A plays important roles in regulating RNA fate, such as RNA degradation [22], nuclear export [23] and so on [24, 25]. Emerging evidence has shown many types of cancers are characterized by abnormal m6A modification due to dysfunctional m6A regulators, especially in AML [26]. It has been reported that m6A writers (METTL3, METTL14, METTL16), erasers (FTO, ALKBH5) and readers (IGF2BP family, YTHDC1) are all involved in regulating AML progression [27,28,29,30,31]. Thus, intervention of key regulators of m6A modification has been recognized as a potential therapeutic approach for cancer therapy [32,33,34,35,36,37].
FTO is one of m6A demethylases and closely related to fat-mass and obesity [38,39,40]. It has been reported that FTO decreased the drug resistance of melanoma and cervical squamous cell carcinoma through mRNA demethylation [41, 42]. However, there is no evidence of the relationship between m6A modification and chemotherapy resistance in AML. Thus, investigating the m6A regulation in chemotherapy resistance AML may provide more comprehensive insights of recurrence after chemotherapy, and help develop more effective targeted therapies to treat AML.
In the present study, we sought to investigate whether dysregulated RNA m6A methylation involves in AML cell resistant to chemotherapy and explore the underlying molecular mechanism. Our results indicate that the elevated expression of FTO plays a critical role in chemotherapy resistance. Combined MeRIP-Seq analysis and functional experiments, we found FOXO3 as a downstream target of FTO. The results suggested that FTO decreased the m6A modification of FOXO3 and enhanced its mRNA degradation rate. In summary, our study provides insights into the multiple molecular mechanisms of chemotherapy resistance in AML, which leads to the recurrence.
Results
Abnormal m6A modification is found in relapsed samples
In order to investigate the impact of dysregulated RNA m6A methylation in AML relapse, we first analyzed the alterations of m6A regulators (10 writers, 2 erasers and 12 readers) between diagnosis and relapse samples based on published article [43]. The expression of m6A regulatory factors changed between diagnosis and relapse of patients, among which METTL3 and FTO were both increased at relapse (Fig. 1A and Fig. S1A).

A RNA expression level comparison of core m6A methyltransferases and demethylases between sequential diagnosis (n = 16) and relapse samples (n = 16) from a public dataset. P-values were calculated using Wilcoxon test, and P < 0.05 was considered as statistically significant. *P < 0.05. B Bar graph showing the total peaks identified in three pairs of sequential samples. C The statistics of the counts of RNAs with m6A peak in three pairs of sequential samples. D Bar graph showing the m6A-modified RNA classification. C represents CR and R represents relapse E, F Gene ontology (GO) analysis of the RNAs with m6A peaks in CR (E) and relapse (F) samples.
Previous studies reported that the resistance of tumor cells to chemotherapy drugs is one of the important reasons for recurrence [44]. Considering the both high AML cell burden at diagnosis and relapse, we picked sequential samples underwent CR after chemotherapy and eventually relapse for subsequent experiments (Table S1). To analyze the change of m6A methylation, we performed MeRIP-seq by using sequential samples at CR and relapse after chemotherapy, respectively (Table S2). In line with previous findings, “UGGAC” was the most conserved motif in the m6A peaks identified from all samples (Fig. S1B), which were unevenly distributed across the whole transcripts as previously reported (Fig. S1C, S1D).
Under standard identification conditions, a higher number of both m6A peaks and RNAs with m6A modifications were identified in the recurrent samples (Fig. 1B, C), which were mainly distributed on mRNA (Fig. 1D). Interestingly, increased m6A modification on lncRNA during relapse suggests that lncRNA also involved in regulating AML relapse (Fig. 1D). Next, Gene ontology (GO) analysis showed that the genes with m6A peaks in CR and relapse samples was mainly enriched in cell differentiation, cell death and apoptosis related pathways (Fig. 1E, F and Table S3), indicates that altered chemotherapy resistance in AML cells may be related to these functions. Taken together, the above results suggest that dysregulated m6A modification plays important roles in AML relapse.
Hypomethylated genes at recurrence are enriched in cell differentiation
Based on the dysregulation of m6A in AML samples during recurrence, we further analyzed differentially methylated m6A modification with three pairs of sequential samples (Table S4), and the results were in consistent with previous (Fig. S2A, S2C). In addition to that, we also found the proportion of differentially methylated peaks also increased in lncRNAs (Fig. 2A), indicates that m6A modification on lncRNA may also be involved in the regulation of AML recurrence.

A Bar graph showing the m6A-modified RNA classification with hypomethylated (down) and hypermethylated (up) sites. B GO analysis of the hypomethylated RNAs in relapse samples.
To investigate the functions affected by abnormal m6A modification, we next performed GO analysis of differentially methylated genes. As Fig. 2B showed that m6A-hypomethylated genes mainly related to cell differentiation, hematopoietic regulation and cell activation, among which “leukocyte differentiation”, “regulation of myeloid cell differentiation” and “regulation of hemopoiesis” these three pathways were enriched in all three paired samples (Fig. 2B and Table S5), while the m6A-hypermethylated genes were not found co-exist pathways (Fig. S2D and Table S5). These results indicate that hypomethylated genes related to cell differentiation play roles in AML relapse.
FTO suppression reduces the chemo-resistance of AML cells through cell differentiation
Based on previous data, we compared the expression level of core m6A methylases and demethylases in sequential samples from 14 patients. Interestingly, we found that FTO was highly expressed in relapse (after chemotherapy) samples (Fig. 3A), which suggests that high expression of FTO may regulate the chemo-resistance and relapse of AML. Therefore, we explored the effect of FTO on chemotherapy resistance of AML cells. Cell viability assay was performed using two human AML cell lines (MV4-11 and THP-1) with high FTO expression (Fig. 3B). In the treatment of cytarabine (Ara-C) or doxorubicin (DOX), FTO knockdown (KD) (Fig. 3C) resulted in a significant reduction of chemotherapy resistance in both cell lines (Fig. 3D–G). Combined the regulation of FTO to chemo-resistance with GO analysis of hypomethylated genes, we hypothesized that overexpression of FTO leads to chemotherapy resistance through abnormal cell differentiation.

A Comparison of RNA expression between complete remission (CR) and relapse samples. B Western blot analysis showing the expression level of FTO in six AML cell lines. Highly FTO expressed cell line were marked by black arrows. C Western blot analysis confirming the effects of FTO knockdown in MV4-11 and THP-1 cells. D, E Cell viability assay showing the effects of FTO on chemotherapy resistance with Ara-C (D) or DOX (E) treatment in MV4-11 cells. F, G Cell viability assay showing the effects of FTO on chemotherapy resistance with Ara-C (F) or DOX (G) treatment in THP-1 cells. H, I Flow cytometry showing the regulation of FTO to cell differentiation with Ara-C treatment, DMSO was used as vehicle (H). The statistical analysis was shown in I. Representative results from three replicates were shown here. P-values were calculated using Two-tailed Student’s t-test, and P < 0.05 was considered as statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
To verify this hypothesis, we performed flow cytometry to identify the effect of FTO on cell differentiation after Ara-C treatment. FTO knockdown cells exhibit stronger capacity of differentiation towards granules and myeloid lineages (Fig. 3H, I). Taken together, the above results indicate that FTO can alter the chemotherapy resistance of AML cells in terms of mediating their differentiation process.
For further verification, we performed functional experiments using Ara-C resistant (Resis) cells. Representative AML cell line MV4-11 was initially exposed to increasing concentrations of the chemotherapeutic drug Ara-C, until they could grow in medium containing high dose of drug (Fig. S3A). The cell viability results showed that knocking down FTO in Ara-C resistant cells could partially reduce drug resistance (Fig. S3B). In addition, drug-resistant cells showed increased expression of CD11b and CD14 (Fig. S3C), and exhibited a weak tendency of continued differentiation after FTO knockdown (Fig. S3D, S3E). Compared the FTO function between sensitive and Ara-C resistant cells, these results suggested that although FTO plays roles in drug resistance of AML cells, inhibition of FTO expression has little significance in delaying the AML progression in drug-resistant cells.
The knockdown of FTO decreases chemotherapy resistance in vivo
To verify the role of FTO in mediating chemotherapy resistance in AML progression, we assessed the effect of FTO-knockdown in NPG mice with two regimens of drug administration: vehicle and Ara-C (100 mg/kg, every two days, intravenous injections). After confirming that the proportion of transplanted tumor cells (GFP+) in mice bone marrow (BM) was higher than that in peripheral blood (PB) through flow cytometry (supplementary Fig. S4A–S4C), we divided thirty mice into three groups randomly and injected tumor cells (negative control (NC), KD-1, KD-2) through tail vein. The mice were treated with Ara-C or PBS when the percentage of GFP+ cells in PB reached approximately 5% without weight change (Fig. 4A and Fig. S4D). Consistent with our previous results in vitro, knockdown of FTO significantly decreased the inhibitory effects of Ara-C treatment in vivo (Fig. 4B, C). Compared the spleen size between administration and control group, we obtained that the spleen size of control group was larger than administration group (Fig. S4E). Moreover, the tumor cell infiltration in NC was stronger than FTO knocked down in the spleen (Fig. 4D). In general, the results in vivo confirmed that the knockdown of FTO significantly decreased chemotherapy resistance.

A Flow cytometry to detect the percentage of GFP+ cells in peripheral blood (PB) before Ara-C treatment. B Flow cytometry to detect the percentage of GFP+ cells in peripheral blood after Ara-C treatment. C Flow cytometry to detect the percentage of GFP+ cells in bone marrow after Ara-C treatment. D Representative images of tumor cell infiltration in spleen. The tumor cells were marked by white arrows. Scale bars represent 100 μm (upper panel) and 50 μm (lower panel), respectively, and the regions of lower panel were marked with white dotted line. P-values were calculated using Two-tailed Student’s t-test, and P < 0.05 was considered as statistically significant. ***P < 0.001 and ****P < 0.0001, ns: not significant.
FOXO3, CREBBP, and SIRPA are potential targets of FTO
To elucidate the molecular mechanism of FTO in regulating chemotherapy resistance of AML cells, we next searched for downstream targets of FTO, which were enriched in the pathways related to cell differentiation (Fig. 2B) and exhibited differential methylation levels in relapsed samples. We screened Integrative Genomics Viewer (IGV) profiles for hypomethylated genes associated with the cell differentiation obtained by MeRIP-seq (Fig. 5A). The m6A-IP-qPCR assay further verified that CREBBP, FOXO3, KMT2E, SIRPA and SYK had m6A modifications in both cell lines (compared to the expression level of CLUC), especially CREBBP and FOXO3, whose modification levels were stronger than the positive control (GLUC) (Fig. 5B, C).

A Genome browser tracks of m6A peaks in the selected transcripts in complete remission and relapse samples, C: complete remission; R: relapse. B, C m6A-IP-qPCR results showing the level of m6A modification in MV4-11 (B) and THP-1 (C) cell lines. GLUC represents positive control and CLUC represents negative control. D, E m6A-IP-qPCR results showing the effects of FTO knockdown on the m6A levels in MV4-11 (D) and THP-1 (E) cell lines of indicated transcripts. Representative results from three replicates were shown here. P-values were calculated using Two-tailed Student’s t-test, and P < 0.05 was considered as statistically significant. *P < 0.05, **P < 0.01.
In addition to that, m6A-IP-qPCR results also showed that compared with other RNAs tested, CREBBP, FOXO3 and SIRPA RNAs exhibited significant and consistent changes in their m6A levels upon FTO knockdown in both MV4-11 (Fig. 5D and Fig. S5A) and THP-1 cell lines (Fig. 5E and Fig. S5B). These results suggested that these three genes are the potential targets of FTO.
FTO regulates the chemotherapy resistance of cells through FOXO3
To further identify the targets affecting differentiation, we examined the m6A modification of FTO targets under low dose Ara-C treatment. FOXO3 and CREBBP remained highly methylated level (Fig. 6A) and elevated methylation in cells with FTO knocked down (Fig. 6B and Fig. S5C), implying that these two genes may be FTO targets involved in regulating differentiation process.

A m6A-IP-qPCR results showing the level of m6A modification in MV4-11 cells with Ara-C treatment. GLUC represents positive control and CLUC represents negative control. B m6A-IP-qPCR results showing the effects of FTO knockdown on the m6A levels in MV4-11 cells with Ara-C treatment. C Western blot analysis to detect the expressions of FOXO3 proteins before and after knocking down FTO in MV4-11 (left) and THP-1 (right) cell lines. GAPDH was used as a loading control. D Flow cytometry to determine the effects of FOXO3 knockdown in rescuing cell differentiation capability of FTO knockdown cells. E RNA stability assay to determine the effect of FTO knockdown on the half-lives (t1/2) of FOXO3 mRNA in MV4-11 (upper panel) and THP-1 (under panel) cells. TI, transcript inhibition. F Working model showing the molecular mechanism of FTO in regulating chemotherapy resistant AML cells. Elevated expression of FTO leads to RNA m6A hypomethylation of FOXO3, which further affected its RNA stability. Downregulation of FOXO3 results in cell differentiation block, which ultimately leads to chemotherapy resistance and AML relapse. Arrows indicate the sequential changes in gene transcription and expression caused by overexpression of FTO. Representative results from three replicates were shown here. P-values were calculated using Two-tailed Student’s t-test, and P < 0.05 was considered as statistically significant. *P < 0.05, **P < 0.01.
As we all known, protein is the final form that exerts biological functions, dysregulated of m6A methylation leads to the change of protein level [33, 45]. The increase of FOXO3 protein level in FTO knockdown cells implied that served as a target of FTO and assisted in regulating differentiation (Fig. 6C). In order to identify whether FTO regulated the cell differentiation through FOXO3, we found the ratio of CD11b+ or CD14+ cells were partially decreased after knocking down of FOXO3 for further rescue (Fig. 6D and Fig. S5D, S5E), suggesting a role of cell differentiation mediated by FTO in regulating FOXO3 expression level.
To investigate the mechanism, the half-life of FOXO3 mRNA was increased in FTO knockdown cells compared to negative control cells after treating with actinomycin D (Fig. 6E). However, there was no significant difference of the FOXO3 mRNA localization (Fig. S5F, S5G), which indicates that the increase of FOXO3 mRNA stability may lead to the increase of its protein level. In conclusion, FTO-m6A-FOXO3 is the main regulatory axis, which affects the chemotherapy resistance of tumor cells by mediating differentiation in the case of abnormal modification of m6A.
Discussion
RNA m6A methylation is the most abundant RNA modification in mammals, abnormal expression of m6A regulatory factors causes dysregulation of m6A modification, which leads to biological dysfunction [46]. It has been proved that dysregulation of epigenetic modifications is a common feature of most human cancers [26]. Currently, the resistance of tumor cells to chemotherapy is still a major obstacle in cancer treatment, the role of m6A modification in chemical stimulus response remains to be studied.
In this study, we found that m6A modification took parts in “remission-relapse” process through MeRIP-seq, and hypomethylated genes were associated with cell differentiation. We next confirmed that m6A demethylase FTO was highly expressed in samples that relapse after chemotherapy, combined with the ability of FTO to regulate cell differentiation [27], we picked FTO to investigate its effect on recurrence. Our results verified that FTO knocked down reduces the resistance of AML cells to chemotherapy at both in vitro and in vivo levels. In terms of mechanism, FOXO3, a factor capable of regulating cell differentiation [47,48,49], has been proved to be a downstream target of FTO and assist in influencing chemotherapy resistance (Fig. 6F).
Currently, recurrence is still the leading cause of death in AML, which usually related to chemotherapy resistance. Yang et al. pointed out the FTO expression level upregulated in human melanoma, knockdown of FTO sensitized melanoma to anti-PD-1 treatment via adaptive immunity [41]. Besides of that, Lin et al. demonstrated FTO played a critical role in sensitivity of chemo-resistant colorectal cancer cells to 5-fluorouracil, which enhanced tolerance through the activity of autophagy [50]. FTO also has been proved contribute to the chemo-radiotherapy resistance of cervical squamous cell carcinoma [42]. Interestingly, the development of resistance phenotype during target therapy with tyrosine kinase inhibitor (TKI) depends on the m6A reduction resulting from FTO overexpression [51]. In consistent with all above findings, here, we demonstrated that FTO took part in sensitivity to chemotherapy in AML, which enhanced resistance via regulating cell differentiation. Similar results were found in both solid and hematologic tumors, so we suggest that m6A, especially FTO, regulates chemotherapy resistance and recurrence in tumor. Besides of that, the combination of FTO inhibitors and conventional chemotherapy may be a more effective strategy for cancer therapy.
From previously published studies, FTO plays tumor-promoting roles in various cancers, such as AML [27], glioblastoma [52], breast cancer [53], gastric cancer [54], etc. Li et al. pointed out that the FTO-knockdown decreased undifferentiated NB4 cells (CD11b-/CD14-) by all-trans retinoic acid (ATRA) treatment, otherwise FTO-overexpression increased CD11b-/CD14- cells, indicating that FTO could inhibit ATRA-induced differentiation of APL cells [27]. In consistent with that, we also found an increased tendency of differentiated cells with FTO knocked down after Ara-C treatment, suggesting that cells with high FTO level perform stronger resistance. It is possible that survival of relapsed cells relies more on the FTO.
Based on bioinformatic analysis and experimental validation, we identified FOXO3 as a regulator connecting m6A modification with differentiation. FOXO3 was shown a critical role in regulating cell differentiation, overexpression of FOXO3 will induce neural stem cell and follicular helper T cells differentiation [47, 49]. Intriguingly, the mechanism of drug resistance to classical chemotherapeutics are invariably linked to FOXO3-FOXM1 axis [48]. In addition, it has been demonstrated that FOXO3 was downstream targets of METTL3 in hepato-cellular carcinoma, hypermethylation of FOXO3 decreased the sorafenib resistance [55]. In this study, we also validated FOXO3 as a downstream target of FTO in AML. FTO reduction increased m6A modification level of FOXO3, which leads to increase in protein level. Our results demonstrate that FOXO3 assists in regulating chemotherapy resistance in AML through cell differentiation.
Previous studies indicated a small distribution of m6A modification on lncRNAs [56]. There are two mainly opinions being clarified in research into m6A and lncRNAs [57]. One is m6A modification can regulate the relationship between lncRNA and specific DNA sites [58,59,60], the other is m6A modification on lncRNAs provides binding sites for readers, which then induces RNA binding proteins to regulate the function of lncRNAs [58, 61]. As previously described, we found that there were also m6A modification on lncRNAs, and the count increased in AML relapse samples, suggesting they were also involved in the recurrence of AML. We further explored the methylation of lncRNAs associated with tumor progression through bioinformatics analysis and IGV [57], and found there was no significant change between CR and relapse samples, such as XIST, MALAT1 and so on (data not shown). Thus, to fully uncover the roles and mechanisms of lncRNAs in promoting AML relapse, further studies about the relationship between m6A and lncRNAs are necessary.
In conclusion, this study demonstrated that m6A modification affects disease recurrence in a chemotherapy resistance manner, providing a new perspective for a comprehensive understanding of AML. We uncovered a critical role of m6A demethylase FTO in regulating AML chemotherapy resistance in vitro and in vivo. Our study suggests that the FTO-m6A-FOXO3 axis acts as a critical regulator in high FTO expression level AML cells, and highlights the functional importance of m6A modification machinery in chemotherapy resistance. More importantly, targeting FTO may represents a potential therapeutic strategy to enhance the treatment response in AML patients suffered from chemotherapy resistance.
Methods and materials
AML patient samples
The sequential AML samples were obtained at the stage of CR and relapse with informed consent at the Hematology Department of Peking University People’s Hospital, and were approved by the local ethics review board (Reference number: 2023PHB039-001). The leukemic samples were stored in liquid nitrogen until used. Total RNA was extracted through TriReagent (T9424, Sigma, Missouri, USA) following the manufacturer’s protocol.
Cell culture
The human AML cell lines MV4-11, THP-1, U937, Molm-13, NB4 and HL60 were maintained in the laboratory. All cell lines have been authenticated and tested as free of mycoplasma contamination. All cell lines were grown in RPMI medium 1640 (C11875500BT, Gibco, Grand Island, USA) containing 10% FBS (Gibco) and 1% penicillin-streptomycin (10378-016, Gibco).
The FTO-knockdown MV4-11 and THP-1 cell lines were established by using lentivirus system. Cells were transfected with lentivirus particles and followed by 5 μg/ml puromycin selection. The virus was purchased from Vigene Biosciences (Shandong, China) and shRNA sequence employed in this experiment is listed in Key Resources Table.
Xenograft mice model of AML
Thirty male NOD.Cg-Prkdcscid Il2rgtm1Vst/Vst (NPG) mice (6 weeks old) were divided into three groups and implanted with 4 × 106 cells of control (MV4-11 NC) or FTO-knockdown (MV4-11 KD-1 or KD-2) by tail vein injection, respectively. Tumor growth was monitored every week through flow cytometry. Each group of mice were further randomized into two groups and treated with cytarabine (Ara-C) (S1648, Selleck, Texas, USA) or PBS (100 mg/kg, once every two days, three times in all) through tail vein injection when the percentage of GFP+ cells in peripheral blood reached ~5%. The mice were sacrificed and detected the proportion of GFP+ cells in bone marrow when chemotherapy finished. The tumor infiltration in spleen was identified through H&E staining. All the animal experiment were performed according to the guidelines of Animal Care and Use Committee. The Institutional Review Board approval number is 2020PHE006.
Western blot analysis
Cellular proteins were extracted with RIPA lysis buffer containing protease inhibitor cocktail (#04693159001, Roche, Basel, Switzerland) and phosphatase inhibitor cocktail (#04906837001, Roche). About 2 × 105 cells per lane was applied to SDS-PAGE and immunoblotting analysis. Detailed information of the primary and secondary antibodies used in this study is shown in Key Resources Table. The quantifications of Western blot were performed through ImageJ.
Cell viability assay
Cells were seeded in density of 2 × 104 per well into 96-well plate and incubate with gradient concentrations of Ara-C (#S1648, Selleck) or doxorubicin (DOX) (#S1208, Selleck) for another 48 h. Cell viability was determined through Cell Counting Kit-8 (CCK8) (#CK04, DOJINDO, Kumamoto Prefecture, Japan) following the manufacturer’s protocol. The absorbance was detected at 450 nm.
Flow cytometry analysis
For in vitro cell differentiation assay, FTO knockdown and control cells were incubated with 250 ng/μl Ara-C (#S1648, Selleck) for 48 h, cells were harvested and washed with chilled PBS and stained with PE-labeled anti-CD11b (#301306, BioLegend, California, USA) and APC-labeled anti-CD14 (#367118, Biolegend) for flow cytometry analysis. The fluorescence intensity was detected using CANTO PLUS (BD Biosciences, New Jersey, USA) and analyzed with the FlowJo.
Construction of Ara-C-resistant cell line
MV4-11 cells were used for constructing drug-resistant cell line. Cells were treated with 1/10 of the wildtype cell IC50 at the initial for 24 h and then transferred to drug-free medium. The concentration of Ara-C was increased and the incubation continued in the same way until the cells were able to grow stably at the high dose of Ara-C.
Subcellular fractionation analysis of RNA
FTO knockdown and control cells were subjected to subcellular fraction using NE-PER Nuclear and Cytoplasmic Extraction Reagents (#78833, Thermo Fisher) according to the manufacturer’s manual. 200 μl of the cytoplasm extract and 100 μl of the nucleus extract were used for total RNA extraction, reverse transcription and qPCR. ND4 was used as separation marker.
RNA stability assay
For RNA stability assay, MV4-11 and THP-1 cells (FTO knockdown and control) were incubated with 5 μg/ml Actinomycin D (#A9415, Sigma). Cells were harvested at 0 h, 1 h, 2 h, and 3 h post treatment and subjected to RT-qPCR for quantitative analysis of target genes. 18S was used as negative control. The degradation rate of RNA was calculated using the following equation:
where t is the duration of transcription inhibition, k is the degradation rate, Nt and N0 are the RNA quantities at time t and time 0. The half-life of RNA (T1/2) can be calculated from the degradation rate as follows:
m6A-seq and m6A-IP-qPCR
For total RNA m6A-seq, it was performed as previously described with miner revision [62]. In brief, total RNA was treated with DNase I (#04716728001, Roche) first to remove genomic DNA. Next, total RNA was fragmented to 100–150 nt by using RNA Fragmentation Reagents (#AM8740, Thermo Fisher, Waltham, USA). At least 10 μg fragmented total RNA was incubated with 5 μg anti-N6-methyladenosine antibody (#202003, Synaptic Systems, Gottingen, Germany) at 4 °C for 6 h, followed by incubation with DynabeadsTM Protein A (#10002D, Thermo Fisher) for another 3 h. Immunoprecipitated RNA was eluted with RLT buffer and recovered through RNeasy Mini Kit (#74106, Qiagen, Germantown, USA). cDNA libraries were constructed from immunoprecipitated RNAs and input RNAs, respectively, by using SMARTer Stranded Total RNA-Seq Kit v2 (#634413, Takara, Kyoto, Japan), and then subjected to next-generation sequencing on NovaSeq platform.
For gene-specific m6A-IP-qPCR, 20 μg of fragmented total RNA (300–500 nt) added 0.1 fmol of positive spike-in control was used for experiment. The m6A-IP was performed by using the N6-methyladenosine Enrichment kit (E1610S, New England Biolabs, Ipswich, USA). Reverse transcription was performed by using ReverTra Ace qPCR RT Master Mix (#FSQ-301, TOYOBO, Osaka, Japan). RT-qPCR was performed by using PowerUP SYBR Green Master Mix (A25742, Invitrogen, California, USA). GAPDH was used as internal control. The sequences of primers used in this study are listed in Key Resources Table.
Processing of high-throughput sequencing data
Trimmomatic software were used to remove adapter and low-quality reads of raw sequencing data [63]. Quality distribution plot and base content distribution were generated by FASTQC [Available: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/]. Clean reads were aligned to the human genome (build 38) using STAR [62], which were used for subsequent analysis.
m6A-seq data analysis
The m6A peak was identified through MetPeak R package [64] in each m6A-IP sample with the corresponding input sample as control. The identification of m6A peaks followed MetPeak; PEAK_CUTOFF_P value = 0.05, FOLD_ENRICHMENT = 1 were considered as high-confidence peaks and used for further analysis. The density plot of peaks in 3'UTR, CDs and 5'UTR was completed with Guitar [65].
Differential methylation analysis
To determine differential m6A methylation modifications between CR and relapse samples, the MeTDiff R package effectively modeled the reads of methylation data and predicted the differential methylation sites by using the maximum likelihood ratio test. High-confidence differential m6A peaks were determined with the criteria of “P-value < 0.05, | log2 (fold-change) | > 1”.
GO analysis
Gene ontology (GO) analysis was performed to facilitate elucidating the biological implications of unique genes in the significant or representative profiles of the gene in the experiment [66]. We downloaded the GO annotations from Ensembl (http://asia.ensembl.org/index.html), and the Gene Ontology (http://www.geneontology.org/). ClusterProfiler was applied to identify the significant GO categories and FDR was used to correct the P-values.
Quantification and statistical analysis
Statistical analyses were performed using Graphpad Prism 8.0 software. P-values were calculated using Two-tailed Student’s t-test or Wilcoxon test, and P < 0.05 was considered as statistically significant. Data are shown as mean ± SD. At least three biological replicates were included in each experiment.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive [67] in National Genomics Data Center [68], China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA-Human: HRA003825) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa-human.
References
Pelcovits A, Niroula R. Acute myeloid leukemia: a review. R I Med J (2013). 2020;103:38–40.
Luppi M, Fabbiano F, Visani G, Martinelli G, Venditti A. Novel Agents for Acute Myeloid Leukemia. Cancers (Basel). 2018;10:429.
Zhang J, Gu Y, Chen B. Mechanisms of drug resistance in acute myeloid leukemia. Onco Targets Ther. 2019;12:1937–45.
Almeida AM, Ramos F. Acute myeloid leukemia in the older adults. Leuk Res Rep. 2016;6:1–7.
Bryan JC, Jabbour EJ. Management of relapsed/refractory acute myeloid leukemia in the elderly: current strategies and developments. Drugs Aging. 2015;32:623–37.
Sanford D, Ravandi F. Management of newly diagnosed acute myeloid leukemia in the elderly: current strategies and future directions. Drugs Aging. 2015;32:983–97.
Sanz MA, Iacoboni G, Montesinos P, Venditti A. Emerging strategies for the treatment of older patients with acute myeloid leukemia. Ann Hematol. 2016;95:1583–93.
Saraceni F, Labopin M, Gorin NC, Blaise D, Tabrizi R, Volin L, et al. Matched and mismatched unrelated donor compared to autologous stem cell transplantation for acute myeloid leukemia in first complete remission: a retrospective, propensity score-weighted analysis from the ALWP of the EBMT. J Hematol Oncol. 2016;9:79.
Tallman MS, Gilliland DG, Rowe JM. Drug therapy for acute myeloid leukemia. Blood. 2005;106:1154–63.
Szer J. The prevalent predicament of relapsed acute myeloid leukemia. Hematol Am Soc Hematol Educ Program. 2012;2012:43–8.
Thanarajasingam G, Kim HT, Cutler C, Ho VT, Koreth J, Alyea EP, et al. Outcome and prognostic factors for patients who relapse after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transpl. 2013;19:1713–8.
Rautenberg C, Germing U, Haas R, Kobbe G, Schroeder T. Relapse of acute myeloid leukemia after allogeneic stem cell transplantation: prevention, detection, and treatment. Int J Mol Sci. 2019;20:228.
Song Y, Chen X, Huang R, Liu J. Dysregulated YAP1/Hippo pathway contributes to doxorubicin (ADM)-resistance in acute myeloid leukemia (AML). Curr Pharm Biotechnol. 2023;24:676–85.
Duy C, Li M, Teater M, Meydan C, Garrett-Bakelman FE, Lee TC, et al. Chemotherapy induces senescence-like resilient cells capable of initiating AML recurrence. Cancer Discov. 2021;11:1542–61.
Zalpoor H, Bakhtiyari M, Akbari A, Aziziyan F, Shapourian H, Liaghat M, et al. Potential role of autophagy induced by FLT3-ITD and acid ceramidase in acute myeloid leukemia chemo-resistance: new insights. Cell Commun Signal. 2022;20:172.
Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell. 2018;22:191–205. e9
Shen C, Sheng Y, Zhu AC, Robinson S, Jiang X, Dong L, et al. RNA demethylase ALKBH5 selectively promotes tumorigenesis and cancer stem cell self-renewal in acute myeloid leukemia. Cell Stem Cell. 2020;27:64–80. e9
Wu Y, Xie L, Wang M, Xiong Q, Guo Y, Liang Y, et al. Mettl3-mediated m(6)A RNA methylation regulates the fate of bone marrow mesenchymal stem cells and osteoporosis. Nat Commun. 2018;9:4772.
Gao Y, Vasic R, Song Y, Teng R, Liu C, Gbyli R, et al. m(6)A Modification prevents formation of endogenous double-stranded RNAs and deleterious innate immune responses during hematopoietic development. Immunity. 2020;52:1007–21. e8
Yin R, Chang J, Li Y, Gao Z, Qiu Q, Wang Q, et al. Differential m(6)A RNA landscapes across hematopoiesis reveal a role for IGF2BP2 in preserving hematopoietic stem cell function. Cell Stem Cell. 2022;29:149–59. e7
Mapperley C, van de Lagemaat LN, Lawson H, Tavosanis A, Paris J, Campos J, et al. The mRNA m6A reader YTHDF2 suppresses proinflammatory pathways and sustains hematopoietic stem cell function. J Exp Med. 2021;218:e20200829.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–20.
Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y, et al. YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife. 2017;6:e31311.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell 2015;161:1388–99.
Huang H, Weng H, Chen J. The biogenesis and precise control of RNA m(6)A methylation. Trends Genet. 2020;36:44–52.
Huang H, Weng H, Chen J. m(6)A modification in coding and non-coding RNAs: roles and therapeutic implications in cancer. Cancer Cell. 2020;37:270–88.
Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, et al. FTO Plays an oncogenic role in acute myeloid leukemia as a N(6)-methyladenosine RNA demethylase. Cancer Cell. 2017;31:127–41.
Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millan-Zambrano G, Robson SC, et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature. 2017;552:126–31.
Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017;23:1369–76.
Su R, Dong L, Li Y, Gao M, He PC, Liu W, et al. METTL16 exerts an m(6)A-independent function to facilitate translation and tumorigenesis. Nat Cell Biol. 2022;24:205–16.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285–95.
Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga G, et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature. 2021;593:597–601.
Zhang ZW, Teng X, Zhao F, Ma C, Zhang J, Xiao LF, et al. METTL3 regulates m(6)A methylation of PTCH1 and GLI2 in Sonic hedgehog signaling to promote tumor progression in SHH-medulloblastoma. Cell Rep. 2022;41:111530.
Niu Y, Wan A, Lin Z, Lu X, Wan G. N (6)-Methyladenosine modification: a novel pharmacological target for anti-cancer drug development. Acta Pharm Sin B. 2018;8:833–43.
Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, et al. Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell. 2019;35:677–91. e10
Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell. 2018;172:90–105. e23
Qing Y, Dong L, Gao L, Li C, Li Y, Han L, et al. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/m(6)A/PFKP/LDHB axis. Mol Cell. 2021;81:922–39. e9
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–7.
Dina C, Meyre D, Gallina S, Durand E, Korner A, Jacobson P, et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet. 2007;39:724–6.
Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–94.
Yang S, Wei J, Cui YH, Park G, Shah P, Deng Y, et al. m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat Commun. 2019;10:2782.
Zhou S, Bai ZL, Xia D, Zhao ZJ, Zhao R, Wang YY, et al. FTO regulates the chemo-radiotherapy resistance of cervical squamous cell carcinoma (CSCC) by targeting beta-catenin through mRNA demethylation. Mol Carcinog. 2018;57:590–7.
Christopher MJ, Petti AA, Rettig MP, Miller CA, Chendamarai E, Duncavage EJ, et al. Immune escape of relapsed AML cells after allogeneic transplantation. N Engl J Med. 2018;379:2330–41.
Ling VY, Straube J, Godfrey W, Haldar R, Janardhanan Y, Cooper L, et al. Targeting cell cycle and apoptosis to overcome chemotherapy resistance in acute myeloid leukemia. Leukemia. 2022;37:143–53.
Jiang X, Liu B, Nie Z, Duan L, Xiong Q, Jin Z, et al. The role of m6A modification in the biological functions and diseases. Signal Transduct Target Ther. 2021;6:74.
Liu J, Jia G. Methylation modifications in eukaryotic messenger RNA. J Genet Genomics. 2014;41:21–33.
Li X, Jiang X, Gao Q, Zhao P. FOXO3 regulates sevoflurane-induced neural stem cell differentiation in fetal rats. Cell Mol Neurobiol. 2022;42:1777–86.
Yao S, Fan LY, Lam EW. The FOXO3-FOXM1 axis: a key cancer drug target and a modulator of cancer drug resistance. Semin Cancer Biol. 2018;50:77–89.
Qi H, Tian D, Li M, Zhang C, Jin H, Liu L, et al. Foxo3 promotes the differentiation and function of follicular helper T cells. Cell Rep. 2020;31:107621.
Lin Z, Wan AH, Sun L, Liang H, Niu Y, Deng Y, et al. N6-methyladenosine demethylase FTO enhances chemo-resistance in colorectal cancer through SIVA1-mediated apoptosis. Mol Ther. 2023;31:517–34.
Yan F, Al-Kali A, Zhang Z, Liu J, Pang J, Zhao N, et al. A dynamic N(6)-methyladenosine methylome regulates intrinsic and acquired resistance to tyrosine kinase inhibitors. Cell Res. 2018;28:1062–76.
Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. m(6)A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18:2622–34.
Niu Y, Lin Z, Wan A, Chen H, Liang H, Sun L, et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol Cancer. 2019;18:46.
Zhou Y, Wang Q, Deng H, Xu B, Zhou Y, Liu J, et al. N6-methyladenosine demethylase FTO promotes growth and metastasis of gastric cancer via m(6)A modification of caveolin-1 and metabolic regulation of mitochondrial dynamics. Cell Death Dis. 2022;13:72.
Lin Z, Niu Y, Wan A, Chen D, Liang H, Chen X, et al. RNA m(6) A methylation regulates sorafenib resistance in liver cancer through FOXO3-mediated autophagy. EMBO J. 2020;39:e103181.
Tang Y, Chen K, Song B, Ma J, Wu X, Xu Q, et al. m6A-Atlas: a comprehensive knowledgebase for unraveling the N6-methyladenosine (m6A) epitranscriptome. Nucl Acids Res. 2021;49:D134–43.
Lan Y, Liu B, Guo H. The role of M(6)A modification in the regulation of tumor-related lncRNAs. Mol Ther Nucleic Acids. 2021;24:768–79.
Jarroux J, Morillon A, Pinskaya M. History, discovery, and classification of lncRNAs. Adv Exp Med Biol. 2017;1008:1–46.
Dossin F, Pinheiro I, Zylicz JJ, Roensch J, Collombet S, Le Saux A, et al. SPEN integrates transcriptional and epigenetic control of X-inactivation. Nature. 2020;578:455–60.
Lence T, Paolantoni C, Worpenberg L, Roignant JY. Mechanistic insights into m(6)A RNA enzymes. Biochim Biophys Acta Gene Regul Mech. 2019;1862:222–9.
Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560–4.
Zeng Y, Wang S, Gao S, Soares F, Ahmed M, Guo H, et al. Refined RIP-seq protocol for epitranscriptome analysis with low input materials. PLoS Biol. 2018;16:e2006092.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Cui X, Meng J, Zhang S, Chen Y, Huang Y. A novel algorithm for calling mRNA m6A peaks by modeling biological variances in MeRIP-seq data. Bioinformatics. 2016;32:i378–i85.
Cui X, Wei Z, Zhang L, Liu H, Sun L, Zhang SW, et al. Guitar: an R/bioconductor package for gene annotation guided transcriptomic analysis of RNA-related genomic features. Biomed Res Int. 2016;2016:8367534.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9.
Chen T, Chen X, Zhang S, Zhu J, Tang B, Wang A, et al. The genome sequence archive family: toward explosive data growth and diverse data types. Genom Proteom Bioinform. 2021;19:578–83.
Members C-N. Partners. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res. 2022;50:D27–38.
Acknowledgements
This work was partially supported by the National Natural Science Foundation of China (81930004 to HX-J); this work was also partially supported by China Postdoctoral Science Foundation (2022M720305 to ZZ-W).
Author information
Authors and Affiliations
Contributions
HX-J conceived and designed the study. ZZ-W performed all the experiments. ZX-S provided AML samples. GH helped perform flow cytometry. HX-J and ZZ-W drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The study was performed in accordance with the Declaration of Helsinki, and were approved by the Peking University People’s Hospital ethics review board (Reference number: 2023PHB039-001).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, ZW., Zhao, XS., Guo, H. et al. The role of m6A demethylase FTO in chemotherapy resistance mediating acute myeloid leukemia relapse. Cell Death Discov. 9, 225 (2023). https://doi.org/10.1038/s41420-023-01505-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-023-01505-y
This article is cited by
-
Exploring the prognostic potential of m6A methylation regulators in low-grade glioma: implications for tumor microenvironment modulation
European Journal of Medical Research (2024)
-
m6A eraser FTO impairs gemcitabine resistance in pancreatic cancer through influencing NEDD4 mRNA stability by regulating the PTEN/PI3K/AKT pathway
Journal of Experimental & Clinical Cancer Research (2023)
