
Abstract
Neutrophils are important effector cells during inflammation, which play complex roles. Therefore, investigating the regulation of neutrophil accumulation during inflammation might provide targets for treating related diseases. In the present study, we generated a ripk3-deficient zebrafish line to study the roles of Ripk3 in neutrophil-related inflammation. The homeostatic hematopoiesis and cytokine expression of the ripk3-deficient larvae were unaltered. The ripk3-deficient larvae with caudal fin fold injury exhibited similar neutrophil enrichment with wild-type larvae, suggesting that Ripk3 is not essential for non-infectious inflammatory responses. When challenged with lipopolysaccharide (LPS), the ripk3-deficient larvae showed significantly less neutrophil accumulation in the injection site and differential expression of several key cytokines. Ripk3 inhibitors could also attenuate neutrophil accumulation in wild-type larvae, indicating that Ripk3 could serve as a candidate target for inflammation treatment. In summary, our study indicated that Ripk3 has an essential role in LPS-induced inflammatory responses. It was suggested that the ripk3-deficient zebrafish might be applied in developing infectious disease models, while Ripk3 also has potential as an inflammation-treatment target.
Similar content being viewed by others


Introduction
Inflammation is an immune response triggered by biological, physical, and chemical inflammatory factors in various tissues and organs of the body. Inflammation initiates as a short-term acute response that mainly involves leukocyte infiltration into the inflammation area, phagocytosis, and tissue repairment [1]. At the onset of inflammation, sentinel innate immune cells such as macrophages and dendritic cells are activated and start to produce a series of mediators, including cytokines and chemokines [2]. Subsequently, neutrophils, natural killer cells, and monocytes are attracted to the inflammation site by these mediators [3]. During inflammation, an important effector cell type is the neutrophil [4], which plays double-sided roles. Generally, neutrophils play essential anti-pathogen roles during infections, as the impairment of neutrophils recruitment would result in pathogen dissemination and even death [5]. On the other hand, neutrophils also induce local tissue damage at the inflammation loci and even result in organ dysfunction [6, 7]. The activation of neutrophils could even trigger some chronic inflammatory diseases, such as atherosclerosis and type II diabetes [8]. Thus, appropriate activation and suppression of neutrophils are critical for inflammatory progress and homeostasis.
Receptor-interacting serine/threonine protein kinase 3 (RIPK3) is a serine/threonine kinase, considered to be a critical element of necroptosis [9, 10]. Inhibitors targeting RIPK3-dependent necroptosis have shown therapeutic potential for multiple diseases such as cancer and metabolic disorders [11]. Accumulating evidence has established that RIPK3 is also an inflammation regulator, which probably has a non-necrotic function [12]. In bone marrow-derived dendritic cells and aortic smooth muscle cells, RIPK3 plays role in inflammation responses by controlling the activation of NF-κB [13, 14]. Inappropriate RIPK3 activity can lead to primary immunodeficiency and autoinflammatory syndrome due to the activation of the NLRP3 inflammasome [15]. RIPK3 plays an essential role in lipopolysaccharide (LPS)-induced acute inflammatory responses, which are not dependent on caspases [16]. Thus, whether RIPK3 could serve as a potential target to release inflammation is worth further exploration.
Zebrafish is an ideal model for studying inflammatory responses and drug evaluation. In addition to in vitro fertilization, fertility, and easy access to microinjection, the hematopoietic systems between mammals and zebrafish share high conserveness [17], and the inflammation responses of zebrafish are also similar to those of mammals [18]. In zebrafish, Ripk3 was found to be essential in regulating necroptosis [19]. In the type 2 diabetes zebrafish model, Ripk3 contributes to islet inflammation by inducing local IL-1β production and recruiting macrophages [20]. However, the potential role of Ripk3 in regulating neutrophil-associated inflammation has never been reported in zebrafish.
In the present study, we utilize the zebrafish model to study the role of Ripk3 in inflammation and evaluate the potential of targeting Ripk3 in treating inflammation. By generating and further characterizing a zebrafish ripk3-deficient line, we found that the ripk3 deficiency perturbed neutrophil enrichment and affected cytokine expressions upon LPS challenge. We did not find obvious changes in homeostatic hematopoiesis, inflammatory cytokines, and neutrophil accumulation during non-infectious inflammation. Importantly, Ripk3 inhibitors had similar effects to ripk3-deficient larvae, indicating that targeting Ripk3 could relieve inflammatory responses. Thus, by using zebrafish as a model, we demonstrate that Ripk3 plays key roles in infectious inflammation and could serve as a therapeutic target for inflammatory-related disease treatment.
Results
Knockout of the zebrafish ripk3 gene by CRISPR-Cas9
To determine the role of ripk3 in inflammation, we generated a ripk3-deficient zebrafish line by targeting the second exon of ripk3 with CRISPR-Cas9 technology (Fig. 1A). The F0 individuals were crossed with wild-type (WT) zebrafish to generate F1 heterozygotes, in which a 17 base pair (bp) deletion was found in the ripk3 gene (−17 bp, +0) (Fig. 1B). The coding of the PKc_like superfamily domain in the Ripk3 protein was predicted to be abolished by a pre-mature stop codon in this mutation (Fig. 1C). To confirm the mutation, the expression levels of ripk3 mRNA were detected with Q-PCR in WT and ripk3-deficient larvae, which showed that the expression of ripk3 was decreased in ripk3-deficient larvae (Fig. 1D, common products). Specific Q-PCR forward primers for WT and mutant ripk3 genes were designed (Mut_FP and WT_FP) (Fig. 1D and Table S1). The expression levels of mutant Q-PCR products were almost undetectable in the WT larvae, while those of WT products were also undetectable in the ripk3-deficient larvae (Fig. 1D), indicating that the WT form was substituted with the mutant form of mRNA in the ripk3-deficient larvae. Therefore, the generation of the ripk3-deficient line (−17 bp, +0) was successful and confirmed to be a loss-of-function mutation.

A CRISPR target of ripk3. The CRISPR/Cas9 target sequence was designed on the second exon of ripk3, and mutations were created using CRISPR-Cas9 technology. B Mutation form of ripk3-deficient larvae. A mutation strain with 17 bp deletion in the ripk3 gene (−17 bp, +0) was applied in this study. C Protein changes upon ripk3 mutation. In the ripk3-deficient larvae, a premature stop codon was generated and disrupted the PKc_like superfamily protein domain. D Validations of the ripk3 mutation. Q-PCR revealed that the WT or mutant product of ripk3 mRNA could only be detected in the WT or ripk3-deficient larvae, respectively. The expression of the common product showed that ripk3 was significantly lower expressed in the ripk3-deficient larvae (n ≥ 10, Student’s t-test).
Deficiency of ripk3 does not affect hematopoiesis
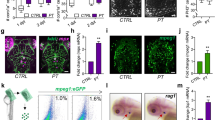
To characterize the effects of ripk3 disruption on homeostatic hematopoiesis, the numbers of myeloid, lymphoid, and erythroid cells in ripk3-deficient larvae were detected by Sudan black (SB) staining and whole-mount in situ hybridization (WISH). The result of SB staining showed no significant difference in the population of neutrophils between ripk3-deficient larvae and WT larvae at 3 days post fertilization (dpf) (Fig. 2A). Based on previous studies, mfap4, rag1, and βe1 were applied as markers of macrophages, lymphocytes, and erythrocytes, respectively [21,22,23]. According to the WISH results, there was no significant difference in the population of macrophages, lymphocytes, and erythrocytes between the ripk3-deficient larvae and WT larvae at 5 dpf (Fig. 2B–D). Thus, the ripk3 deficiency had no significant effect on the homeostatic hematopoiesis in zebrafish.

A SB staining showing unchanged neutrophils (n = 8, Student’s t-test). B WISH of mfap4 showing unchanged macrophages (n = 8, Student’s t-test). C WISH of rag1 showing normal T lymphocytes (red circle showing the thymus) (Fisher’s exact tests, n = 14). D WISH of βe1 showing normal erythrocytes (red box showing the caudal hematopoietic tissue) (Fisher’s exact tests, n > 12).
Deficiency of ripk3 does not affect cytokines expression
To examine whether the ripk3 deficiency would affect inflammation status, we detected the expression levels of several pro-inflammatory (il1b, il6, and cxcl8a) [24] and anti-inflammatory (il4, il10, and il13) [25] cytokines by Q-PCR. The expression of all these cytokines was unaltered upon ripk3 deficiency compared with their WT controls (Fig. 3A, B). The results demonstrated that the ripk3 deficiency does not affect the basal expression of cytokines during normal physiological conditions.

A Normal pro-inflammatory cytokines expression upon ripk3 deficiency. The expression levels of il1b, il6, and cxcl8a were unchanged in 3 dpf ripk3-deficient larvae (n ≥ 10, Student’s t-test). B Normal anti-inflammatory cytokines expression upon ripk3 deficiency. The expression levels of il4, il10, and il13 were unchanged in 3 dpf ripk3-deficient larvae (n ≥ 10, Student’s t-test).
Deficiency of ripk3 had limited effects on non-infectious inflammatory responses
Caudal fin fold injury is a well-established methodology to induce non-infectious inflammatory responses in zebrafish [26]. WT larvae first received an injury (Fig. 4A) and were then collected at different time points to optimize the most robust inflammation response. SB staining revealed that increasing neutrophils migrated to the injury transection site until 5 h post-injury (hpi), and neutrophils exited the caudal fin fold after 5 hpi (Fig. S1). Therefore, we applied the caudal fin fold injury to ripk3-deficient larvae and collected the samples at 5 hpi to detect their inflammatory responses (Fig. 4A). It was found that both ripk3-deficient and WT larvae exhibited almost no neutrophil at the caudal fin fold region before treatment, while similar neutrophil accumulation was found at 5 hpi with no significant difference (Fig. 4B, C). Therefore, in non-infectious inflammatory responses, the ripk3 deficiency does not affect the recruitment of neutrophils.

A Experimental design. The caudal fin folds of 3 dpf larvae were injured by sterile syringe needles, and the local accumulation of neutrophils and cytokines expression were detected at 5 hpi. B, C Normal neutrophils attraction towards the injury site. At 0 hpi, few neutrophils could be found in the injury area (red circle). At 5 hpi, similar numbers of neutrophils (red arrows pointed) were attracted to the caudal fin folds (n ≥ 12, Student’s t-test). D The expression levels of pro-inflammatory cytokines (il1b, il6, and cxcl8a) (n ≥ 10, One-way ANOVA). E The expression levels of anti-inflammatory cytokines (il4, il10, and il13) (n ≥ 10, one-way ANOVA).
We also detected the expression of cytokines through Q-PCR. It was found that the untreated ripk3-deficient and WT larvae shared similar cytokine expression levels (Fig. 4D, E), which was identical with the basal expression data (Fig. 3). Upon caudal fin fold injury, the expression levels of all detected pro-inflammatory cytokines (il1b, il6, and cxcl8a) were upregulated in WT (Fig. 4D). However, the injury-induced upregulation of il1b and il6 expression is attenuated in the ripk3-deficient larvae, whereas the induction of il-8 shows no differences between WT and ripk3-deficient larvae (Fig. 4D), suggesting that the ripk3 deficiency attenuated some of the pro-inflammatory responses during injury. For the expression of anti-inflammatory cytokines, the injury could induce il4 upregulation in both WT and ripk3-deficient larvae (Fig. 4E). Interestingly, the injury-induced expression of il10 was significantly blocked in ripk3-deficient larvae, while the injury-unaffected expression of il13 was slightly induced in the ripk3-deficient larvae (Fig. 4E), suggesting that injury stimulated anti-inflammatory cytokines might be affected diversely by ripk3 deficiency.
Therefore, the above results showed that the deficiency of ripk3 affected the expression of several cytokines upon non-infectious inflammation, with little effect on the recruitment of neutrophils.
Deficiency of ripk3 attenuates LPS-induced inflammatory responses
To verify whether Ripk3 plays a role in the infectious inflammatory response, we treated zebrafish larvae with LPS, a gram-negative bacterial component. WT larvae were first injected with LPS or PBS (as control) at 3 dpf in the yolk and collected at different time points to optimize the most robust infectious inflammation response (Fig. 5A). We found that the LPS-injected larvae showed apparent local neutrophil accumulation at 2 hpi compared with the control group (Fig. S2A, B). The accumulation of neutrophils peaked at 5 hpi and started to decrease afterward (Fig. S2A, B). We thus detected the expression of pro-inflammatory cytokines in LPS- and PBS-injected groups at 5 hpi. The pro-inflammatory factors in WT zebrafish were generally upregulated after the LPS challenge (Fig. S2C). The above data demonstrated that the microinjection of LPS into 3 dpf zebrafish larvae could successfully induce a strong infectious inflammatory response.

A Experimental design. The 3 dpf WT and ripk3-deficient larvae were injected with LPS, and collected at 5 hpi for the detection of cytokines. The neutrophil accumulation was evaluated by SB staining at 2, 5, and 8 hpi. B Fewer neutrophils migrated to the injection sites. At 2, 5, and 8 hpi, fewer neutrophils (red arrows pointed) could be found in the yolk sac of the ripk3-deficient larvae. C Quantification of (B) (n ≥ 6, one-way ANOVA). D Quantification of (B), showing the trends of the accumulated neutrophils. E The expression levels of pro-inflammatory cytokines (il1b, il6, and cxcl8a) at 5 hpi (n ≥ 10, one-way ANOVA). F The expression levels of anti-inflammatory cytokines (il4, il10, and il13) at 5 hpi (n ≥ 10, one-way ANOVA).
Next, we treated the ripk3-deficient and WT larvae with LPS and collected them at 2, 5, and 8 hpi to detect their neutrophil enrichment status (Fig. 5A). Interestingly, although WT larvae exhibited a robust accumulation of neutrophils, the neutrophils of ripk3-deficient larvae remained unchanged in the yolk region (Fig. 5B–D). Thus, this phenomenon demonstrated that Ripk3 is essential for the LPS-induced accumulation of neutrophils in inflammatory responses.
We also detected the expression levels of cytokines at 5 hpi. Upon LPS treatment, the expression of pro-inflammatory cytokines il1b and il6 were markedly upregulated in both WT and ripk3-deficient larvae, although the upregulated level in ripk3-deficient larvae was not as high as in WT (Fig. 5E), suggesting that the ripk3 deficiency might weaken the pro-inflammatory process. However, LPS induced only slight changes of cxcl8a expression in WT and ripk3-deficient larvae (Fig. 5E), suggesting that cxcl8a might not be critical for LPS induced inflammation responses. For the anti-inflammatory cytokines, il4 and il13 were significantly downregulated in WT larvae upon LPS treatment, whereas the LPS induced downregulation of the two cytokines was blocked in the challenged ripk3-deficient larvae (Fig. 5F). However, the expression of il10 remained unchanged in WT larvae but upregulated robustly in ripk3-deficient larvae after LPS induction (Fig. 5F). Thus, during the LPS challenge process, ripk3-deficient larvae showed the attenuation of pro-inflammatory cytokine expression and the maintenance of relatively high anti-inflammatory cytokine levels.
Taken together, we demonstrated that in infection-induced inflammation, the ripk3 deficiency could block neutrophil accumulation, prevent pro-inflammatory cytokine over-production, and maintain relatively high anti-inflammatory cytokine levels, suggesting that ripk3 perturbation might diminish inflammation in infectious disorders.
Targeting Ripk3 could inhibit LPS-induced inflammation
To evaluate whether Ripk3 could serve as a candidate target for treating inflammatory responses, we applied Ripk3 inhibitors, namely GSK872, GSK840, GSK843, and RIPK3-IN-1, to WT larvae and injected them with LPS. The concentrations of Ripk3 inhibitors were tested with 2 dpf WT larvae, in which the teratogenic concentrations were identified (Table 1). The final treatment concentrations were selected as around half the teratogenic concentrations (Table 1).
We next investigated whether the LPS-induced SB+ neutrophil accumulation could be blocked by RIPK3 inhibitors. When treated with these inhibitors, the PBS-injected groups showed no significant difference compared with the non-treated control group, indicating that these inhibitors would not affect the basal development of neutrophils (Fig. 6A, B). When LPS injection was performed along with the treatment of RIPK3 inhibitors, no significant difference could be found between the LPS- and PBS-injected larvae in each group (Fig. 6A, B), indicating that the LPS-induced neutrophil enrichment was abolished by inhibiting RIPK3. Therefore, the results validated the essential role of Ripk3 in neutrophil-related inflammatory responses and also implied that targeting RIPK3 to diminish inflammation could have potential application in clinical treatments.

A Attenuated LPS-induced neutrophil accumulation upon Ripk3 inhibition. The WT larvae were pre-treated with Ripk3 inhibitors and then injected with LPS at 3 dpf. The neutrophil accumulation (red arrows pointed) was significantly inhibited by Ripk3 inhibitors at 5 hpi. B Quantification of (A) (n ≥ 5, one-way ANOVA).
Discussion
In the present research, we generated a ripk3-deficient zebrafish line and investigated the role of Ripk3 in inflammatory responses. The results showed that ripk3-deficient zebrafish could serve as an animal model to further monitor inflammatory-related diseases with impairment of neutrophil responses. As we demonstrated that targeting ripk3 could block neutrophil accumulation in the model, we believe that Ripk3 could be a potential therapeutic target to treat neutrophil-associated inflammatory diseases.
Since leukocytes are the main effector cells involved in inflammation, we first sought to detect whether the deficiency of ripk3 affects hematopoiesis in homeostatic status, especially the development of leukocytes. It was found that myeloid, lymphoid, and erythroid hematopoiesis were all unaltered in the ripk3-deficient larvae. In mice, RIPK1, another member of the RIPK family, was found to regulate embryonic hematopoiesis [27]. When the RIPK1 suppressors, LUBAC and Caspase-8, are absent, RIPK3 could prevent RIPK1 from inducing lethal hematopoietic defects [27]. Therefore, only the triple mutations of Ripk3, Caspase-8, and Hoil-1 (LUBAC component) would trigger severe RIPK1-dependent hematopoietic defects [27], while a single mutation of the Ripk3 gene was not sufficient to affect hematopoiesis. In another study, bone-marrow-specific knockout of the Ripk3 gene induced higher cell death rates of the bone-marrow nucleated cells [28]. However, the overall hematopoietic lineages exhibited no defect, probably due to the stronger expansion activities of hematopoietic stem/progenitor cells [28]. Combined with our research in zebrafish, it could be concluded that the deficiency of Ripk3 is not sufficient to affect homeostatic hematopoiesis.
Subsequently, to characterize the roles of Ripk3 in non-infectious inflammatory responses, we induced caudal fin fold injury in the ripk3-deficient larvae. In the caudal fin fold transected larvae, both ripk3-deficient and WT individuals exhibited similar neutrophil recruitment and cytokine expression. It was thus suggested that the physical, non-infectious inflammatory responses were likely to be independent of Ripk3. During tissue injuries, damage-associated molecular pattern proteins (DAMPs) were released [29]. DAMPs can activate classical pattern recognition receptors (PRRs) [30]. Besides, DAMPs can also be sensed by various non-PRR receptors, including the receptor for advanced glycation end products (RAGE) [31], triggering receptors expressed on myeloid cells (TREMs) [32] and N-formyl peptide receptors (FPRs) [33]. Noteworthily, RAGE, TREMs, and FPRs are all expressed on neutrophils [29]. Therefore, the highly complicated DAMP-related inflammatory pathways, triggering neutrophil recruitment to the injured caudal fin fold, are not likely to depend on Ripk3 function in zebrafish.
To characterize the roles of Ripk3 in infectious inflammatory responses, we microinjected the ripk3-deficient larvae with LPS. Interestingly, it was found that the deficiency of ripk3 abolished the enrichment of neutrophils in the injection site. To decipher the underlying mechanisms of this phenomenon, the expression of pro-inflammatory cytokines was detected, and il1b was found to be expressed significantly higher in LPS-injected WT larvae. The well-known pro-inflammatory cytokine IL-1β is mainly released by macrophages [24] and neutrophils are recruited by IL-1β during inflammation [34]. In mammals, it has been reported that RIPK3 plays indispensable roles in activating the NLRP3 inflammasome, which mediates the maturation and secretion of IL-1β [15]. Therefore, RIPK3 might play conserved roles in inducing the production of IL-1β in both zebrafish and mammals. The insufficiency of IL-1β production, probably in macrophages, might be responsible for the impairment of neutrophil enrichment.
The expression levels of anti-inflammatory cytokines (il4, il10, and il13) were also highly expressed in ripk3-deficient larvae during LPS-induced inflammation. IL-4 could dramatically inhibit the secretion of IL-1β, IL-6, and TNF-α by monocytes [35]. IL-13 could also suppress the production of cytokines such as IL-1β, IL-6, and IL-8 by LPS-treated monocytes [36]. IL-10 is the most important anti-inflammatory cytokine that could inhibit the pro-inflammatory cytokines derived by monocytes, neutrophils, and natural killer cells [25]. Therefore, the upregulation of these anti-inflammatory cytokines might exert their functions by suppressing the release of pro-inflammatory cytokines, thus inhibiting the recruitment of neutrophils. The relationship between Ripk3 and these anti-inflammatory cytokines has barely been reported, it is worth further investigation and might reveal novel regulation networks in inflammatory responses.
Since the ripk3-deficient larvae exhibited attenuated neutrophil accumulation upon LPS infections, this zebrafish line could serve as a disease model to simulate diseases in which neutrophil enrichment was dysfunctional. For example, sepsis is a syndrome secondary to infections in which the neutrophil migration toward infection sites was severely impaired [5]. Therefore, when a bacterial or fungal infection was induced in ripk3-deficient zebrafish, the defected neutrophil responses would probably trigger sepsis-like disorders. It is worth developing more disease models based on the ripk3-deficient zebrafish, as the zebrafish model is quite suitable for drug evaluation and screening.
On the other hand, since neutrophil roles in different diseases vary largely, we also sought the possible applications of targeting Ripk3 in treating neutrophilia-related diseases. When we applied Ripk3 inhibitors to LPS-treated larvae, the enrichment of neutrophils in the injection site was strongly inhibited by these inhibitors. Therefore, targeting Ripk3 could also serve as a possible clinical treatment to contain diseases with neutrophilic inflammation. Diseases such as Alzheimer’s disease, epilepsy, type II diabetes, systemic lupus erythematosus, and atherosclerosis are all associated with local accumulation of pro-inflammatory neutrophils [8], and thus might be potential applications for Ripk3-targeted treatments.
In summary, our study revealed an indispensable role of Ripk3 in LPS-induced inflammatory responses, accumulating neutrophils in the inflammation site. The ripk3-deficient zebrafish line could serve as a disease model of impaired neutrophil enrichment in infectious inflammation responses. In addition, targeting Ripk3 could attenuate the accumulation of neutrophils to the inflammation sites, providing valuable insights into developing relevant therapeutic strategies for diminishing inflammation in clinical applications.
Materials and methods
Zebrafish maintenance
AB strain WT zebrafish were maintained according to standard protocols [37]. The adult zebrafish were maintained in a recirculating system at 28 °C under a 14/10 h light/dark cycle. Before the experiment, zebrafish were intercrossed in a male-to-female ratio of 1:2, and their embryos were incubated in egg water containing 0.003% 1-phenyl-2-thiourea (PTU).
Generation of the ripk3-deficient line
Mutagenesis was performed with CRISPR/Cas9 technology as described [38, 39]. The ripk3 gRNA (5′-CCGTGTGCTCTCGCCCTCC-3′) was synthesized with T7 RNA polymerase (Thermo Fisher, USA), mixed with Cas9 protein (New England Biolabs, USA), and injected into 1-cell stage WT zebrafish embryos. The embryos were raised to adulthood as F0 generation and crossed with WT zebrafish to obtain F1 generation embryos with ripk3 deficiency. Finally, an F1 founder line with 17 bp deletion of the ripk3 gene was obtained and applied in further experiments.
WISH
The mfap4, rag1, and βe1 probes labeled with Digoxigenin (Roche, Switzerland) were transcribed in vitro with T7 RNA polymerase (Thermo Fisher, USA). The 4% paraformaldehyde (PFA, Sigma-Aldrich, USA) fixed zebrafish larvae were hybridized with either a rag1 or βe1 probes according to the standard protocol [40]. For the WISH of mfap4 probe, we used maleic acid buffer in antibody blocking and washing, for a better signal-to-noise ratio [41].
RNA extraction and Q-PCR
For each Q-PCR sample, at least 10 larvae were collected as a pool. The total RNA of each sample was isolated using TRIzol reagent (Thermo Fisher, USA), and then the cDNA was generated using a HiScript II Q Select RT SuperMix kit (Vazyme, China) according to the manufacturer’s instructions. Three samples were applied in each Q-PCR group. Q-PCR was performed using a SYBR Green PCR Core Reagent kit on a LightCycler 96 system (Roche, Switzerland). Fold changes were determined by the ΔΔ comparative threshold method. Primers are detailed in Table S1.
In addition, for WT and ripk3-deficient larvae, a common Q-PCR forward primer (co_FP) and a reverse primer (RP) were designed to detect the expression of ripk3. On the other hand, a WT-specific forward primer (wt_FP) and a mutant-specific forward primer (mut_FP) were respectively designed to detect the expression of WT or mutant form of ripk3 mRNA.
LPS microinjection and RIPK3 inhibitor treatment
To establish the inflammatory model, 3 dpf zebrafish larvae were anesthetized with 0.02% tricaine and yolk-microinjected with 1 nL LPS (1 mg/mL) per larva on an agarose plate as reported previously [42]. PBS-injected larvae were used as the control group.
Four different Ripk3 inhibitors (GSK872, GSK840, GSK843, and RIPK3-IN-1) [43, 44] were purchased from MedChemExpress, USA. The concentrations of these inhibitors used were listed in Table 1. When treated with Ripk3 inhibitors, WT larvae were soaked in egg water with inhibitors from 2 dpf. The larvae were injected with PBS or LPS at 3 dpf and further soaked in egg water with inhibitors until collected.
Caudal fin fold injury
The 3 dpf larvae were first anesthetized with 0.02% tricaine. Then, their caudal fin folds were transected with a disposable syringe needle on a sterile plastic Petri dish to induce the injury [45].
SB staining
SB is a lipid stain that strongly favors the staining of the granules of granulocytes [46], which are all neutrophils in 3 dpf zebrafish larvae. Therefore, using SB staining, we could specifically mark the neutrophils and study their behaviours. After LPS microinjection or caudal fin fold injury, 3 dpf larvae were fixed with 4% PFA for 2 h at room temperature, washed twice with PBST, and then stained with SB solution for 30 min as described [46]. After that, the larvae were washed extensively in 70% ethanol, and their SB positive signals were observed by a Zeiss stereomicroscope. The SB signals (black coloured) were counted manually, with the Z-plane of every signal carefully examined to avoid miss counting.
Statistical analysis
The differences between categorical variables were analyzed by Fisher’s exact tests. The continuous variables were analyzed using Student’s t-test or one-way analysis of variance (ANOVA), depending on whether the comparisons were between two groups or among multiple groups, respectively. The figures were expressed as mean ± SEM from at least three independent experiments with duplicate samples and analyzed with the statistical software GraphPad Prism 7.0. The values were considered statistically significant when P < 0.05 (ns indicates not significant; * indicates p < 0.05; ** indicates p < 0.01; *** indicates p < 0.001, and **** indicates p < 0.0001) and shown as means ± SEM.
Data availability
Data sharing is not applicable as no dataset was generated.
References
Tauber AI. Metchnikoff and the phagocytosis theory. Nat Rev Mol Cell Biol. 2003;4:897–901.
Amit I, Garber M, Chevrier N, Leite AP, Donner Y, Eisenhaure T, et al. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science. 2009;326:257–63.
Kang S-J, Liang H-E, Reizis B, Locksley RM. Regulation of hierarchical clustering and activation of innate immune cells by dendritic cells. Immunity. 2008;29:819–33.
Castanheira FVS, Kubes P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood. 2019;133:2178–85.
Sônego F, Castanheira FVES, Ferreira RG, Kanashiro A, Leite CAVG, Nascimento DC, et al. Paradoxical roles of the neutrophil in sepsis: Protective and deleterious. Front Immunol. 2016;7:155.
Brandes M, Klauschen F, Kuchen S, Germain RN. A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell. 2013;154:197–212.
Uderhardt S, Martins AJ, Tsang JS, Lämmermann T, Germain RN. Resident macrophages cloak tissue microlesions to prevent neutrophil-driven inflammatory damage. Cell. 2019;177:541–55.
Nicolás-Ávila JÁ, Adrover JM, Hidalgo A. Neutrophils in homeostasis, immunity, and cancer. Immunity. 2017;46:15–28.
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–9.
Christofferson DE, Yuan J. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol. 2010;22:263–8.
Liu Y, Liu T, Lei T, Zhang D, Du S, Girani L, et al. RIP1/RIP3-regulated necroptosis as a target for multifaceted disease therapy (Review). Int J Mol Med. 2019;44:771–86.
Moriwaki K, Chan FK-M. The inflammatory signal adaptor RIPK3: Functions beyond necroptosis. Int Rev Cell Mol Biol. 2017;328:253–75.
Moriwaki K, Balaji S, McQuade T, Malhotra N, Kang J, Chan FK-M. The necroptosis adaptor RIPK3 promotes injury-induced cytokine expression and tissue repair. Immunity. 2014;41:567–78.
Wang Q, Liu Z, Ren J, Morgan S, Assa C, Liu B. Receptor-interacting protein kinase 3 contributes to abdominal aortic aneurysms via smooth muscle cell necrosis and inflammation. Circ Res. 2015;116:600–11.
Speir M, Lawlor KE. RIP-roaring inflammation: RIPK1 and RIPK3 driven NLRP3 inflammasome activation and autoinflammatory disease. Semin Cell Dev Biol. 2021;109:114–24.
Najjar M, Saleh D, Zelic M, Nogusa S, Shah S, Tai A, et al. RIPK1 and RIPK3 kinases promote cell-death-independent inflammation by Toll-like receptor 4. Immunity. 2016;45:46–59.
Jagannathan-Bogdan M, Zon LI. Hematopoiesis. Development. 2013;140:2463–7.
Novoa B, Figueras A. Zebrafish: Model for the study of inflammation and the innate immune response to infectious diseases. Adv Exp Med Biol. 2012;946:253–75.
Yu T, Kuang H, Chen J, Lin X, Wu Y, Chen K, et al. Tripartite-motif family protein 35-28 regulates microglia development by preventing necrotic death of microglial precursors in zebrafish. J Biol Chem. 2020;295:8846–56.
Yang B, Maddison LA, Zaborska KE, Dai C, Yin L, Tang Z, et al. RIPK3-mediated inflammation is a conserved β cell response to ER stress. Sci Adv. 2020;6:eabd7272.
Jessen JR, Willett CE, Lin S. Artificial chromosome transgenesis reveals long-distance negative regulation of rag1 in zebrafish. Nat Genet. 1999;23:15–6.
Walton EM, Cronan MR, Beerman RW, Tobin DM. The macrophage-specific promoter mfap4 allows live, long-term analysis of macrophage behavior during mycobacterial infection in zebrafish. PLoS One. 2015;10:e0138949.
Quinkertz A, Campos-Ortega JA. A new beta-globin gene from the zebrafish, betaE1, and its pattern of transcription during embryogenesis. Dev Genes Evol. 1999;209:126–31.
Zhang J-M, An J. Cytokines, inflammation, and pain. Int Anesthesiol Clin. 2007;45:27–37.
Opal SM, DePalo VA. Anti-inflammatory cytokines. Chest. 2000;117:1162–72.
Renshaw SA, Loynes CA, Trushell DMI, Elworthy S, Ingham PW, Whyte MKB. A transgenic zebrafish model of neutrophilic inflammation. Blood. 2006;108:3976–8.
Peltzer N, Darding M, Montinaro A, Draber P, Draberova H, Kupka S, et al. LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature. 2018;557:112–7.
Chen Y, Wu Z, Luo X, Bai S, Zhao L. Effect of the conditional knockout of bone marrow specific RIPK3 gene on bone marrow hematopoiesis in mice. Int J Clin Exp Pathol. 2018;11:568–76.
Pandolfi F, Altamura S, Frosali S, Conti P. Key role of DAMP in inflammation, cancer, and tissue repair. Clin Ther. 2016;38:1017–28.
Cao X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat Rev Immunol. 2016;16:35–50.
Hudson BI, Lippman ME. Targeting RAGE signaling in inflammatory disease. Annu Rev Med. 2018;69:349–64.
Ford JW, McVicar DW. TREM and TREM-like receptors in inflammation and disease. Curr Opin Immunol. 2009;21:38–46.
Weiß E, Kretschmer D. Formyl-peptide receptors in infection, inflammation, and cancer. Trends Immunol. 2018;39:815–29.
Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, et al. IL-1α and IL-1β recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol. 2011;187:4835–43.
te Velde AA, Huijbens RJ, Heije K, de Vries JE, Figdor CG. Interleukin-4 (IL-4) inhibits secretion of IL-1 beta, tumor necrosis factor alpha, and IL-6 by human monocytes. Blood. 1990;76:1392–7.
de Waal Malefyt R, Figdor CG, Huijbens R, Mohan-Peterson S, Bennett B, Culpepper J, et al. Effects of IL-13 on phenotype, cytokine production, and cytotoxic function of human monocytes. Comparison with IL-4 and modulation by IFN-gamma or IL-10. J Immunol. 1993;151:6370–81.
Westerfield M. The zebrafish book: A guide for the laboratory use of zebrafish (Danio rerio). 4th ed. Eugene: University of Uregon Press; 2000.
Xiao A, Wang Z, Hu Y, Wu Y, Luo Z, Yang Z, et al. Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic Acids Res. 2013;41:e141.
Chang N, Sun C, Gao L, Zhu D, Xu X, Zhu X, et al. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res. 2013;23:465–72.
Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc. 2008;3:59–69.
Narayanan R, Oates AC. Detection of mRNA by whole mount in situ hybridization and DNA extraction for genotyping of zebrafish embryos. Bio-Protoc. 2019;9:e3193.
Yang L-L, Wang G-Q, Yang L-M, Huang Z-B, Zhang W-Q, Yu L-Z. Endotoxin molecule lipopolysaccharide-induced zebrafish inflammation model: a novel screening method for anti-inflammatory drugs. Molecules 2014;19:2390–409.
Hart AC, Abell L, Guo J, Mertzman ME, Padmanabha R, Macor JE, et al. Identification of RIPK3 type II inhibitors using high-throughput mechanistic studies in hit triage. ACS Med Chem Lett. 2020;11:266–71.
Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell. 2014;56:481–95.
Li L, Yan B, Shi Y-Q, Zhang W-Q, Wen Z-L. Live imaging reveals differing roles of macrophages and neutrophils during zebrafish tail fin regeneration. J Biol Chem. 2012;287:25353–60.
Le Guyader D, Redd MJ, Colucci-Guyon E, Murayama E, Kissa K, Briolat V, et al. Origins and unconventional behavior of neutrophils in developing zebrafish. Blood. 2008;111:132–41.
Acknowledgements
We thank Dr. Jingwei Xiong and Dr. Bo Zhang for sharing CRISPR/Cas9-related material (gRNA-pMD19-T) and protocols.
Funding
This work was supported by the National Natural Science Foundation of China (31922023), the National Key R&D Program of China (2018YFA0800200), the Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (2019), and the China Postdoctoral Science Foundation (2018M643071).
Author information
Authors and Affiliations
Contributions
Y Zhang supervised this study; WW performed most experiments and analyzed the data; Y Zhang, GL, and WW wrote the manuscript; JC and Y Zhou initiated the knockout of ripk3.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
All work involving zebrafish was reviewed and approved by the Animal Research Advisory Committee of South China University of Technology, Guangzhou, China.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wen, W., Chen, J., Zhou, Y. et al. Loss of Ripk3 attenuated neutrophil accumulation in a lipopolysaccharide-induced zebrafish inflammatory model. Cell Death Discov. 8, 88 (2022). https://doi.org/10.1038/s41420-022-00891-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-022-00891-z
