
Abstract
Phosphodiesterase 2A (Pde2A) is a dual-specific PDE that breaks down both cAMP and cGMP cyclic nucleotides. We recently highlighted a direct relationship between Pde2A impairment, a consequent increase of cAMP, and the appearance of mouse congenital heart defects (CHDs). Here we aimed to characterize the pathways involved in the development of CHDs and in their prevention by pharmacological approaches targeting cAMP and cGMP signaling. Transcriptome analysis revealed a modulation of more than 500 genes affecting biological processes involved in the immune system, cardiomyocyte development and contractility, angiogenesis, transcription, and oxidative stress in hearts from Pde2A−/− embryos. Metoprolol and H89 pharmacological administration prevented heart dilatation and hypertabeculation in Pde2A−/− embryos. Metoprolol was also able to partially impede heart septum defect and oxidative stress at tissue and molecular levels. Amelioration of cardiac defects was also observed by using the antioxidant NAC, indicating oxidative stress as one of the molecular mechanisms underpinning the CHDs. In addition, Sildenafil treatment recovered cardiac defects suggesting the requirement of cAMP/cGMP nucleotides balance for the correct heart development.

Similar content being viewed by others
Introduction
Phosphodiesterase 2A (Pde2A) hydrolyzes both cyclic nucleotides (cAMP and cGMP) and is activated by cGMP. Knockout mouse models for the different PDE isoforms have been generated (Supplementary Table S1) and up to date, the Pde2A knockout mouse is the only embryonically lethal, most likely due to an extremely reduced liver size, impairment of the hepatic niche, and elevated anemia [1, 2].
We recently reported that hearts of Pde2A−/− embryos and hearts of embryos treated with a selective Pde2A inhibitor show ventricular and atrial septum defects, hypertrabeculation, heart dilatation, and non-compaction defects [3, 4].
We also demonstrated a direct relationship between Pde2A deficiency, the consequent increase in cAMP, and the occurrence of mouse congenital heart defects (CHDs). Indeed, cAMP has been shown to significantly transform fetal cardiomyocytes into cardiac pacemaker-like cells during cardiogenesis, increasing gap-junction markers [5, 6], promoting phosphorylation of targets such as L-type calcium channel via Protein Kinase A (PKA), and decreasing its concentration during heart development [7, 8].
On the contrary, an increase in local pools of cAMP-regulated by pharmacological Pde2A inhibition showed an anti-hypertrophic phenotype in adult mouse hearts [9].
Pde2A is localized at the plasma membrane, in the cytoplasm, and in the nucleus of neonatal murine cardiomyocytes [4, 10]. The Pde2A2 isoform was also described to be localized in the mitochondrial matrix and its inhibition to stimulate oxidative phosphorylation [11]. Recently, cAMP/PKA signaling domains localized at mitochondrial membranes and regulated by Pde2A2 were described to be involved in mitochondrial morphological change and apoptosis [12]. Because of this specific localization and function, we might hypothesize a contribution of Pde2A in reactive oxygen species (ROS) production. Indeed, ROS have traditionally been regarded as byproducts of aerobic metabolism, with mitochondrial respiration being the major intracellular source of accidental ROS production [13].
The stimulation of β-1 adrenergic receptor (β1-AdR) is the primary source of cAMP in the heart through adenylyl cyclase (AC) activation [14]. Several uncontrolled and controlled clinical studies provided the first evidence that Metoprolol, a selective β1-adrenoceptor blocking agent (β-blocker), had beneficial clinical and hemodynamic effects in patients with heart failure due to idiopathic dilated cardiomyopathy (DCM) [15,16,17,18,19]. Metoprolol was able to extend survival in DCM mice and prevent cardiac remodeling and dysfunction [20, 21].
The cAMP target PKA plays several functions in the normal heart such as contraction, metabolism, and gene transcription regulation. However, increased PKA activity and protein levels were described to aggravate heart failure [22,23,24]. PKA activators induced ROS and consequent electrical disturbance in heart failure, whereas PKA inhibition with H89 prevented it in humans [25]. H89 administration resulted also in the attenuation of DCM and the prevention of heart failure [24]. Moreover, it has been demonstrated to decrease cell death in heart disease and to improve cardiac function after myocardial infarction in mice [26].
It is also well known that cGMP plays a protective role in cardiac physiology and sildenafil, a selective inhibitor of PDE5A, acts in heart remodeling by increasing cGMP levels [27, 28].
Here, investigating the transcriptome of Pde2A deficient embryonic hearts by RNA sequencing (RNA-seq) analysis, we found a significant modulation of more than 500 genes associated with biological processes such as immune response, cardiomyocyte development, and contractility, angiogenesis, control of gene transcription and oxidative stress. Testing pharmacological treatments that blunt cAMP/PKA signaling, we observed that Metoprolol and H89 can robustly prevent CHDs in Pde2A deficient embryos similarly to N-acetyl-cysteine (NAC) antioxidant administration. Moreover, among the pathways severely up-regulated, we found that Metoprolol treatment reestablishes the expression levels of oxidative stress-related genes and NAC reduces in vivo heart oxidative stress. Finally, a pharmacological treatment affecting cGMP signaling was found to ameliorate cardiac defects to a similar extent.
Results
RNA-seq analysis reveals alteration of pathways involved in immune system, angiogenesis, oxidative stress, gene transcription and cardiogenesis
We previously showed downregulation of critical genes involved in cardiac development in Pde2A knockout embryos when compared to wild types [3]. To better define the global transcriptional profile of the two genotypes, RNA-seq was performed on isolated hearts from Pde2A+/+ and Pde2A−/− embryos at E14.5.
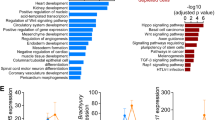
Principal component analysis (PCA) indicated that Pde2A−/− was consistently different from Pde2A+/+, with the first component showing more than 50% of the global variance (Fig. 1A). The extensive alteration in the expression profile between Pde2A−/− and Pde2A+/+ was also displayed by the heatmap (Fig. 1B): a stringent analysis (fold change ≥ 2; p-value < 0.05) found 55 downregulated genes and 460 upregulated genes in the Pde2A−/− with respect to Pde2A+/+embryonic hearts (Supplementary Table S2). A subsequent gene ontology (GO) analysis using the DAVID functional enrichment on biological processes revealed that upregulated genes were involved in pathways such as immune system and inflammatory response, chemokine-mediated signaling, angiogenesis, cellular response to hypoxia, and positive regulation of ERK1 and ERK2 cascades (Fig. 1C). The differentially expressed genes were investigated using the gene set enrichment analysis (GSEA) and compared to gene sets from Molecular Signatures Database (MSigDB) to characterized different cellular populations enriched in Pde2A−/− hearts. In line with our previous data, the cardiomyocyte lineage negatively correlated with Pde2A−/− (Fig. 1D), whereas the other lineages such as endothelial, fibroblast, smooth muscle cells, macrophages, are strongly and positively correlated with Pde2A−/− (Supplementary Fig. S1).

A Plots of samples on first (PC1) and second (PC2) principal components of RNA-seq data. The PC percentage of variance is reported. B Heatmap representation of 515 differentially expressed genes considering a log2-fold change >+1 and <−1 and p-adjusted < 0.05. C Gene ontology biological processes with a significant enrichment (p-value < 0.05 and gene count > 10) in the list of Pde2A−/− differentially expressed genes (n = 515). D Gene set enrichment analysis (GSEA) of Pde2A−/− differentially expressed genes (n = 515) by the cardiomyocyte gene set of the Molecular Signatures Database (MSigDB); n = 6 Pde2A+/+ and n = 5 Pde2A−/− hearts.
Some genes, associated with enriched pathways found in DAVID analysis, were validated by RT-qPCR (Fig. 2). We selected genes linked to the immune system/inflammation (Fig. 2A), genes involved in angiogenesis and hypoxia (Fig. 2B, C), genes involved in survival, proliferation, and transcription (Fig. 2D, E). Some genes, such as Ccl2, Vegfa, Il1a, and Fgf23, were robustly upregulated and expressed in different biological processes. Real-time PCR fully confirmed the RNA-seq analysis. A deeper investigation of RNA-seq results highlighted that also several genes involved in oxidative stress were altered in Pde2A−/− embryonic hearts (Supplementary Table S2). Real-time PCR analysis confirmed that most of them were upregulated in knockout hearts at E14.5 (Fig. 2F). These results indicated that in Pde2A−/− hearts there is a high level of inflammation, oxidative stress, angiogenesis, and a reduction of cardiomyocyte differentiation, at least at the transcriptional level.

Histograms of relative mRNA expression of genes up-regulated in RNA-seq analysis and enriched in biological processes for immune response (A), hypoxia (B), angiogenesis (C), ERK1/2 cascade (D), trancription regulation (E), oxidative stress (F). Each dot represents an embryo; n = 5 Pde2A+/+ and n = 5 Pde2A−/− hearts; unpaired Student’s t-test *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Metoprolol treatment prevents CHDs of Pde2A −/− embryos
Pde2A can hydrolyze both cAMP and cGMP and, moreover, its catalytic activity rises when cGMP binds allosterically to the Pde2A GAF-B domain.
We measured the cyclic nucleotide levels in embryonic hearts from Pde2A+/+ and Pde2A−/− and we observed that the cAMP level is almost doubled in knockout compared to wild-type hearts at E14.5 (Supplementary Fig. S2A), as previously published [3], whereas negligible differences were found in cGMP content (Supplementary Fig. S2B).
In addition, referring to the KEGG PATHWAY database, the RNA-seq data were queried in order to compare the expression of genes involved in cAMP (KEGG ID: mmu04024) and cGMP (KEGG ID: mmu04022) pathways in the heart of Pde2A−/− and Pde2A+/+ embryos (Supplementary Fig. S3A, B). We found a total of 63 genes for the cAMP pathway and 50 genes for the cGMP pathway with a significative up- or down-regulation in Pde2A−/−. Among these, 24 genes were common to both cyclic nucleotides (Supplementary Fig. S3C). Specific highly modulated cAMP and cGMP-dependent genes were 15 and 5, respectively (Supplementary Fig. S3C).
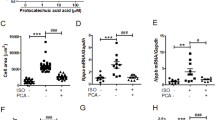
Initially, we focused on pharmacological treatments that directly impact cAMP signaling to test whether they could rescue cardiac malformations in Pde2A−/− embryos. Metoprolol, a selective antagonist of β1-adrenergic receptor and consequently of cAMP synthesis, was administered after implantation to pregnant female mice every day, starting at E5.5 from the plug until E13.5, the day before sacrifice (Fig. 3A).

A Scheme of Metoprolol (MET) treatment. B Picture of Pde2A+/+ and Pde2A−/− embryos at E14.5 treated with Metoprolol. Scale bar = 0.5 cm. C cAMP level in Pde2A+/+ and Pde2A−/− in hearts of Metoprolol treated. n = 4 for each condition and genotype; ANOVA two-way was used to compare Pde2A−/− versus the relative Pde2A+/+ in both conditions and Pde2A−/− not treated versus Pde2A−/− Metoprolol samples; **P ≤ 0.01. D Micro-CT picture of the hearts in Metoprolol-treated Pde2A+/+ and Pde2A−/− embryos. E–G Ratio between total heart (E), atrial (F), and ventricular (G) volumes relative to embryo volumes obtained by micro-CT analyses of Metoprolol treated or not treated (NT) Pde2A+/+ and Pde2A−/− embryos. At least n = 4 for each genotype/treatment. ANOVA two-way was used to compare Pde2A−/− versus the relative Pde2A+/+ in both conditions and Pde2A−/− not treated versus Pde2A−/− Metoprolol samples; *P ≤ 0.05, **P ≤ 0.01.
Morphological examination of Metoprolol-treated E14.5 Pde2A−/− embryos revealed anemia, hemorrhages, and reduced liver size, as shown in Pde2A−/− untreated embryos [3] (Fig. 3B and Supplementary Fig. S4A, B). The level of cAMP was restored and became similar in Pde2A+/+ and Pde2A−/− hearts after Metoprolol treatment, validating the treatment efficacy (Fig. 3C). In addition, micro-CT analysis revealed similar parameters between Pde2A−/− embryonic hearts treated with Metoprolol and Pde2A+/+ hearts; total heart volume, atrial volume, and ventricular volume normalized to embryos volume did not show difference in wild-type versus knockout treated embryos contrary to wild-type versus knockout untreated embryos (Fig. 3D–G). Moreover, half of the Pde2A−/− treated embryos had no interventricular septum defects (Fig. 3D and Supplementary Video S1–S3). Morphological and morphometric analyses of the hearts were then performed on hematoxylin and eosin-stained sections of paraffin-embedded E14.5 embryos. Contrary to untreated embryos, no differences in the contralateral axis between Pde2A+/+ and Pde2A−/− treated hearts were observed (Fig. 4A, B), confirming the ability of Metoprolol to prevent cardiac enlargement. Similarly, the hypertrabeculation of Pde2A−/− hearts was improved by Metoprolol administration, and wild-type and knockout embryos showed similar thickness and number of cardiac trabeculae, respectively (Fig. 4A, C, D). Furthermore, immunofluorescence for endomucin, which marks the endocardium, revealed trabecular network reconstitution after Metoprolol treatment in Pde2A−/− embryos (Supplementary Fig. S5). However, a significant thinning of the ventricular myocardium wall persisted in Pde2A−/− heart after Metoprolol treatment, even though a trend of thickness increase was observed (Fig. 4A–E). Hematoxylin and Eosin staining confirmed that half of treated Pde2A−/− hearts did not show ventricular septum defects (Fig. 4A). These results suggest that Metoprolol, reestablishing the cAMP levels, can partially revert Pde2A−/− cardiac defects, probably acting on some of the pathways specifically controlled by the Pde2A/cAMP system.

A Haematoxylin and Eosin staining of transversal sections of Metoprolol treated (MET) embryos, the heart is shown. Left and right ventricles (LV, RV), atria (LA, RA), and interventricular septum (IVS) are indicated. Insets show magnification of trabeculae and myocardial wall (MW). B–E Graphs of contralateral axis (B), trabeculae thickness (C), trabeculae number (D), and myocardial wall (E) analyses in treated and not treated (NT) samples. n = 9 Pde2A+/+ and n = 11 Pde2A−/− not treated embryos and n = 4 Pde2A+/+ and n = 3 Pde2A−/− Metoprolol treated embryos; ANOVA two-way was used to compare Pde2A−/− versus the relative Pde2A+/+ in both conditions and Pde2A−/− not treated versus Pde2A−/− Metoprolol samples. *P ≤ 0.05, ***P ≤ 0.001, ****P ≤ 0.0001.
Metoprolol treatment restores oxidative stress response and the antioxidant NAC ameliorates cardiac defects in hearts of Pde2A −/− embryos
We then evaluated the expression level of genes that resulted more upregulated in Pde2A knockout hearts by RNA-seq analysis and common to different pathways, in Metoprolol treated and untreated embryos. We observed that except for Fgf23, Metoprolol treatment did not affect the upregulation of Gata1 and Bmp10 found in Pde2A−/− as well as the expression increase of Epor, Ccl2, and Ccl12 genes. (Fig. 5A). Vice versa, the expression of several genes involved in oxidative stress, was reverted by Metoprolol treatment (Fig. 5B). These results suggest that oxidative stress is counteracted by reestablishing the functional cAMP/PKA signaling.

A Histograms of relative mRNA expression of most up-regulated genes involved in different biological processes evaluated by RT-qPCR. B Histograms of relative mRNA expression of critical oxidative stress genes evaluated by RT-qPCR. n = 9 Pde2A+/+ and n = 11 Pde2A−/− not treated (NT) embryos and n = 4 Pde2A+/+ and n = 3 Pde2A−/− Metoprolol (MET) treated embryos; ANOVA two-way was used to compare Pde2A−/− versus the relative Pde2A+/+ in both conditions and Pde2A−/− not treated versus Pde2A−/− Metoprolol samples. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Based on previous results, we checked whether these alterations could be rescued by the antioxidant NAC treatments (Fig. 6). Phenotypically the NAC-treated mutant embryos appeared like the Pde2A−/− embryos (Fig. 6A). The CM-H2DCFDA staining revealed that ROS positive cells were augmented in Pde2A−/− isolated cardiac cells, compared to Pde2A+/+ cells confirming an increase of oxidative stress in the absence of Pde2A (Fig. 6B). NAC administration significantly counteracted ROS production in Pde2A−/− cardiac samples (Fig. 6B). Metoprolol treatment was also evaluated for ROS production and a slight but significant reduction was observed (Fig. 6B).

A Picture of Pde2A+/+ and Pde2A−/− embryos at E14.5 treated with NAC. Scale bar = 0.5 cm. B Flow cytometry analysis of ROS level in cardiac cells isolated from Pde2A+/+ and Pde2A−/− heart embryos in the presence or absence of NAC or Metoprolol (MET). At least n = 3 for each genotype and treatment. Student t-test was used to compare Pde2A−/− versus Pde2A+/+ CM-H2DCFD+ cells and ANOVA one-way to compare the Pde2A−/−/Pde2A+/+ ratio of NAC or MET treatment versus untreated samples. *P ≤ 0.05, **P ≤ 0.01. C Micro-CT picture of the hearts in NAC-treated Pde2A+/+ and Pde2A−/− embryos. Scale bar = 0.5 cm. D and E Ratio between total heart (D), ventricular (E) volumes relative to embryo volumes obtained by micro-CT analyses of NAC treated or not treated (NT) Pde2A+/+ and PDE2A−/− embryos. At least n = 3 for each genotype /treatment. F Haematoxylin and Eosin staining of transversal sections of NAC treated or NT embryos, the heart is shown. Scale bar = 400 µm. G Graphs of contralateral axis, trabeculae thickness, trabeculae number, and myocardial analyses in treated and not-treated samples. At least n = 6 Pde2A+/+ and n = 6 Pde2A−/− NT embryos and n = 4 Pde2A+/+ and n = 5 Pde2A−/− NAC treated embryos. ANOVA two-way was used to compare Pde2A−/− versus the relative Pde2A+/+ in both conditions and Pde2A−/− not treated versus Pde2A−/− NAC samples. *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001.
Micro-CT analysis did not show major rescue in Pde2A−/− embryos treated with NAC, except for total heart volume (Fig. 6C–E and Supplementary Videos S4, S5). However, histological analysis revealed improvement in parameters such as contralateral axis and trabecular number and thickness (Fig. 6F–G). To be noted NAC appeared to decrease some of these values also in Pde2A+/+embryos, suggesting an effect on NAC in the basal homeostasis of cardiac oxidative stress during development.
Overall, these results indicate that antioxidants can be a valuable treatment for CHDs caused by cAMP unbalance.
H89 treatment partially rescues CHDs of Pde2A −/− embryos
To validate the cause–effect of the increase of cAMP level in the onset of CHDs, we investigated downstream pathways by using a PKA inhibitor. Several PKA inhibitors have been developed, however, none of them is specific. We decided to use H89 that is more potent as a PKA inhibitor compared to its ability to inhibit various other protein kinases, such as S6K1, MSK1, PKA, ROCKII, PKBα, and MAPKAP-K1b [29, 30]. Moreover, H89 is largely employed in in vivo studies and, most importantly, already tested and well-tolerated during pregnancy [31]. Morphological examination of H89-treated Pde2A−/− E14.5 embryos showed anemia, hemorrhages, and reduced liver size like not-treated embryos (Supplementary Fig. S6A). Micro-CT analysis revealed that H89 treatment was able to prevent the increase of total heart, atrial, and ventricular volumes observed in Pde2A−/− respect to Pde2A+/+ embryos (Supplementary Fig. S6B, C). However, Pde2A−/− H89-treated hearts maintained ventricular septum defects (Supplementary Fig. S6B and Video S6-S7).
Heart microscopic examination of hematoxylin and eosin-stained embryonic sections revealed that H89 was able to prevent the increase of the contralateral axis, the increase of trabecular number and thickness, but not the ventricular septum defect or the reduction of the myocardial wall in Pde2A−/− (Supplementary Fig. S6D, E). These data strenghten the hypothesis that inhibition of the cAMP/PKA system, although partially, can rescue CHDs of Pde2A−/− embryos.
Sildenafil treatment prevents CHDs of Pde2A −/− embryos
We next investigated whether the pharmacological treatment that directly impacts cGMP signaling could also prevent cardiac malformations in Pde2A−/− embryos. Pharmacological treatment of pregnant Pde2A+/− females with Sildenafil, a drug preserving cGMP levels by inhibiting Pde5A, was performed.
Macroscopic examination revealed no recovery of gross phenotypes of Pde2A−/− embryos (Fig. 7A). On the contrary, micro-CT (Fig. 7B and Supplementary video S8, S9) and histological analyses (Fig. 7D, E) showed recovery of heart defects in terms of contralateral axis, trabecular number and thickness and ventricular septum defects (Fig. 7D, E), but not the heart volumes (Fig. 7C) or wall thickness (Fig. 7E). These data indicate that prevention of cGMP degradation leads to improvement of CHDs in Pde2A−/− embryos.

A Picture of Sildenafil (SILD) treated Pde2A+/+ and Pde2A−/− E14.5. embryos. Scale bar = 0.5 cm. B Micro-CT picture of the hearts in Sildenafil-treated Pde2A+/+ and Pde2A−/− embryos. C Ratio between total hearts and ventricular volumes relative to embryo volumes obtained by micro-CT analyses of Sildenafil treated or not treated (NT) Pde2A+/+ and Pde2A−/− embryos. At least n = 5 for each genotype/treatment. D Haematoxylin and Eosin staining of transversal sections of Pde2A+/+ and Pde2A−/− Sildenafil treated embryos, the heart is shown. Left and right ventricles (LV, RV), atria (LA, RA), and interventricular septum (IVS) are indicated. Inset shows a magnification of trabeculae and myocardial wall (MW). E Graphs of contralateral axis, trabeculae thickness and trabeculae number, and myocardial wall analyses in treated and not treated samples. At least n = 5 for each condition/genotype. ANOVA two-way was used to compare Pde2A−/− versus the relative Pde2A+/+ in both conditions and Pde2A−/− not treated versus Pde2A−/− Sildenafil samples. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 ****P ≤ 0.0001.
Discussion
In this study, RNA-seq was performed on isolated hearts from Pde2A+/+ and Pde2A−/− at E14.5. Gene expression analysis revealed extensive alterations in the transcriptional profile of Pde2A−/− hearts and 515 genes showed strong gene expression alteration. Notably, the transcriptome analysis revealed the deregulation of many pathways, including those involved in the immune response, angiogenesis, hypoxia, oxidative stress, and transcription factors involved in the ERK signaling cascade, by performing a stringent analysis with DAVID functional enrichment. All these pathways are important for correct cardiac development and their alterations have also been reported in various cardiovascular diseases [32].
Some of the genes found altered in Pde2A−/− hearts, such as Epor, Ccl2, and Fgf23 are described to be involved in mitochondrial metabolism rather than in many other functions [33,34,35]. The binding of erythropoietin to its receptor, Epor, activates endothelial nitric oxide synthase (eNOS) which is involved in the regulation of mitochondrial biogenesis, turnover, and proliferation [33]. Ccl2 overexpression may be linked to alterations in mitochondrial dynamics that regulate anabolic and catabolic pathways, indicating a relationship between mitochondrial dysfunction, autophagy, and chronic disease [34]. High levels of Fgf23 are able to induce oxidative stress through the activation of NADPH oxidase complex [35].
Although it is widely known that inflammatory cytokines and oxidative, stress levels are elevated in numerous cardiovascular diseases [36, 37], only a few inconclusive studies have been published on the function of inflammation and oxidative stress in CHDs in neonates and adults [38,39,40,41].
Altogether, the RNA-seq results indicated an alteration of mitochondrial function in the knockout model for Pde2A. A deeper investigation confirmed that genes associated with oxidative stress such as Nox2, Ncf1, Ncf2, Nrf2, Ucp2, Hspa1a, and Gpx1 are significantly upregulated in Pde2A−/− embryonic hearts.
Nox2 is the catalytic subunit of the mitochondrial NADPH oxidase (Nox) that, upon activation, binds various regulatory subunits such as neutrophil cytosolic factor 1 (Ncf1) and neutrophil cytosolic factor 2 (Ncf2) and plays an important role in the production of ROS [42]. Physiologically, the cells have developed various mechanisms to protect themselves from the accumulation of ROS. These mechanisms include the prevention of the accumulation of ROS through uncoupling protein 2 (Ucp2), the reduction of hydrogen peroxide mediated by the nuclear factor erythroid 2-related factor 2 (Nrf2), and ROS degradation via glutathione peroxidase 1 (Gpx1) [43, 44]. In case of stress, it is also important to maintain adequate protein homeostasis inside the cell. Indeed, the chaperon heat shock protein 1A (Hspa1a), during cellular stress, might activate two opposite mechanisms, the folding, and the degradation of proteins depending on its acetylation/deacetylation state [45].
We found increased levels of cAMP, but not cGMP in the heart of Pde2A−/− embryos. However different local compartmentalization of cGMP in Pde2A deficient embryos could contribute to heart defects. In addition, the expression of genes specific to the cGMP pathway resulted to be highly altered in the RNA-seq analysis.
The Metoprolol and H89 used in this study are drugs of clinical use that act upstream and downstream of the cAMP cascade, respectively. Metoprolol was also administrated to pregnant women [46] and was reported to influence cGMP signaling [47].
Both drugs were not able to prevent the gross phenotypes of Pde2A knockout embryos such as liver size, anemia, and hemorrhages. However, E14.5 embryos from mothers who had Metoprolol treatment regained cardiac dimensions, and hypertrabeculation defect, and partially restored the interventricular septal defect. On the other hand, embryos harvested from mothers treated with H89 showed a recovery of cardiac dimension and hypertrabeculation, but not the interventricular septum. H89 clearly is effective in having an in vivo action, however, the exact identity of the targeted kinase(s) remains to be determined, this is because H89, as well as inhibiting PKA also inhibits various other kinases [29, 30].
Molecular analyses carried out on hearts of E14.5 Pde2A−/− embryos showed that Metoprolol can reduce the level of cAMP and the expression of genes that induce oxidative stress (Nox2, Ncf1, Ncf2), whereas preserves the high expression of genes that protect from oxidative stress (Ucp2, Gpx1). It also decreased oxidative stress in the Pde2A−/− cardiac cells, evaluated by CM-H2DFCDA staining, which specifically detects peroxidase formation. Vice versa, Metoprolol treatment does not affect the upregulation of genes implicated in the inflammatory response possibly because they trigger oxidative stress or because the left heart defects maintain an inflammatory state.
NAC is a safe and well-tolerated compound displaying antioxidant properties; it acts mainly by providing the intracellular cysteine necessary to increase glutathione levels. Our results indicate that NAC attenuated just some CHDs carried by Pde2A−/− embryos, possibly because of the low drug bioavailability from the mother to the embryos. Dose increase, modification of drug administration procedures, or combination of NAC with other antioxidants and/or anti-inflammatory compounds may be more effective in preventing CHDs.
To investigate whether CHDs could be also due to cAMP/PKA independent effects such as altered local cGMP-dependent pathways or cyclic nucleotide-independent pathways, Sildenafil was used to increase cGMP levels and to reestablish the cAMP/cGMP balance in the Pde2A−/− hearts. Indeed, Sildenafil ameliorates cardiac defects comparably to other pharmacological treatments suggesting the relevance of the cAMP/cGMP ratio for correct heart development.
Up to now, it is not yet clear to what extent the different types of cardiac cells are differentially affected in the heart of Pde2A−/− versus Pde2A+/+ embryos and how the used drugs impact these cell populations. Future studies will address this topic by using flow cytometry and spatial digital transcriptomic analyses. To date, human CHDs related to the complete absence of PDE2A have not been found, probably because, as in the mouse model, homozygotes are embryonic lethal, or because other PDEs compensate for the lack of PDE2A.
Few recent reports were published describing patients with PDE2A mutations; in all these cases, the patients presented alterations related to the nervous system but no evaluation of cardiac performance with electrocardiograms, arrhythmias, and cardiac function was performed [48,49,50].
In conclusion, this study highlighted the involvement of Pde2A in new biological processes such as inflammation, angiogenesis, transcription, and oxidative stress and how pharmacological treatments, restoring the cyclic nucleotide balancing, can counteract congenital heart defects. Overall, the information obtained from our study will be useful to investigate human CHDs associated with impairment/imbalance of the cAMP/cGMP/PDE2A system and to investigate therapies suitable for their recovery.
Methods
Mouse breeding and in vivo treatments
Pde2A+/− mice (B6; 129P2-Pde2A<tm1Dgen>/H; EM: 02366) were obtained from EMMA (UK). Timed mating was set up between Pde2A+/− females and Pde2A+/− males. Genotyping for Pde2A was performed as previously reported [3]. To test if pharmacological treatments, that affect directly or indirectly cAMP, can rescue cardiac malformations of Pde2A−/− mice we chose two drugs that inhibit β1-adrenergic receptor signaling (Metoprolol) and PKA activity (H89), respectively. The drugs were administered every day starting at E5.5 from the plug until E13.5. Embryos were collected at E14.5.
Metoprolol (Sigma-Aldrich, MO, USA) was administered by gavage at 10 mg/kg dissolved in drinking water [20, 51]. H89 (H-89 dihydrochloride hydrate, Sigma-Aldrich, MO, USA) was administered intraperitoneal at 1 mg/kg [30, 52].
Sildenafil (Pfizer, NY, USA) was administered by gavage at Sildenafil 70 mg/kg/day dissolved in drinking water [53]. NAC (Sigma-Aldrich, MO, USA) was administered at 1 g/kg/day to Pde2A+/− females in drinking water starting the first day of the plug and until day E14.5 [54]. Hearts of the resulting E14.5 embryos both males and females were isolated in Hanks balanced solution as previously described [3, 55] and processed for the following analyses. Only alive Pde2A+/+ and Pde2A−/− were included in the study whereas abortions and Pde2A+/− embryos were excluded. Plugged mothers and their embryos were randomly distributed in the not-treated or drug-treated groups. For each type of experiment, the minimal group size was estimated by the power analysis performed with G*Power 3.0.10 software using the following parameters: α = 0.05, 1−β = 0.85, and d = 1.
Micro-CT imaging and volume measurements
Imaging specimen preparation: Embryos were fixed in 10% neutral buffered formalin (Bio-Optica, Italy) or 4% paraformaldehyde (PAF, Sigma-Aldrich, MO, USA) overnight at room temperature (RT) and embedded in paraffin following standard procedures.
Micro-CT scanning: Computed tomography images were acquired by a high-resolution 3D micro-CT imaging system (Skyscan 1172G Bruker, Kontich—Belgium), using a L7901-20 Microfocus X-ray Source (Hamamatsu). The acquisition of volumes was performed in 1.5 mL micro-tubes, with a camera pixel/size of 7.9 µm, camera binning 2 × 2, tube voltage peak of 39 kV, tube current of 240 µA, and exposure time of 450 ms. Reconstructions of tomographic datasets were performed using built-in NRecon Skyscan Software (Version:1.6.6.0; Bruker). The 3D volumes were analyzed using 3D Visualization Software CTvox v. 2.5 (Bruker). Tissue segmentation: Manual image-by-image segmentation aimed at the calculation of heart volume was applied, using Bruker micro-CT Analyzer Version 1.13 software. A histological atlas of mouse development [56] was used for guidance to accurately identify, demarcate, and segment each embryo (n = at least 3) in a specific volume of interest (VOI) for automated volume measurements [56, 57].
Hematoxylin and eosin staining and immunofluorescence
Embryos at E14.5 were fixed in 10% neutral buffered formalin (Sigma-Aldrich) overnight at RT and included in paraffin (Sigma-Aldrich, MO, USA) following standard procedures. Serial paraffin sections of whole embryos (5 μm of thickness) were obtained, deparaffinized, and stained with hematoxylin and eosin (Sigma-Aldrich, MO, USA). Wall and trabeculae thickness, contralateral axis length, and trabeculae number were evaluated as previously described [3]. All histological analyses were performed in a blinded way by two different authors. The images were collected on a ZEISS axioskop 2 plus microscope mounting Axiocam 503 CCD camera and analyzed with the ImageJ Software (version 1.52t, NIH-Bethesda, MD, USA).
For immunofluorescence, 5 μm E14.5 paraffine sections underwent deparaffinization and microwave antigen retrieval in pH 6.0 sodium citrate with 0.05% Tween-20 solution (Sigma-Aldrich, MO, USA), followed by gradual chilling. Samples were permeabilized with 0.2% NP40 (Sigma-Aldrich, MO, USA) in phosphate-buffered saline (PBS) for 30 min and blocked with 1% bovine serum albumin (Sigma-Aldrich, MO, USA) in PBS for 1 h at room temperature. Sections were incubated overnight at 4 °C with anti-Endomucin antibody (Ab106100, Abcam, UK). After 3 × 5 min washes with PBS, sections were stained for 1 h at room temperature with anti-rat secondary antibody (Alexa Fluor 488; Thermo Fisher Scientific, MA, USA) followed by 2 min of Sudan Black (Sigma-Aldrich, MO, USA; 0.03g/10 ml 70% EtOH) to reduce the background and nuclei were counterstained with Hoechst (Sigma-Aldrich, MO, USA) for 5 min. Slides were mounted with Fluoromount Aqueous Mounting Medium (Sigma-Aldrich, MO, USA). The images were collected on Nikon Eclipse Ti-S microscope equipped with a Photometric CoolSNAP EZ turbo 1394 camera and analyzed on ImageJ (version 1.52t, NIH-Bethesda, MD, USA).
RNA-sequencing and RT-qPCR
Total RNA was isolated from embryonic cardiac tissue using the Total RNA Purification kit (Norgen Biotek Corporation, ON, CA) according to the manufacturer’s instructions. For RNA-sequencing, the integrity and purity of the RNA were initially checked at the Bioanalyzer (Agilent 2100 bioanalyzer, Santa Clara, CA, USA) by using an RNA 6000 Nano Agilent kit. The RNA seq analysis was done at the EMBL service facility, Heidelberg. mRNA-Seq libraries were prepared from total RNA samples treated with DNase and single-end sequenced on the specified Illumina NextSeq 500 sequencing instrument. RNA-seq data are available online at the geo platform (GSE234392).
RNA-seq data (~37.7M reads per sample) were processed following the Illumina data processing pipeline. STAR aligner was used to map reads on the GRCm38-mm10 mouse reference genome. Subsequent data analysis was performed in R. Briefly, unexpressed genes were pre-filtered using a cut-off of 10 read counts. Count data of genes with non-zero expression (n = 24,697) were input into the DESeq2 package for differential expression analysis between Pde2A−/− and Pde2A+/+ samples. The variance stabilizing transformation of data was done before PCA and hierarchical clustering of the heatmap. Functional enrichment analysis was performed using GSEA software and DAVID web tools.
cAMP and cGMP pathway analysis and comparison were performed using the annotations of the KEGG PATHWAY database.
For quantitative RT-PCR, mRNA was reverse transcribed to cDNA through Maxima H minus reverse transcriptase (Thermo Fischer Scientific, MA, USA). RT-qPCR reaction was carried out by using PowerUp SYBR Green Master Mix (Thermo Fisher Scientific, MA, USA). Target transcripts were analyzed using the Applied 7500 sequence detector system (Applied Biosystems, Carlsbad, CA, USA).
For the quantification analysis, the comparative threshold cycle (Ct) method was used. The Ct values of each gene were normalized to the Ct value of β-Act in the same RNA sample. The gene expression levels were evaluated by fold change using the equation 2−ddCt.
Primers used in mRNA analyses are reported in Supplementary Table S3.
cAMP and cGMP assay
Embryonic hearts were homogenized and sonicated in 10 volumes of 0.1 M HCl with 0.1% Triton X-100 and processed with the Direct cAMP/cGMP ELISA kit (Enzo Life Science Inc., NY, USA) following the manufacturer’s instruction. Heart samples and standards were acetylated before being loaded onto the plate by using Acetic Anhydride and Triethylamine 1:2. Samples were incubated overnight at 4 °C on a plate shaker. A substrate solution was added and incubated for 1 hour at room temperature. The reaction was stopped, and samples were read by the spectrophotometer at an optical density of 405 nm. Results are referred to as cAMP and cGMP standard curves performed together with the cAMP and cGMP Assay.
Reactive oxidative stress detection
Oxidative stress was quantified by measuring total reactive oxygen species (ROS) using 2 μM 2’,7’-dichlorodihydrofluorescein diacetate (CM-H2DCFDA; Thermo Fisher, MO, USA) staining and FACS analysis. Briefly, heart embryos, from untreated or NAC or Metaprolol treated mice, were dissected, washed in PBS, and dissociated with 5 mg/ml Collagenase Type II (Sigma-Aldrich, MO, USA) at 37 °C for 30 min. After washing with PBS, cells were resuspended in PBS and stained with CM-H2DCFDA for 30 min at 37 °C in the dark. At the end of the incubation, to remove the dye, cells were centrifuged and washed with PBS. The final cell suspensions were filtered through a 70 μm strainer, stained with Sytox Blue Dead Cell Stain (Thermo Fisher Scientific, MA, USA), and analyzed on a Cytoflex (Beckman Coulter, CA, USA). Flow cytometry data were analyzed with CytExpert software.
Statistical analysis
All data are expressed as mean ± SEM and analyzed with Student’s t-test with two tails or ANOVA-one way or ANOVA-two way with Tukey correction. Differences were considered significant if *P < 0.05.
Data availability
The data supporting this study are presented in the paper and in the supplementary information. Other information is available upon request. RNAseq data have been deposited in the Gene Expression Omnibus and are accessible through the accession number GSE234392.
References
Stephenson DT, Coskran TM, Wilhelms MB, Adamowicz WO, O’donnell MM, Muravnick KB, et al. Immunohistochemical localization of phosphodiesterase 2A in multiple mammalian species. J Histochem Cytochem. 2009;57:933–49.
Barbagallo F, Rotilio V, Assenza MR, Aguanno S, Orsini T, Putti S, et al. PDE2A is indispensable for mouse liver development and hematopoiesis. Int J Mol Sci. 2020;21:2902.
Assenza MR, Barbagallo F, Barrios F, Cornacchione M, Campolo F, Vivarelli E, et al. Critical role of phosphodiesterase 2A in mouse congenital heart defects. Cardiovasc Res. 2018;114:830–45.
Isidori AM, Cornacchione M, Barbagallo F, di Grazia A, Barrios F, Fassina L, et al. Inhibition of type 5 phosphodiesterase counteracts β2-adrenergic signalling in beating cardiomyocytes. Cardiovasc Res. 2015;106:408–20.
Ruhparwar A, Er F, Martin U, Radke K, Gruh I, Niehaus M, et al. Enrichment of cardiac pacemaker-like cells: neuregulin-1 and cyclic AMP increase If-current density and connexin 40 mRNA levels in fetal cardiomyocytes. Med Biol Eng Comput. 2007;45:221–7.
Tsai SY, Maass K, Lu J, Fishman GI, Chen S, Evans T. Efficient generation of cardiac Purkinje cells from ESCs by activating cAMP signaling. Stem Cell Rep. 2015;4:1089–102.
Yan X, Gao S, Tang M, Xi J, Gao L, Zhu M, et al. Adenylyl cyclase/cAMP-PKA-mediated phosphorylation of basal L-type Ca2+ channels in mouse embryonic ventricular myocytes. Cell Calcium. 2011;50:433–43.
Carvalho TMDCS, Cardarelli S, Giorgi M, Lenzi A, Isidori AM, Naro F. Phosphodiesterases expression during murine cardiac development. Int J Mol Scie. 2021;22:2593.
Zoccarato A, Surdo NC, Aronsen JM, Fields LA, Mancuso L, Dodoni G, et al. Cardiac hypertrophy is inhibited by a local pool of cAMP regulated by phosphodiesterase 2. Circ Res. 2015;117:707–19.
Weber S, Zeller M, Guan K, Wunder F, Wagner M, El-Armouche A. PDE2 at the crossway between cAMP and cGMP signalling in the heart. Cell Signal. 2017;38:76–84.
Acin-Perez R, Russwurm M, Günnewig K, Gertz M, Zoidl G, Ramos L, et al. A phosphodiesterase 2A isoform localized to mitochondria regulates respiration. J Biol Chem. 2011;286:30423–32.
Monterisi S, Lobo MJ, Livie C, Castle JC, Weinberger M, Baillie G, et al. PDE2A2 regulates mitochondria morphology and apoptotic cell death via local modulation of cAMP/PKA signalling. Elife. 2017;6:e21374.
Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat Rev Mol Cell Biol. 2007;8:722–8.
Madamanchi A. β-Adrenergic receptor signaling in cardiac function and heart failure. McGill J Med. 2007;10:99.
Heilbrunn SM, Bristow MR, Valantine HA, Ginsburg R, Fowler MB. Increased a-receptor density and improved hemodynamic response to catecholamine stimulation during long-term metoprolol therapy in heart failure from dilated cardiomyopathy. Circulation. 1986;79:483–90.
Waagstein F, Caidahl K, Wallentin I, Bergh CH, Hjalmarson A. Long-term beta-blockade in dilated cardiomyopathy. Effects of short- and long-term metoprolol treatment followed by withdrawal and readministration of metoprolol. Circulation. 1989;80:551–63.
Waagstein F, Hjalmarson A, Swedberg K, Bristow MR, Gilbert EM, Camerini F, et al. Beneficial effects of metoprolol in idiopathic dilated cardiomyopathy. Lancet. 1993;342:1441–6.
Hjalmarson Å, Goldstein S, Fagerberg B, Wedel H, Waagstein F, Kjekshus J, et al. Effects of controlled-release metoprolol on total mortality, hospitalizations, and well-being in patients with heart failure: the metoprolol CR/XL randomized intervention trial in congestive heart failure (MERIT-HF). J Am Med Assoc. 2000;283:1295–302.
White M, Yusuf S, McKelvie RS, Pericak D, Young J, Latini R, et al. Effects of metoprolol CR in patients with ischemic and dilated cardiomyopathy: the randomized evaluation of strategies for left ventricular dysfunction pilot study. Circulation. 2000;101:378–84.
Zhan DY, Morimoto S, Du CK, Wang YY, Lu QW, Tanaka A, et al. Therapeutic effect of β-adrenoceptor blockers using a mouse model of dilated cardiomyopathy with a troponin mutation. Cardiovasc Res. 2009;84:64–71.
Martínez-Milla J, Raposeiras-Roubín S, Pascual-Figal DA, Ibáñez B. Role of beta-blockers in cardiovascular disease in 2019. Rev Esp Cardiolía (Engl Ed). 2019;72:844–52.
Wang J, Liu X, Arneja AS, Dhalla NS. Alterations in protein kinase A and protein kinase C levels in heart failure due to genetic cardiomyopathy. Can J Cardiol. 1999;15:683–90.
Antos CL, Frey N, Marx SO, Reiken S, Gaburjakova M, Richardson JA, et al. Dilated cardiomyopathy and sudden death resulting from constitutive activation of protein kinase a. Circ Res. 2001;89:997–1004.
Saad NS, Elnakish MT, Ahmed AAE, Janssen PML. Protein kinase A as a promising target for heart failure drug development. Arch Med Res. 2018;49:530–7.
Du Y, Yan L, Wang J, Zhan W, Song K, Han X, et al. β1-Adrenoceptor autoantibodies from DCM patients enhance the proliferation of T lymphocytes through the β1-AR/cAMP/PKA and p38 MAPK pathways. PLoS ONE. 2012;7:e52911.
Zhang X, Szeto C, Gao E, Tang M, Jin J, Fu Q, et al. Cardiotoxic and cardioprotective features of chronic β-adrenergic signaling. Circ Res. 2013;112:498–509.
Dunkerly-Eyring B, Kass DA. Myocardial phosphodiesterases and their role in cGMP regulation. J Cardiovasc Pharm. 2020;75:483–93.
Tzoumas N, Farrah TE, Dhaun N, Webb DJ. Established and emerging therapeutic uses of PDE type 5 inhibitors in cardiovascular disease. Br J Pharm. 2020;177:5467–88.
Lochner A, Moolman JA. The many faces of H89: a review. Cardiovasc Drug Rev. 2006;24:261–74.
Murray AJ. Pharmacological PKA inhibition: all may not be what it seems. Sci Signal. 2008;1:re4.
Sahir N, Mas C, Bourgeois F, Simonneau M, Evrard P, Gressens P. Caffeine-induced telencephalic vesicle evagination in early post-implantation mouse embryos involves cAMP-dependent protein kinase (PKA) inhibition. Cereb Cortex. 2001;11:343–9.
Nakamura T, Colbert M, Krenz M, Molkentin JD, Hahn HS, Dorn GW 2nd, et al. Mediating ERK1/2 signaling rescues congenital heart defects in a mouse model of Noonan syndrome. J Clin Investig. 2007;117:2123–32.
Carraway MS, Suliman HB, Jones WS, Chen CW, Babiker A, Piantadosi CA. Erythropoietin activates mitochondrial biogenesis and couples red cell mass to mitochondrial mass in the heart. Circ Res. 2010;106:1722–30.
Luciano-Mateo F, Cabré N, Fernández-Arroyo S, Baiges-Gaya G, Hernández-Aguilera A, Rodríguez-Tomàs E, et al. Chemokine C–C motif ligand 2 overexpression drives tissue-specific metabolic responses in the liver and muscle of mice. Sci Rep. 2020;10:11954.
McCarty MF, Lerner A, Dinicolantonio JJ, Iloki-Assanga SB. High intakes of bioavailable phosphate may promote systemic oxidative stress and vascular calcification by boosting mitochondrial membrane potential—is good magnesium status an antidote? Cells. 2021;10:1744.
Redon A, Oliva MR, Tormos C, Giner V, Chaves J, Iradi A, et al. Antioxidant activities and oxidative stress byproducts in human hypertension. Hypertension. 2003;41:1096–101.
Senoner T, Dichtl W. Oxidative stress in cardiovascular diseases: still a therapeutic target? Nutrients. 2019;11:2090.
Rokicki W, Strzałkowski A, Kłapcińska B, Danch A, Sobczak A. Antioxidant status in newborns and infants suffering from congenital heart defects. Wiad Lek. 2003;56:337–40.
Ercan S, Çakmak A, Kösecik M, Erel Ö. The oxidative state of children with cyanotic and acyanotic congenital heart disease. Anadolu Kardiyol Derg. 2009;9:486–90.
Pirinccioglu AG, Alyan Ö, Kizil G, Kangin M, Beyazit N. Evaluation of oxidative stress in children with congenital heart defects. Pediatr Int. 2012;54:94–98.
Vanreusel I, Vermeulen D, Goovaerts I, Stoop T, Ectors B, Cornelis J, et al. Circulating reactive oxygen species in adults with congenital heart disease. Antioxidants. 2022;11:2369.
Kawahara T, Lambeth D. Molecular evolution of Phox-related regulatory subunits for NADPH oxidase enzymes. BMC Evol Biol. 2007;17:178.
Pierelli G, Stanzione R, Forte M, Migliarino S, Perelli M, Volpe M, et al. Uncoupling protein 2: a key player and a potential therapeutic target in vascular diseases. Oxid Med Cell Longev. 2017;2017:1–11.
Brigelius-Flohé R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta. 2013;1830:3289–303.
Seo JH, Park JH, Ji Lee E, Thuy Lu Vo T, Choi H, Yong Kim J, et al. ARD1-mediated Hsp70 acetylation balances stress-induced protein refolding and degradation. Nat Commun. 2016;7:12882.
Katsi V, Papakonstantinou IP, Papazachou O, Makris T, Tsioufis K. Beta-blockers in pregnancy: clinical update. Curr Hypertens Rep. 2023;25:13–24.
Trappanese DM, Liu Y, McCormick RC, Cannavo A, Nanayakkara G, Baskharoun MM, et al. Chronic β1-adrenergic blockade enhances myocardial β3-adrenergic coupling with nitric oxide-cGMP signaling in a canine model of chronic volume overload: new insight into mechanisms of cardiac benefit with selective β1-blocker therapy. Basic Res Cardiol. 2015;110:456.
Salpietro V, Perez-Dueñas B, Nakashima K, San Antonio-Arce V, Manole A, Efthymiou S, et al. A homozygous loss-of-function mutation in PDE2A associated to early-onset hereditary chorea. Mov Disord. 2018;33:482–8.
Doummar D, Dentel C, Lyautey R, Metreau J, Keren B, Drouot N, et al. Biallelic PDE2A variants: a new cause of syndromic paroxysmal dyskinesia. Eur J Hum Genet. 2020;28:1403–13.
Yousaf H, Rehmat S, Jameel M, Ibrahim R, Hashmi SN, Makhdoom EUH, et al. A homozygous founder variant in PDE2A causes paroxysmal dyskinesia with intellectual disability. Clin Genet. 2023;104:324–33.
Blain A, Greally E, Laval S, Blamire A, Straub V, MacGowan GA. Beta-blockers, left and right ventricular function, and in-vivo calcium influx in muscular dystrophy cardiomyopathy. PLoS ONE. 2013;8:e57260.
Yu M, Liu T, Chen Y, Li Y, Li W. Combination therapy with protein kinase inhibitor H89 and Tetrandrine elicits enhanced synergistic antitumor efficacy. J Exp Clin Cancer Res. 2018;37:114.
Dilworth MR, Andersson I, Renshall LJ, Cowley E, Baker P, Greenwood S, et al. Sildenafil citrate increases fetal weight in a mouse model of fetal growth restriction with a normal vascular phenotype. PLoS ONE. 2013;8:e77748.
Moazzen H, Lu X, Ma NL, Velenosi TJ, Urquhart BL, Wisse LJ, et al. N-Acetylcysteine prevents congenital heart defects induced by pregestational diabetes. Cardiovasc Diabetol. 2014;13:46.
Di Siena S, Gimmelli R, Nori SL, Barbagallo F, Campolo F, Dolci S, et al. Activated c-Kit receptor in the heart promotes cardiac repair and regeneration after injury. Cell Death Dis. 2016;7:e2317.
Baldock R, Bard J, Davidson D, Morriss-Kay G, editors. Kaufman’s atlas of mouse development supplement with coronal sections. Academic Press, UK: Elsevier; 2016. p. 177–91.
Dorr AE, Lerch JP, Spring S, Kabani N, Henkelman RM. High resolution three-dimensional brain atlas using an average magnetic resonance image of 40 adult C57Bl/6J mice. Neuroimage. 2008;42:60–69.
Acknowledgements
We thank the EMMA service project that funded the EC FP7 Capacities Specific Program and allowed us to obtain Pde2A+/− mice. We are particularly thankful to Dr. V. Benes, EMBL Head of Genomics Core Facility, EMBL Heidelberg, Germany for the RNA-seq processing, Dr. E. Perlas, Head of Histology Core Facility, EMBL Rome, Italy for helpful suggestions, to Prof. M. Giorgi, University of Sapienza, Italy, and Dr. G. Ruberti and G.D. Tocchini-Valentini, IBBC-CNR, Monterotondo Rome, Italy for critical reading. This work was supported by StHs [2019 FN, LdA, MP], the University of Sapienza [Ateneo 2020 to LdA], and AIRC 2019 [IG 23329 to MP]. VF was supported by the Lazio Innova POR FESR Lazio 2014 to 2020 HDACiPLAT Research Project [A0375-2020-36575 granted to Dr. Giovina Ruberti, IBBC-CNR]. This paper is dedicated to the memory of Flavia Ferrari.
Author information
Authors and Affiliations
Contributions
SC and MB performed the experiments in vivo, analyzed the histological samples, and performed RT-qPCR; TO acquired, analyzed, and interpreted the Micro-CT data; VF performed the RNA-seq analysis, LM performed experiments, contributed to the interpretation of the results and to the draft of the manuscript, AGdeO contributed to the pharmacological in vivo treatments and RT-qPCR experiments, FL, SM, and FF designed, performed, analyzed and interpreted the Flow cytometry experiments, FN contributed to the conception of the study and critically revised the manuscript, LDA and MP conceptualized, designed and coordinated the study, interpreted the data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All our experimental animal procedures were carried out in accordance with Directive 2010/63/EU and with the approval of the Italian Ministry of Health n 919/2020-PR dated 28/9/2020. There were no human subjects used in this study, so human ethics approval and consent to participate are not applicable in this study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Sergio Lavandero
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cardarelli, S., Biglietto, M., Orsini, T. et al. Modulation of cAMP/cGMP signaling as prevention of congenital heart defects in Pde2A deficient embryos: a matter of oxidative stress. Cell Death Dis 15, 169 (2024). https://doi.org/10.1038/s41419-024-06549-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-024-06549-1
