
Abstract
Abnormal activation of telomerase occurs in most cancer types, which facilitates escaping from cell senescence. As the key component of telomerase, telomerase reverse transcriptase (TERT) is regulated by various regulation pathways. TERT gene changing in its promoter and phosphorylation respectively leads to TERT ectopic expression at the transcription and protein levels. The co-interacting factors play an important role in the regulation of TERT in different cancer types. In this review, we focus on the regulators of TERT and these downstream functions in cancer regulation. Determining the specific regulatory mechanism will help to facilitate the development of a cancer treatment strategy that targets telomerase and cancer cell senescence.

As the most important catalytic subunit component of telomerase, TERT is rapidly regulated by transcriptional factors and PTM-related activation. These changes directly influence TERT-related telomere maintenance by regulating telomerase activity in telomerase-positive cancer cells, telomerase assembly with telomere-binding proteins, and recruiting telomerase to the telomere. Besides, there are also non-canonical functions that are influenced by TERT, including the basic biological functions of cancer cells, such as proliferation, apoptosis, cell cycle regulation, initiating cell formation, EMT, and cell invasion. Other downstream effects are the results of the influence of transcriptional factors by TERT. Currently, some small molecular inhibitors of TERT and TERT vaccine are under research as a clinical therapeutic target. Purposeful work is in progress.
Similar content being viewed by others
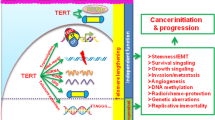
Facts
-
As the most important catalytic subunit component of telomerase, TERT is regulated by transcriptional factors and PTM-related activation.
-
Telomere maintenance by regulating telomerase activity and assembly with telomere-binding proteins, as well as recruiting telomerase to telomere, are regulated by TERT in telomerase-positive cancer cells.
-
The non-canonical biological function of TERT and its influence on different components of the tumor microenvironment in cancer cells, including angiogenesis, vascularization, EMT, proliferation, inflammatory factors, and immune response, are influenced by TERT.
-
Small molecular inhibitors of TERT and TERT vaccine are under research as potential therapeutic targets.
Introduction
Telomeres, the repetitive ends of chromosomes, comprise ca. 10–15 kb of tandem double-stranded 5′-TTAGGG-3′ repeats. Based on recent research by the ICGC/TCGA Pan-Cancer Analysis of Whole Genomes (PCAWG) Consortium, while telomere sequences comprise long stretches of 5′-TTAGGG-3′ (G-type) repeat arrays, there is increasing evidence that telomeric-associated sequences include variants—most commonly 5′-TGAGGG-3′ (G-type), 5′-TCAGGG-3′ (C-type), and 5′-TTGGGG-3′ (J-type) repeats, toward the proximal, subtelomeric regions [1]. These DNA sequences are coated with shelterin, a specialized protein complex that plays a fundamental role in regulating telomere protection [2,3,4]. Telomere structure and function serve to maintain chromosomal integrity and regulate cell division.
In most cancers, germline and stem cells, long-term proliferation requires telomerase activity to overcome the telomere shortening, which occurs with cell division. This shortening eventually arrests cellular growth, potentially providing an initial proliferative barrier to tumor formation in humans [5]. In adults, most stem cells exhibit telomerase activity, although at substantially lower levels. Although such reduced levels are sufficient for slowing down telomere shortening and increasing the replicative lifespan [6], they cannot prevent replicative senescence. Telomeres maintain DNA by acting on its 3′ overhang via the telomerase RNA component (TERC or TR) template; the telomerase adds single-stranded 5′-TTAGGG-3′ repeats to the lagging strand of the newly synthesized DNA. Finally, new primer RNA is added, and this is subsequently filled in with DNA polymerase alpha. In cancer, abnormal telomerase activation causes cancer cells to overcome cell growth arrest, resulting in cell immortalization [7].
Telomerase reverse transcriptase (TERT) is a key component of telomerase. Human TERT was first cloned in 1997 [5, 8,9,10], eight years after telomerase was first identified in HeLa cells [11]. The catalytic component of the telomerase holoenzyme complex comprises single molecules of TERT, WRAP53 (also known as TCAB1), two molecules of H/ACA ribonucleoprotein complex subunits DKC1, NOP10, NHP2, and GAR1, and a telomerase RNA template [12,13,14]. While TERT is rarely detected in normal human cells, except in embryonic cells, germ cells, and stem cells, TERC is universally expressed as a lncRNA in normal cells [5, 12, 15,16,17]. However, when acting as part of the telomerase holoenzyme [18], TERT can regulate telomere elongation, ultimately participating in tumor formation in a telomere-dependent or -independent manner.
Mutant TERT promoters, commonly detected in various types of cancer, induce TERT overexpression by recruiting transcription factors that do not normally regulate TERT gene expression and promoting abnormal telomerase activation [19,20,21,22,23]. Functionally, TERT affects cancer formation mostly by maintaining telomere length and enabling cells to avert cell senescence [24]. TERT has other non-canonical activity independently of its telomere-lengthening function, in particular, affecting the DNA repair response [25], gene transcription [24], and ubiquitin-proteasomal function [26, 27]. In this review, we focus on the regulators of TERT and the specific regulatory in cancer. These contents will help to facilitate the development of the cancer treatment strategy that targets telomerase and cancer cell senescence.
Structural properties of TERT
TERT contains 1132 amino acids and comprises 4 telomerase-specific motifs, including the telomerase essential N-terminal (TEN) domain, the TERT RNA-binding domain (TRBD), the reverse transcriptase (RT) domain, and the C-terminal extension (CTE) [8, 28,29,30]. A new TERT domain, TRAP, was recently characterized via cryoelectron microscopy mapping of the Tetrahymena telomerase; TRAP is a large insertion of RT fingers [31, 32]. TERT and telomerase RNA (hTR) are composed of telomerase catalytic core of telomerase; TERT is associated with the pseudoknot/template (PK/t) domain and conserved regions 4 and 5 (CR4/5) of hTR. A H/ACA ribonucleoprotein (RNP) lobe is essential for telomerase biogenesis, which forms a telomerase holoenzyme with a TERT-related catalytic core [33].
Telomerase access to and activity at telomeres for telomere maintenance is tightly regulated. In normal conditions, a six-numbered protein complex, called shelterin, is composed of POT1, TPP1, TIN2, TRF1, TRF2, and RAP1 and protects the telomeric DNA ends. As a main component of shelterin, TPP1 has been implicated in telomerase recruitment to telomeres. Structurally, the TEN domain of TERT provides an anchor site for binding to telomeric DNA, while this binding initiates the primer-template interaction for telomere elongation. Furthermore, the TEN domain provides binding sites for TPP1 and contributes indirectly to telomerase activity by forming a complex with TRAP that stabilizes the active fold of TRAP, which participates directly in telomere repeat-addition processivity [30, 32]. TRBD interacts with the RT thumb subdomain, resulting in a closed ring-like tertiary structure with a large cavity at its center that binds to the primer-template duplex [34]. The fingers and palms of the TERT’s RT domain interact with the RNA backbone to place the template in the active site [35]. The RT motifs secure the hTR to the protein to maintain ribonucleoprotein stability and provide an active site for catalysis [36]. The CTE and TRBD domains of TERT interact simultaneously with the CR4/5 domain of hTR. Although the hTR binding sites are not active TERT sites, this interaction ensures the correct positioning of the TERT domain following TPP1 binding to the TEN domain, which allows the anchor site to engage the DNA substrate [37]. Moreover, TPP1, POT1, and TIN2 form a complex within Shelterin. POT1 binds to both the DNA substrate and TEN domain of TERT, forming a gate in front of the active telomerase site, which stabilizes DNA binding [38]. As a result, TPP1 and POT1 provide more space to accommodate the G-quadruplex structures formed by the telomeric-DNA product, reduce DNA dissociation during repeat-synthesis, and cooperatively increase telomerase repeat-addition processivity [5, 16, 37, 38].
Effects on TERT in cancer formation
Transcriptional regulation
It is suggested that the transcription of hTERT is the dominant step in the regulation of telomerase activity. Recurrent mutations and chromosomal rearrangements in TERT promoters are frequent in human cancers.
Alteration in the TERT gene includes genomic gains/amplifications, promoter rearrangements, and “hotspot” promoter mutations at a position relative to the transcription start site [39]. These alterations have been reported in multiple cancer types, especially TERT promoter mutations, which are associated with increased TERT expression [20, 21, 40,41,42]. In a pan-cancer study of somatic TERT promoter mutations and amplification, Sounak et al. found that among 30,733 specimens, almost 11.3% of cases with TERT promoter mutation were accompanied by 2.3% TERT amplification and 86.3% with no TERT amplification or promoter mutation [40]. It is indicated that TERT overexpression, caused by promoter mutation, is attributed to telomerase abnormal activation in cancer, although only a small portion of cancer patients are recognized as having TERT mutation.
hTERT promoter mutations are recognized in melanomas, while different types of cancer, including astrocytic glioma [43], sebaceous neoplasms [41], thyroid cancer [44], bladder cancers [45], and chondrosarcoma [46] have also been detected. The promoter mutation −124 C > T has been reported in almost half of patients with chondrosarcoma [46]; together, −124 C > T and −146 C > T are potentially important biomarkers in malignant melanoma linked to poor prognosis [42]. In 2020, members of PCAWG characterized the genomic footprint of the telomere mechanism and reported C228T or C250T promoter mutations in different cancer types [1], consistent with findings obtained in thyroid cancer and sebaceous neoplasms [41, 44]. However, TERT promoter mutations are not frequently detected in leukemias and colorectal cancers [47, 48]. Therefore, it has been hypothesized that in cancers originating in tissues with low self-renewal rates and limited ability to maintain tissue homeostasis, hTERT promoter mutations occur more often than those in cancer that already display high telomerase expression [47, 49]. Allelic DNA methylation within the TERT promoter further contributes to the regulation of TERT expression in cancer. Differential allelic expression occurs in almost 50% of cancer cell lines [50], which is more common than in human normal tissues.
Hypermethylation of the TERT hypermethylated oncological region enhances TERT expression when coupled with a hypomethylated proximal core promoter. However, this impact is abolished when the core TERT promoter is hypermethylated, which is often the case when TERT promoter mutations are present [51, 52].
Mechanistically, mutant TERT promoters enable “enhancer hijacking” near the TERT transcription start site [1, 53], which eventually leads to telomerase activation by abrogating transcription silencing of TERT [47]. In glioblastoma multiforme (GBM) cells, the NF-κB signal pathway selectively induces TERT expression with mutant TERT promoters, which is due to the p52-binding sites formation after specific mutation in TERT promoters [54]. In BRAFV600E-driven human cancer cells, including melanoma cell line A375, thyroid cancer cell lines BCPAP and 8305 C, and breast cancer cell line MDA-MB-231, MAPK/ERK signaling affects the binding capacity of GABP to mutant TERT promoter and eventually results in TERT activation. Specifically, the downstream factor Sp1 is phosphorylated, resulting in HDAC1 dissociation from the zinc finger DNA-binding domain of Sp1 and constantly promotes GABPA binding to the mutant TERT promoter [55]. Similarly, in BRAFV600E papillary thyroid cancer (PTC) cells, MAPK pathway activation by the BRAFV600E mutation upregulates E-twenty six (ETS) transcription factors, including ETV1, ETV4, ETV5, and ELF3, and increases TERT expression by binding to the ETS-binding site which generated by the TERT promoter mutations [56]. Phosphorylation of FOS, another downstream factor of BRAF V600E, promotes FOS binding to the GABPB 5′ UTR, constantly activating GABPB and promoting its binding to the mutant TERT promoter in PTC cells and melanoma cells [57, 58]. In summary, these studies demonstrate that BRAF V600E and TERT promoter mutations cooperatively upregulate TERT expression via the activation of the MAPK signal pathway dependent on FOS and GABP.
Post-translational modifications (PTMs) of TERT
Following translation, PTM occurs via the addition of various chemical groups to one or more amino acid residues. PTM, which is especially common in cancer, can change the protein’s physicochemical properties by altering its spatial conformation and activity state, subcellular localization, and protein–protein interactions [59]. PTMs increase the biochemical diversity of TERT functions, thereby affecting telomerase regulation in cancer cells. TERT phosphorylation is the most important type, which plays an important role in regulating telomerase holoenzyme integrity. Thr249 [60], Ser824 [61], Ser227 [61], Ser457 [62], and Tyr707 [63] are known sites of phosphorylation by different kinases; the specific impacts are listed in Table 1.
In the human melanoma SK-MEL 28 cell line, Akt (also known as protein kinase B) phosphorylates TERT at serine 824 and 277, enhancing telomerase activity [61]. Akt inactivation via the phosphatidylinositol-Akt kinase pathway inhibitor wortmannin diminishes TERT phosphorylation and inhibits human telomerase activity [61]. In H1299 cells, phosphorylation at serine 227 by Akt is required for efficient nuclear translocation of hTERT and cellular immortalization [64]. Genistein, an isoflavone abundant in soy, has multiple molecular effects on cancer propagation [65]. In prostate cancer cells, genistein reduces telomerase activity by inhibiting Akt, thereby dephosphorylating TERT [66, 67]. The dietary component of genistein is a potential therapeutic molecule within a TERT-targeting cancer treatment strategy.
Protein kinase C (PKC), a serine/threonine kinase, participates directly or indirectly in various signal transduction pathways via target-protein phosphorylation [68]. PKC phosphorylation of TERT promotes the association between hsp90 and TERT, thus maintaining telomerase holoenzyme integrity [69]. The PKC activator (e.g., phorbol myristate acetate) enhances telomerase activity, whereas the PKC inhibitors (BIC, H-7, and bisindolylmaleimide I) inhibit it [69,70,71]. PKC isoenzymes α, β, δ, ε, and ζ are overexpressed in cancer patients and differentially correlated with high telomerase activity in different cancer types. In head and neck cancer cells, OEC-M1, PKC α, β, δ, ε, and ζ are involved in telomerase regulation through phosphorylation [69]. In nasopharyngeal carcinoma cells, among these isoenzymes, only PKC-ζ participates in activating telomerase activity [72]. In human breast cancer cell PMC42, PKC-α is implicated in TERT phosphorylation [73], while protein phosphatase 2 A (PP2A) directly dephosphorylates TERT, affecting telomerase assembly and disrupting its activity on the contrary [74]. These findings reveal the various functions of different PKC isoenzymes.
CDK1, a cell-cyclin-dependent serine/threonine kinase, plays an important role in telomere maintenance [75, 76]. During mitosis, CDK1 phosphorylates TERT at threonine 249, and this posttranslational modification is essential for tumor formation in pancreatic cancer and hepatocellular carcinoma [60]. hTERT phosphorylation at threonine 249 is positively associated with aggressive cancer phenotypes with high proliferative activity, high pathological grade, and severe nuclear atypia [77]. Phosphorylated hTERT expression is not associated with telomere length, suggesting that this phosphorylation might influence tumor formation dependent on RdRP activity rather than telomerase activity in the HeLa cell line.
Under oxidative stress, the Scr kinase family phosphorylates TERT at tyrosine 707, triggering TERT export from the nucleus to mitochondria in a GTPase Ran-dependent manner [63, 78]. In addition, H2O2 treatment downregulates mitochondrial TERT via phosphorylation by Src kinase in human embryonic kidney cells [79], which indicated that Scr kinase-related TERT phosphorylation could result in the translocation of TERT protein, which might influence telomerase activity both in cancer cells and human embryonic kidney cells.
Dual-specificity tyrosine-(Y)-phosphorylation-regulated kinase 2 (Dyrk2), also a cyclin-dependent kinase [80], binds directly to TERT, phosphorylating it at Ser457 [62], following which the Dyrk2–EDVP E3 ligase complex ubiquitinates TERT for degradation, downregulating it [62].
Hypoxia increases telomerase activity, possibly by activating the mitogen-activated protein kinase (MAPK) signal. In the A2780 and HT-29 cell lines, MAPK inhibitors diminish the upregulation of telomerase activity but do not influence basal telomerase activity [81]. In many cancers, inhibiting MAPK pathway effectors inhibits TERT mRNA expression [82]. However, evidence showing that MAPK directly phosphorylates TERT remains lacking.
c-Abl, a crucial prototypic non-receptor tyrosine kinase, is commonly activated by DNA damage and implicated in various cellular processes. c-Abl binds with and phosphorylates DNA-damage response proteins such as P73, and activates p53, often inducing cell death [83, 84]. Following exposure to ionizing radiation, c-Abl is activated by DNA damage and tyrosine phosphorylation of TERT increases; this is abolished by c-Abl deletion [85]. Following genotoxic stress, TERT phosphorylation inhibits telomerase activity and negatively regulates telomere length [85].
The oncogenic counterpart of c-Abl, the BCR-ABL fusion protein, causes certain human leukemias [86]. The translocation of chromosomes 22 and 9 results in the formation of the Philadelphia chromosome (Ph), the cytogenetic characteristic of chronic myeloid leukemia [87]. In this process, part of the breakpoint cluster region (BCR) gene on chromosome 22 fuses with the ABL gene on chromosome 9, forming BCR-ABL fusion protein with tyrosine kinase [88]. TERT expression and telomerase activity are higher in BCR-ABL positive cells (the K562 cell line) than BCR-ABL deficient cells (the HL60 cell line), as a result of TERT phosphorylation at tyrosine residues by BCR-ABL [87]. Gleevec inhibits TERT phosphorylation and downregulates TERT mRNA in BCR-ABL positive cells by inhibiting BCR-ABL kinase activity [88]. In BCR-ABL positive cells, Gleevec (imatinib), an inhibitor of receptor tyrosine kinase, induces telomerase translocation out of the nucleolus, indicating that TERT phosphorylation can influence telomerase activity by altering its location in cancer cells, consistent with other findings [64, 89, 90].
Ubiquitination of TERT, another form of PTM, also occurs in human cancers. Makorin Ring finger protein 1, encoded by MKRN1- a ubiquitin ligase, is a TERT-binding protein that regulates TERT degradation [91, 92]. The proteasome inhibitor MG132 reverses the loss of telomerase activity by promoting MKRN1 accumulation during cell cycle arrest [92]. Sphingosine kinase 2 generates lysophospholipid sphingosine 1-phosphate, a factor that binds to TERT and inhibits the interaction of TERT with MKRN1 [93]. Human double minute 2 (HDM2), an E3 ubiquitin-protein ligase, induces ubiquitination of target genes involved in tumorigenesis, including p53 [94]. As with MKRN1, HDM2 binds to TERT and induces TERT polyubiquitination [95]. In U2OS cells, HDM2 polyubiquitinates TERT principally at the N-terminus at five potential sites. Depletion of HDM2 strengthens cellular protective effects against apoptosis by stabilizing TERT [95]. HDM2 is also required in the p73-dependent relief of p53-mediated TERT suppression on a transcriptional level [96].
Polo-like kinase 1 (Plk1) is a Ser/Thr kinase that participates primarily in various aspects of mitotic events. Plk1 interacts directly with TERT, independently of its kinase activity, and enhances the stability of TERT by inhibiting its ubiquitin-mediated degradation by MKRN1 and HDM2, thereby increasing cancer cell telomerase activity [97, 98]. Hsc70-interacting protein (CHIP), which interacts with Hsc/Hsp70, is physically associated with TERT and downregulates telomerase activity via ubiquitin-mediated degradation, independent of Hsp90 binding. This degradation results in cytoplasmic accumulation of TERT, inhibiting its nuclear localization [99]. However, CHIP does not influence telomerase activity because the interaction between TERT and CHIP occurs in the cytoplasm, suggesting that CHIP may be associated only with intermediate or immature nonfunctional TERT [99].
Dyrk2 can directly phosphorylate TERT, which results in TERT degradation. This degradation is associated with the EDD-DDB1-VprBP E3 ligase complex, while Dyrk2 acts as a scaffolding protein for ubiquitination substrates [62].
Small ubiquitin-related modifier (SUMO) modification (SUMOylation) is a key regulatory modification of eukaryotic cells that may alter target protein activity, interactions, or longevity, ultimately influencing physiological processes, especially those in cancer cells [100,101,102,103,104,105]. SUMO1 directly SUMOylates TERT via CBX4, a SUMO E3 ligase. The SUMOylation of TERT enhances the migration and invasiveness of MCF7 cells [106].
TERT-related regulations in telomere maintenance
TERT is regulated by specific modulators, and these regulations influence the telomerase assembly, trafficking, blocking association, and mainly telomerase activity. Some positive regulators of telomerase promote telomere elongation by influencing telomerase holoenzyme assembly. In contrast, negative regulators inhibit telomerase activity, particularly by preventing telomerase from binding with the long telomere-end or with telomere-binding proteins (Table 2 and Fig. 1).

The activators include NVL2 and EST1 lengthen telomere by influencing telomerase holoenzyme assembly and activating telomerase, respectively. Some inhibitors such as PinX1, MCRS2, and PML inhibit telomerase activity through unknown mechanism. MKRN1 influences protein stability of TERT and decreases telomerase activity eventually. PIF1 could prevent the combination of telomerase with long telomere ends which leads to telomere shortening. CST complex which combined with CTC1, TEN1, and STN1 could destroy the interaction of the telomere with TPP1-POT1 and also leads to telomere shortening.
Factors that influence telomerase holoenzyme assembly
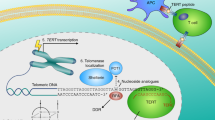
Different domains and unique spatial structures provide a structural basis for TERT to assemble telomerase holoenzymes. Therefore, modulators that contribute to the intracellular distribution of TERT also influence this tightly regulated process. The 70-kDa heat shock protein (HSP70), a ubiquitous molecular chaperone that controls various cellular protein-folding and remodeling processes [107], transiently binds to TERT in the absence of TR. The 90-kDa heat shock protein (HSP90), accompanied by chaperone p23, binds specifically with TERT and ensures its correct assembly with the template RNA [108]. HSP90 and p23 load TERT into the Cajal body, generating an enzymatically active telomerase complex. Once an active assembled complex is formed, the combination of HSP70 and TERT is abolished [108] and Telomerase Cajal body protein 1 (TCAB1) then binds to TERT and facilitates telomerase translocation from the Cajal bodies into the nucleoplasm, where TPP1 facilitates the recruitment of the complex to the telomere (Fig. 2) [12, 109].

Transportation of TERT from the Cajal body to nuclear following telomerase construction due to Hsp90 and p23 through the activation of TCAB. Other factors participating in TERT translocations are described, and the interaction between TERT and shelterin complex is shown.
In HeLa cells, Nuclear valosin-containing protein-like (NVL), which was first recognized as an interactor with TERT in yeast, could interact with TERT and is positively associated with telomerase holoenzyme assembly. Knockdown of endogenous NVL2 downregulates TERT, reducing telomerase activity [110]. This suggests that NVL2 is essential for telomerase enzymatic activity.
Factors that influence the recruitment of telomerase to the telomere
After telomerase holoenzyme assembly, the recruitment of telomerase to the telomere is a limiting step to promoting telomere addition. The modulators of TERT also participate in this critical process. hEST1A and hEST1B are human homologs of the yeast telomerase subunit Est1p, which is a functional factor in telomere maintenance. hEST1A and hEST1B bind to TERT independently of the RNA subunit, participating in telomere maintenance by activating or recruiting telomerase to the telomere [111, 112].
During the S phase of the cell cycle, telomerase is recruited to the telomere via the interaction of TPP1 with the TEN domain of TERT; telomerase then extends the G-rich strand of the telomere via TPP1–POT1 complex-promoted translocation of telomerase. In the late S/G2 phase, the CST complex (comprising CTC1, TEN1, and STN1) restricts the interaction between telomerase and TPP1–POT1, preventing telomerase from accessing the newly synthesized G-rich telomere strand, thus enabling it to bind to telomeric single-stranded DNA [113,114,115,116,117,118,119].
The PIF1 family helicases, which have multiple roles in eukaryotes, negatively regulate telomerase activity by preventing it from combining with the long telomere ends [120, 121]. In addition, PIF1 binds directly to TERT and influences proliferation and apoptosis independent of telomerase activity, although the mechanisms remain unclear in cervical cancer cells [122, 123].
Activating transcription factor 7 (ATF7), the primary factor in stress-induced telomere shortening, binds to TERT and leads to the release of ATF7 and telomerase from telomeres, eventually resulting in telomere shortening [124]. However, in HeLa cells, shRNA-induced alteration of ATF7 expression did not affect telomerase activity, which indicated a telomerase-independent telomere length regulation [124].
Factors that influence telomerase activity
As the main component subunit of telomerase, the regulations of TERT result in changes in telomerase activity. The oncogene PinX1, first identified in a yeast two-hybrid screen as a TRF1-binding protein, is a major haploinsufficient tumor suppressor essential for maintaining telomerase activity and chromosome stability [125]. PinX1 binds to the 74 aa C-terminal fragment of TERT in its telomerase inhibitory domain, inhibiting telomerase activity and thereby inducing telomere shortening [126]. PinX1–TERT binding maintains PinX1-mediated TRF1 stability and influences the TRF1 nucleolar localization, which is important in preserving telomere integrity [127, 128].
MCRS2, a novel isoform of human micro-spherule protein 1 (MCRS1), interacts with PinX1. MCRS2 binds to TERT, forming a stable complex that inhibits telomerase activity, thereby shortening the telomere [129].
The gene encoding promyelocytic leukemia (PML), a tumor suppressor primarily expressed in the nucleoplasm, interacts directly or indirectly with TERT in HepG2, U2OS, and HFF cell lines, negatively regulating telomerase activity and eventually resulting in telomere shortening [130].
TERT-related regulations facilitate oncogenesis independent of telomerase
TERT exhibits non-canonical functions involved in tumor oncogenesis independent of telomerase activity, which is detailed in the following sections (Table 2 and Fig. 3).

Instead of telomerase regulation, TERT also participate in proliferation, apoptosis, cell cycle controlling, tumor-initiating cell formation, EMT, cell invasion, and the regulation of transcriptional factor-related gene expression changing processes. These changings in cancer also influence the components in tumor microenvironment.
Factors that influence telomere maintenance independent of telomerase
In human cancer cell lines, TERT physically forms a complex with BRG1, a SWI/SNF-related chromatin remodeling protein, and nucleostemin, a nucleolar GTP-binding protein. The TERT–BRG1–nucleostemin complex drives tumor-initiating cell formation by regulating BRG1 activity rather than contributing directly to telomere maintenance [131, 132].
Factors that influence different components of the tumor microenvironment
According to the heterogeneity of different types of cancers, the tumor microenvironment (TME) is composed of neoplastic, stromal, endothelial, and infiltrating immune cells, and the interactions of these elements determine the pathological process in cancer progression [133, 134]. Previous studies have shown that the interactions between TERT and other factors, including c-MET [135], nuclear factor κB (NF-κB) p65 [136, 137], Myc [138], and Sp1 [139], influence downstream gene expression and signal transduction. These telomere-independent activities contribute to the regulations of other cell processes in TME, including angiogenesis, vascularization, epithelial-to-mesenchymal transition (EMT) [135, 140], and inflammation [136, 137], eventually promoting cancer cell proliferation [122].
Effects on blood vessels: angiogenesis and vascularization
In human gastric tumor samples, TERT expressions are positively associated with VEGF expressions. Mechanistically, Sp1, the transcription factor of VEGF, interacts with TERT and facilitates angiogenesis in cancer via VEGF activation [139]. In hepatocellular carcinoma cells, TERT binding to Sp1 stimulates DNMT3B transcription and induces aberrant cancer-specific global DNA methylation and AKT hyperactivation [141]. The activation of AKT modulates the increased expression of VEGF and activates angiogenesis both in normal tissues and in cancers [142]. The binding of TERT to MYC in cells with high MYC expression can stabilize MYC protein by inhibiting MYC ubiquitination, thereby activating the downstream targets of MYC, such as EIF2A and NCL [138]. In breast cancer cells, VEGF expression is increased by c-MYC and eventually induces tumor angiogenesis [143]. TERT also regulates MMP expression, especially MMP9, in an NF-κB-dependent manner [137]. MMP9 knockdown inhibits angiogenesis in prostate cancer cells via the release of angiostatin, which serves as a key regulator of angiogenesis [144]. Besides, in an NSCLC mouse model, TERT deficiency decreased the positive signal of PanCK/CD31 double staining and CD34 staining, suggesting the involvement of TERT in vascularization [145].
Effects on mesenchymal tissue
In A549 and H1299 lung cancer cells, hTERT overexpression upregulates c-MET and promotes the expression of mesenchymal markers, including vimentin and N-cadherin [135]. In the NSCLC mouse model, TERT deficiency inhibits the expression of c-MET and reduces EMT by influencing the expression of mesenchymal markers, including vimentin and fibronectin [145]. Similarly, a TERT promoter variant enhances EMT in prostate cancer and promotes the development of castration-resistant prostate cancer [140]. In HPV-immortalized cervical cancer cells, overexpression of TERT alters EMT characteristics [146]. The authors showed that TERT is a critical factor in the EMT process in cancer progression.
Effects on inflammatory factors and immune response
The NF-κB family comprises inducible transcription factors with important roles in cancer progression [147] and acts as a pro-inflammatory transcription factor involved in the regulation of inflammation [147, 148]. TERT binding to NF-κB (also known as p65) is required for the promotion of NF-κB-dependent gene expression such as IL-6 and TNF-α [136]; these factors are critical for inflammation in cancer progression. In an NSCLC mouse model, TERT promotes lung inflammation by influencing the expression of Ifng, Tnf, Il10, and PD-1, markers of inflammation, and tumor immunosuppression [145].
TERT also affects the immune system and is considered a self-antigen constitutively expressed in many tumors, with potential therapeutic value for anticancer immunotherapy [149]. TERT is recognized by the adaptive immune system; in particular, CD4+ and CD8+ T cells recognize TERT as short peptides processed inside the cell before being exported to and presented at the cell surface by major histocompatibility complex I/II molecules, which induces an adaptive immune response associated with the inhibition of tumor growth [150,151,152,153,154]. These findings provide the theoretical basis for the discovery and the clinical application of the TERT vaccine in cancer treatment.
The current and future pharmacological strategies targeting TERT or telomerase
Owing to their good anti-cancer properties, TERT and telomerase are used as therapeutic targets for telomerase-positive cancer. Small molecule inhibitors and telomerase-targeted vaccines are the most frequent types in drug development.
TMPyP4, RHPS4, BRACO-19, and telomestatin are typical ligands that fold the single-stranded telomeric DNA (the telomeric substrate) into a four-stranded quadruplex structure, inhibiting telomerase catalytic activity [155]. In humans, TMPyP4 downregulates TERT transcription [156], inhibiting telomerase activity and TERT expression [157, 158]. However, the suppression proliferative of RHPS4 in glioblastoma stem-like cells is independent of telomeric dysfunction [159], which indicates a differential influence in cancer treatment. Significantly, a high level of G-rich DNA exists throughout the genome, especially in the promoter regions of oncogenes. Therefore, G4 ligands have several risks when used in telomerase inhibition and may have a significant off-target effect [160,161,162].
6-thio-2’-deoxyguanosine (6-thio-dG, also named THIO) is a nucleoside analog that could induce telomere dysfunction by incorporating into telomeric DNA in telomerase-positive cells [163]. In NSCLC xenografts, 6-thio-dG treatment reduces tumor growth by increasing telomere damage [145]. The therapeutic effect has also been validated in Gliomas [164] and Melanoma [165, 166] both in vivo and in vitro. Ilgen Mender et al. found that 6-thio-dG mitigates chemotherapy resistance to ECFG inhibitors and is commonly used in chemotherapy combinations (e.g., gemcitabine and cisplatin) in NSCLC cell lines [167]. In xenografts from neuroblastoma cell lines, pediatric high-risk group-3 medulloblastoma xenografts, and an orthotopic patient-derived xenograft model of diffuse intrinsic pontine glioma, 6-thio-dG inhibits tumor growth by inducing telomere dysfunction, which showed a promising approach to mitigate therapy-resistant telomerase-positive pediatric brain tumors [168, 169]. Similarly, 6-thio-dG significantly improves the efficacy of the combination of anti-PD-L1 and anti-VEGF based on CD8+ T cells in Hepatocellular Carcinoma [170], consistent with earlier research [171].
Directly targeting active TERT sites is a more popular therapeutic strategy than using compounds that inhibit TERT DNA-binding [172, 173]. 2-[[(E)-3-naphthalen-2-ylbut-2-enoyl] amino] benzoic acid (BIBR1532) non-competitively binds to the active sites of TERT, inhibiting its telomerase activity [24, 30]. Amin et al. found that short-term BIBR1532 impairs cellular proliferation in a zebrafish model, which is a potential anticancer treatment strategy [174]. Recent research on new BIBR1532-based analogs, designed to overcome ineligible pharmacokinetics, showed greater antitumor activity in the Ehrlich carcinoma model [175].
Imetelstat (GRN163L), a specific telomerase inhibitor that directly binds to the TR component in the catalytic site of the telomerase enzyme, has been used in a clinical study [176]. Imetalstat can induce cell death and improve the treatment efficacy of chemotherapy in patient-derived xenografts of Acute myeloid leukemia (AML) [177, 178]. A phase II study of lower-risk myelodysplastic patients reported meaningful effects with Imetelstat [179], and a phase III study of lower-risk myelodysplastic syndromes who have relapsed or are refractory to erythropoiesis-stimulating agents reported that approximately 40% patients achieved transfusion independency at least 8 weeks [180]. Therefore, almost 91% of patients had grade 3–4 treatment-emergent adverse events [180]. These results indicate that telomerase-targeting therapy has considerable prospects but warrants further investigations to reduce adverse drug reactions.
Telomerases are HLA class-I antigens, hence TERT-derived vaccines have also been considered as an approach to stimulate immune response in targeting cancers [181]. To date, GV1001 single-use [182] or combined with HR2822 [183], UV1 [184] single-use or combined with Ipilimumab (checkpoint inhibitor) [185], Vx-001 [186], and Gx-301 [187] have undergone clinical trials in patients with different types of cancer. Based on CD4+/CD8+ T cell recognition, the TERT vaccine presents a promising therapeutic efficacy in a proportion of cancer, although its safety and universal applicability in different types of cancer requires further research and verification [153, 188, 189].
Conclusions and perspectives
Telomerase reactivation is essential for cancer progression, but it does not occur during normal cell senescence. Telomerase-mediated stress responses in cancer result in telomere escape from shortening with every division. As the most important catalytic subunit component of telomerase, TERT is regulated by various regulation pathways, including transcriptional factors, PTM-related activation, and inhibition by co-interacting proteins.
The most common TERT gene changing is promoter mutant, which results in the binding of the ETS family transcription factor on “enhance hijacking” near the TERT transcription start sites. As for PTMs of TERT protein, phosphorylation has been extensively studied, and ubiquitination and SUMOylation have been reported in various cancer cells. The phosphorylation of TERT mainly results in telomerase activity changing by influencing the stability of TERT with the regulation of ubiquitination, maintaining telomerase holoenzyme integrity, and changing the translocation of TERT in cancer cells between nuclear and cytoplasmic. Besides, a small portion of the phosphorylation of TERT does not influence telomerase activity more than RdRP activity, which indicates that the functions of TERT in cancers should be considered.
The modulators of TERT, which regulate telomerase assembly, trafficking, blocking association, especially telomerase activity have been well-researched in recent years. Some positive regulators of telomerase promote telomere elongation by influencing telomerase holoenzyme assembly, recruiting telomerase to telomere, and stabilizing TERT protein. Negative regulators inhibit telomerase activity, particularly by preventing telomerase from binding with the long telomere-end or with telomere-binding proteins. Besides, the modulators that induce the non-canonical function of TERT that regulates gene expression by some transcription factors, such as c-MET, NF-κB p65, Myc, and Sp1, also participate in cancer progression.
To date, various small molecular drugs, including G4 ligands representative of TMPyP4 and telomerstatin, 6-thio-dG, BIBR1532, Imetelastat, and TERT vaccine, have been investigated. However, due to its limited effect and undetermined drug safety, further research remains warranted before clinical use.
To summarize, the TERT modulators discussed in this review provide potential targets for cancer treatment. Interventions targeting atypical telomere-related molecules influence cancer-associated cellular processes, exerting synergistic anticancer effects. However, further research remains warranted to identify novel treatment strategies.
References
Sieverling L, Hong C, Koser SD, Ginsbach P, Kleinheinz K, Hutter B, et al. Genomic footprints of activated telomere maintenance mechanisms in cancer. Nat Commun. 2020;11:733.
Amano H, Chaudhury A, Rodriguez-Aguayo C, Lu L, Akhanov V, Catic A, et al. Telomere dysfunction induces sirtuin repression that drives telomere-dependent disease. Cell Metab. 2019;29:1274–90.e9.
Srinivas N, Rachakonda S, Kumar R. Telomeres and telomere length: a general overview. Cancers (Basel). 2020;12:558.
Amir M, Khan P, Queen A, Dohare R, Alajmi MF, Hussain A, et al. Structural features of nucleoprotein CST/shelterin complex involved in the telomere maintenance and its association with disease mutations. Cells. 2020;9:359.
Shay JW, Wright WE. Telomeres and telomerase: three decades of progress. Nat Rev Genet. 2019;20:299–309.
Allsopp RC, Morin GB, DePinho R, Harley CB, Weissman IL. Telomerase is required to slow telomere shortening and extend replicative lifespan of HSCs during serial transplantation. Blood. 2003;102:517–20.
Claude E, Decottignies A. Telomere maintenance mechanisms in cancer: telomerase, ALT or lack thereof. Curr Opin Genet Dev. 2020;60:1–8.
Nakamura TM, Morin GB, Chapman KB, Weinrich SL, Andrews WH, Lingner J, et al. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997;277:955–9.
Harrington L, McPhail T, Mar V, Zhou W, Oulton R, Bass MB, et al. A mammalian telomerase-associated protein. Science. 1997;275:973–7.
Harrington L, Zhou W, McPhail T, Oulton R, Yeung DS, Mar V, et al. Human telomerase contains evolutionarily conserved catalytic and structural subunits. Genes Dev. 1997;11:3109–15.
Morin GB. The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell. 1989;59:521–9.
Venteicher AS, Abreu EB, Meng Z, McCann KE, Terns RM, Veenstra TD, et al. A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science. 2009;323:644–8.
Nguyen THD, Tam J, Wu RA, Greber BJ, Toso D, Nogales E, et al. Cryo-EM structure of substrate-bound human telomerase holoenzyme. Nature. 2018;557:190–5.
Egan ED, Collins K. Specificity and stoichiometry of subunit interactions in the human telomerase holoenzyme assembled in vivo. Mol Cell Biol. 2010;30:2775–86.
Nguyen THD, Collins K, Nogales E. Telomerase structures and regulation: shedding light on the chromosome end. Curr Opin Struct Biol. 2019;55:185–93.
Turner KJ, Vasu V, Griffin DK. Telomere biology and human phenotype. Cells. 2019;8:73.
Podlevsky JD, Bley CJ, Omana RV, Qi X, Chen JJ. The telomerase database. Nucleic Acids Res. 2008;36:D339–43.
Maida Y, Yasukawa M, Furuuchi M, Lassmann T, Possemato R, Okamoto N, et al. An RNA-dependent RNA polymerase formed by TERT and the RMRP RNA. Nature. 2009;461:230–5.
Heidenreich B, Kumar R. TERT promoter mutations in telomere biology. Mutat Res Rev Mutat Res. 2017;771:15–31.
Yuan X, Dai M, Xu D. TERT promoter mutations and GABP transcription factors in carcinogenesis: more foes than friends. Cancer Lett. 2020;493:1–9.
Hafezi F, Jaxel L, Lemaire M, Turner JD, Perez-Bercoff D. TERT promoter mutations increase sense and antisense transcription from the TERT promoter. Biomedicines. 2021;9:1773.
Bell RJ, Rube HT, Xavier-Magalhães A, Costa BM, Mancini A, Song JS, et al. Understanding TERT promoter mutations: a common path to immortality. Mol Cancer Res. 2016;14:315–23.
Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–61.
Dratwa M, Wysoczańska B, Łacina P, Kubik T, Bogunia-Kubik K. TERT-regulation and roles in cancer formation. Front Immunol. 2020;11:589929.
Masutomi K, Possemato R, Wong JM, Currier JL, Tothova Z, Manola JB, et al. The telomerase reverse transcriptase regulates chromatin state and DNA damage responses. Proc Natl Acad Sci USA. 2005;102:8222–7.
Im E, Yoon JB, Lee HW, Chung KC. Human telomerase reverse transcriptase (hTERT) positively regulates 26S proteasome activity. J Cell Physiol. 2017;232:2083–93.
Hu C, Ni Z, Li BS, Yong X, Yang X, Zhang JW, et al. hTERT promotes the invasion of gastric cancer cells by enhancing FOXO3a ubiquitination and subsequent ITGB1 upregulation. Gut. 2017;66:31–42.
Lingner J, Hughes TR, Shevchenko A, Mann M, Lundblad V, Cech TR. Reverse transcriptase motifs in the catalytic subunit of telomerase. Science. 1997;276:561–7.
Collins K. The biogenesis and regulation of telomerase holoenzymes. Nat Rev Mol Cell Biol. 2006;7:484–94.
Liu B, He Y, Wang Y, Song H, Zhou ZH, Feigon J. Structure of active human telomerase with telomere shelterin protein TPP1. Nature. 2022;604:578–83.
Wang Y, Gallagher-Jones M, Sušac L, Song H, Feigon J. A structurally conserved human and Tetrahymena telomerase catalytic core. Proc Natl Acad Sci USA. 2020;117:31078–87.
Jiang J, Wang Y, Sušac L, Chan H, Basu R, Zhou ZH, et al. Structure of telomerase with telomeric DNA. Cell. 2018;173:1179–90.e13.
Egan ED, Collins K. An enhanced H/ACA RNP assembly mechanism for human telomerase RNA. Mol Cell Biol. 2012;32:2428–39.
Gillis AJ, Schuller AP, Skordalakes E. Structure of the Tribolium castaneum telomerase catalytic subunit TERT. Nature. 2008;455:633–7.
Mitchell M, Gillis A, Futahashi M, Fujiwara H, Skordalakes E. Structural basis for telomerase catalytic subunit TERT binding to RNA template and telomeric DNA. Nat Struct Mol Biol. 2010;17:513–8.
Bryan TM, Goodrich KJ, Cech TR. Telomerase RNA bound by protein motifs specific to telomerase reverse transcriptase. Mol Cell. 2000;6:493–9.
Ghanim GE, Fountain AJ, van Roon AM, Rangan R, Das R, Collins K, et al. Structure of human telomerase holoenzyme with bound telomeric DNA. Nature. 2021;593:449–53.
Sekne Z, Ghanim GE, van Roon AM, Nguyen THD. Structural basis of human telomerase recruitment by TPP1-POT1. Science. 2022;375:1173–6.
Gupta S, Won H, Chadalavada K, Nanjangud GJ, Chen YB, Al-Ahmadie HA, et al. TERT copy number alterations, promoter mutations and rearrangements in adrenocortical carcinomas. Endocr Pathol. 2022;33:304–14.
Gupta S, Vanderbilt CM, Lin YT, Benhamida JK, Jungbluth AA, Rana S, et al. A pan-cancer study of somatic TERT promoter mutations and amplification in 30,773 tumors profiled by clinical genomic sequencing. J Mol Diagn. 2021;23:253–63.
Muñoz-Jiménez MT, Blanco L, Ruano Y, Carrillo R, Santos-Briz Á, Riveiro-Falkenbach E, et al. TERT promoter mutation in sebaceous neoplasms. Virch Arch. 2021;479:551–8.
Guo Y, Chen Y, Zhang L, Ma L, Jiang K, Yao G, et al. TERT promoter mutations and telomerase in melanoma. J Oncol. 2022;2022:6300329.
Fujimoto K, Arita H, Satomi K, Yamasaki K, Matsushita Y, Nakamura T, et al. TERT promoter mutation status is necessary and sufficient to diagnose IDH-wildtype diffuse astrocytic glioma with molecular features of glioblastoma. Acta Neuropathol. 2021;142:323–38.
Liu R, Xing M. TERT promoter mutations in thyroid cancer. Endocr Relat Cancer. 2016;23:R143–55.
Weyerer V, Eckstein M, Strissel PL, Wullweber A, Lange F, Tögel L, et al. TERT promoter mutation analysis of whole-organ mapping bladder cancers. Genes (Basel). 2021;12:230.
Zhang Y, Chen Y, Yang C, Seger N, Hesla AC, Tsagkozis P, et al. TERT promoter mutation is an objective clinical marker for disease progression in chondrosarcoma. Mod Pathol. 2021;34:2020–7.
Chiba K, Johnson JZ, Vogan JM, Wagner T, Boyle JM, Hockemeyer D. Cancer-associated TERT promoter mutations abrogate telomerase silencing. Elife. 2015;4:e07918.
Heidenreich B, Rachakonda PS, Hemminki K, Kumar R. TERT promoter mutations in cancer development. Curr Opin Genet Dev. 2014;24:30–7.
Ramlee MK, Wang J, Toh WX, Li S. Transcription regulation of the human telomerase reverse transcriptase (hTERT) gene. Genes. 2016;7:50.
Huang FW, Bielski CM, Rinne ML, Hahn WC, Sellers WR, Stegmeier F, et al. TERT promoter mutations and monoallelic activation of TERT in cancer. Oncogenesis. 2015;4:e176.
Lee DD, Komosa M, Sudhaman S, Leão R, Zhang CH, Apolonio JD, et al. Dual role of allele-specific DNA hypermethylation within the TERT promoter in cancer. J Clin Invest. 2021;131:e146915.
Lee DD, Leão R, Komosa M, Gallo M, Zhang CH, Lipman T, et al. DNA hypermethylation within TERT promoter upregulates TERT expression in cancer. J Clin Invest. 2019;129:223–9.
Barthel FP, Wei W, Tang M, Martinez-Ledesma E, Hu X, Amin SB, et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat Genet. 2017;49:349–57.
Li Y, Zhou QL, Sun W, Chandrasekharan P, Cheng HS, Ying Z, et al. Non-canonical NF-κB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat Cell Biol. 2015;17:1327–38.
Wu Y, Shi L, Zhao Y, Chen P, Cui R, Ji M, et al. Synergistic activation of mutant TERT promoter by Sp1 and GABPA in BRAF(V600E)-driven human cancers. NPJ Precis Oncol. 2021;5:3.
Song YS, Yoo SK, Kim HH, Jung G, Oh AR, Cha JY, et al. Interaction of BRAF-induced ETS factors with mutant TERT promoter in papillary thyroid cancer. Endocr Relat Cancer. 2019;26:629–41.
Liu R, Zhang T, Zhu G, Xing M. Regulation of mutant TERT by BRAF V600E/MAP kinase pathway through FOS/GABP in human cancer. Nat Commun. 2018;9:579.
Akıncılar SC, Khattar E, Boon PL, Unal B, Fullwood MJ, Tergaonkar V. Long-range chromatin interactions drive mutant TERT Promoter Activation. Cancer Discov. 2016;6:1276–91.
Liu X, Zhang Y, Wang Y, Yang M, Hong F, Yang S. Protein phosphorylation in cancer: role of nitric oxide signaling pathway. Biomolecules. 2021;11:1009.
Yasukawa M, Ando Y, Yamashita T, Matsuda Y, Shoji S, Morioka MS, et al. CDK1 dependent phosphorylation of hTERT contributes to cancer progression. Nat Commun. 2020;11:1557.
Kang SS, Kwon T, Kwon DY, Do SI. Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. J Biol Chem. 1999;274:13085–90.
Jung HY, Wang X, Jun S, Park JI. Dyrk2-associated EDD-DDB1-VprBP E3 ligase inhibits telomerase by TERT degradation. J Biol Chem. 2013;288:7252–62.
Haendeler J, Hoffmann J, Brandes RP, Zeiher AM, Dimmeler S. Hydrogen peroxide triggers nuclear export of telomerase reverse transcriptase via Src kinase family-dependent phosphorylation of tyrosine 707. Mol Cell Biol. 2003;23:4598–610.
Chung J, Khadka P, Chung IK. Nuclear import of hTERT requires a bipartite nuclear localization signal and Akt-mediated phosphorylation. J Cell Sci. 2012;125:2684–97.
Mukund V, Mukund D, Sharma V, Mannarapu M, Alam A. Genistein: its role in metabolic diseases and cancer. Crit Rev Oncol Hematol. 2017;119:13–22.
Li Y, Liu L, Andrews LG, Tollefsbol TO. Genistein depletes telomerase activity through cross-talk between genetic and epigenetic mechanisms. Int J Cancer. 2009;125:286–96.
Eitsuka T, Nakagawa K, Kato S, Ito J, Otoki Y, Takasu S, et al. Modulation of telomerase activity in cancer cells by dietary compounds: a review. Int J Mol Sci. 2018;19:478.
Kang JH, Toita R, Kim CW, Katayama Y. Protein kinase C (PKC) isozyme-specific substrates and their design. Biotechnol Adv. 2012;30:1662–72.
Chang JT, Lu YC, Chen YJ, Tseng CP, Chen YL, Fang CW, et al. hTERT phosphorylation by PKC is essential for telomerase holoprotein integrity and enzyme activity in head neck cancer cells. Br J Cancer. 2006;94:870–8.
Ku WC, Cheng AJ, Wang TC. Inhibition of telomerase activity by PKC inhibitors in human nasopharyngeal cancer cells in culture. Biochem Biophys Res Commun. 1997;241:730–6.
Bodnar AG, Kim NW, Effros RB, Chiu CP. Mechanism of telomerase induction during T cell activation. Exp Cell Res. 1996;228:58–64.
Yu CC, Lo SC, Wang TC. Telomerase is regulated by protein kinase C-zeta in human nasopharyngeal cancer cells. Biochem J. 2001;355:459–64.
Li H, Zhao L, Yang Z, Funder JW, Liu JP. Telomerase is controlled by protein kinase Calpha in human breast cancer cells. J Biol Chem. 1998;273:33436–42.
Li H, Zhao LL, Funder JW, Liu JP. Protein phosphatase 2A inhibits nuclear telomerase activity in human breast cancer cells. J Biol Chem. 1997;272:16729–32.
Teixeira MT, Gilson E. When CDK1 rides the telomere cycle. Mol Cell. 2006;24:491–2.
Liu CC, Gopalakrishnan V, Poon LF, Yan T, Li S. Cdk1 regulates the temporal recruitment of telomerase and Cdc13-Stn1-Ten1 complex for telomere replication. Mol Cell Biol. 2014;34:57–70.
Matsuda Y, Yamashita T, Ye J, Yasukawa M, Yamakawa K, Mukai Y, et al. Phosphorylation of hTERT at threonine 249 is a novel tumor biomarker of aggressive cancer with poor prognosis in multiple organs. J Pathol. 2022;257:172–85.
Ahmed S, Passos JF, Birket MJ, Beckmann T, Brings S, Peters H, et al. Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J Cell Sci. 2008;121:1046–53.
Büchner N, Zschauer TC, Lukosz M, Altschmied J, Haendeler J. Downregulation of mitochondrial telomerase reverse transcriptase induced by H2O2 is Src kinase dependent. Exp Gerontol. 2010;45:558–62.
Tandon V, de la Vega L, Banerjee S. Emerging roles of DYRK2 in cancer. J Biol Chem. 2021;296:100233.
Seimiya H, Tanji M, Oh-hara T, Tomida A, Naasani I, Tsuruo T. Hypoxia up-regulates telomerase activity via mitogen-activated protein kinase signaling in human solid tumor cells. Biochem Biophys Res Commun. 1999;260:365–70.
Stern JL, Hibshman G, Hu K, Ferrara SE, Costello JC, Kim W, et al. Mesenchymal and MAPK expression signatures associate with telomerase promoter mutations in multiple cancers. Mol Cancer Res. 2020;18:1050–62.
Bohio AA, Wang R, Zeng X, Ba X. c‑Abl‑mediated tyrosine phosphorylation of DNA damage response proteins and implications in important cellular functions (Review). Mol Med Rep. 2020;22:612–9.
Pan B, Yang L, Wang J, Wang Y, Wang J, Zhou X, et al. C-Abl tyrosine kinase mediates neurotoxic prion peptide-induced neuronal apoptosis via regulating mitochondrial homeostasis. Mol Neurobiol. 2014;49:1102–16.
Kharbanda S, Kumar V, Dhar S, Pandey P, Chen C, Majumder P, et al. Regulation of the hTERT telomerase catalytic subunit by the c-Abl tyrosine kinase. Curr Biol. 2000;10:568–75.
Hantschel O, Superti-Furga G. Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat Rev Mol Cell Biol. 2004;5:33–44.
Chai JH, Zhang Y, Tan WH, Chng WJ, Li B, Wang X. Regulation of hTERT by BCR-ABL at multiple levels in K562 cells. BMC Cancer. 2011;11:512.
Shtivelman E, Lifshitz B, Gale RP, Roe BA, Canaani E. Alternative splicing of RNAs transcribed from the human abl gene and from the bcr-abl fused gene. Cell. 1986;47:277–84.
Liu K, Hodes RJ, Weng N. Cutting edge: telomerase activation in human T lymphocytes does not require increase in telomerase reverse transcriptase (hTERT) protein but is associated with hTERT phosphorylation and nuclear translocation. J Immunol. 2001;166:4826–30.
Minamino T, Mitsialis SA, Kourembanas S. Hypoxia extends the life span of vascular smooth muscle cells through telomerase activation. Mol Cell Biol. 2001;21:3336–42.
Kim JH, Park SM, Kang MR, Oh SY, Lee TH, Muller MT, et al. Ubiquitin ligase MKRN1 modulates telomere length homeostasis through a proteolysis of hTERT. Genes Dev. 2005;19:776–81.
Salvatico J, Kim JH, Chung IK, Muller MT. Differentiation linked regulation of telomerase activity by Makorin-1. Mol Cell Biochem. 2010;342:241–50.
Panneer Selvam S, De Palma RM, Oaks JJ, Oleinik N, Peterson YK, Stahelin RV, et al. Binding of the sphingolipid S1P to hTERT stabilizes telomerase at the nuclear periphery by allosterically mimicking protein phosphorylation. Sci Signal. 2015;8:ra58.
Wang H, Zhao D, Nguyen LX, Wu H, Li L, Dong D, et al. Targeting cell membrane HDM2: a novel therapeutic approach for acute myeloid leukemia. Leukemia. 2020;34:75–86.
Oh W, Lee EW, Lee D, Yang MR, Ko A, Yoon CH, et al. Hdm2 negatively regulates telomerase activity by functioning as an E3 ligase of hTERT. Oncogene. 2010;29:4101–12.
Toh WH, Kyo S, Sabapathy K. Relief of p53-mediated telomerase suppression by p73. J Biol Chem. 2005;280:17329–38.
Huang Y, Sun L, Liu N, Wei Q, Jiang L, Tong X, et al. Polo-like kinase 1 (Plk1) up-regulates telomerase activity by affecting human telomerase reverse transcriptase (hTERT) stability. J Biol Chem. 2015;290:18865–73.
Kishi K, van Vugt MA, Okamoto K, Hayashi Y, Yaffe MB. Functional dynamics of Polo-like kinase 1 at the centrosome. Mol Cell Biol. 2009;29:3134–50.
Lee JH, Khadka P, Baek SH, Chung IK. CHIP promotes human telomerase reverse transcriptase degradation and negatively regulates telomerase activity. J Biol Chem. 2010;285:42033–45.
Flotho A, Melchior F. Sumoylation: a regulatory protein modification in health and disease. Annu Rev Biochem. 2013;82:357–85.
Eifler K, Vertegaal ACO. SUMOylation-mediated regulation of cell cycle progression and cancer. Trends Biochem Sci. 2015;40:779–93.
Han ZJ, Feng YH, Gu BH, Li YM, Chen H. The post-translational modification, SUMOylation, and cancer (Review). Int J Oncol. 2018;52:1081–94.
Xie M, Yu J, Ge S, Huang J, Fan X. SUMOylation homeostasis in tumorigenesis. Cancer Lett. 2020;469:301–9.
Yau TY, Molina O, Courey AJ. SUMOylation in development and neurodegeneration. Development. 2020;147:dev175703.
Sheng Z, Zhu J, Deng YN, Gao S, Liang S. SUMOylation modification-mediated cell death. Open Biol. 2021;11:210050.
Sanyal S, Mondal P, Sen S, Sengupta Bandyopadhyay S, Das C. SUMO E3 ligase CBX4 regulates hTERT-mediated transcription of CDH1 and promotes breast cancer cell migration and invasion. Biochem J. 2020;477:3803–18.
Rosenzweig R, Nillegoda NB, Mayer MP, Bukau B. The Hsp70 chaperone network. Nat Rev Mol Cell Biol. 2019;20:665–80.
Forsythe HL, Jarvis JL, Turner JW, Elmore LW, Holt SE. Stable association of hsp90 and p23, but Not hsp70, with active human telomerase. J Biol Chem. 2001;276:15571–4.
Arndt GM, MacKenzie KL. New prospects for targeting telomerase beyond the telomere. Nat Rev Cancer. 2016;16:508–24.
Her J, Chung IK. The AAA-ATPase NVL2 is a telomerase component essential for holoenzyme assembly. Biochem Biophys Res Commun. 2012;417:1086–92.
Snow BE, Erdmann N, Cruickshank J, Goldman H, Gill RM, Robinson MO, et al. Functional conservation of the telomerase protein Est1p in humans. Curr Biol. 2003;13:698–704.
Reichenbach P, Höss M, Azzalin CM, Nabholz M, Bucher P, Lingner J. A human homolog of yeast Est1 associates with telomerase and uncaps chromosome ends when overexpressed. Curr Biol. 2003;13:568–74.
Roake CM, Artandi SE. Regulation of human telomerase in homeostasis and disease. Nat Rev Mol Cell Biol. 2020;21:384–97.
Latrick CM, Cech TR. POT1-TPP1 enhances telomerase processivity by slowing primer dissociation and aiding translocation. EMBO J. 2010;29:924–33.
Wang F, Podell ER, Zaug AJ, Yang Y, Baciu P, Cech TR, et al. The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature. 2007;445:506–10.
Lim CJ, Barbour AT, Zaug AJ, Goodrich KJ, McKay AE, Wuttke DS, et al. The structure of human CST reveals a decameric assembly bound to telomeric DNA. Science. 2020;368:1081–5.
Miyake Y, Nakamura M, Nabetani A, Shimamura S, Tamura M, Yonehara S, et al. RPA-like mammalian Ctc1-Stn1-Ten1 complex binds to single-stranded DNA and protects telomeres independently of the Pot1 pathway. Mol Cell. 2009;36:193–206.
Wang F, Stewart JA, Kasbek C, Zhao Y, Wright WE, Price CM. Human CST has independent functions during telomere duplex replication and C-strand fill-in. Cell Rep. 2012;2:1096–103.
Chen LY, Redon S, Lingner J. The human CST complex is a terminator of telomerase activity. Nature. 2012;488:540–4.
Li JR, Yu TY, Chien IC, Lu CY, Lin JJ, Li HW. Pif1 regulates telomere length by preferentially removing telomerase from long telomere ends. Nucleic Acids Res. 2014;42:8527–36.
Phillips JA, Chan A, Paeschke K, Zakian VA. The pif1 helicase, a negative regulator of telomerase, acts preferentially at long telomeres. PLoS Genet. 2015;11:e1005186.
Wang J, Zhu X, Ying P, Zhu Y. PIF1 affects the proliferation and apoptosis of cervical cancer cells by influencing TERT. Cancer Manag Res. 2020;12:7827–35.
Gagou ME, Ganesh A, Phear G, Robinson D, Petermann E, Cox A, et al. Human PIF1 helicase supports DNA replication and cell growth under oncogenic-stress. Oncotarget. 2014;5:11381–98.
Maekawa T, Liu B, Nakai D, Yoshida K, Nakamura KI, Yasukawa M, et al. ATF7 mediates TNF-α-induced telomere shortening. Nucleic Acids Res. 2018;46:4487–504.
Zhou XZ, Huang P, Shi R, Lee TH, Lu G, Zhang Z, et al. The telomerase inhibitor PinX1 is a major haploinsufficient tumor suppressor essential for chromosome stability in mice. J Clin Invest. 2011;121:1266–82.
Zhou XZ, Lu KP. The Pin2/TRF1-interacting protein PinX1 is a potent telomerase inhibitor. Cell. 2001;107:347–59.
Yoo JE, Park YN, Oh BK. PinX1, a telomere repeat-binding factor 1 (TRF1)-interacting protein, maintains telomere integrity by modulating TRF1 homeostasis, the process in which human telomerase reverse Transcriptase (hTERT) plays dual roles. J Biol Chem. 2014;289:6886–98.
Soohoo CY, Shi R, Lee TH, Huang P, Lu KP, Zhou XZ. Telomerase inhibitor PinX1 provides a link between TRF1 and telomerase to prevent telomere elongation. J Biol Chem. 2011;286:3894–906.
Song H, Li Y, Chen G, Xing Z, Zhao J, Yokoyama KK, et al. Human MCRS2, a cell-cycle-dependent protein, associates with LPTS/PinX1 and reduces the telomere length. Biochem Biophys Res Commun. 2004;316:1116–23.
Oh W, Ghim J, Lee EW, Yang MR, Kim ET, Ahn JH, et al. PML-IV functions as a negative regulator of telomerase by interacting with TERT. J Cell Sci. 2009;122:2613–22.
Okamoto N, Yasukawa M, Nguyen C, Kasim V, Maida Y, Possemato R, et al. Maintenance of tumor initiating cells of defined genetic composition by nucleostemin. Proc Natl Acad Sci USA. 2011;108:20388–93.
Maida Y, Masutomi K. Telomerase reverse transcriptase moonlights: Therapeutic targets beyond telomerase. Cancer Sci. 2015;106:1486–92.
Cangelosi D, Morini M, Zanardi N, Sementa AR, Muselli M, Conte M, et al. Hypoxia predicts poor prognosis in neuroblastoma patients and associates with biological mechanisms involved in telomerase activation and tumor microenvironment reprogramming. Cancers (Basel). 2020;12:2343.
Petrova V, Annicchiarico-Petruzzelli M, Melino G, Amelio I. The hypoxic tumour microenvironment. Oncogenesis. 2018;7:10.
Prasad RR, Mishra DK, Kumar M, Yadava PK. Human telomerase reverse transcriptase promotes the epithelial to mesenchymal transition in lung cancer cells by enhancing c-MET upregulation. Heliyon. 2022;8:e08673.
Ghosh A, Saginc G, Leow SC, Khattar E, Shin EM, Yan TD, et al. Telomerase directly regulates NF-κB-dependent transcription. Nat Cell Biol. 2012;14:1270–81.
Ding D, Xi P, Zhou J, Wang M, Cong YS. Human telomerase reverse transcriptase regulates MMP expression independently of telomerase activity via NF-κB-dependent transcription. FASEB J. 2013;27:4375–83.
Koh CM, Khattar E, Leow SC, Liu CY, Muller J, Ang WX, et al. Telomerase regulates MYC-driven oncogenesis independent of its reverse transcriptase activity. J Clin Invest. 2015;125:2109–22.
Liu N, Ding D, Hao W, Yang F, Wu X, Wang M, et al. hTERT promotes tumor angiogenesis by activating VEGF via interactions with the Sp1 transcription factor. Nucleic Acids Res. 2016;44:8693–703.
Dong X, Zhang Q, Hao J, Xie Q, Xu B, Zhang P, et al. Large multicohort study reveals a prostate cancer susceptibility allele at 5p15 regulating TERT via androgen signaling-orchestrated chromatin binding of E2F1 and MYC. Front Oncol. 2021;11:754206.
Yu J, Yuan X, Sjöholm L, Liu T, Kong F, Ekström TJ, et al. Telomerase reverse transcriptase regulates DNMT3B expression/aberrant DNA methylation phenotype and AKT activation in hepatocellular carcinoma. Cancer Lett. 2018;434:33–41.
Karar J, Maity A. PI3K/AKT/mTOR pathway in angiogenesis. Front Mol Neurosci. 2011;4:51.
Gao FY, Li XT, Xu K, Wang RT, Guan XX. c-MYC mediates the crosstalk between breast cancer cells and tumor microenvironment. Cell Commun Signal. 2023;21:28.
Gupta A, Zhou CQ, Chellaiah MA. Osteopontin and MMP9: associations with VEGF expression/secretion and angiogenesis in PC3 prostate cancer cells. Cancers (Basel). 2013;5:617–38.
Piñeiro-Hermida S, Bosso G, Sánchez-Vázquez R, Martínez P, Blasco MA. Telomerase deficiency and dysfunctional telomeres in the lung tumor microenvironment impair tumor progression in NSCLC mouse models and patient-derived xenografts. Cell Death Differ. 2023;30:1585–600.
Wang A, Zhou D, Krawczyk E, Li T, Simic V, Lu J, et al. Overexpression of the telomerase holoenzyme induces EMT and tumorigenesis of HPV-immortalized keratinocytes. J Med Virol. 2023;95:e28681.
Aggarwal BB. Nuclear factor-kappaB: the enemy within. Cancer Cell. 2004;6:203–8.
Gupta SC, Kunnumakkara AB, Aggarwal S, Aggarwal BB. Inflammation, a Double-Edge Sword for Cancer and Other Age-Related Diseases. Front Immunol. 2018;9:2160.
Zanetti M. A second chance for telomerase reverse transcriptase in anticancer immunotherapy. Nat Rev Clin Oncol. 2017;14:115–28.
Schroers R, Huang XF, Hammer J, Zhang J, Chen SY. Identification of HLA DR7-restricted epitopes from human telomerase reverse transcriptase recognized by CD4+ T-helper cells. Cancer Res. 2002;62:2600–5.
Schroers R, Shen L, Rollins L, Rooney CM, Slawin K, Sonderstrup G, et al. Human telomerase reverse transcriptase-specific T-helper responses induced by promiscuous major histocompatibility complex class II-restricted epitopes. Clin Cancer Res. 2003;9:4743–55.
Godet Y, Fabre E, Dosset M, Lamuraglia M, Levionnois E, Ravel P, et al. Analysis of spontaneous tumor-specific CD4 T-cell immunity in lung cancer using promiscuous HLA-DR telomerase-derived epitopes: potential synergistic effect with chemotherapy response. Clin Cancer Res. 2012;18:2943–53.
Dosset M, Godet Y, Vauchy C, Beziaud L, Lone YC, Sedlik C, et al. Universal cancer peptide-based therapeutic vaccine breaks tolerance against telomerase and eradicates established tumor. Clin Cancer Res. 2012;18:6284–95.
Adotévi O, Mollier K, Neuveut C, Dosset M, Ravel P, Fridman WH, et al. Targeting human telomerase reverse transcriptase with recombinant lentivector is highly effective to stimulate antitumor CD8 T-cell immunity in vivo. Blood. 2010;115:3025–32.
Balasubramanian S, Hurley LH, Neidle S. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy? Nat Rev Drug Discov. 2011;10:261–75.
Grand CL, Han H, Muñoz RM, Weitman S, Von Hoff DD, Hurley LH, et al. The cationic porphyrin TMPyP4 down-regulates c-MYC and human telomerase reverse transcriptase expression and inhibits tumor growth in vivo. Mol Cancer Ther. 2002;1:565–73.
Konieczna N, Romaniuk-Drapała A, Lisiak N, Totoń E, Paszel-Jaworska A, Kaczmarek M, et al. Telomerase Inhibitor TMPyP4 Alters Adhesion and Migration of Breast-Cancer Cells MCF7 and MDA-MB-231. Int J Mol Sci. 2019;20:2670.
Cheng MJ, Cao YG. TMPYP4 exerted antitumor effects in human cervical cancer cells through activation of p38 mitogen-activated protein kinase. Biol Res. 2017;50:24.
Berardinelli F, Tanori M, Muoio D, Buccarelli M, di Masi A, Leone S, et al. G-quadruplex ligand RHPS4 radiosensitizes glioblastoma xenograft in vivo through a differential targeting of bulky differentiated- and stem-cancer cells. J Exp Clin Cancer Res. 2019;38:311.
Tsai R, Fang K, Yang P, Hsieh Y, Chiang I, Chen Y, et al. TERRA regulates DNA G-quadruplex formation and ATRX recruitment to chromatin. Nucleic Acids Res. 2022;50:12217–34.
Yan C, Chang Y, Gao H, Zhang Q, Peng S, Wang D, et al. G-quadruplex apurinic site-programmed chiral cyanine assemblies for specifically recognizing guanosine and guanine. Analyst. 2021;146:5866–72.
Shankar U, Mishra SK, Jain N, Tawani A, Yadav P, Kumar A. Ni(+2) permease system of Helicobacter pylori contains highly conserved G-quadruplex motifs. Infect Genet Evol. 2022;101:105298.
Mender I, Gryaznov S, Dikmen ZG, Wright WE, Shay JW. Induction of telomere dysfunction mediated by the telomerase substrate precursor 6-thio-2’-deoxyguanosine. Cancer Discov. 2015;5:82–95.
Yu S, Wei S, Savani M, Lin X, Du K, Mender I, et al. A modified nucleoside 6-Thio-2’-deoxyguanosine exhibits antitumor activity in gliomas. Clin Cancer Res. 2021;27:6800–14.
Zhang G, Wu LW, Mender I, Barzily-Rokni M, Hammond MR, Ope O, et al. Induction of telomere dysfunction prolongs disease control of therapy-resistant melanoma. Clin Cancer Res. 2018;24:4771–84.
Reyes-Uribe P, Adrianzen-Ruesta MP, Deng Z, Echevarria-Vargas I, Mender I, Saheb S, et al. Exploiting TERT dependency as a therapeutic strategy for NRAS-mutant melanoma. Oncogene. 2018;37:4058–72.
Mender I, LaRanger R, Luitel K, Peyton M, Girard L, Lai TP, et al. Telomerase-mediated strategy for overcoming non-small cell lung cancer targeted therapy and chemotherapy resistance. Neoplasia. 2018;20:826–37.
Sengupta S, Sobo M, Lee K, Senthil Kumar S, White AR, Mender I, et al. Induced telomere damage to treat telomerase expressing therapy-resistant pediatric brain tumors. Mol Cancer Ther. 2018;17:1504–14.
Fischer-Mertens J, Otte F, Roderwieser A, Rosswog C, Kahlert Y, Werr L, et al. Telomerase-targeting compounds Imetelstat and 6-thio-dG act synergistically with chemotherapy in high-risk neuroblastoma models. Cell Oncol (Dordr). 2022;45:991–1003.
Mender I, Siteni S, Barron S, Flusche AM, Kubota N, Yu C, et al. Activating an adaptive immune response with a telomerase-mediated telomere targeting therapeutic in hepatocellular carcinoma. Mol Cancer Ther. 2023;22:737–50.
Mender I, Zhang A, Ren Z, Han C, Deng Y, Siteni S, et al. Telomere stress potentiates STING-dependent anti-tumor immunity. Cancer Cell. 2020;38:400–11.e6.
Sullivan HJ, Readmond C, Radicella C, Persad V, Fasano TJ, Wu C. Binding of telomestatin, TMPyP4, BSU6037, and BRACO19 to a telomeric G-quadruplex-duplex hybrid probed by all-atom molecular dynamics simulations with explicit solvent. ACS Omega. 2018;3:14788–806.
Mulholland K, Wu C. Binding of telomestatin to a telomeric G-quadruplex DNA probed by all-atom molecular dynamics simulations with explicit solvent. J Chem Inf Model. 2016;56:2093–102.
Amin A, Morello M, Petrara MR, Rizzo B, Argenton F, De Rossi A, et al. Short-term TERT inhibition impairs cellular proliferation via a telomere length-independent mechanism and can be exploited as a potential anticancer approach. Cancers (Basel). 2023;15:2673.
Al-Karmalawy AA, Nafie MS, Shaldam MA, Elmaaty AA, Antar SA, El-Hamaky AA, et al. Ligand-based design on the dog-bone-shaped BIBR1532 pharmacophoric features and synthesis of novel analogues as promising telomerase inhibitors with in vitro and in vivo evaluations. J Med Chem. 2023;66:777–92.
Carloni LE, Wechselberger R, De Vijlder T. Characterization of in vitro G-quadruplex formation of imetelstat telomerase inhibitor. Nucleic Acid Ther. 2021;31:341–50.
Barwe SP, Huang F, Kolb EA, Gopalakrishnapillai A. Imetelstat induces leukemia stem cell death in pediatric acute myeloid leukemia patient-derived xenografts. J Clin Med. 2022;11:1923.
Bruedigam C, Porter AH, Song A, Vroeg In de Wei G, Stoll T, Straube J, et al. Imetelstat-mediated alterations in fatty acid metabolism to induce ferroptosis as a therapeutic strategy for acute myeloid leukemia. Nat Cancer. 2023.
Steensma DP, Fenaux P, Van Eygen K, Raza A, Santini V, Germing U, et al. Imetelstat achieves meaningful and durable transfusion independence in high transfusion-burden patients with lower-risk myelodysplastic syndromes in a phase II study. J Clin Oncol. 2021;39:48–56.
Platzbecker U, Santini V, Fenaux P, Sekeres MA, Savona MR, Madanat YF, et al. Imetelstat in patients with lower-risk myelodysplastic syndromes who have relapsed or are refractory to erythropoiesis-stimulating agents (IMerge): a multinational, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2023;S0140-6736:01724–5.
Kailashiya C, Sharma HB, Kailashiya J. Telomerase based anticancer immunotherapy and vaccines approaches. Vaccine. 2017;35:5768–75.
Jo JH, Kim YT, Choi HS, Kim HG, Lee HS, Choi YW, et al. Efficacy of GV1001 with gemcitabine/capecitabine in previously untreated patients with advanced pancreatic ductal adenocarcinoma having high serum eotaxin levels (KG4/2015): an open-label, randomised, Phase 3 trial. Br J Cancer. 2024;130:43–52.
Brunsvig PF, Aamdal S, Gjertsen MK, Kvalheim G, Markowski-Grimsrud CJ, Sve I, et al. Telomerase peptide vaccination: a phase I/II study in patients with non-small cell lung cancer. Cancer Immunol Immunother. 2006;55:1553–64.
Ellingsen EB, O’Day S, Mezheyeuski A, Gromadka A, Clancy T, Kristedja TS, et al. Clinical activity of combined telomerase vaccination and pembrolizumab in advanced melanoma: results from a phase I trial. Clin Cancer Res. 2023;29:3026–36.
Ellingsen EB, Bounova G, Kerzeli I, Anzar I, Simnica D, Aamdal E, et al. Characterization of the T cell receptor repertoire and melanoma tumor microenvironment upon combined treatment with ipilimumab and hTERT vaccination. J Transl Med. 2022;20:419.
Gridelli C, Ciuleanu T, Domine M, Szczesna A, Bover I, Cobo M, et al. Clinical activity of a htert (vx-001) cancer vaccine as post-chemotherapy maintenance immunotherapy in patients with stage IV non-small cell lung cancer: final results of a randomised phase 2 clinical trial. Br J Cancer. 2020;122:1461–6.
Filaci G, Fenoglio D, Nolè F, Zanardi E, Tomasello L, Aglietta M, et al. Telomerase-based GX301 cancer vaccine in patients with metastatic castration-resistant prostate cancer: a randomized phase II trial. Cancer Immunol Immunother. 2021;70:3679–92.
Jansons J, Skrastina D, Kurlanda A, Petkov S, Avdoshina D, Kuzmenko Y, et al. Reciprocal inhibition of immunogenic performance in mice of two potent DNA immunogens targeting HCV-related liver cancer. Microorganisms. 2021;9:1073.
Negrini S, De Palma R, Filaci G. Anti-cancer immunotherapies targeting telomerase. Cancers (Basel). 2020;12:2260.
Jakob S, Schroeder P, Lukosz M, et al. Nuclear protein tyrosine phosphatase Shp-2 is one important negative regulator of nuclear export of telomerase reverse transcriptase. J Biol Chem. 2008;283:33155–61.
Maekawa T, Liu B, Nakai D, et al. ATF7 mediates TNF-α-induced telomere shortening. Nucleic Acids Res. 2018;46:4487–504.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (Nos. 82020108024 and 81772924), the Health Commission of Jilin Province (No. 2020J033), and the Graduate Innovation Fund of Jilin University (No. 101832020CX260). The funders had no role in the design of the study and collection, analysis, interpretation of data, decision to publish, or preparation of the paper.
Author information
Authors and Affiliations
Contributions
ML developed the initial idea and the application of net benefit theory. ML, YZ, and LG review previous related publications. Draft conceived and designed by ML and YZ. The paper was revised and approved by YJ, LG, HS, YL, and DZ. All authors read and approved the final paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests. The figures were created with BioRender software, ©biorender.com.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Anastasis Stephanou
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, M., Zhang, Y., Jian, Y. et al. The regulations of telomerase reverse transcriptase (TERT) in cancer. Cell Death Dis 15, 90 (2024). https://doi.org/10.1038/s41419-024-06454-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-024-06454-7
