
Abstract
Chronic obstructive pulmonary disease (COPD) is a risk factor for lung cancer development. COPD induces activation of hypoxia-induced signaling, causing remodeling of surrounding microenvironmental cells also modulating the release and cargo of their extracellular vesicles (EVs). We aimed to evaluate the potential role of circulating EVs from COPD subjects in lung cancer onset. Plasma-EVs were isolated by ultracentrifugation from heavy smoker volunteers with (COPD-EVs) or without (heavy smoker-EVs, HS-EV) COPD and characterized following MISEV guidelines. Immortalized human bronchial epithelial cells (CDK4, hTERT-HBEC3-KT), genetically modified with different oncogenic alterations commonly found in lung cancer (sh-p53, KRASV12), were used to test plasma-EVs pro-tumorigenic activity in vitro. COPD-EVs mainly derived from immune and endothelial cells. COPD-EVs selectively increased the subset of CD133+CXCR4+ metastasis initiating cells (MICs) in HBEC-sh-p53-KRASV12high cells and stimulated 3D growth, migration/invasion, and acquisition of mesenchymal traits. These effects were not observed in HBEC cells bearing single oncogenic mutation (sh-p53 or KRASV12). Mechanistically, hypoxia-inducible factor 1-alpha (HIF-1α) transferred from COPD-EVs triggers CXCR4 pathway activation that in turn mediates MICs expansion and acquisition of pro-tumorigenic effects. Indeed, HIF-1α inhibition or CXCR4 silencing prevented the acquisition of malignant traits induced by COPD-EVs alone. Hypoxia recapitulates the effects observed with COPD-EVs in HBEC-sh-p53-KRASV12high cells. Notably, higher levels of HIF-1α were observed in EVs from COPD subjects who subsequently developed cancer compared to those who remained cancer-free. Our findings support a role of COPD-EVs to promote the expansion of MICs in premalignant epithelial cells through HIF-1α-CXCR4 axis activation thereby potentially sustaining lung cancer progression.
Similar content being viewed by others


Introduction
Cigarette smoking can be considered the main risk factor for the development of chronic obstructive pulmonary disease (COPD) and for the onset of lung cancer [1]. However, only 20% of smokers develop COPD [2], whereas the incidence of lung cancer is higher in subjects with COPD [3]. Since COPD and lung cancer display the activation of some common pathways related to inflammation or growth factors [4], it is likely that the presence of COPD could be a potent driver for lung cancer progression. In NSCLC, we and other shown that CD133 represents a robust marker to identify cells endowed with stem like properties and cancer initiating potential (cancer-initiating cells-CICs) [5,6,7]. It is now well-recognized that CICs represent an heterogeneous population comprised of cells deputed to specific function, endowed with different properties and characterized by specific markers [8]. In lung cancer for instance different markers have been described to possibly identify CIC subset, suggesting a great phenotypic heterogeneity of such population [9,10,11,12]. We also provided evidence for functional heterogeneity of lung CICs, demonstrating that CD133+ CICs co-expressing the chemokine receptor CXCR4 possess increased disseminating and mesenchymal properties and are defined metastasis initiating cells (MICs) [13]. Epithelial to Mesenchymal Transition (EMT) is an important mechanism for the acquisition of migration and pro-metastatic traits and it is also associated with the induction of MICs phenotype [13].
The pronounced inflammation in lung microenvironment of COPD subjects, caused by the recruitment of several immune cells such as neutrophils [14] and macrophages [15], represents the major source of reactive nitrogen and oxygen species that lead to cell DNA damage. Furthermore, it has been shown that the reduced air flow in COPD subjects stimulated the formation of an hypoxic microenvironment in the lungs [16]. In particular, the activation of hypoxia-inducible factor 1-alpha (HIF-1α) signaling pathway has been associated with a reduction in lung functionality [17]. Notably, HIF-1α has also been described to regulate self-renewal of CICs, supporting their maintenance and cancer progression [18] and also directly regulates CXCR4 expression [19].
An hypoxic condition has been shown to increase the extracellular vesicles (EVs) production in several lung inflammatory diseases [20] and cancers [21, 22]. EVs are a heterogeneous group of membrane-limited particles produced by almost all cell types. Since these vesicles transfer bioactive molecules (proteins, lipids, and nucleic acids), they have been recognized as key mediators of cell-to-cell communication. Indeed, growing evidence suggested that alterations in extracellular vesicles production are closely associated with chronic airway inflammatory diseases like COPD [23, 24]. In particular, a high number of microvesicles released by platelets and immune cells was observed in the plasma of COPD subjects [25] and the pro-inflammatory content of EVs secreted by monocytes induced the destruction of alveolar wall [26]. Interestingly, endothelial cells contribute to the release of CD31+-EVs that stimulate COPD progression by inducing apoptosis in the neighboring endothelial cells [27]. Furthermore, epithelial cells damaged by cigarette smoking release a high number of EVs that promote inflammation through IL-8 and VEGF [28].
Although COPD and lung cancer share common features, the processes behind the development of lung cancer in COPD subjects are yet to be understood, in particular regarding the potential role of circulating EVs in promoting the progression of premalignant epithelial lesions.
In this study, we characterized circulating EVs isolated from heavy smoker volunteers with or without COPD to investigate their ability to induce pro-tumorigenic features in different subtypes of bronchial epithelial cells and in the subset of CICs. We analyzed the effect of hypoxia on EVs release and the role of the transcription factor HIF-1α in the induction of protumorigenic phenotype caused by COPD-EVs. Finally, we showed an increased amount of HIF-1α inside EVs from COPD subjects who have developed lung cancer.
Results
Isolation and characterization of EVs from heavy smoker volunteers and COPD subjects
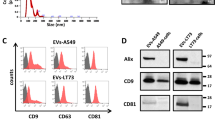
EVs from heavy-smoker volunteers (HS-EVs) and from COPD subjects (COPD-EVs) were isolated from 1 ml of plasma and characterized in accordance with Minimal Information for Studies of Extracellular Vesicles (MISEV) guidelines [29]. Nanoparticle tracking analysis showed that HS-EVs and COPD-EVs had a mean size of 81.02 nm (SEM = 5.945 nm) and 91.88 nm (SEM = 4.176 nm), respectively, without any significant differences between the two groups and a similar total number of EVs was counted in the plasma (Fig. 1A). Plasma EVs displayed a spherical shape and a relatively wide size distribution by TEM (Fig. 1B). Flow cytometry analysis detected the presence of canonical EVs markers such as tetraspanins CD9, CD63 and CD81 on their surface with comparable levels between HS-EVs and COPD-EVs groups (Fig. 1C). Western blotting further confirmed the presence of EVs’ markers and the presence of lipoprotein contaminants in our preparation mainly due to the isolation method adopted and the complexity of the starting biofluid (Fig. 1D).

A Concentration and size distribution of plasma-derived EVs from subjects with (COPD-EVs) or without (HS-EVs) COPD using nanoparticle tracking analysis (n = 5 per group). B Representative TEM images showing the spherical morphology and size distribution of plasma-derived EVs. C Flow cytometry analysis of conventional EV markers on COPD- and HS-EVs showing the presence conventional markers (CD9, CD81, CD63) (n = 3 per group). D Western blot of EVs from COPD and HS volunteers for EV markers (CD9, CD81, Tsg101) and a marker of lipoprotein contamination (ApoA1). E Profiles of cell surface markers of plasma-derived EVs determined by flow cytometry. The values are expressed as median fluorescence intensity (n = 5 per group) *p < 0.05, ***p < 0.001 versus HS-EVs. Data are expressed as mean and SEM.
To identify the cellular origin of plasma EVs, we performed a profiling analysis of lineage-specific markers using the MACSPlex Kit. Interestingly, we observed that COPD-EVs had higher levels of endothelial markers such as CD31, CD105, and CD146 than HS-EVs (Fig. 1E). Moreover, the higher abundance of CD3, CD45, HLA type I and II markers on EVs of COPD subjects suggested a derivation from immune cells.
COPD-EVs increase the number of CD133+CXCR4+ cells
To investigate the role of plasma EVs isolated from COPD subjects in fostering tumorigenic progression, we first evaluated the uptake of PKH26-labeled EVs on human immortalized bronchial epithelial cells HBEC-1, and differently transformed epithelial cell lines as HBEC-3, HBEC-5, HBEC-6, and HBEC-KRASV12high that model sequential steps of the neoplastic transformation process. In flow cytometry analysis, we observed a similar uptake of HS-EVs and COPD-EVs by all cell lines (Fig. 2A).

A Flow cytometry analysis of the percentage of fluorescent HBEC-1, HBEC-3, HBEC-5, HBEC-6 and HBEC-KRASV12high cells after treatment with PKH26-labeled COPD- and HS-EVs (1 µg) (n = 5 per group). B Gating strategy used for the identification of CD133+ CICs and CD133+CXCR4+ MICs in HBEC cells after treatment with HS-EVs and COPD-EVs evaluated via flow cytometry. C Bar charts show the relative amount of CD133+ CICs and CD133+CXCR4+ MICs in HBEC-1 (n = 5), HBEC-3 (n = 3), HBEC-5 (n = 5), HBEC-6 (n = 5) and HBEC-KRASV12high after EVs treatment (n = 7).
We next explored how COPD-EVs promote the acquisition of tumorigenic potential through the modulation of CIC phenotype in immortalized and transformed epithelial cells. Flow cytometry analysis proved a differential modulation of CD133+ cell subsets induced by COPD-EVs according to the specific genetic background of treated cells (Fig. 2B, C). In particular, COPD-EVs compared to HS-EVs significantly increased the subset of CD133+ CICs and specifically the subset of metastatic CD133+CXCR4+ cells only in HBEC-6 cells, harboring p53 and KRASV12 mutation, and in HBEC-KRASV12high cells, whereas modest effect on CICs was observed in HBEC 1 immortalized cells and in transformed HBEC lines harboring sh-p53 or KRASV12 oncogenic mutation only (HBEC 3 and 5) (Fig. 2C). These data suggest that COPD-EVs are more prone to foster the expansion of aggressive metastasis initiating cells in premalignant cells accumulating multiple oncogenic hits.
HBEC-KRASV12high acquire tumorigenic properties following COPD-EVs treatment
To further characterize the acquisition of a more aggressive phenotype induced by COPD-EVs, we analyzed the migratory and invasive abilities of EV-treated HBEC subtypes using a transwell assay. After 24 h, the treatment with both HS-EVs and COPD-EVs did not affect the migration of HBEC-1, HBEC-3, and HBEC-5 neither towards EGF, chemoattractant for epithelial cells (Fig. 3A and Supplementary Fig. S1A), nor towards SDF-1, the specific chemoattractant for CXCR4+ cells (Fig. 3B and Supplementary Fig. S1B). Instead, the treatment with COPD-EVs significantly increased the migration of both HBEC-6 and HBEC-KRASV12high compared to controls in the two experimental conditions. Invasion assay revealed an increased invasive ability of COPD-EVs treated HBEC-KRASV12high (Fig. 3C and Supplementary Fig. S1C, D). Along with the acquisition of CICs phenotype and migrating/invasive properties, COPD-EVs treatment of HBEC-KRASV12high significantly up-regulated the expression of some stemness and mesenchymal genes, as Integrin-α6, fibronectin and SNAI2, with a concomitant down-modulation of E-cadherin (CHD1) compared to controls, indicating a possible induction of EMT (Fig. 3D and Supplementary Fig. S2A). We confirmed a similar trend in the modulation of these EMT markers induced by COPD-EVs also at protein levels, using flow cytometry (Fig. 3E and Supplementary Fig. S2B) and immunofluorescence (Fig. 3F). Conversely, no modulation in these genes was observed in the other tested cell lines (Supplementary Fig. S2C).

A The migratory ability of HBEC cells after the indicated treatments with EVs towards an EGF gradient was evaluated in Transwell assay (n = 4). Migration data are expressed as the number of migrated cells compared to the number of migrated control cells. B Evaluation of the migration towards SDF-1 factor in HBEC cells after the treatment with HS-EVs or COPD-EVs. C The acquisition of invasive properties after EVs treatment was analyzed in HBEC cells following the gradient constituted by using EGF or SDF-1. Invasion data are expressed as the number of invaded cells versus the number of invaded control cells. D Expression level of genes involved in stemness maintenance and in EMT process (n = 5). B2m was used as housekeeping control. E Flow cytometry confirmed the induction of EMT in HBEC-KRASV12high after COPD-EVs considering E-cadherin (CDH1) and fibronectin (FN1) expression (n = 3). F Representative images of E-cadherin (green) and Snail2 (red) immunofluorescent staining of HBEC-KRASV12high after EV administration. G HBEC-KRASV12high migration ability after EVs administration (wound healing assay, n = 4). The Bar chart shows the percentage of wound compared to controls. H Analysis of 2D proliferation of HBEC-KRASV12high cells treated with EVs (n = 4). I 3D proliferation assay using VitroGel. Images show the colony formation ability of HBEC-KRASV12high cells treated with 15 µg of COPD- or HS-EVs. In all experiments, untreated cells (NT) were used as control. Data are expressed as mean and SEM.
Based on these preliminary results, we focused our attention on HBEC-KRASV12high in which a more striking effect in the acquisition of a malignant phenotype was observed after COPD-EVs treatment. We further observed that the treatment with COPD-EVs significantly increased the percentage of wound closure compared to HS-EVs (% wound closure: COPD-EVs= 75.8%, SEM = 5.1%; HS-EVs= 58.2%, SEM = 4.8%; NT = 60.6%, SEM = 3.1%. p = 0.0451 vs. HS-EVs and p = 0.042 vs. NT) (Fig. 3G and Supplementary Fig. S3). Instead, proliferation assay showed that COPD-EVs did not affect the growth rate of HBEC-KRASV12high, cultured as monolayer (Fig. 3H); however, COPD-EVs but not HS-EVs fostered the 3D-colony forming ability of HBEC-KRASV12high, providing and indirect indication on the pro-tumorigenic features of COPD-EVs treated cells (Fig. 3I). Overall, these results suggest that COPD-EVs promote the increase of CD133+ cancer-initiating cells, in particular through the expansion of CD133+CXCR4+ MICs subset, along with the acquisition of pro-tumorigenic and invasive properties in HBEC-KRASV12high cells.
HIF-1α is transferred from COPD-EVs to HBEC-KRASV12high
To investigate the factors causing the increase of MICs subset and the acquisition of pro-tumorigenic properties induced by COPD-EVs we focused on SDF-1, the cytokine that activates CXCR4 downstream signaling pathways, also known to regulate CICs phenotype [30]. Treatment of HBEC-KRASV12high cells with neutralizing antibody against SDF-1 did not abrogate the modulation of CICs cells induced by COPD-EVs, indicating that SDF-1-CXCR4 activation axis was not central in COPD-EVs induced expansion of CICs (Fig. 4A). Since it has been demonstrated that COPD causes a hypoxic lung microenvironment, we decided to analyze inside EVs the amount of the transcription factor HIF-1α, the master regulator of the cellular response to hypoxia. HIF-1α level, evaluated via ELISA, was significantly lower in HS-EVs (2.1 pg/ml, SEM = 1.5 pg/ml) than in COPD-EVs (7.5 pg/ml, SEM = 2.0 pg/ml) (*p = 0.03) (Fig. 4B). The transfer of HIF-1α into recipient cells via EVs was evaluated by treating HBEC-KRASV12high with PKH-labeled-EVs. Intracellular level of HIF-1α in PKH-fluorescent positive cells treated with COPD-EVs was 2.2-fold increased compared to the HS-EV treatment (p < 0.01) (Fig. 4C). To further elucidate whether the increase of HIF-1α in recipient cells was actually due to EVs protein shuttling or to the induction of endogenous transcript, levels of HIF-1α mRNA were evaluated after EVs treatment of HBEC-KRASV12high. No differences in HIF-1α transcription were observed between the two groups (Fig. 4D), indicating that HIF-1α was not induced into recipient cells but shuttled by EVs. Moreover, the transfer of HIF-1α from COPD-EVs induced the transcription of known downstream targets such as VEGF and CAIX in recipient cells (Fig. 4D). To assess the role of HIF-1α in the induction of MICs phenotype caused by COPD-EVs, we exploited a selective HIF-1α inhibitor, PX-478, before EVs treatment. HIF-1α inhibition completely abrogates the expansion of HBEC-KRASV12high CD133+ CICs induced by COPD-EVs, especially acting on CD133+CXCR4+ MICs (Fig. 4E). Coherently with effects on tumorigenic CICs subset, the blockade of HIF-1α transfer after COPD-EVs treatment reduced the number of 3D-colonies (Fig. 4F). Furthermore, the inhibition of HIF-1α reversed the pro-migratory effect, towards both SDF-1 and EGF chemoattractants, induced by COPD-EVs (Fig. 4G).

A Analysis of CD133 modulation in HBEC-KRASV12high treated with EVs in the presence of SDF-1 neutralizing antibody (n = 3). B Quantification (pg/µl) of HIF-1α carried by COPD or HS-EVs via ELISA (n = 10). C On the left, representative dot plots show the higher positivity for HIF-1α in HBEC-KRASV12high treated with COPD-EVs compared to HS-EVs. On the right, the bar chart shows the percentage of HIF-1α in PKH26-labeled EV-treated HBEC-KRASV12high using flow cytometry (n = 7). D mRNA expression level of HIF-1α, VEGF and CAIX in EV-treated cells (n = 4). E Flow cytometry analysis of CD133+ and CD133+CXCR4+ cells after concomitant HIF-1α inhibition and EVs administration in HBEC-KRASV12high (n = 5). F Quantification of colonies constituted by HBEC-KRASV12high cells treated with 15 µg of COPD- or HS-EVs and concomitant HIF-1α inhibition in 3D culture condition. Untreated cells (NT) were used as a negative control. G Evaluation of migratory capabilities of HBEC-KRASV12high towards EGF and SDF-1 gradients after the treatment with HS- and COPD-EVs in the presence or not of HIF-1α inhibitor (n = 3). Data are expressed as mean and SEM.
HIF-1α shuttle induces CXCR4 modulation and CD133+ stem cells
Since HIF-1α controls directly CXCR4 expression that in turn modulates CICs phenotype, we investigated its possible involvement as mediator of COPD-EVs induced-CICs effect. We confirmed that CXCR4 transcription was significantly increased in HBEC-KRASV12high cells after COPD-EVs treatment and that the inhibition of HIF-1α completely abrogated this effect (Fig. 5A). Notably, no CXCR4 modulation was observed after HS-EVs administration. Furthermore, CXCR4 silencing in HBEC-KRASV12high recipient cells completely prevented the increase of CD133+ and CD133+CXCR4+ subsets caused by COPD-EVs (Fig. 5B). Of note, the blockade of CXCR4 in HBEC-KRASV12high reverted the capacity of COPD-EVs to promote migration (Fig. 5C) and invasion (Fig. 5D) of these cells. Furthermore, the number of 3D colonies induced by COPD-EVs was reduced after CXCR4 silencing in recipient cells (Fig. 5E).

A The increase of mRNA of CXCR4 in HBEC-KRASV12high induced by COPD-EVs was abrogated by the use of HIF-1α inhibitor. B The silencing of CXCR4 reversed the modulation of CD133+ CICs and CD133+CXCR4+ MICs in COPD-EVs treated. In all experiments, untreated cells were used as control. C The silencing of CXCR4 in HBEC-KRASV12high cells reversed the migratory capability gained by cells treated with COPD-EVs. D Pro-invasive properties given by the treatment with COPD-EVs to HBEC-KRASV12high cells were abolished by the silencing of CXCR4. E Colonies formation induced by COPD-EVs treatment in HBEC-KRASV12high cells was inhibited by CXCR4 silencing. *p < 0.05. Data are expressed as mean and SEM.
All together these data support the crucial role of CXCR4 as a mediator of hypoxia-induced modulation of MIC phenotype.
Hypoxia plays a key role in MIC induction and in cancer development
To further demonstrate the central role of hypoxia in the regulation of MIC phenotype, HBEC-1, HBEC-3, HBEC-5, HBEC-6, and HBEC-KRASV12high were incubated for 72 h at 2% oxygen and the modulation of CD133+CXCR4+ was evaluated. Hypoxia induced a 4-fold increase of MIC cells only in HBEC-KRAS V12high cells (*p = <0.05) unlike other cell lines tested (Fig. 6A). Once again, the HIF-1α blocking (Fig. 6B) or the CXCR4 silencing (Fig. 6C) prevented the expansion of MICs in HBEC-KRASV12high in hypoxic conditions. Finally, we demonstrated that hypoxia promoted migratory and invasive capacities of HBEC-KRASV12high that are abrogated by the inhibition of HIF-1α or CXCR4 in recipient cells towards an EGF gradient (Fig. 6D, E and Supplementary Fig. S4). These results corroborate the observation that the cellular response to hypoxia is central in promoting MIC expansion but only in transformed epithelial cells accumulating multiple oncogenic hits, similar to what we previously observed after COPD-EVs treatment.

A Relative amount of CD133+CXCR4+ MICs in HBEC-1, HBEC-3, HBEC-5, HBEC-6, and HBEC-KRASV12high in normoxic or hypoxic condition (n = 3). B In hypoxic condition, the treatment with anti-HIF-1α of HBEC-KRASV12high inhibits the expansion of CD133+CXCR4+ MICs compared to untreated cells cultured in hypoxia (n = 4). C) MICs expansion in HBEC-KRASV12high was abrogated by inducing the silencing of CXCR4 (n = 4). D Migration assay towards an EGF gradient using HBEC-KRASV12high pre-treated with HIF-1α inhibitor or CXCR4 siRNA and cultured 48 h in hypoxic condition prior to perform the assay. E Invasion assay towards an EGF gradient using HBEC-KRASV12high pre-treated with HIF-1α inhibitor or CXCR4 siRNA and cultured 48 h in hypoxic condition prior to seed cells for the assay. F Evaluation of endothelial markers by using MACSPlex kit on EVs released by endothelial cells cultured in both normoxic and hypoxic conditions (n = 3). G Flow cytometry analysis on HBEC-KRASV12high to evaluate the possible expansion of CIC and MIC subsets following the treatment with END-EVs Normoxia and END-EVs Hypoxia. H ELISA performed to evaluate the level of HIF-1α within EVs isolated from COPD volunteers that subsequently developed (n = 15) or not (n = 13) lung cancer. *p < 0.05, **p < 0.01, ***p < 0.001. Data are expressed as mean and SEM.
Our MACSPlex analysis in Fig. 1E indicated a significant enrichment of endothelial markers in circulating EVs obtained from COPD subjects, suggesting their endothelial cell origin. To assess whether hypoxic environment occurring in COPD subjects promotes EVs release by endothelial cells, we cultured endothelial cells in hypoxic and standard oxygen culture conditions and we next recovered and analyzed EVs via MACSPlex. Hypoxia induced an increase in endothelial markers on the surface of EVs compared to normal condition (Fig. 6F), confirming that hypoxia boosted the release of EVs from endothelial cells.
We verified that EVs released by endothelial cells, maintained both in normoxic (END-EVs Normoxia) and in hypoxic conditions (END-EVs Hypoxia), have a similar ability to increase the total number of CD133+ cells in HBEC-KRASV12high recipient cells, but EVs released by endothelial cells under hypoxia can specifically expand the pool of CD133+CXCR4+ cells, suggesting their unique ability to activate CXCR4 pathway (Fig. 6G).
Finally, to confirm the involvement of hypoxia in cancer development, we analyzed the level of HIF-1α inside EVs from COPD volunteers, who subsequently developed or not lung cancer. We found significant higher levels of HIF-1α (125.8 pg/ml, SEM = 48.66 pg/ml) in EVs from COPD subjects who subsequently developed cancer (i.e. before CT scan detection) than COPD subjects who remained cancer free for at least three years (12.34 pg/ml, SEM = 2.835 pg/ml) (*p = 0.04) (Fig. 6H).
Taken together, these results highlight the strong involvement of COPD-derived hypoxia in inducing the release of EVs by endothelial cells and the potential role of HIF-1α within EVs in fostering lung cancer development that may be exploited as biomarker of lung cancer risk.
Discussion
Smoking leads to an increase of inflammation in the lungs that modifies the phenotype of epithelial and stromal cells [28]. COPD is associated with persistent respiratory symptoms and elevated inflammation levels [31]. Several evidences link the persistence of COPD with a higher incidence of lung cancer [32]. However, only 1 % of COPD subjects develop lung cancer indicating the existence of a complex mechanism behind the increased risk of lung carcinogenesis [33].
Recently, EVs emerged as important regulators of COPD and lung cancer [34]. It has been reported that COPD and inflammation modifies the phenotype of circulating EVs either considering surface markers or cargo [23]. Indeed, this pathological condition stimulates the release of EVs from endothelial cells and monocytes in the blood [35], whereas in the broncho-alveolar lavage vesicles expressing endothelial and macrophages markers were detected in COPD smokers compared to non smoker subjects [36]. In both of these studies the amount of vesicles correlates with the severity of COPD. Here, we confirmed a higher presence of endothelial-derived EVs in COPD subjects but we also found a general increase of surface markers derived from immune cells suggesting a possible involvement of adaptive immune response in the pathogenesis of the disease.
Hypoxia-inducible factors (HIFs), strongly deregulated in COPD disease, were also deregulated in several solid tumors and modulated different signaling pathways such as proliferation, EMT, metastatization and also resistance to therapy [37]. It has been demonstrated in experimental mouse models that COPD-like airway inflammation can lead to lung cancer promotion in a context of KRAS mutant epithelial cells [38, 39]. Interestingly, we proved that both in vitro hypoxic conditions and HIF-1α shuttled from COPD-EVs to recipient cells induce aggressive cancer phenotype and malignant progression through the increase of MICs population, but only in the context of an already activated genetic background of epithelial cells carrying different oncogenic hits, including amplification of KRASV12.
HIF-1α and hypoxia are involved in EV biogenesis both in normal and pathological conditions and are also implicated in the regulation of EV cargo such as protein and miRNAs [40]. It has been shown that nasopharyngeal carcinoma-derived vesicles induced EMT phenotype in recipient cell through the direct transfer of HIF-1α and this modulation increase the migratory and pro-metastatic ability of these cells [41]. Moreover, HIF-1α directly regulates the transcription of CXCR4, a chemokine receptor involved in cancer cell migration, invasion and in the maintenance of CICs in different solid tumors [42, 43]. In NSCLC, we found that CXCR4 pathway is activated in the specific subset of CD133+ CICs endowed with the highest metastasis-initiating ability [13]. We also previously showed that microenvironmental stimuli modulate lung MICs, through the induction of the EMT process [13]. Here, we demonstrated that the shuttle of HIF-1α contained in EVs from cancer-free individuals activates the transcription of CXCR4 that in turn increases the frequency of mesenchymal and disseminating CD133+CXCR4+ MICs. This increase was linked to the acquisition of tumorigenic, disseminating and mesenchymal properties by already transformed epithelial cells as functionally evaluated through in vitro assays.
We have already demonstrated that the transfer of plasma EV’s cargo from lung cancer subjects [44] or from individuals with a high risk of lung cancer [45] modulated the phenotype of premalignant epithelial cells through shuttling of c-Myc. Here, we show that COPD alters the protein content of EVs and that HIF-1α/CXCR4 axis activation caused by COPD-EVs in non-tumorigenic epithelial cells leads to the acquisition of malignant features and cancer progression.
Of clinical relevance, we observed that levels of HIF-1α in EVs from COPD subjects who subsequently developed cancer are significantly higher than in COPD subjects who remained cancer-free. These data suggest that in the context of COPD, analysis of HIF-1α in EVs cargo could be exploited to provide a novel biomarker to identify individuals at the highest risk of developing lung cancer.
Materials and methods
Cell lines
Immortalized human bronchial epithelial cells (HBEC-1: hTERT+Cdk4; HBEC-3: hTERT+Cdk4 + KRASV12; HBEC-5: hTERT+Cdk4+sh-p53; HBEC-6: hTERT+Cdk4+sh-p53 + KRASV12; HBEC-KRASV12high: hTERT+Cdk4+sh-p53+KRASV12high) were provided by Prof. J.D. Minna (UT Southwestern, TX) [46]. Cells were cultured in Keratinocyte–Serum-Free Medium supplemented with 5 ng/ml human recombinant EGF and 50 μg/ml bovine pituitary extract (K-SFM; Thermo Fisher Scientific, Waltham Massachusetts, USA).
More details on cells treatments are provided in Supplementary Methods.
Clinical specimens
Plasma samples were collected from heavy smoker volunteers (age 50–75 years) enrolled in a low dose CT screening trial performed at our institution (BioMild Trial, ClinicalTrials.gov: NCT02247453) [47]. Clinical characteristics of heavy smokers volunteers cancer free and heavy smokers volunteers who developed lung cancer are summarized in Table 1. Plasma collection was approved by Internal Review and Ethics Boards of Istituto Nazionale Tumori of Milan (INT 11-21).
EVs isolation and characterization
EVs were isolated from the plasma of heavy smokers and COPD volunteers by ultracentrifugation at 120,000 × g at 4 °C for 90 min using a TLA-100.3 fixed-angle rotor in a TL-100 ultracentrifuge (Beckman Coulter, Brea, CA, USA). EV-enriched pellet was washed in filtered phosphate-buffered saline (PBS; Thermo Fisher Scientific) centrifuged at 120,000 × g for 60 min at 4 °C to eliminate impurities and later resuspended in PBS. The protein content of purified EVs was verified by Bradford assay.
For EVs isolation from conditioned media (CM), HUVEC were maintained in serum-free medium for 72 h in normal oxygen condition or in 2 % oxygen condition at 37 °C. Then, the CM was collected and centrifuged at 3200 × g for 25 min in order to remove cell debris. EVs isolation was performed following the same protocol used for plasma.
EVs concentration and size distribution were evaluated by using a NanoSight NS300 instrument (Malvern Panalytical) whereas surface markers profile of plasma EVs was characterized by MACSPlex Kit (Milteny Biotec), as detailed in Supplementary Methods.
Flow cytometry
EVs analysis
EVs (15 µg) were incubated with 10 µl of Aldehyde/Sulfate latex beads, 4% w/v 4 µm (Invitrogen, Thermo Fisher Scientific) for 15 min at room temperature (RT) and later incubated overnight at 4 °C in PBS. After incubation, 100 mM glycine was added for 30 min at RT and then samples were stained with antibodies, as detailed in Supplementary Methods.
HBEC analysis
Quantification of CICs in different cell lines was performed 72 h post-treatment with EVs. Cells were resuspended in staining buffer (PBS 1X + 0.5 % BSA) and incubated with PE-conjugate anti-human CD133 antibody (Miltenyi Biotec) and APC-conjugate anti-human CXCR4 (BD Pharmingen) antibody for 20 min at 4 °C. After washing, cells were incubated with 7-AAD live/dead solution (Thermo Fisher Scientific) and the acquisition was performed using a FACSCanto II flow cytometer (BD Pharmingen).
Wound-healing assay
Wound-healing assay was performed to evaluate the migratory ability of HBEC-KRASV12high. 5×104 HBEC-KRASV12high, pre-treated with 15 µg of HS- or COPD-EVs, were seeded on both sides of Culture Insert 2-well in µ-Dish 35 mm (Ibidi). After 24 h, the culture insert was removed and 1 ml of Keratynocyte-SFM was added in each well. The cell-free gap was monitored and photographed immediately and after 3 h using a JuLI Stage Real-Time Cell History Recorder (NanoEntek). The percentage of wound closure was evaluated in 5 random fields using the ImageJ Software.
3D Proliferation assay
The 3D proliferation assay was performed to evaluate the ability of HBEC-KRASV12high to grow as colonies. The assay was performed using Vitrogel 3D (TheWell Bioscience, USA). A 1:1 VitroGel mix was added to 24-well plates and incubated for 20 min at 37 °C. Then, 5 × 104 of pretreated HBEC-KRASV12high were seeded on the VitroGel’s layer. After 14 days, the number of colonies was counted in 5 random fields per condition.
Statistical analysis
The statistical analysis was performed using the Graphpad Prism 5 software. Statistically significant differences were determined with Student’s t‐test when comparing two groups or the ANOVA test for multiple comparisons.
Data availability
All datasets generated and analyzed during this study are included in this published article and its Supplementary Information files. Additional data are available from the corresponding author on reasonable request.
References
Blanco I, Diego I, Bueno P, Casas-Maldonado F, Miravitlles M. Geographic distribution of COPD prevalence in the world displayed by Geographic Information System maps. Eur Respir J. 2019;54:1900610.
Terzikhan N, Verhamme KM, Hofman A, Stricker BH, Brusselle GG, Lahousse L. Prevalence and incidence of COPD in smokers and non-smokers: the Rotterdam Study. Eur J Epidemiol. 2016;31:785–92.
Durham AL, Adcock IM. The relationship between COPD and lung cancer. Lung Cancer. 2015;90:121–7.
Moermans C, Heinen V, Nguyen M, Henket M, Sele J, Manise M, et al. Local and systemic cellular inflammation and cytokine release in chronic obstructive pulmonary disease. Cytokine. 2011;56:298–304.
Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15:504–14.
Bertolini G, Roz L, Perego P, Tortoreto M, Fontanella E, Gatti L, et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc Natl Acad Sci USA. 2009;106:16281–6.
Tirino V, Camerlingo R, Franco R, Malanga D, La RA, Viglietto G, et al. The role of CD133 in the identification and characterisation of tumour-initiating cells in non-small-cell lung cancer. Eur J Cardiothorac Surg. 2009;36:446–53.
Prasetyanti PR, Medema JP. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol Cancer. 2017;16:41.
Sullivan JP, Spinola M, Dodge M, Raso MG, Behrens C, Gao B, et al. Aldehyde dehydrogenase activity selects for lung adenocarcinoma stem cells dependent on notch signaling. Cancer Res. 2010;70:9937–48.
Leung EL, Fiscus RR, Tung JW, Tin VP, Cheng LC, Sihoe AD, et al. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS ONE. 2010;5:e14062.
Liu J, Xiao Z, Wong SK, Tin VP, Ho KY, Wang J, et al. Lung cancer tumorigenicity and drug resistance are maintained through ALDH(hi)CD44(hi) tumor initiating cells. Oncotarget. 2013;4:1698–711.
Zhang WC, Shyh-Chang N, Yang H, Rai A, Umashankar S, Ma S, et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell. 2012;148:259–72.
Bertolini G, D’Amico L, Moro M, Landoni E, Perego P, Miceli R, et al. Microenvironment-modulated metastatic CD133+/CXCR4+/EpCAM-lung cancer-initiating cells sustain tumor dissemination and correlate with poor prognosis. Cancer Res. 2015;75:3636–49.
Genschmer KR, Russell DW, Lal C, Szul T, Bratcher PE, Noerager BD, et al. Activated PMN exosomes: pathogenic entities causing matrix destruction and disease in the lung. Cell. 2019;176:113–26.
Vlahos R, Bozinovski S. Role of alveolar macrophages in chronic obstructive pulmonary disease. Front Immunol. 2014;5:435.
Pullamsetti SS, Mamazhakypov A, Weissmann N, Seeger W, Savai R. Hypoxia-inducible factor signaling in pulmonary hypertension. J Clin Invest. 2020;130:5638–51.
Wu H, Ma H, Wang L, Zhang H, Lu L, Xiao T, et al. Regulation of lung epithelial cell senescence in smoking-induced COPD/emphysema by microR-125a-5p via Sp1 mediation of SIRT1/HIF-1a. Int J Biol Sci. 2022;18:661–74.
Zhang Q, Han Z, Zhu Y, Chen J, Li W. Role of hypoxia inducible factor-1 in cancer stem cells (Review). Mol Med Rep. 2021;23:17.
Korbecki J, Kojder K, Kapczuk P, Kupnicka P, Gawrońska-Szklarz B, Gutowska I. The effect of hypoxia on the expression of CXC chemokines and CXC chemokine receptors—a review of literature. Int J Mol Sci. 2021;22:843.
Mohan A, Agarwal S, Clauss M, Britt NS, Dhillon NK. Extracellular vesicles: novel communicators in lung diseases. Respir Res. 2020;21:175.
Herrmann A, Rice M, Lévy R, Pizer BL, Losty PD, Moss D, et al. Cellular memory of hypoxia elicits neuroblastoma metastasis and enables invasion by non-aggressive neighbouring cells. Oncogenesis. 2015;4:e138.
Zonneveld MI, Keulers TGH, Rouschop KMA. Extracellular vesicles as transmitters of hypoxia tolerance in solid cancers. Cancers (Basel). 2019;11:154.
O’Farrell HE, Bowman RV, Fong KM, Yang IA. Plasma extracellular vesicle miRNAs can identify lung cancer, current smoking status, and stable COPD. Int J Mol Sci. 2021;22:5803.
Cañas JA, Sastre B, Rodrigo-Muñoz JM, Del Pozo V. Exosomes: a new approach to asthma pathology. Clin Chim Acta. 2019;495:139–47.
Kadota T, Fujita Y, Yoshioka Y, Araya J, Kuwano K, Ochiya T. Extracellular vesicles in chronic obstructive pulmonary disease. Int J Mol Sci. 2016;17:1801.
Li M, Yu D, Williams KJ, Liu ML. Tobacco smoke induces the generation of procoagulant microvesicles from human monocytes/macrophages. Arterioscler Thromb Vasc Biol. 2010;30:1818–24.
Kubo H. Extracellular vesicles in lung disease. Chest. 2018;153:210–6.
Feller D, Kun J, Ruzsics I, Rapp J, Sarosi V, Kvell K, et al. Cigarette smoke-induced pulmonary inflammation becomes systemic by circulating extracellular vesicles containing Wnt5a and inflammatory cytokines. Front Immunol. 2018;9:1724.
Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7:1535750.
Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010;16:2927–31.
Oudijk EJ, Lammers JW, Koenderman L. Systemic inflammation in chronic obstructive pulmonary disease. Eur Respir J Suppl. 2003;46:5s–13s.
Sekine Y, Katsura H, Koh E, Hiroshima K, Fujisawa T. Early detection of COPD is important for lung cancer surveillance. Eur Respir J. 2012;39:1230–40.
Parris BA, O’Farrell HE, Fong KM, Yang IA. Chronic obstructive pulmonary disease (COPD) and lung cancer: common pathways for pathogenesis. J Thorac Dis. 2019;11:S2155–S2172.
Szalontai K, Gémes N, Furák J, Varga T, Neuperger P, Balog JÁ, et al. Chronic obstructive pulmonary disease: epidemiology, biomarkers, and paving the way to lung cancer. J Clin Med. 2021;10:2889.
Nieri D, Daniele M, Lombardi S, Bazzan E, Santerini S, De Cusatis G, et al. Circulating extracellular vesicles are associated with disease severity and interleukin-6 levels in COPD: a pilot study. J Clin Med. 2021;10:5014.
Bazzan E, Radu CM, Tinè M, Neri T, Biondini D, Semenzato U, et al. Microvesicles in bronchoalveolar lavage as a potential biomarker of COPD. Am J Physiol Lung Cell Mol Physiol. 2021;320:L241–L245.
Hapke RY, Haake SM. Hypoxia-induced epithelial to mesenchymal transition in cancer. Cancer Lett. 2020;487:10–20.
Moghaddam SJ, Li H, Cho SN, Dishop MK, Wistuba II, Ji L, et al. Promotion of lung carcinogenesis by chronic obstructive pulmonary disease-like airway inflammation in a K-ras-induced mouse model. Am J Respir Cell Mol Biol. 2009;40:443–53.
Gong L, Cumpian AM, Caetano MS, Ochoa CE, De la Garza MM, Lapid DJ, et al. Promoting effect of neutrophils on lung tumorigenesis is mediated by CXCR2 and neutrophil elastase. Mol Cancer. 2013;12:154.
Kucharzewska P, Christianson HC, Welch JE, Svensson KJ, Fredlund E, Ringnér M, et al. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc Natl Acad Sci USA. 2013;110:7312–7.
Aga M, Bentz GL, Raffa S, Torrisi MR, Kondo S, Wakisaka N, et al. Exosomal HIF1α supports invasive potential of nasopharyngeal carcinoma-associated LMP1-positive exosomes. Oncogene. 2014;33:4613–22.
Romain B, Hachet-Haas M, Rohr S, Brigand C, Galzi JL, Gaub MP, et al. Hypoxia differentially regulated CXCR4 and CXCR7 signaling in colon cancer. Mol Cancer. 2014;13:58.
Bao Y, Wang Z, Liu B, Lu X, Xiong Y, Shi J, et al. A feed-forward loop between nuclear translocation of CXCR4 and HIF-1α promotes renal cell carcinoma metastasis. Oncogene. 2019;38:881–95.
Borzi C, Calzolari L, Ferretti AM, Caleca L, Pastorino U, Sozzi G, et al. c-Myc shuttled by tumour-derived extracellular vesicles promotes lung bronchial cell proliferation through miR-19b and miR-92a. Cell Death Dis. 2019;10:759.
Pontis F, Roz L, Mensah M, Segale M, Moro M, Bertolini G, et al. Circulating extracellular vesicles from individuals at high-risk of lung cancer induce pro-tumorigenic conversion of stromal cells through transfer of miR-126 and miR-320. J Exp Clin Cancer Res. 2021;40:237.
Sato M, Larsen JE, Lee W, Sun H, Shames DS, Dalvi MP, et al. Human lung epithelial cells progressed to malignancy through specific oncogenic manipulations. Mol Cancer Res. 2013;11:638–50.
Pastorino U, Boeri M, Sestini S, Sabia F, Milanese G, Silva M, et al. Baseline computed tomography screening and blood microRNA predict lung cancer risk and define adequate intervals in the BioMILD trial. Ann Oncol. 2022;33:395–405.
Funding
The study was supported by grants from the Italian Association for Cancer Research (Investigator Grant 23244 to G.S.; 21431 to L.R.), Italian Ministry of Health (RF-2018-12367824 to G.S.; GR-2019-12369047 to O.F; RF-2016-02362946 to L.R.), Fondazione Regionale per la Ricerca Biomedica (Regione Lombardia) (1731093 to G.B.), Italian Ministry of Health “Ricerca Corrente” funds.
Author information
Authors and Affiliations
Contributions
IP, PG, FP, MB performed all the experiments under the supervision of OF and GB; AMF performed electronic microscopy analysis; OF, GB, LR, GS conceived the project and designed the experiments; OF, GB analyzed and interpreted the data; UP run the lung cancer screening trial, enrolled volunteers and collected clinical information; PS collected subjects samples; OF, GB, LR, GS wrote the manuscript. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics
Human samples were obtained under protocols approved by the Independent Ethics Committee of the Istituto Nazionale dei Tumori (INT 11-21).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Dr Giovanni Blandino
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Petraroia, I., Ghidotti, P., Bertolini, G. et al. Extracellular vesicles from subjects with COPD modulate cancer initiating cells phenotype through HIF-1α shuttling. Cell Death Dis 14, 681 (2023). https://doi.org/10.1038/s41419-023-06212-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-023-06212-1
