
Abstract
Parkinson’s disease (PD) is characterised by selective death of dopaminergic (DA) neurons in the midbrain and motor function impairment. Gastrointestinal issues often precede motor deficits in PD, indicating that the gut-brain axis is involved in the pathogenesis of this disease. The features of PD include both mitochondrial dysfunction and activation of the unfolded protein response (UPR) in the endoplasmic reticulum (ER). PINK1 is a mitochondrial kinase involved in the recycling of defective mitochondria, and PINK1 mutations cause early-onset PD. Like PD patients, pink1 mutant Drosophila show degeneration of DA neurons and intestinal dysfunction. These mutant flies also lack vital proteins due to sustained activation of the kinase R-like endoplasmic reticulum kinase (dPerk), a kinase that induces the UPR. Here, we investigated the role of dPerk in intestinal dysfunction. We showed that intestinal expression of dPerk impairs mitochondrial function, induces cell death, and decreases lifespan. We found that suppressing dPerk in the intestine of pink1-mutant flies rescues intestinal cell death and is neuroprotective. We conclude that in a fly model of PD, blocking gut-brain transmission of UPR-mediated toxicity, is neuroprotective.
Similar content being viewed by others

Introduction
Parkinson’s disease (PD) is a neurodegenerative disease and one of the most common movement disorders. At present, over 10 million people have PD (reviewed in [1]). The motor disturbances experienced by PD patients are linked to the selective loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc). One of the most common and disabling nonmotor symptoms of PD is constipation, a form of gut impairment [2]. There are substantive data suggesting that alterations in the intestine may precede neurodegeneration in patients with PD (reviewed in [3]). Postmortem studies have suggested that α-synuclein aggregates that contribute to PD travel from the enteric nervous system in the gut to the central nervous system and cause disease pathology [4]. Studies in which α-synuclein pathology was modelled in mice showed that gut-to-brain transmission of this toxic protein causes PD-associated neurodegeneration in the CNS [5], supporting the concept that alterations in the gastrointestinal tract can cause neurodegeneration. Mitochondrial dysfunction is a hallmark of PD (reviewed in [6]). Mitochondria accumulate damage with age, and cells have essential quality control mechanisms to ensure mitochondrial health (reviewed in [7]). Damaged mitochondria can be cleared and recycled via mitophagy, a specialised form of autophagy. PINK1 codes for a mitochondrial kinase that promotes mitophagy and ensures mitochondrial health (reviewed in [6]). Mutations in PINK1 cause autosomal recessive PD [8], and the fruit fly (Drosophila melanogaster) has been used to model PINK1-dependent PD. Pink1 mutant Drosophila show degeneration of DA neurons and global activation of R-like endoplasmic reticulum kinase (dPerk) signalling. In pink1 mutant flies, dPerk activates the unfolded protein response (UPR), leading to shutdown of protein synthesis [9]. Genetic or pharmacological inhibition of dPerk in pink1 mutants is neuroprotective [9]. The UPR normally acts to protect cells against stress that results from the accumulation of unfolded or damaged proteins. However, several studies indicate that when this pathway becomes constitutively activated, it reduces mRNA translation and promotes neurodegeneration (reviewed in [10]). In pink1 mutant flies, preventing cell death in the gut blocks the degeneration of DA neurons in the CNS, further highlighting the potential role of gut-brain communication in PD models ([11] and reviewed in [12]). dPerk is also important for intestinal homeostasis [13]. As dPerk also plays a role in neurodegeneration in fly models of PD, we explore the role of this kinase in gut-brain signalling. We show that dPerk expression in the gut causes intestinal dysfunction and that silencing dPerk in the gut protects neurons in the CNS of pink1 mutant flies. We provide further evidence for gut-brain axis signalling in a model of PD associated with mitochondrial defects.
Results
dPerk expression results in intestinal dysfunction and decreased lifespan
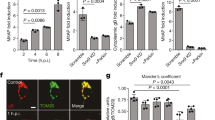
Pink1 mutant flies exhibit compromised mitochondrial function, characterised by loss of the mitochondrial membrane potential (Δψm) and activation of the kinase dPerk [9]. They also show intestinal dysfunction characterised by an increase in cell death in the midgut [11]. We first tested whether prolonged expression of dPerk in intestinal cells is sufficient to trigger mitochondrial dysfunction. To assess mitochondrial function in the intestine, we measured Δψm using tetramethylrhodamine methyl ester (TMRM). We observed that the expression of dPerk caused loss of the Δψm in the fly midgut (Fig. 1a–c), indicating that sustained activation of dPerk in the intestine is sufficient to impair mitochondrial function. Mitochondria play a key role in controlling apoptosis in Drosophila (reviewed in [14]), and intestinal apoptosis in flies can be inhibited by the expression of Buffy, a member of the Bcl-2 family of apoptosis antagonists [11]. We therefore tested whether dPerk-dependent loss of the Δψm is linked to cellular death in the intestine. We found that the expression of dPerk, but not its kinase-dead counterpart (dPerk-KD), caused a significant increase in the levels of active death caspase-1 (Dcp-1), an effector caspase in Drosophila that is important for apoptosis (Fig. 1d, e). Cell death in the gut results in a proliferative response by intestinal stem cells (ISCs) [15]. We next measured the degree of cellular proliferation in dPerk-expressing flies by monitoring the number of phospho-histone H3 (PH3)-positive cells. Histone H3 is phosphorylated during mitotic cell division, and we found that dPerk expression causes an increase in the number of cells positive for this proliferation marker (Fig. 2a, b). Taken together, these data indicate that dPerk expression causes intestinal damage by compromising mitochondrial function and inducing cell death and that the gut then compensates for this loss of cells by increasing proliferation.

a An illustration of the imaged region of the adult fly gut. b, c Expression of dPerk leads to a reduction in the Δψm in the fly intestine. b Representative confocal images showing whole-mounted intestine loaded with TMRM. The intensity of TMRM is visualised using a five-tone heatmap. c Quantitative data (asterisks, one-way ANOVA with Dunnett’s multiple comparison test, comparison of control flies with dPerk and dPerk-KD flies, n indicates the number of guts). The data are shown as the average of 8 ROIs per gut. d, e dPerk expression in the intestine causes caspase (Dcp-1) activation. d Representative confocal images showing Dcp-1-positive cells in the midgut. Both dPerk constructs (dPerk and dPerk-KD) were tagged with HA. Green, Dcp-1; red, HA; blue, cell nuclei. e Quantification of Dcp-1-positive cells in the midgut (asterisks, Kruskal‒Wallis with Dunn’s multiple comparison test, comparison of control flies with dPerk and dPerk-KD flies, n indicates the number of guts). Data from 10-day-old male flies were analysed. Genotypes: control: w; NP3048Gal4/ + ; + , dPerk: w; NP3048Gal4/UASdPerk; +, dPerk-KD: w; NP3048Gal4/UASdPerk-K671R; +.

a, b Cell proliferation was measured by counting the number of PH3-positive cells. a Representative confocal images showing PH3-positive cells in the midgut. Red, PH3; blue, cell nuclei. b Quantification of PH3-positive cells in the midgut (asterisks, Kruskal‒Wallis with Dunn’s multiple comparison test, comparison of control flies with dPerk and dPerk-KD flies, n indicates the number of guts). The number of PH3-positive cells in the anterior and posterior midgut (from the proventriculus region to the posterior midgut-hindgut junction) was determined. Data from 10-day-old male flies were analysed. Genotypes: control: w; NP3048Gal4/ + ; + , dPerk: w; NP3048Gal4/UASdPerk; +, dPerk-KD: w; NP3048Gal4/UASdPerk-K671R; +.
Next, we assessed the physiological consequences of intestinal damage caused by dPerk expression in the gut. We performed quantitative analysis of fly faecal deposits using the method described by Cognigni and colleagues [16]. dPerk-expressing flies showed a reduction in the number of deposits per plate, suggesting that intestinal dysfunction leads to reduced faecal output (Fig. 3a, b). dPerk expression further leads to a reduction in the circularity of the faeces (Fig. 3a, c). Together, these findings show that dPerk-dependent intestinal damage leads to physiological changes in fly defecation.

a Representative images of fly faeces over a 20-hour feeding period. Data from 10-day-old male flies fed on food supplemented with blue dye to aid the visualisation of the faeces were analysed. b Expression of dPerk reduces the number of faecal deposits. The data are shown as the number of deposits per plate (means ± sem, asterisks, Kruskal‒Wallis with Dunn’s multiple comparison test, comparison of control flies with dPerk and dPerk-KD flies). c Expression of dPerk alters the shape of faecal deposits (asterisks, Kruskal‒Wallis with Dunn’s multiple comparison test, comparison of control flies with dPerk and dPerk-KD flies, n indicates the number of faecal deposits). Genotypes: control: w; NP3048Gal4/ + ; + , dPerk: w; NP3048Gal4/UASdPerk; +, dPerk-KD: w; NP3048Gal4/UASdPerk-K671R; +.
Ageing leads to functional decline of individual organs, such as the gut. In flies, intestinal defects predict age-onset mortality [17]. Therefore, we tested whether dPerk-dependent intestinal toxicity can lead to premature death. Our analysis showed a reduction in the lifespan of flies in which intestinal toxicity was triggered by dPerk expression (Fig. 4a).

a Lifespan analysis showed that the intestinal expression of dPerk in flies reduces lifespan (asterisks, log-rank test) but b does not affect the development of the flies (asterisks Chi-square two-tailed, 95% confidence intervals). Temporal control expression of dPerk using Gal80ts in the flies c reduces lifespan (asterisks, log-rank test) and in 13 day old males, d decreases Δψm (asterisks, Mann-Whitney two-tailed test), e increase caspase (Dcp-1) activation (asterisks, two-tailed Student’s t-test) and f increases the number of PH3-positive cells (asterisks, Mann-Whitney two-tailed test) in the fly intestine. Genotypes: a, b control: w; NP3048Gal4/ + ; + , dPerk: w; NP3048Gal4/UASdPerk; +, c–f control: w; NP3048Gal4/ + ; Gal80ts/+, dPerk: w; NP3048Gal4/UASdPerk; Gal80ts/+.
The NP3084Gal4 driver can induce the expression of dPerk throughout fly development. We therefore examined the potential effect on development of expressing dPerk using this driver, by evaluating the eclosion of adult flies. We observed comparable eclosion rates between control flies and flies expressing dPerk (Fig. 4b) showing that the decrease in fly lifespan following its intestinal upregulation is not due to a developmental abnormality.
Therefore, to further prove that the damaging effects of dPerk observed is not due to expression throughout development, we used the Gal4-UAS gene expression system modified with a temperature-sensitive Gal80 protein (Gal80ts), ubiquitously expressed from the tubulin 1α promoter [18]. At 18 °C, the ubiquitously produced Gal80 protein is functional and acts as a repressor of the Gal4 transcriptional activator by binding to the Gal4 protein. At restrictive temperatures above 29 °C, Gal80 loses its ability to bind Gal4 and its repressive role, allowing the target gene to express under the control of the upstream activation sequence (UAS). Flies were grown at 18 °C and the eclosed adults were kept at 29 °C from 1 day post-eclosion, allowing for temporally regulated intestinal expression of dPerk in adult flies with the NP3084Gal4 driver. We found that the expression of dPerk in the adult intestine alone, caused a decrease in lifespan (Fig. 4c), a decrease in mitochondrial function in the intestinal cells (Fig. 4d), as well as an increase in both intestinal apoptosis (Fig. 4e) and cell proliferation (Fig. 4f).
Silencing of dPerk suppresses intestinal defects in pink1 mutant flies
Studies on pink1 mutant Drosophila, which are used to model PD, have indicated that mitochondrial dysfunction is an important feature of the pathogenesis of PD (reviewed in [6]). Mitochondrial impairment in pink1 mutant flies compromises neuronal function. In pink1 mutant young adults, mitochondrial impairment causes defective secretion of neuropeptides, leading to sleep defects [19]. Pink1 mutations also cause loss of DA neurons in aged flies [20], which can be prevented by the inhibition of dPerk [9]. Pink1 mutant flies also show intestinal dysfunction and blocking cell death in the intestine of these flies is neuroprotective [11]. We show here that the expression of dPerk in the gut is sufficient to cause mitochondrial dysfunction (Fig. 1) and compromise gut health. Additionally, suppression of dPerk in pink1 mutants is neuroprotective [9]. Therefore, we next asked whether suppressing dPerk in the intestine of pink1 mutant flies is sufficient to restore neuronal heath. We assessed mitochondrial function in the midguts of pink1 mutant flies (Fig. 5a–c) and observed loss of the Δψm. Next, we suppressed dPerk expression in the gut by RNA interference (RNAi) using the NP3084 midgut specific Gal4 driver. Downregulation of dPerk increased the Δψm in the intestine of pink1 mutant flies (Fig. 5d, e). We confirmed that cell death is increased in pink1 mutant flies, as previously reported [11], by assessing active Dcp-1 levels. We found that RNAi-mediated downregulation of dPerk suppressed intestinal apoptosis in pink1 mutants (Fig. 6a, b). We also measured the levels of PH3, a marker for proliferation of intestinal stem cells (ISCs), to assess if the reduction of intestinal cell death modulates the proliferative response in these cells [21]. We confirmed that the number of PH3-positive cells was increased in the pink1 mutants, as previously reported [11]. However, RNAi-mediated downregulation of dPerk did not affect the number of PH3-positive cells in the guts of these mutants (Fig. 6c).

a An illustration of the imaged region in the posterior midgut of the adult fly. c pink1 mutant flies show loss of the Δψm in the midgut (mean ± sem; asterisks, unpaired t test). d–e Recovery of the Δψm upon intestinal RNAi-mediated suppression of dPerk, with two independent RNAi lines, in pink1 mutant flies. b Representative confocal images showing the midguts of adult flies loaded with TMRM. The intensity of TMRM is visualised using a five-tone heatmap. Quantitative data (mean ± sem; asterisks, unpaired t test) with RNAi#1 d and RNAi#2 e. The flies were 10 days old in b and 20 days old in c–e. The data are shown as the average of 8 ROIs per gut. Genotypes: control: w; NP3048Gal4/ + ; + , Pink1: pink1B9; NP3048Gal4/ + ; + , Pink1, dPerk-RNAi#1: pink1B9; NP3048Gal4/UAS dPerk RNAi#1; +, Pink1, dPerk-RNAi#2: pink1B9; NP3048Gal4/UAS dPerk RNAi#2; +.

a, b Caspase activation in the intestine of pink1 mutant flies was blocked by dPerk RNAi. a Representative confocal images showing Dcp-1-positive cells in the posterior midgut region. b Quantitation of Dcp-1-positive cells in the posterior midgut showing that silencing dPerk reduced apoptosis of enterocytes of pink1 mutant flies (asterisks, one-way ANOVA with Dunnett’s multiple comparison test, all genotypes are compared to the control, n indicates the number of guts). c The number of PH3-positive cells in pink1 mutants was not affected by RNAi-mediated suppression of dPerk (asterisks, Kruskal‒Wallis with Dunn’s multiple comparison test, all genotypes are compared to the control, n indicates the number of guts). Data from 30-day-old male flies were analysed. Genotypes: control: w; NP3048Gal4/ + ; + , Pink1: pink1B9; NP3048Gal4/ + ; + , Pink1, dPerk-RNAi#1: pink1B9; NP3048Gal4/UAS dPerk RNAi#1; +, Pink1, dPerk-RNAi#2: pink1B9; NP3048Gal4/UAS dPerk RNAi#2; +.
We conclude that silencing dPerk in the intestine of pink1 mutant flies restores mitochondrial health and blocks apoptosis in midgut enterocytes, while having a negligible effect on the proliferation of ISCs.
Suppressing dPerk in the intestine of pink1 mutant flies is neuroprotective
Constipation, a form of gut dysfunction, is one of the most common and disabling nonmotor symptoms of PD [2].
In pink1 mutant flies, gut dysfunction is associated with neurodegeneration and sleep defects [11]; therefore, we evaluated whether RNAi-mediated silencing of dPerk in the guts of pink1 mutant flies can rescue these neuronal defects. Downregulation of dPerk reversed the defects in total activity of pink1 flies (Fig. 7a). We then examined the effects of dPerk suppression in the intestine on the degeneration of DA neurons in the brains of pink1 mutant flies. We found that RNAi-mediated suppression of dPerk in the intestine rescued the loss of DA neurons in pink1 mutants (Fig. 7b, c). To confirm that the NP3084Gal4 driver only silenced dPerk in the gut, we measured the transcript levels of dPerk in the guts and heads of pink1 mutant and pink1 mutant with RNAi suppression of dPerk. The results showed that suppression of dPerk in the guts of pink1 mutant or pink1 mutant with RNAi suppression of dPerk, decreased the level of this transcript in the gut but not in the head (Fig. 7d).

a Intestinal suppression of dPerk rescued activity defects in pink1 mutants (asterisks, Kruskal‒Wallis with Dunn’s multiple comparison test, comparison of pink1 mutant flies with control flies and dPerk-RNAi-treated pink1 mutant flies, n indicates the number of flies). b, c Intestinal suppression of dPerk prevented loss of the PPL1 cluster of DA neurons in pink1 mutants. b Top: schematic representation of the PPL1 anatomical location. Bottom: representative confocal images showing loss (and rescue) of PPL1 neurons in pink1 mutant and dPerk-RNAi-treated pink1 mutant flies. c RNAi-mediated suppression of dPerk in the intestine prevented loss of the PPL1 cluster of DA neurons in pink1 mutants (asterisks, Kruskal‒Wallis with Dunn’s multiple comparison test, comparison of pink1 mutant flies with control flies and dPerk-RNAi-treated pink1 mutant flies, n indicates the number of brains). d RNAi-mediated suppression of dPerk using a gut-specific driver reduced the mRNA levels of dPerk in the intestine (left, means ± sem; asterisks, unpaired t test, n indicates the number of biological replicates) but not in the heads (right) of pink1 mutants (means ± sem; NS, unpaired t test, n indicates the number of biological replicates). Data from 30-day-old male flies were analysed. Genotypes: a–c control: w;NP3084Gal4/ + ; + , Pink1: pink1B9; NP3048Gal4/ + ; + , Pink1, dPerk-RNAi#1: pink1B9; NP3048Gal4/UAS dPerk RNAi#1; +, Pink1, dPerk-RNAi#2: pink1B9; NP3048Gal4/UAS dPerk RNAi#2; +.
In addition to gut specific expression, the NP3084Gal4 driver also has some off-target expression in a small region of the brain [22]. The detection of dPerk levels when silencing dPerk by RNAi in the brain may be diluted if the driver only drives expression in a subset of neurons. Hence, we examined the expression of the NP3084Gal4 driver at the cellular level in the adult fly brain. We used NP3084Gal4 to drive GFP in adult flies and as expected we found that this driver induces GFP expression throughout the fly midgut (Fig. 8a).

a A representative image showing NP3084Gal4 drives GFP expression throughout the adult fly midgut. b NP3084Gal4 does not drive expression in the PPL1 cluster region of the adult fly brain. We used either the elav-Gal4 (top) or the NP3084Gal4 (bottom) to assay GFP fluorescence in adult fly brains in the PPL1 cluster of DA neurons. c RNAi-mediated suppression of dPerk in the intestine, using the mex1Gal4 driver prevented loss of the PPL1 cluster of DA neurons in pink1 mutants (asterisks, Mann-Whitney two-tailed test, n indicates the number of brains). d, e Analysis of the effect of the RNAi-mediated suppression of dPerk on the transcript levels of Relish in the intestine of pink1 mutants (asterisks, one-way ANOVA with Dunnett’s multiple comparison test, comparison of pink1 mutant with control and each dPerk-RNAi line. Genotypes: a w; NP3048Gal4/UASGFP; + , b top: w; elavGal4/UASGFP and bottom: w; NP3048Gal4/UASGFP c Pink1: pink1B9; mex1Gal4/+; +, Pink1, dPerk-RNAi#1: pink1B9; mex1Gal4/UAS dPerk RNAi#1 d control: w; NP3048Gal4/ + ; + , Pink1: pink1B9; NP3048Gal4/ + ; +, Pink1, dPerk-RNAi#1: pink1B9; NP3048Gal4/UAS dPerk RNAi#1; +, Pink1, dPerk-RNAi#2: pink1B9; NP3048Gal4/UAS dPerk RNAi#2; +.
We also analysed the cluster of PPL1 neurons in flies where GFP expression was driven by either elavGal4, a pan-neuronal driver, or NP3084Gal4, the gut driver. As anticipated, we found that GFP is detected in the PPL1 and other clusters of neurons with elavGal4 but not with NP3084Gal4 driver (Fig. 8b). This demonstrates that the NP3084Gal4 driver does not induce off-target expression in this subset of neurons that are lost in pink1 mutant flies. Compared to the NP3084Gal4, another enterocyte driver, mex1Gal4 [23] was reported to have no off-target expression in other organs [22]. We also found that RNAi-mediated suppression of dPerk in the intestine, using mex1Gal4, rescued the loss of DA neurons in pink1 mutant flies (Fig. 8c).
We have previously shown that the intestinal suppression of the NF-κB-like transcription factor, Relish, is neuroprotective in pink1 mutant flies [11]. We next measured the expression of relish in the gut of pink1 and pink1, dPerk RNAi lines #1 and #2. We found an increase in the expression of relish in pink1 mutants that was suppressed in one of the dPerk RNAi lines (Fig. 8d, e).
We therefore conclude that suppressing intestinal dysfunction by downregulating dPerk specifically in the midgut of pink1 mutant flies is sufficient to rescue neurotoxicity.
Discussion
We observed that sustained dPerk expression in the intestine led to loss of the Δψm and apoptotic cell death. During the first 24 h of sustained UPR activation, PERK signalling promotes the fission and fusion of mitochondria and enhances their function (reviewed in [24]). Following UPR activation for 24 h, mammalian cells eventually “give up the ghost”, and mitochondrial fragmentation leads to cellular suicide thorough apoptosis, a form of programmed cell death [25]. Our observations in the fly intestine extend these findings obtained in mammalian models by showing that sustained expression of dPerk causes loss of the Δψm. Previously, we showed that dPerk expression in flies reduces the levels of several mitochondrial components [26]; thus, we propose that loss of these components of the mitochondrial proteome causes a decline in mitochondrial function and loss of the Δψm.
We found that the suppression of dPerk in the enterocytes of pink1 mutant flies prevented their apoptotic demise. Stress in enterocytes of adult Drosophila causes the proliferation of ISCs. This proliferation can be measured by counting PH3-positive cells. Upon cell division, each ISC produces one daughter cell that preserves the ISC fate and one that can develop into an enterocyte to replace damaged cells [21]. We observed that the suppression of dPerk by RNAi did not suppress the increase in PH3-positive cells found in pink1 mutant flies (Fig. 6c). This signal presumably originates from stressed enterocytes to ISCs in pink1 mutants and involves JNK and JAK-STAT as well as other signalling pathways [21] likely acting upstream of the apoptotic execution phase. Thus, even though the suppression of dPerk can prevent enterocyte cell death, this may not prevent cell-cell communication between enterocytes and ISCs in pink1 mutants. It is also possible that the proliferation of ISCs is occurring in pink1 mutants independently of enterocyte damage. The ISCs in pink1 mutants are also likely to have mitochondrial damage. Mutations in pink1 increases the levels of mitochondrial reactive oxygen species (ROS) in flies [27]. ROS can control the proliferation of ISCs (reviewed in [28]) and therefore act as a cell autonomous mechanism that triggers their proliferation, independent of damaged enterocytes.
Both the intestinal suppression of Relish (our previous work [11]) and dPerk (this study), using a suite of three independent enterocyte Gal4 drivers, NP3084Gal4, NP1Gal4 [11] and mex1Gal4, confer neuroprotection to pink1 mutant flies. To understand the relationship between the regulation of dPerk and relish signalling, we measured the expression of relish following the downregulation of dPerk in pink1 mutant flies. We found that downregulating dPerk using one of the two dPerk RNAi lines significantly lowered expression levels of relish in pink1 mutants. These findings indicate that dPerk may alter intestinal homeostasis by inhibiting an inflammatory response within the gut. Decreasing dPerk levels in the intestine of pink1 mutants might improve mitochondrial function (as observed by measuring Δψm, Fig. 5), by decreasing the amount of DNA released by mitochondria and dampening the inflammatory response. However, it is still unclear first, why the transcript levels of relish are increased in the intestine of pink1 mutant flies and second, why suppressing dPerk reduces relish transcript levels.
The downregulation of relish in the intestine of pink1 mutant flies was not observed with dPerkRNAi#2 (Fig. 8d). However, we noted that the suppression of dPerk levels using RNAi#2 is not as efficient as those with RNA#1 line (Fig. 7d). It is therefore possible that the lack of the downregulation of relish, conferred by the expression of dPerk RNAi#2 in the intestine of pink1 mutants, is a consequence of the moderate loss of dPerk expression in this line.
Our results build on prior observations [11] to show that a gut-brain pathway causes neurotoxicity linked to mitochondrial dysfunction in Drosophila. In mice, this gut-brain route involves the vagus nerve [5]. In our model, it is unclear how mitochondrial toxicity is passed from the gut to the CNS. We reason that this transmission is indirect, as there are currently no known connections between the gut and DA neurons. Nevertheless, there are connections between the CNS and the gut that involve neurons with cell bodies in the posterior segments of the abdominal ganglion of the ventral nerve cord that send axons to the posterior portion of the midgut. These connections are relayed to the CNS by insulin-producing neurons [29]. It is possible that these insulin-producing neurons form a “wired” route to transmit mitochondrial toxicity from the gut to the brain in pink1 mutant flies. An alternate indirect mechanism through which mitochondrial toxicity can be transmitted from the gut to the brain could involve a “wireless” mechanism. Crewe and colleagues [30] reported that mammalian cells can communicate mitochondrial stress via a “wireless” humoral mechanism. This mechanism involves the release of mitochondrial particles (small extracellular vesicles) into the circulation and the transport of these particles between the fat body and heart. Similar mechanisms may occur in flies, where mitochondrial vesicles communicate stress between the intestine and the brain. Our study using fruit flies highlights the importance of the gut-brain axis in the onset and progression of PD. This supports Hippocrates’s 2000-year-old claim that “all disease begins in the gut”.
Methods
Genetics and Drosophila strains
Fly stocks and crosses (unless otherwise stated) were maintained on standard cornmeal agar media at 25 °C. The following strains were used: pink1B9 as previously described (Tufi et. al., 2014), dPerk RNAi lines (RNAi#1, ID:16427; RNAi#2, ID: 110278) (Vienna Drosophila RNAi Centre, Vienna, Austria), w; P{GawB}NP3084; + (KSC_113094), w; UAS dPerk; +,w; UAS dPerk-KD; + as previously described [26], w; tubP-Gal80ts/TM2 (Bloomington 7017) and w; P{mex1-Gal4} (Bloomington 91368). All crosses involving UAS dPerk and UAS dPerk-KD were kept at 18 °C, and progenies with selected genotypes were kept at 25 °C until assayed. All crosses involving Gal80ts were kept at 18 °C, and the hatched progenies with selected genotypes were kept at 29 °C until assayed. All the experiments on adult flies were performed using males.
Analysis of faecal output and the T.U.R.D. assay
To aid the visualisation of fly excreta, 0.5% Brilliant Blue FCF (80717, Sigma‒Aldrich) was added to standard cornmeal agar media. The dye-containing food was offered ad libitum as a wedge of solid food placed on a Petri dish, and five 10-day-old males were allowed to excrete on the clear flat plastic surface of the dish for 20 hours at 25 °C. A total of twenty-five flies per genotype were examined. At the end of the experiment, the flies and food were removed from the Petri dishes, and the dishes containing fly deposits were scanned with a transparency scanner (Epson Perfection, V750 Pro). Images were cropped and prepared for analysis using Adobe Photoshop 2022 (23.4.1). Images of fly faecal deposits were analysed using T.U.R.D. software, a tool designed to assess the colour and morphology of excreta produced by flies fed on a dye-supplemented diet [31].
Locomotor assays
Twenty-five-day-old males were individually loaded into Drosophila Activity Monitors (DAM5) within 8 × 65-mm2 glass Pyrex tubes (Trikinetics, Waltham) containing normal fly food. The flies were maintained at 25 °C on a 12/12-h light-dark cycle for at least 8 days. Sleep and activity data were analysed using the Sleep and Circadian Analysis MATLAB Program (SCAMP) developed by the Griffith laboratory [32]. Analyses were performed for 7 days starting at the first Zeitgeber time (ZT0) to allow acclimation. At least 40 flies of each genotype were used. Flies with rhythmic index scores < 1 were removed from the analyses.
Lifespan analysis and eclosion
Groups of 12 newly eclosed males of each genotype were placed into separate vials containing food and maintained at 25 °C (or at 29 °C for Gal80ts experiments). The flies were transferred to vials containing fresh food every 2 to 3 days, and the number of dead flies was recorded. The data are presented as Kaplan‒Meier survival curves, and significance was determined by log-rank tests. For eclosion assays, crosses were set in multiple vials with 10 virgins and 4 males for all conditions and allowed to lay for 2 days and the number of empty and total pupae scored at the end of eclosion. The data are presented as percentages, and significance was determined by using the Chi-squared test.
Microscopy-based assessment of mitochondrial function and morphology
The Δψm in the fly gut was measured as previously described [33]. Briefly, adult fly midguts were incubated for 40 minutes at room temperature with 25 nM TMRM in 1× Hank’s Balanced Salt Solution (HBSS) (14175-053, Gibco). TMRM was used in redistribution mode to assess the Δψm; therefore, a reduction in the TMRM fluorescence intensity indicated mitochondrial depolarization. Confocal images were acquired using a Zeiss LSM 880 or 980 confocal microscope equipped with a 20× objective. The illumination intensity was kept at a minimum (at 0.1–0.2% of the laser output) to avoid phototoxicity, and a pinhole was used to obtain a 1 μm thick optical slice. The fluorescence intensity of TMRM was quantified at an excitation wavelength of 565 nm and emission wavelength greater than 580 nm. A single Z-stack per gut was acquired, and 8 10–20 μm2 ROIs were selected. The mean maximal fluorescence intensity in these ROIs was measured.
Immunofluorescence and confocal microscopy
To image the intestine, guts were dissected from live flies in ice-cold PBS and fixed in 4% PFA (for anti-HA, anti-PH3, anti-TH and anti-GFP antibody incubation) or heptane-methanol fixative (for anti-Dcp1 antibody incubation) for 30 minutes and blocked overnight in 10% normal goat serum PBS/0.5% Triton. The samples were then incubated with primary antibodies (anti-cleaved Drosophila Dcp-1 (CST, cat no. 9678, 1:100), anti-HA (Roche Applied, no. 11583816001, 1:1000) and anti-PH3 (mix 1-1 of CST, cat no. 9701 (Ser10) and cat no. 9713 (Ser28), 1:100), anti-GFP (ab13970, 1:1000) and anti-TH (Immunostar, 22941, 1:50) at 4 °C overnight and then with secondary antibodies (Alexa Flour™ 488-conjugated F(ab’)2 fragment of goat anti-rabbit IgG (H + L) (Invitrogen, A11070), Alexa Flour™ 488-conjugated goat anti-chicken IgG (H + L) (ab 150169) or Alexa Flour™ 568-conjugated goat anti-mouse IgG (H + L) (Invitrogen, A11031) and Hoechst 33342 (1:500, Invitrogen, H3570) diluted 1:500 for 2 hours at room temperature.
Analysis of DA neurons
Brains from 30-day-old flies were dissected and stained with anti-TH antibody (Immumostar, no. 22941, 1:50) as previously described [34]. The brains were placed in PBS + 0.1% Triton in a coverslip clamp chamber (ALA Scientific Instruments) using a harp composed of platinum wire and nylon string and imaged by confocal microscopy. The numbers of TH-positive neurons in the PPL1 cluster in each brain hemisphere were determined.
RNA extraction and reverse transcription quantitative real-time PCR
Total RNA was extracted from 25 heads or 15–20 freshly dissected guts using TRIzol (Ambion) and quantified by spectrophotometric analysis (Nanodrop, Thermo Scientific). RT‒qPCR was performed with a real-time cycler (Applied Biosystems 75000, Fast Real-Time PCR Systems) using the SensiFAST SYBR Lo-ROX One-Step Kit (Bioline). The fold change in expression was calculated using the comparative Ct method [35]. For RT‒qPCR, we measured the coefficient of variance (CV) of the technical replicates, and any samples with a CV > 3% was excluded from statistical analysis. The gene-specific primers were Dm_PEK_2_SG QuantiTect Primer Assay (Qiagen, QT00968618), and Relish (forward, 5′-CATCAGGAGACAGAGCGTGA-3′; reverse, 5′-CCGACTTGCGGTTATTGATT-3′) and rp49 (forward, 5′-TGTCCTTCCAGCTTCAAGATGACCATC-3′; reverse, 5′-CTTGGGCTTGCGCCATTTGTG-3′) (Sigma‒Aldrich).
Statistical analyses
Statistical analyses were performed using GraphPad Prism (www.graphpad.com). The data are presented as the mean values, and the error bars indicate ± sem. In the violin plots, the solid line represents the median, while the dashed lines represent the quartiles. The individual data points in the figures correspond to biological replicates. The number of biological replicates per experimental variable (n) is indicated in either the figure or respective figure legend. For all statistical analyses, the Shapiro–Wilk test was used to test if the data followed a normal distribution. Based on normality test results, parametric or nonparametric analysis was used. Samples were excluded from the analysis based on the Prism outlier test, ROUT, Q = 1%. Blinding was not performed. Significance is indicated as * for P < 0.05, ** for P < 0.01, *** for P < 0.001, **** for P < 0.0001 and NS for P ≥ 0.05.
Digital image processing
Fluorescence and transmission electron microscope images were acquired as uncompressed bitmapped digital data (TIFF format) and processed using Adobe Photoshop, employing established scientific imaging workflows.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article.
References
Abou-Sleiman PM, Hanna MG, Wood NW. Genetic association studies of complex neurological diseases. J Neurol Neurosurg Psychiatry. 2006;77:1302–4.
Frazzitta G, Ferrazzoli D, Folini A, Palamara G, Maestri R. Severe constipation in Parkinson’s disease and in parkinsonisms: prevalence and affecting factors. Front Neurol. 2019;10:621.
Harsanyiova J, Buday T, Kralova Trancikova A. Parkinson’s Disease and the Gut: Future Perspectives for Early Diagnosis. Front Neurosci. 2020;14:626.
Braak H, Rub U, Gai WP, Del, Tredici K. Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm (Vienna). 2003;110:517–36.
Kim S, Kwon SH, Kam TI, Panicker N, Karuppagounder SS, Lee S, et al. Transneuronal propagation of pathologic alpha-synuclein from the gut to the brain models Parkinson’s disease. Neuron. 2019;103:627–641.e627.
Celardo I, Martins LM, Gandhi S. Unravelling mitochondrial pathways to Parkinson’s disease. Br J Pharmacol. 2014;171:1943–57.
de Castro IP, Martins LM, Loh SH. Mitochondrial quality control and Parkinson’s disease: a pathway unfolds. Mol Neurobiol. 2011;43:80–86.
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–60.
Celardo I, Costa AC, Lehmann S, Jones C, Wood N, Mencacci NE, et al. Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease. Cell Death Dis. 2016;7:e2271.
Halliday M, Mallucci GR. Targeting the unfolded protein response in neurodegeneration: A new approach to therapy. Neuropharmacology. 2014;76 Pt A:169–74.
Fedele G, Loh SHY, Celardo I, Santos Leal N, Lehmann S, Costa AC, et al. Suppression of intestinal dysfunction in a Drosophila model of Parkinson’s disease is neuroprotective. Nat Aging. 2022;2:1–15.
Aparicio R, Schmid ET, Walker DW. Gut mitochondrial defects drive neurodegeneration. Nat Aging. 2022;2:277–9.
Wang L, Ryoo HD, Qi Y, Jasper H. PERK limits drosophila lifespan by promoting intestinal stem cell proliferation in response to ER stress. PLoS Genet. 2015;11:e1005220.
Clavier A, Rincheval-Arnold A, Colin J, Mignotte B, Guenal I. Apoptosis in Drosophila: which role for mitochondria? Apoptosis. 2016;21:239–51.
Naszai M, Carroll LR, Cordero JB. Intestinal stem cell proliferation and epithelial homeostasis in the adult Drosophila midgut. Insect Biochem Mol Biol. 2015;67:9–14.
Cognigni P, Bailey AP, Miguel-Aliaga I. Enteric neurons and systemic signals couple nutritional and reproductive status with intestinal homeostasis. Cell Metab. 2011;13:92–104.
Biteau B, Hochmuth CE, Jasper H. JNK activity in somatic stem cells causes loss of tissue homeostasis in the aging Drosophila gut. Cell Stem Cell. 2008;3:442–55.
McGuire SE, Le PT, Osborn AJ, Matsumoto K, Davis RL. Spatiotemporal rescue of memory dysfunction in Drosophila. Science. 2003;302:1765–8.
Valadas JS, Esposito G, Vandekerkhove D, Miskiewicz K, Deaulmerie L, Raitano S, et al. ER lipid defects in neuropeptidergic neurons impair sleep patterns in Parkinson’s disease. Neuron. 2018;98:1155–1169.e1156.
Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–61.
Buchon N, Broderick NA, Poidevin M, Pradervand S, Lemaitre B. Drosophila intestinal response to bacterial infection: activation of host defense and stem cell proliferation. Cell Host Microbe. 2009;5:200–11.
Weaver LN, Ma T, Drummond-Barbosa D. Analysis of Gal4 expression patterns in adult drosophila females. G3 (Bethesda). 2020;10:4147–58.
Phillips MD, Thomas GH. Brush border spectrin is required for early endosome recycling in Drosophila. J Cell Sci. 2006;119:1361–70.
Almeida LM, Pinho BR, Duchen MR, Oliveira JMA. The PERKs of mitochondria protection during stress: insights for PERK modulation in neurodegenerative and metabolic diseases. Biol Rev Camb Philos Soc. 2022;97:1737–48.
Hom JR, Gewandter JS, Michael L, Sheu SS, Yoon Y. Thapsigargin induces biphasic fragmentation of mitochondria through calcium-mediated mitochondrial fission and apoptosis. J Cell Physiol. 2007;212:498–508.
Popovic R, Celardo I, Yu Y, Costa AC, Loh SHY, Martins LM. Combined transcriptomic and proteomic analysis of perk toxicity pathways. Int J Mol Sci. 2021;22:4598.
Travaglio M, Michopoulos F, Yu Y, Popovic R, Foster E, Coen M, et al. Increased cysteine metabolism in PINK1 models of Parkinson’s disease. Dis Model Mech. 2023;16:dmm049727.
Morris O, Jasper H. Reactive Oxygen Species in intestinal stem cell metabolism, fate and function. Free Radic Biol Med. 2021;166:140–6.
Nassel DR, Kubrak OI, Liu YT, Luo JN, Lushchak OV. Factors that regulate insulin producing cells and their output in Drosophila. Front Physiol. 2013;4:252.
Crewe C, Funcke JB, Li S, Joffin N, Gliniak CM, Ghaben AL, et al. Extracellular vesicle-based interorgan transport of mitochondria from energetically stressed adipocytes. Cell Metab. 2021;33:1853–1868.e1811.
Wayland MT, Defaye A, Rocha J, Jayaram SA, Royet J, Miguel-Aliaga I, et al. Spotting the differences: probing host/microbiota interactions with a dedicated software tool for the analysis of faecal outputs in Drosophila. J Insect Physiol. 2014;69:126–35.
Donelson NC, Kim EZ, Slawson JB, Vecsey CG, Huber R, Griffith LC. High-resolution positional tracking for long-term analysis of Drosophila sleep and locomotion using the “tracker” program. PLoS ONE. 2012;7:e37250.
Tufi R, Gandhi S, de Castro IP, Lehmann S, Angelova PR, Dinsdale D, et al. Enhancing nucleotide metabolism protects against mitochondrial dysfunction and neurodegeneration in a PINK1 model of Parkinson’s disease. Nat Cell Biol. 2014;16:157–66.
Whitworth AJ, Theodore DA, Greene JC, Benes H, Wes PD, Pallanck LJ. Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson’s disease. Proc Natl Acad Sci USA. 2005;102:8024–9.
Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8.
Acknowledgements
We thank the Bloomington Drosophila Stock Centre and Vienna Drosophila RNAi Centre for fly stocks. We also thank the Fly Facility, Department of Genetics, University of Cambridge for generating the transgenic lines. We thank Dr G. Fedele for guidance with laboratory experiments.
Funding
This work was funded by the UK Medical Research Council, intramural project MC_UU_00025/3 (RG94521).
Author information
Authors and Affiliations
Contributions
LMM and RP initiated the project. LMM, RP, AM, and SHYL designed the study, coordinated the experiments, and provided conceptual input for the paper. LMM, SHYL, and RP wrote the manuscript. RP, AM, SHYL, NSL, LM, and YY performed the experiments and analysed the data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Eric Baehrecke
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Popovic, R., Mukherjee, A., Leal, N.S. et al. Blocking dPerk in the intestine suppresses neurodegeneration in a Drosophila model of Parkinson’s disease. Cell Death Dis 14, 206 (2023). https://doi.org/10.1038/s41419-023-05729-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-023-05729-9
