
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is one of the most aggressive and deadliest cancer worldwide. The primary reasons for this are the lack of early detection methods and targeted therapy. Emerging evidence highlights the metabolic addiction of cancer cells as a potential target to combat PDAC. Oncogenic mutations of KRAS are the most common triggers that drive glucose uptake and utilization via metabolic reprogramming to support PDAC growth. Conversely, high glucose levels in the pancreatic microenvironment trigger genome instability and de novo mutations, including KRASG12D, in pancreatic cells through metabolic reprogramming. Here, we review convergent and diverse metabolic networks related to oncogenic KRAS mutations between PDAC initiation and progression, emphasizing the interplay among oncogenic mutations, glucose metabolic reprogramming, and the tumor microenvironment. Recognizing cancer-related glucose metabolism will provide a better strategy to prevent and treat the high risk PDAC population.
Similar content being viewed by others


Facts
-
Although the association between diabetes and PDAC has been revealed, whether diabetes is a predisposing factor or an early manifestation of malignancy remains unsettled.
-
One of the potential therapeutic strategies for PDAC is to target the metabolic addiction of cancer cells.
-
KRAS proto-oncogene mutations shut glycolysis hexosamine biosynthesis and pentose phosphate pathways.
-
High glucose initiates genome instability and de novo mutations, including KRASG12D, in nontumorigenic pancreatic cells.
-
Alternation of O-linked-N-acetylglucosaminylation changes cellular and physiological homeostasis fueling PDAC initiation and progression.
PDAC: a growing silent killer
Pancreatic ductal adenocarcinoma (PDAC), the seventh leading cause of cancer-related death worldwide in 2020, accounts for ~95% of all pancreatic cancers as well as 4.9% and 4.5% of estimated age-standardized incidence and mortality rates, respectively, with almost as many deaths as the number of cases [1]. PDAC will foreseeably become the second leading cause of cancer-related deaths by 2026 [2], with an ~11.5% 5-year relative survival rate in the United States [3]. Due to the lack of an early detection method and effective therapeutics, diagnosis generally occurs at an advanced stage, when patients already have locoregional extensions or metastases that render surgical resection ineffective [4]. PDAC stems from abnormal acinar-to-ductal metaplasia (ADM) and pancreatic intraepithelial neoplasia (PanIN) of grades I–III [5]. One of the potential therapeutic strategies for PDAC is to target the unique nutrient availability and utilization in cancer cells [6, 7]. Significant advances have been made in understanding metabolic adaptations to KRAS hyperactivation. Efforts are ongoing to design metabolism-targeted diagnostic and therapeutic strategies. Nevertheless, the most potent strategy should aim to prevent PDAC initiation and enhance early detection.
Considering genetic and personal risk factors [8], novel findings have suggested aberrant metabolites not only as promoters [9] but also as initiators [10] for PDAC, and common metabolic reprogramming patterns have gained a hotspot for prevention or early detection [11]. This review provides a synopsis of metabolic dependence supporting oncogenic mutation-driven PDAC progression, emphasizing that aberrant nutrient availability and utilization may also cause oncogenic mutations. This review also offers an outlook of potential targets for PDAC therapeutics, prevention, or early detection.
KRAS mutation and metabolic alterations
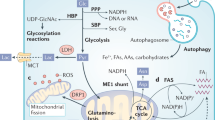
Cancer cells become dependent on activated oncogenes or their downstream metabolic processes for survival and proliferation [12]. Inhibiting oncogenes or their downstream mediators is expected to be lethal to metabolically addicted cells without harming normal cells. Advances include dependence on poly(ADP-ribose) polymerase activity in the context of BRCA deficiency [13, 14]. The somatic mutation of oncogenic KRAS is the most recognized genetic alteration and is thus the most attractive drug target in PDAC [15, 16]. Mutant KRAS increases the expression of glucose transporter 1 (GLUT1) and rate-limiting glycolytic enzymes, including hexokinases, phosphofructokinase 1 (PFK1), and lactate dehydrogenase A (LDHA), promoting glycolytic activity and increasing lactate production [17, 18]. By upregulating these enzymes, mutant KRAS triggers the shunting of glycolytic intermediates into the hexosamine biosynthesis pathway (HBP) to generate UDP-N-acetylglucosamine (UDP-GlcNAc) for glycoprotein, glycolipid, proteoglycan, and glycosylphosphatidylinositol anchor biosynthesis in cancer cells [5, 19]. The shunting of glycolytic intermediates into the nonoxidative arm of the pentose phosphate pathway (PPP) generates the ribose 5-phosphate necessary for nucleic acid biosynthesis and nicotinamide adenine nucleotide phosphate (NADPH) for regenerating glutathione (GSH) from oxidized GSH (GSSG) to support ROS scavenging for redox balance. GSH biosynthesis depends on glutamine. While most cells convert glutamine-derived glutamate to α-ketoglutarate via glutamate dehydrogenase (GLUD1) to fuel the tricarboxylic acid (TCA) cycle, PDAC relies on glutamine-derived aspartate via glutamic oxaloacetate transaminase 1 (GOT1), which is catalyzed by the mitochondrial uncoupling protein 2 (UCP2) [20]. Glutamine-derived aspartate can be converted into oxaloacetate, which is subsequently converted into malate and then pyruvate, increasing the NADPH/NADP+ ratio, which could additionally maintain the cellular redox balance for PDAC progression [21]. Other metabolic adaptations to KRAS hyperactivation include the dependence of amino acids such as serine [21, 22], glycine [23], branched-chain keto acid [24, 25], and lipid metabolism [26] in PDAC to facilitate survival [16, 27], which mirrors genetic heterogeneity in PDAC [28,29,30], are summarized in BOX1. Efforts on designing suitable metabolism-targeted diagnostic and therapeutic strategies are ongoing.
In addition, nutrient deprivation in the PDAC tumor microenvironment (TME) [31] may drive KRAS-mutated cancer cells to be more dependent on lysosomal nutrient scavenging pathways, such as macropinocytosis [27] and autophagy [32], to recycle energy and biosynthetic building blocks from the microenvironment [16]. Inactivating oncogenic KrasG12D in an inducible KrasG12D murine model of PDAC reversed metabolic reprogramming and rapid proliferation, supporting that Kras mutation is essential for PDAC initiation and progression [18, 33,34,35]. Figure 1 illustrates the current understanding of metabolic addictions driven by KRAS mutations. However, withdrawal of transgene expression may have differential effects from the loss of endogenous oncogenic KRAS expression caused by inhibitors. PDAC cells survive even genetic ablation of Kras both in vitro and in vivo [36, 37], indicating that reversing the oncogenic behaviors driven by mutant KRAS might not completely halt all PDAC development. Additional non-KRAS genetic heterogeneity and other genetic alterations may dysregulate metabolism in PDAC, which are summarized in BOX 1. However, how the polygenic risk of the mutations that shape the metabolism and phenotype of cancer cells remains to be elucidated [38].

The active oncogenic KRAS mutant triggers glycolysis for energy generation and building block biosynthesis and shunting into the nonoxidative arm of the pentose phosphate pathway for nucleic acid biosynthesis and the hexosamine biosynthesis pathway for UDP-N-acetylglucosamine biosynthesis through upregulation of key enzymes in glycolysis. In addition, micropinocytosis and autophagy ensure that mutant KRAS cancer cells obtain energy and biosynthetic building blocks from the microenvironment. GLUT1 glucose transporter 1, HK1/2 hexokinases 1 and 2, PFK1 phosphofructokinase 1, LDHA lactate dehydrogenase A, ASCT2, also known as solute carrier family 1, member 5 (SLC1A5); SN2, also known as solute carrier family 38, member 5 (SLC38A5); MCT monocarboxylic transporters, GOT aspartate transaminase, GLUD1 glutamate dehydrogenase 1, GLS1 glutaminase 1, G6P glucose 6-phosphate, F6P fructose 6-phosphate, FBP fructose 1,6-phosphate, DHAP dihydroxyacetone phosphate, G3P glyceraldehyde 3-phosphate, 3PG 3-phosphoglycerate, αKG α-ketoglutarate, Asp aspartate, OAA oxaloacetate, TCA cycle tricarboxylic acid cycle. The graph was created with BioRender.com.
Association between diabetes and PDAC
The association between diabetes and PDAC has been recognized. The prevalence of diabetes is higher in PDAC than in other cancers [11]. Diabetic patients with PDAC frequently have larger tumors, reduced median survival [39], and higher mortality [40, 41]. Unlike insulin and proinsulin levels, which are independently associated with an increased risk of PDAC among nondiabetic patients, circulating markers, including baseline fasting blood glucose and glycated hemoglobin, are associated with the increased risk of PDAC a dose-dependent manner among patients with diabetes [42, 43]. However, ~45–65% of patients with PDAC have diabetes at diagnosis, with new-onset diabetes (within <3 years) accounting for ~25% [44], implying that hyperglycemia is not essential for PDAC development. Although whether diabetes is an early manifestation of malignancy [11, 45] or a predisposing factor for the development of PDAC [46,47,48] is unsettled, the association of diabetes with PDAC has gained sufficient support from different studies.
High glucose-triggered DNA damage and de novo mutations, including KRAS G12D , drive proliferating acinar cell transformation
Oncogenic KRAS mutations are thought to be an early genetic mutation that exerts dramatic metabolic reprogramming in PDAC. However, oncogenic KRAS mutations are not sufficient to initiate PDAC [49, 50]. Other factors are required to orchestrate tumor formation. The physiological roles of the pancreas are in modulating carbohydrate, protein, and lipid metabolism, as well as the availability and utilization of metabolites such as lactate and glutamine [51, 52]. Interestingly, glucose is not used as the primary energy source for the pancreas, unlike other organs [51, 52], implying that excess metabolites such as glucose may be a risk factor for PDAC initiation [53]. Oncogenic Kras and a chronic high-fat diet synergistically promote PDAC development [9]. Although with a lower tumorigenic capacity than a chronic high-fat diet, a chronic high-carbohydrate diet promoted ADM and PanIN lesions with increased inflammation and fibrosis compared to a normal diet in KrasG12D/+ mice [54]. In contrast, a low glycemic diet impairs tumor growth by altering cellular lipid composition [55]. Sugar consumption contributes to the development of metabolic diseases, including type 2 diabetes [56, 57], and some cancers, partly through weight gain but also through independent metabolic effects of glucose and fructose [58]. Epidemiological studies have highlighted a potential association between sugar consumption and PDAC incidence and mortality [59,60,61]. However, it remains inconclusive in part because of the limitations of study design and evaluation tools to appropriately weigh the strength of genetic predisposition- or sugar consumption-induced obesity and metabolic outcomes [58, 60, 61].
Comparing nonmalignant pancreatic samples from PDAC individuals with and without a history of diabetes revealed that pancreases with diabetes sustained significantly more DNA damage and DNA mutation than other organs, suggesting a pancreas-specific effect [10]. Acinar cells or nontumorigenic pancreatic cells in long-term high glucose culture increased the average KRAS mutation rate, although to a small extent, supporting the selective advantage of glucose for KRAS hotspot mutations [10]. The increases in the de novo KRASG12D mutation rate were consistent regardless of the different measurement tools, including immunofluorescence staining and flow cytometry followed by sequencing. The lack of a significant increase in pyruvate/lactate production under high glucose in nontumorigenic pancreatic cells under aerobic conditions supported that metabolic reprogramming is not energy-consuming [62]. The increase in unscheduled glycolysis may lead to DNA damage at least partially by reducing the dNTP pool by O-GlcNAcylation of ribonucleotide reductase catalytic subunit M1(RRM1), which may subsequently cause adaptive DNA mutations to maintain cell survival [10].
In addition, since KRAS mutation alone is insufficient to generate PDAC [49, 50], specific pancreatic cells containing KRAS mutations may require additional alterations to exhibit subtle phenotypic changes at the beginning. It remains to be explored whether metabolic factors in the glucose pathway and microenvironmental nutrients may play key roles in this regard.
Glycosylation and KRAS mutation
O-linked-N-acetylglucosaminylation (O-GlcNAcylation) is a unique nutrient- and stress-responsive glycosylation that involves the addition of N-acetylglucosamine (GlcNAc) moieties from UDP-GlcNAc to serine and threonine residues of proteins. The addition and removal of O-GlcNAc rely on O-GlcNAc transferase (OGT), which adds UDP-GlcNAc to target proteins, and O-GlcNAcase (OGA), which removes O-GlcNAc from O-GlcNAcylated proteins [63]. The generation of UDP-GlcNAc is not only for O-GlcNAcylation but also for O-glycosylation and N-glycosylation of proteins to maintain cellular survival under stress [63]. As a nutrient sensor, HBP is essential for amino sugar biosynthesis by integrating core metabolic intermediates from glycolysis, fatty acids, amino acids, and nucleotide metabolism [64, 65]. However, either cancer cells under glucose deprivation, hypoxia, or oncogenic KRAS mutation [8] or nontumorigenic pancreatic cells under high glucose levels [10] cause hyper-O-GlcNAcylation of cellular proteins, challenging the regulation of O-GlcNAcylation via the versatile UDP-GlcNAc.
Metabolic reprogramming may be dynamic, unsynchronized, and coordinated with neighboring cells in an organ in response to extracellular and intracellular stresses. Several O-GlcNAcylation targets related to PDAC metabolism and progression, such as notch receptor 1 promoting cancer development [66], SRY-box transcription factor 2 regulating self-renewal [67], sirtuin 7 (SIRT7) triggering cancer progression by blocking the SIRT7-proteasome activator subunit 3 (PAME3) interaction [68], and nuclear factor kappa B modulating cancer-associated inflammation [69], have been reported. Although hyper-O-GlcNAcylation of insulin receptor substrate 1 and AKT serine/threonine kinase 2 inhibits their phosphorylation and induces insulin resistance in peripheral cells [70], the corresponding regulation of endocrine and exocrine secretion from the pancreas remains unclear. Protein O-GlcNAcylation in response to high glucose-driven metabolic reprogramming may potentially have multiple targets beyond PFK1 and RRM1 in nontumorigenic pancreatic cells [10]. The precise control and crucial targets of O-GlcNAcylation and how O-GlcNAcylation helps adapt and maintain homeostasis in a specific organ under stress remain to be explored. The safe use of glucosamines, the UDP-GlcNAc precursor, as a dietary supplement for osteoarthritis under variable O-GlcNAcylation due to UDP-GlcNAc imbalance or OGT/OGA dysregulation has to be reconsidered [62, 71].
Thus, unlike glycated proteins such as carbohydrate antigen 19-9 (CA19-9) and hemoglobin A1c (Hb A1c) with mutually correlated levels [72], glycosylated proteins that help cancer cells adapt to stress and malignant phenotypes could serve as potential diagnostic and therapeutic targets [63, 71].
Potential factors regulating HBP in a high glucose status
HBP is essential for versatile UDP-GlcNAc synthesis. Regulations of enzymes critical for HBP and controlling high glucose-induced protein O-GlcNAcylation should be discussed. PFK1, a 340 kDa heterotetrameric allosteric enzyme composed of PFKL, PFKM, and PFKP, works with a concerted symmetric transition from an enzymatically inactive T-state to the active R-state and can be dissociated into inactive dimers and monomers [73]. Increased PFK activity may help pancreatic cells maintain their survival advantage during adaptation to the poor oxygen and nutrient supply microenvironment [18]. In contrast, high glucose-induced O-GlcNAcylation inactivates PFK1 and RRM1 and subsequently inhibits glycolysis and dNTP generation, respectively, resulting in high-frequency DNA damage, specifically in pancreatic cells [10]. Modulation of PFK activity during adaptation or hyperproliferation may result from different posttranslational modifications and/or subunit assembly, which warrants further investigation.
Another crucial target for rewiring glycolysis in the HBP is GFAT. GFAT, the first and rate-limiting enzyme of HBP, catalyzes glucosamine 6-phosphate by integrating glucose and glutamine metabolism. High levels of GFAT predict a poor prognosis in patients with PDAC [74]. GFAT1 depletion diminished high glucose-induced DNA damage and colony formation in pancreatic cells [10]. GFAT2 upregulation positively correlates with hyaluronan synthesis and epithelial-to-mesenchymal transition [75]. Overexpression of both GFAT1 and GFAT2 is associated with poor survival of PDAC (TGCA dataset, unpublished results). The bidirectional PFK2 isozymes (PFKFB1, PFKFB2, PFKFB3, and PFKFB4) and TP53-induced glycolysis and apoptosis regulator (TIGAR), a fructose 2,6 bisphosphatase that shapes the metabolic profile of cells by modulating HBP, have yet to be explored. Understanding how to balance PFK and GFAT enzymatic activities during adaptation and hyperproliferation may provide new insights into the modulation of HBP (Figs. 2, 3).

Approximately 2–5% of cellular glucose enters the HBP to generate the end product UDP-GlcNAc. Glutamine:fructose-6-phosphate amidotransferase (GFAT) is the rate-limiting enzyme for the HBP. O-GlcNAc transferase (OGT) adds UDP-GlcNAc to target protein serine and threonine residues, and O-GlcNAcase (OGA) removes O-GlcNAc from O-GlcNAcylated proteins. The balance between the enzyme activities of phosphofructokinase (PFK) and GFAT through regulating GFAT may be crucial to direct the pathways. G6P glucose 6-phosphate, F6P fructose 6-phosphate, FBP fructose 1,6-phosphate, GlcN6P glucosamine-6-phosphate, GlcNAc6P N-acetylglucosamine-6-phosphate, GlcNAc1P N-acetylglucosamine-1-phosphate, GAT acetyl-CoA:D-glucosamine-6-phosphate N-acetyltransferase, AGM phosphor-N-acetylglucosamine mutase, AGX1 UDP-GlcNAc pyrophosphorylase, GNK GlcNAc kinase. The graph was created with BioRender.com.

A During resting, approximately 2–5% of cellular glucose enters the hexosamine biosynthesis pathway (HBP) to generate the end product UDP-GlcNAc. B During adaptation, high glucose induces genome instability through O-linked-N-acetylglucosaminylation (O-GlcNAcylation) of phosphofructokinase 1 (PFK1) and ribonucleotide reductase catalytic subunit M1 (RRM1) to direct glycolysis and the pentose phosphate pathway (PPP) into the HBP. C During hyperproliferation, such as in pancreatic cancer cells, both high glucose and high glutamine are required through overactivation/overexpression of PFK1, shunting glycolysis into the PPP and HBP to generate energy and biosynthesis precursors to meet the needs of cancer cells. G6P glucose 6-phosphate, F6P fructose 6-phosphate, FBP fructose 1,6-phosphate, GFAT glutamine:fructose-6-phosphate amidotransferase, R5P ribose 5-phosphate, PRPP phosphoribosyl pyrophosphate, NDP nucleoside diphosphate, dNDP deoxynucleoside diphosphates, GlcN6P glucosamine-6-phosphate, UDP-GlcNAc uridine diphosphate-N-acetylglucosamine.
Since epigenetics plays a key role in regulating gene expression in different tissues and cell types [76], its contribution to the causal relationship of metabolite preference toward lower PFK activity in pancreatic cells is of great interest. Furthermore, ER stress may trigger HBP overactivation. The unfolded protein response (UPR)-HBP axis is partially triggered via GFAT1, a direct transcriptional target of a spliced form of X-box binding protein 1 (XBP1s), to protect cells under stress [77]. XBP1s also promotes pro-survival signaling during acinar cell differentiation [78]. In addition, ER stress sensors such as glutathione peroxidase 7 relieve ER oxidative stress by promoting 78 kDa glucose-regulated protein chaperone activity [79], linking the stress sensor to HBP activity.
Glucosamine-phosphate N-acetyltransferase 1 (GNPNAT1) is a key enzyme associated with glucose and fatty acid catabolism and UDP-GlcNAc biosynthesis. Although GNPNAT1 overexpression is associated with poor survival of patients with PDAC, the mechanisms underlying the link remain to be explored (TGCA dataset, unpublished results). Overexpression of phosphoacetylglucosamine mutase (PGM3, a phospho-N-acetylglucosamine mutase) has been linked to gemcitabine resistance. The PGM3 inhibitor FR054 synergizes with gemcitabine to suppress PDAC growth by promoting the UPR and inhibiting EGFR-AKT signaling [80]. The expression of UDP-N-acetylglucosamine phosphorylase (UAP1), GlcNAc kinase (GNK), OGT, or OGA was not associated with poor survival of PDAC (TGCA dataset, unpublished results). These results suggest that multiple intertwined factors may connect the regulation of HBP pathway.
Nutrient sharing and competition: how KRAS-mutated cells prevail in competition
Stress from the pancreatic cell microenvironment
The TME of PDAC is composed mainly of stroma with primary fibroblasts and immune cells [31, 81] and is a physical and oxidative stress source. Fibroblast-induced cell dysfunction may occur through extracellular matrix (ECM)-induced physical destruction, resulting in pancreatic fibrosis [82] and PDAC progression [83]. Those tissue injuries lead fibroblasts to produce extensive ECM to increase interstitial stresses [84]. The stresses in PDAC may exceed ten times of those observed in a normal pancreas [85, 86]. Although stromal components create a metabolic niche for cancer cells to maintain tumor survival, they might also restrain cancer progression [87,88,89,90].
Oxidative stress from TME can be resulted from nutrient imbalances. Consistent with this notion, metabolic imbalance-induced genomic instability and DNA damage may involve oxidative stress and DDR inefficiency. Using nucleotide supplements to reverse high glucose-induced DNA damage supports this potential [10, 62]. Interestingly, similar example is that BRCA2 DNA repair-associated (BRCA2)-deficient cells also experience endogenous oxidative stress-blocked mtDNA replication and stability, which could be ameliorated by the ROS scavenger N-acetylcysteine [48]. The redox shuttle enables NADPH transfer from the cytosol to the mitochondria to support cellular homeostasis. However, a severely inefficient DDR from the challenge of metabolite imbalance may lead to cell death. BCL-2 family proteins control cell death or differentiation primarily through the irreversible release of intermembrane space proteins, caspase activation, and apoptosis through direct interaction-regulated mitochondrial outer membrane permeabilization (MOMP). Aberrant oxidative stress alters the affinities and relative abundance of BCL-2 family proteins, affecting BCL-2 family protein interactions [46]. However, no consistent trends in ROS levels have been detected between two nontumorigenic pancreatic cell lines upon high glucose-induced DNA damage [10]. Other cellular antioxidants, including superoxide dismutases, glutathione S-transferases, glutathione peroxidases, and periaxin, may also protect against ROS-induced cell death, partly through nuclear factor erythroid 2–related factor 2 [91]. Personalized nutrient guidelines for restraining specific tumor growth will be an interesting subject to explore.
PDAC associated microbiome: an enigma
The microbiota may be another risk factor for PDAC. Differences in oral, gut, and pancreatic microbiota have been reported among patients with PDAC. Oral microbiota dysbiosis may contribute to PDAC pathogenesis [92]. PDAC pathogenesis has been reported to link to an increased abundance of periodontal disease-associated Porphyromonas gingivalis and Fusobacterium sp. but reduced abundances of Neisseria elongata and Streptococcus mitis [93]. High levels of plasma antibodies against P. gingivalis have been correlated with a reduced risk of PDAC [94].
The interaction between microbiota and host, nutrient/drug efficacy, and toxicity directly alter or indirectly modify host physiology and pharmacodynamics. Current gut microbiome-cancer associations are witnessed in bacteria causing tumor progression and bacteria modulating antitumor immune responses [95, 96], which are partially dependent on cometabolites derived from the host and microbiota, such as short-chain fatty acids, bile acids, and indole derivatives, which are greatly influenced by nutrients [97]. For gut dysbiosis, an increased abundance of Bacteroidetes but a reduced abundance of Firmicutes in patients with PDAC have been found [98]. A lower α-diversity in the gut microbiome in patients with PDAC was detected. Increased abundance of Veillonella, Klebsiella, and Selenomonas species and lipopolysaccharide-producing bacteria but decreased abundance of Bifidobacterium species and butyrate-producing bacteria have been reported [99]. The association between Helicobacter pylori, a gastric pathogen that colonizes ~50% of individuals worldwide, and PDAC pathogenesis has been noted [100] but remains controversial.
The human tumor microbiome includes tumor type-specific intratumoral bacteria [101,102,103]. The α-diversity of the tumor microbiome was higher among individuals with increased long-term survival. An intratumoral microbiome signature (Pseudoxanthomonas-Streptomyces-Saccharopolyspora-Bacillus clausii) was associated with long-term patient survival [102]. Altered gut microbiome composition and changes in host processing of bacteria-derived metabolites may imply the link between diabetes and PDAC initiation and progression with geographical, racial, dietary, and lifestyle-related differences [104].
KRAS-mutated cells prevail in competition under high-fat-induced inflammation
Not all pancreatic cells develop PanIN in genetically engineered mice with the KrasG12D mutation, suggesting that additional factors are needed for the initial transformation. Figure 4 illustrates the current understanding of high glucose-triggered cancer initiation, which may be further promoted to become PDAC. Mutant cells are often recognized and passively eliminated from epithelial tissues through epithelial defense against cancer [105]. Kras-mutant cells drive metabolic reprogramming that enables them to rapidly grow and outcompete their adjacent regular counterparts to coexist with a large proportion of healthy cells in the preexisting general population [106]. High-fat diet feeding-induced inflammation promotes the coexistence of Kras-mutant cells with epithelia [107,108,109]. KrasG12D maintains an irreversible ADM through MAPK constitutive signaling to protect against inflammation-induced tissue damage [110]. The evidence provides certain clues for how KRAS-mutated cancer cells may succeed in a nutrient competition.

In response to DNA damage, cells may activate cell cycle checkpoints, process epigenetic and transcriptional programs or DNA repair (with epithelial memory), or undergo apoptosis when the damage is severe. Cells with adapted DNA mutation (e.g., oncogenic KRAS mutation) survived apoptosis and extrusion, with histologic and genetic progression, may transform into cancer cells and further grow as pancreatic ductal adenocarcinoma (PDAC) in adaptive to exocrine microenvironment-induced genome instability. Histologically, PDAC stems from microscopic abnormal acinar-to-ductal metaplasia (ADM) and pancreatic intraepithelial neoplasias (PanINs) of grades I–III to PDAC with increasing disorganization and nuclear abnormalities. High percentages of patients with PDAC carry somatic mutations, including KRAS proto-oncogene, GTPase (KRAS; ~95%) cyclin-dependent kinase inhibitor 2A (CDKN2A; ~90%), tumor protein 53 (TP53; ~70%), and SMAD family member 4 (SMAD4; ~55%). The pancreatic exocrine microenvironment is composed of exocrine cells at different stages, fibroblasts, immune cells, and microbes. The role of these microenvironmental components in adaptation, cancer initiation and progression remains to be elucidated. The graph was created with BioRender.com.
Functional genomics to uncover the metabolic dependence of PDAC
Studies on the in vivo metabolic dependence of PDAC have been challenging due to the complexity and heterogeneity of the TME. Although there are limitations regarding intercellular nutrient sharing, in vivo metabolism-focused CRISPR screening in PDAC is expected to determine the essential global metabolic dependence for PDAC tumor growth [111, 112]. Current findings revealed that tumor growth depends on the crosstalk between the immune system and heme biosynthesis [111]. Genetic or pharmacological inhibition of farnesyl diphosphate farnesyl transferase delayed tumor growth and promoted CD8+ T-cell infiltration through PI3K/AKT signaling, indicating the potential targeting of cholesterol biosynthesis and autophagy to combat PDAC [112]. A new platform for characterizing metabolic dependencies under distinct genetic drivers or different PDAC statuses, such as initiation and metastasis, will provide a breakthrough for the field.
BOX1: Complexity of genetic alterations and metabolism in PDAC
In addition to the most frequent oncogenic somatic mutation KRAS, a high percentage of patients carry inactivating somatic mutations in the tumor suppressors cyclin-dependent kinase inhibitor 2 A (CDKN2A), tumor protein 53 (TP53), and SMAD family member 4 (SMAD4) [113,114,115], reinforcing their roles during PanIN-to-PDAC [116]. Other mutations, such as those in Gα protein subunits (such as GNAS), MYC, TP53, and PTEN, drive metabolic shifts in PDAC. GNAS-activating mutations increase lipid utilization and fatty acid oxidation to promote PDAC tumor progression [117]. MYC orchestrates metabolic reprogramming of PDAC progression through extrinsic and intrinsic factors via its natural role as a transcription factor [118,119,120,121]. TP53 controls cellular metabolism by directly modulating different transcriptional programs, such as the autophagy network [122], or redox control through the p53 target TIGAR [123, 124]. Restoration of wild-type p53 induces α-ketoglutarate accumulation to increase chromatin accessibility and tumor suppression [125]. Loss of the tumor suppressor PTEN hyperactivates phosphoinositide-3-kinase (PI3K)–AKT signaling and metabolic processes such as glucose metabolism, de novo lipid synthesis, and redox balance [126] and cooperates with mutant KRAS-driven events in multiple PDAC models [127,128,129,130].
There is still much room for metabolic efforts for early diagnosis and prevention. Approximately 3–10% of patients with PDAC carry inherited germline mutations in genes such as BRCA1 DNA repair-associated (BRCA1), BRCA2, and ATM serine/threonine kinase [131]. The direct functional link of BRCA1 and BRCA2 to the DNA damage response was first demonstrated [132, 133]. The discovery of more sensitive poly(ADP-ribose) polymerase (PARP) inhibition in BRCA-mutant cancer cells has led to the development of new biomarker-driven synthetic lethal treatment strategies for different cancers [134, 135]. However, in a mouse model of PDAC initiation, loss of heterozygosity (LOH) of BRCA2 while promoting chromosomal instability may not be essential for PDAC initiation [136]. Intriguingly, even in the presence of KRAS oncogenic mutations, BRCA2 LOH inhibits tumor formation when wild-type TP53 remains. BRCA2 LOH can accelerate PDAC tumorigenesis only after TP53 is mutated [137, 138]. PDAC metabolism shifts in response to the BRCAness phenotype remain to be elucidated. These findings postulated that the selected population sharing the BRCAness phenotype would be more sensitive to DNA damaging agents and DDR inhibitors. The appropriate subtype, therapeutic window, potential combination strategies, and functional differences of specific variants in the DDR pathway remain defined.
Approximately 5% of patients with PDAC carry RB mutations or deletions. RB was cloned and sequenced in the 1980s [139] and has been well characterized as a tumor suppressor for inhibiting the G0/G1 to S phase transition during cell cycle progression [140]. RB silences gene transcription by recruiting corepressor complexes, including histone deacetylases, to E2F transcription factors specifically targeting gene promotors [141]. Loss of RB inhibits glucose oxidation by directly inducing the expression of a glucose homeostasis sensor and modulator pyruvate dehydrogenase kinase 4 [141], promotes glutamine uptake via increased expression of the glutamine transporter and GLS1, perturbing redox homeostasis by reducing GSH levels [142]. However, the role and functional alteration of the loss of RB in PDAC development remain to be characterized.
Concluding remarks
PDAC remains a difficult-to-treat cancer. Despite state-of-the-art comprehensive detection approaches, such as ultrasound, computed tomography scans, magnetic resonance imaging, and positron emission tomography scans, as well as treatment approaches such as surgery, radiation, chemotherapy, and immunotherapy, overall survival has not improved over the past several decades. Knowledge about metabolic dysregulation promoted by the RAS protein family, particularly mutant KRAS, has advanced substantially. Oncogenic KRAS mutations and their downstream reprogrammed metabolic pathways have been attractive therapeutic targets. Since metabolites such as glucose may trigger DNA damage and mutation through glucose metabolic reprogramming, a new niche for PDAC management has been emerged from these findings for early detection, prevention and treatment.
Data availability
There are no experimental datasets given that this is a review article prepared based on the literature review. All the data supporting the findings of this review are available from the corresponding author upon reasonable request.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Rahib L, Wehner MR, Matrisian LM, Nead KT. Estimated projection of US cancer incidence and death to 2040. JAMA Netw Open. 2021;4:e214708.
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72:7–33.
Singhi AD, Wood LD. Early detection of pancreatic cancer using DNA-based molecular approaches. Nat Rev Gastroenterol Hepatol. 2021;18:457–68.
Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014;39:91–100.
Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Disco. 2011;10:671–84.
Hanahan D. Hallmarks of cancer: new dimensions. Cancer Disco. 2022;12:31–46.
Kenner BJ, Abrams ND, Chari ST, Field BF, Goldberg AE, Hoos WA, et al. Early detection of pancreatic cancer: applying artificial intelligence to electronic health records. Pancreas 2021;50:916–22.
Chung KM, Singh J, Lawres L, Dorans KJ, Garcia C, Burkhardt DB, et al. Endocrine-exocrine signaling drives obesity-associated pancreatic ductal adenocarcinoma. Cell 2020;181:832.
Hu CM, Tien SC, Hsieh PK, Jeng YM, Chang MC, Chang YT, et al. High glucose triggers nucleotide imbalance through O-GlcNAcylation of key enzymes and induces KRAS mutation in pancreatic cells. Cell Metab. 2019;29:1334–49.e1310.
Aggarwal G, Kamada P, Chari ST. Prevalence of diabetes mellitus in pancreatic cancer compared to common cancers. Pancreas 2013;42:198–201.
Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 2009;136:823–37.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005;434:913–7.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434:917–21.
Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012;491:399–405.
Encarnacion-Rosado J, Kimmelman AC. Harnessing metabolic dependencies in pancreatic cancers. Nat Rev Gastroenterol Hepatol. 2021;18:482–92.
Yun JY, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009;325:1555–9.
Ying HQ, Kimmelman AC, Lyssiotis CA, Hua SJ, Chu GC, Fletcher-Sananikone E, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012;149:656–70.
Papke B, Der CJ. Drugging RAS: know the enemy. Science 2017;355:1158–63.
Raho S, Capobianco L, Malivindi R, Vozza A, Piazzolla C, De Leonardis F, et al. KRAS-regulated glutamine metabolism requires UCP2-mediated aspartate transport to support pancreatic cancer growth. Nat Metab. 2020;2:1373–81.
Baksh SC, Todorova PK, Gur-Cohen S, Hurwitz B, Ge Y, Novak JSS, et al. Extracellular serine controls epidermal stem cell fate and tumour initiation. Nat Cell Biol. 2020;22:779–90.
Baksh SC, Todorova PK, Gur-Cohen S, Hurwitz B, Ge Y, Novak JSS, et al. Author Correction: Extracellular serine controls epidermal stem cell fate and tumour initiation. Nat Cell Biol. 2020;22:1396.
Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 2012;336:1040–4.
Zhu Z, Achreja A, Meurs N, Animasahun O, Owen S, Mittal A, et al. Tumour-reprogrammed stromal BCAT1 fuels branched-chain ketoacid dependency in stromal-rich PDAC tumours. Nat Metab. 2020;2:775–92.
Falcone M, Maddocks ODK. The KRAS-BCAA-BCAT2 axis in PDAC development. Nat Cell Biol. 2020;22:139–40.
Rozeveld CN, Johnson KM, Zhang LZ, Razidlo GL. KRAS controls pancreatic cancer cell lipid metabolism and invasive potential through the lipase HSL. Cancer Res. 2020;80:4932–45.
Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013;497:633–7.
Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531:47–52.
Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu SD, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17:500–U140.
Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. 2015;47:1168–78.
Ho WJ, Jaffee EM, Zheng L. The tumour microenvironment in pancreatic cancer—clinical challenges and opportunities. Nat Rev Clin Oncol. 2020;17:527–40.
Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–29.
Kapoor A, Yao WT, Ying HQ, Hua SJ, Liewen A, Wang QY, et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 2014;158:185–97.
Collins MA, Bednar F, Zhang YQ, Brisset JC, Galban S, Galban CJ, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Investig. 2012;122:639–53.
Rajbhandari N, Lin WC, Wehde BL, Triplett AA, Wagner KU. Autocrine IGF1 signaling mediates pancreatic tumor cell dormancy in the absence of oncogenic drivers. Cell Rep. 2017;18:2243–55.
Chen PY, Muzumdar MD, Dorans KJ, Robbins R, Bhutkar A, Del Rosario A, et al. Adaptive and reversible resistance to Kras inhibition in pancreatic cancer cells. Cancer Res. 2018;78:985–1002.
Muzumdar MD, Dorans KJ, Chung KM, Robbins R, Tammela T, Gocheva V, et al. Clonal dynamics following p53 loss of heterozygosity in Kras-driven cancers. Nat Commun. 2016;7:12685.
Kerk SA, Papagiannakopoulos T, Shah YM, Lyssiotis CA. Metabolic networks in mutant KRAS-driven tumours: tissue specificities and the microenvironment. Nat Rev Cancer. 2021;21:510–25.
Chu CK, Mazo AE, Goodman M, Egnatashvili V, Sarmiento JM, Staley CA, et al. Preoperative diabetes mellitus and long-term survival after resection of pancreatic adenocarcinoma. Ann Surg Oncol. 2010;17:502–13.
Coughlin SS, Calle EE, Teras LR, Petrelli J, Thun MJ. Diabetes mellitus as a predictor of cancer mortality in a large cohort of US adults. Am J Epidemiol. 2004;159:1160–7.
Carstensen B, Read SH, Friis S, Sund R, Keskimaki I, Svensson AM, et al. Cancer incidence in persons with type 1 diabetes: a five-country study of 9,000 cancers in type 1 diabetic individuals. Diabetologia 2016;59:980–8.
Kim NH, Chang Y, Lee SR, Ryu S, Kim HJ. Glycemic status, insulin resistance, and risk of pancreatic cancer mortality in individuals with and without diabetes. Am J Gastroenterol. 2020;115:1840–8.
Wolpin BM, Bao Y, Qian ZR, Wu C, Kraft P, Ogino S, et al. Hyperglycemia, insulin resistance, impaired pancreatic beta-cell function, and risk of pancreatic cancer. J Natl Cancer Inst. 2013;105:1027–35.
Sharma A, Kandlakunta H, Nagpal SJS, Feng Z, Hoos W, Petersen GM, et al. Model to determine risk of pancreatic cancer in patients with new-onset diabetes. Gastroenterology 2018;155:730–9.e733.
Pannala R, Leirness JB, Bamlet WR, Basu A, Petersen GM, Chari ST. Prevalence and clinical profile of pancreatic cancer-associated diabetes mellitus. Gastroenterology 2008;134:981–7.
Everhart J, Wright D. Diabetes mellitus as a risk factor for pancreatic cancer. A meta-analysis. JAMA. 1995;273:1605–9.
Chow WH, Gridley G, Nyren O, Linet MS, Ekbom A, Fraumeni JF Jr., et al. Risk of pancreatic cancer following diabetes mellitus: a nationwide cohort study in Sweden. J Natl Cancer Inst. 1995;87:930–1.
Ben QW, Xu MJ, Ning XY, Liu J, Hong SY, Huang W, et al. Diabetes mellitus and risk of pancreatic cancer: a meta-analysis of cohort studies. Eur J Cancer. 2011;47:1928–37.
Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112–26.
Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–50.
Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017;551:115–8.
Rabinowitz JD, Enerback S. Lactate: the ugly duckling of energy metabolism. Nat Metab. 2020;2:566–71.
Wellen KE, Thompson CB. Cellular metabolic stress: considering how cells respond to nutrient excess. Mol Cell. 2010;40:323–32.
Zhu L, Ji JT, Ma JJ, Wang D, Liu MY, Du JX, et al. Differential effects of dietary macronutrients on the development of oncogenic KRAS-mediated pancreatic ductal adenocarcinoma. Cancers 2022;14:2723.
Lien EC, Westermark AM, Zhang Y, Yuan C, Li Z, Lau AN, et al. Low glycaemic diets alter lipid metabolism to influence tumour growth. Nature 2021;599:302–7.
Bhupathiraju SN, Tobias DK, Malik VS, Pan A, Hruby A, Manson JE, et al. Glycemic index, glycemic load, and risk of type 2 diabetes: results from 3 large US cohorts and an updated meta-analysis. Am J Clin Nutr. 2014;100:218–32.
Livesey G, Taylor R, Livesey HF, Buyken AE, Jenkins DJA, Augustin LSA, et al. Dietary glycemic index and load and the risk of Type 2 diabetes: a systematic review and updated meta-analyses of prospective cohort studies. Nutrients 2019;11:1280.
Malik VS, Hu FB. The role of sugar-sweetened beverages in the global epidemics of obesity and chronic diseases. Nat Rev Endocrinol. 2022;18:205–18.
Genkinger JM, Li R, Spiegelman D, Anderson KE, Albanes D, Bergkvist L, et al. Coffee, tea, and sugar-sweetened carbonated soft drink intake and pancreatic cancer risk: a pooled analysis of 14 cohort studies. Cancer Epidemiol Biomark Prev. 2012;21:305–18.
Schernhammer ES, Hu FB, Giovannucci E, Michaud DS, Colditz GA, Stampfer MJ, et al. Sugar-sweetened soft drink consumption and risk of pancreatic cancer in two prospective cohorts. Cancer Epidemiol Biomark Prev. 2005;14:2098–105.
Navarrete-Munoz EM, Wark PA, Romaguera D, Bhoo-Pathy N, Michaud D, Molina-Montes E, et al. Sweet-beverage consumption and risk of pancreatic cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC). Am J Clin Nutr. 2016;104:760–8.
Hsu YS, Wu PJ, Jeng YM, Hu CM, Lee WH. Differential effects of glucose and N-acetylglucosamine on genome instability. Am J Cancer Res. 2022;12:1556–76.
Slawson C, Hart GW. O-GlcNAc signalling: implications for cancer cell biology. Nat Rev Cancer. 2011;11:678–84.
Denzel MS, Antebi A. Hexosamine pathway and (ER) protein quality control. Curr Opin Cell Biol. 2015;33:14–18.
Denzel MS, Storm NJ, Gutschmidt A, Baddi R, Hinze Y, Jarosch E, et al. Hexosamine pathway metabolites enhance protein quality control and prolong life. Cell 2014;156:1167–78.
Yang C, Hu JF, Zhan Q, Wang ZW, Li G, Pan JJ, et al. SHCBP1 interacting with EOGT enhances O-GlcNAcylation of NOTCH1 and promotes the development of pancreatic cancer. Genomics 2021;113:827–42.
Sharma NS, Gupta VK, Dauer P, Kesh K, Hadad R, Giri B, et al. O-GlcNAc modification of Sox2 regulates self-renewal in pancreatic cancer by promoting its stability. Theranostics 2019;9:3410–24.
He X, Li Y, Chen Q, Zheng L, Lou J, Lin C, et al. O-GlcNAcylation and stablization of SIRT7 promote pancreatic cancer progression by blocking the SIRT7-REGgamma interaction. Cell Death Differ. 2022. https://doi.org/10.1038/s41418-022-00984-3.
Ma Z, Vocadlo DJ, Vosseller K. Hyper-O-GlcNAcylation is anti-apoptotic and maintains constitutive NF-kappaB activity in pancreatic cancer cells. J Biol Chem. 2013;288:15121–30.
Yang X, Ongusaha PP, Miles PD, Havstad JC, Zhang F, So WV, et al. Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature 2008;451:964–9.
Thomas D, Rathinavel AK, Radhakrishnan P. Altered glycosylation in cancer: a promising target for biomarkers and therapeutics. Biochim Biophys Acta Rev Cancer. 2021;1875:188464.
Noy A, Bilezikian JP. Clinical review-63—diabetes and pancreatic-cancer—clues to the early diagnosis of pancreatic malignancy. J Clin Endocr Metab. 1994;79:1223–31.
Uyeda K. Phosphofructokinase. Adv Enzymol Relat Areas Mol Biol. 1979;48:193–244.
Yang C, Peng P, Li L, Shao M, Zhao J, Wang L, et al. High expression of GFAT1 predicts poor prognosis in patients with pancreatic cancer. Sci Rep. 2016;6:39044.
Oikari S, Makkonen K, Deen AJ, Tyni I, Karna R, Tammi RH, et al. Hexosamine biosynthesis in keratinocytes: roles of GFAT and GNPDA enzymes in the maintenance of UDP-GlcNAc content and hyaluronan synthesis. Glycobiology 2016;26:710–22.
Lokk K, Modhukur V, Rajashekar B, Martens K, Magi R, Kolde R, et al. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns. Genome Biol. 2014;15:r54.
Wang ZV, Deng Y, Gao N, Pedrozo Z, Li DL, Morales CR, et al. Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell 2014;156:1179–92.
Hess DA, Humphrey SE, Ishibashi J, Damsz B, Lee AH, Glimcher LH, et al. Extensive pancreas regeneration following acinar-specific disruption of Xbp1 in mice. Gastroenterology 2011;141:1463–72.
Wei PC, Hsieh YH, Su MI, Jiang X, Hsu PH, Lo WT, et al. Loss of the oxidative stress sensor NPGPx compromises GRP78 chaperone activity and induces systemic disease. Mol Cell. 2012;48:747–59.
Kim EJ, Bond MR, Love DC, Hanover JA. Chemical tools to explore nutrient-driven O-GlcNAc cycling. Crit Rev Biochem Mol Biol. 2014;49:327–42.
Wang Y, Liang Y, Xu H, Zhang X, Mao T, Cui J, et al. Single-cell analysis of pancreatic ductal adenocarcinoma identifies a novel fibroblast subtype associated with poor prognosis but better immunotherapy response. Cell Discov. 2021;7:36.
Omary MB, Lugea A, Lowe AW, Pandol SJ. The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest. 2007;117:50–59.
Apte M, Pirola RC, Wilson JS. Pancreatic stellate cell: physiologic role, role in fibrosis and cancer. Curr Opin Gastroenterol. 2015;31:416–23.
Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 2020;20:174–86.
Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–29.
Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015;75:544–53.
Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2015;28:831–3.
Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735–47.
Halbrook CJ, Lyssiotis CA. Employing metabolism to improve the diagnosis and treatment of pancreatic cancer. Cancer Cell. 2017;31:5–19.
Chung KM, Singh J, Lawres L, Dorans KJ, Garcia C, Burkhardt DB, et al. Endocrine-exocrine signaling drives obesity-associated pancreatic ductal adenocarcinoma. Cell 2020;181:832–47.e818.
Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science 2020;368:eaaw5473.
Michaud DS, Joshipura K, Giovannucci E, Fuchs CS. A prospective study of periodontal disease and pancreatic cancer in US male health professionals. J Natl Cancer Inst. 2007;99:171–5.
Jacob JA. Study links periodontal disease bacteria to pancreatic cancer risk. JAMA. 2016;315:2653–4.
Michaud DS, Izard J, Wilhelm-Benartzi CS, You DH, Grote VA, Tjonneland A, et al. Plasma antibodies to oral bacteria and risk of pancreatic cancer in a large European prospective cohort study. Gut 2013;62:1764–70.
Steele CW, Jamieson NB, Evans TR, McKay CJ, Sansom OJ, Morton JP, et al. Exploiting inflammation for therapeutic gain in pancreatic cancer. Br J cancer. 2013;108:997–1003.
Cordon-Cardo C, Prives C. At the crossroads of inflammation and tumorigenesis. J Exp Med. 1999;190:1367–70.
Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010;140:883–99.
Half E, Keren N, Reshef L, Dorfman T, Lachter I, Kluger Y, et al. Fecal microbiome signatures of pancreatic cancer patients. Sci Rep. 2019;9:16801.
Ren Z, Jiang J, Xie H, Li A, Lu H, Xu S, et al. Gut microbial profile analysis by MiSeq sequencing of pancreatic carcinoma patients in China. Oncotarget 2017;8:95176–91.
Guo Y, Liu W, Wu J. Helicobacter pylori infection and pancreatic cancer risk: a meta-analysis. J Cancer Res Ther. 2016;12:C229–C232.
Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020;368:973.
Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W, et al. Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell 2019;178:795–806.e712.
Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017;357:1156–60.
Metwaly A, Reitmeier S, Haller D. Microbiome risk profiles as biomarkers for inflammatory and metabolic disorders. Nature Rev Gastroenterol Hepatol. 2022;19:383–97.
Kajita M, Fujita Y. EDAC: Epithelial defence against cancer-cell competition between normal and transformed epithelial cells in mammals. J Biochem. 2015;158:15–23.
Kon S, Ishibashi K, Katoh H, Kitamoto S, Shirai T, Tanaka S, et al. Cell competition with normal epithelial cells promotes apical extrusion of transformed cells through metabolic changes. Nat Cell Biol. 2017;19:530–41.
Sasaki A, Nagatake T, Egami R, Gu GQ, Takigawa I, Ikeda W, et al. Obesity suppresses cell-competition-mediated apical elimination of RasV12-transformed cells from epithelial tissues. Cell Rep. 2018;23:974–82.
Sato N, Yako Y, Maruyama T, Ishikawa S, Kuromiya K, Tokuoka SM, et al. The COX-2/PGE2 pathway suppresses apical elimination of RasV12-transformed cells from epithelia. Commun Biol. 2020;3:132.
Abt ER, Le TM, Dann AM, Capri JR, Poddar S, Lok V, et al. Reprogramming of nucleotide metabolism by interferon confers dependence on the replication stress response pathway in pancreatic cancer cells. Cell Rep. 2022;38:110236.
Del Poggetto E, Ho IL, Balestrieri C, Yen EY, Zhang SJ, Citron F, et al. Epithelial memory of inflammation limits tissue damage while promoting pancreatic tumorigenesis. Science 2021;373:1326.
Zhu XG, Chudnovskiy A, Baudrier L, Prizer B, Liu Y, Ostendorf BN, et al. Functional genomics in vivo reveal metabolic dependencies of pancreatic cancer cells. Cell Metab. 2021;33:211–21.e216.
Biancur DE, Kapner KS, Yamamoto K, Banh RS, Neggers JE, Sohn ASW, et al. Functional genomics identifies metabolic vulnerabilities in pancreatic cancer. Cell Metab. 2021;33:199–210.e198.
Dong DL, Hart GW. Purification and characterization of an O-GlcNAc selective N-acetyl-beta-D-glucosaminidase from rat spleen cytosol. J Biol Chem. 1994;269:19321–30.
Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501.
Kanda M, Matthaei H, Wu J, Hong SM, Yu J, Borges M, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial Neoplasia. Gastroenterology 2012;142:730–U129.
Ying HQ, Dey P, Yao WT, Kimmelman AC, Draetta GF, Maitra A, et al. Genetics and biology of pancreatic ductal adenocarcinoma. Gene Dev. 2016;30:355–85.
Patra KC, Kato Y, Mizukami Y, Widholz S, Boukhali M, Revenco I, et al. Mutant GNAS drives pancreatic tumourigenesis by inducing PKA-mediated SIK suppression and reprogramming lipid metabolism. Nat Cell Biol. 2018;20:811–22.
Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, metabolism, and cancer. Cancer Disco. 2015;5:1024–39.
Sodir NM, Kortlever RM, Barthet VJA, Campos T, Pellegrinet L, Kupczak S, et al. MYC instructs and maintains pancreatic adenocarcinoma phenotype. Cancer Disco. 2020;10:588–607.
Muthalagu N, Monteverde T, Raffo-Iraolagoitia X, Wiesheu R, Whyte D, Hedley A, et al. Repression of the Type I interferon pathway underlies MYC- and KRAS-dependent evasion of NK and B cells in pancreatic ductal adenocarcinoma. Cancer Disco. 2020;10:872–87.
Bhattacharyya S, Oon C, Kothari A, Horton W, Link J, Sears RC, et al. Acidic fibroblast growth factor underlies microenvironmental regulation of MYC in pancreatic cancer. J Exp Med. 2020;217:e20191805.
Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA, et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013;27:1016–31.
Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009;9:691–700.
Cheung EC, DeNicola GM, Nixon C, Blyth K, Labuschagne CF, Tuveson DA, et al. Dynamic ROS control by TIGAR regulates the initiation and progression of pancreatic cancer. Cancer Cell. 2020;37:168–82.e164.
Morris JPT, Yashinskie JJ, Koche R, Chandwani R, Tian S, Chen CC, et al. alpha-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 2019;573:595–9.
Hoxhaj G, Manning BD. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer. 2020;20:74–88.
Ying H, Elpek KG, Vinjamoori A, Zimmerman SM, Chu GC, Yan H, et al. PTEN is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF-kappaB-cytokine network. Cancer Disco. 2011;1:158–69.
Hill R, Calvopina JH, Kim C, Wang Y, Dawson DW, Donahue TR, et al. PTEN loss accelerates KrasG12D-induced pancreatic cancer development. Cancer Res. 2010;70:7114–24.
Baer R, Cintas C, Dufresne M, Cassant-Sourdy S, Schonhuber N, Planque L, et al. Pancreatic cell plasticity and cancer initiation induced by oncogenic Kras is completely dependent on wild-type PI 3-kinase p110alpha. Genes Dev. 2014;28:2621–35.
Wu CY, Carpenter ES, Takeuchi KK, Halbrook CJ, Peverley LV, Bien H, et al. PI3K regulation of RAC1 is required for KRAS-induced pancreatic tumorigenesis in mice. Gastroenterology 2014;147:1405–16.e1407.
Petersen GM. Familial pancreatic cancer. Semin Oncol. 2016;43:548–53.
Zhong Q, Chen CF, Li S, Chen Y, Wang CC, Xiao J, et al. Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science 1999;285:747–50.
Li S, Ting NS, Zheng L, Chen PL, Ziv Y, Shiloh Y, et al. Functional link of BRCA1 and ataxia telangiectasia gene product in DNA damage response. Nature 2000;406:210–5.
Pilie PG, Tang C, Mills GB, Yap TA. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol. 2019;16:81–104.
Lord CJ, Tutt AN, Ashworth A. Synthetic lethality and cancer therapy: lessons learned from the development of PARP inhibitors. Annu Rev Med. 2015;66:455–70.
Skoulidis F, Cassidy LD, Pisupati V, Jonasson JG, Bjarnason H, Eyfjord JE, et al. Germline Brca2 heterozygosity promotes Kras(G12D) -driven carcinogenesis in a murine model of familial pancreatic cancer. Cancer Cell. 2010;18:499–509.
Rowley M, Ohashi A, Mondal G, Mills L, Yang L, Zhang LZ, et al. Inactivation of Brca2 promotes Trp53-associated but inhibits KrasG12D-dependent pancreatic cancer development in mice. Gastroenterology 2011;140:1303.
Gerdes B, Ramaswamy A, Ziegler A, Lang SA, Kersting M, Baumann R, et al. p16INK4a is a prognostic marker in resected ductal pancreatic cancer: an analysis of p16INK4a, p53, MDM2, an Rb. Ann Surg. 2002;235:51–9.
Lee WH, Bookstein R, Hong F, Young LJ, Shew JY, Lee EY. Human retinoblastoma susceptibility gene: cloning, identification, and sequence. Science 1987;235:1394–9.
DiCiommo D, Gallie BL, Bremner R. Retinoblastoma: the disease, gene and protein provide critical leads to understand cancer. Semin Cancer Biol. 2000;10:255–69.
Hsieh MCF, Das D, Sambandam N, Zhang MQ, Nahle Z. Regulation of the PDK4 isozyme by the Rb-E2F1 complex. J Biol Chem. 2008;283:27410–7.
Reynolds MR, Lane AN, Robertson B, Kemp S, Liu Y, Hill BG, et al. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 2014;33:556–66.
Acknowledgements
This work was supported by funds from the peak project and thematic project and the short-term visiting program for domestic scholars from Academia Sinica, Taiwan, funds from the National Science and Technology Council, Taiwan (104-0210-01-09-02, 105-0210-01-13-01, 106-0210-01-15-02, 107-2320-B-039-032-MY3, and 110-2320-B-039-018), and higher education sprout project by the Ministry of Education, Taiwan via the Drug Development Center of China Medical University from the Featured Areas Research Center Program, and funds from Philips Morris Foundation, USA.
Author information
Authors and Affiliations
Contributions
YHL and WHL wrote the manuscript with input from all authors and were in charge of overall direction and planning. CMH and YSH assisted with literature analyses and comments. All authors discussed the results and provided critical feedback on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Massimiliano Agostini
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, YH., Hu, CM., Hsu, YS. et al. Interplays of glucose metabolism and KRAS mutation in pancreatic ductal adenocarcinoma. Cell Death Dis 13, 817 (2022). https://doi.org/10.1038/s41419-022-05259-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-022-05259-w
This article is cited by
-
Exploring the logic and conducting a comprehensive evaluation of AdipoRon-based adiponectin replacement therapy against hormone-related cancers—a systematic review
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
