
Abstract
Adenoid cystic carcinoma (ACC) is an aggressive salivary gland malignancy with limited treatment options for recurrent or metastatic disease. Due to chemotherapy resistance and lack of targeted therapeutic approaches, current treatment options for the localized disease are limited to surgery and radiation, which fails to prevent locoregional recurrences and distant metastases in over 50% of patients. Approximately 20% of patients with ACC carry NOTCH-activating mutations that are associated with a distinct phenotype, aggressive disease, and poor prognosis. Given the role of NOTCH signaling in regulating tumor cell behavior, NOTCH inhibitors represent an attractive potential therapeutic strategy for this subset of ACC. AL101 (osugacestat) is a potent γ-secretase inhibitor that prevents activation of all four NOTCH receptors. While this investigational new drug has demonstrated antineoplastic activity in several preclinical cancer models and in patients with advanced solid malignancies, we are the first to study the therapeutic benefit of AL101 in ACC. Here, we describe the antitumor activity of AL101 using ACC cell lines, organoids, and patient-derived xenograft models. Specifically, we find that AL101 has potent antitumor effects in in vitro and in vivo models of ACC with activating NOTCH1 mutations and constitutively upregulated NOTCH signaling pathway, providing a strong rationale for evaluation of AL101 in clinical trials for patients with NOTCH-driven relapsed/refractory ACC.
Similar content being viewed by others


Introduction
Adenoid cystic carcinoma (ACC) is a relatively uncommon secretory gland malignancy with a high propensity for perineural invasion, locoregional recurrence, and distant metastasis despite curative-intent treatment. Due to its insidious infiltrative growth pattern, ACC is often advanced by the time of clinical recognition. The therapeutic management for patients with locoregional and recurrent/metastatic ACC is limited to surgery and radiation, as no systemic agent has been found to be effective in improving long-term disease control [1]. The mortality rate of ACC remains high, with over 60% of the patients succumbing to the disease within 15 years of diagnosis. Therefore, new therapeutic approaches for treating ACC are urgently needed.
Like other malignancies, ACC is thought to arise through genetic and epigenetic aberrations that lead to the mis-expression of tumor suppressor genes and oncogenes. Molecular and genetic characterization of ACC has begun to reveal the most common driver mutations in this still incompletely understood cancer [2,3,4,5]. One common driver mutation in ACC consists of chromosomal rearrangements that produce MYB-NFIB (~70% of the tumors) or MYBL1-NFIB fusion genes, which appear to have a central role in the genesis of ACC. While representing useful markers in the diagnosis of ACC, MYB family members, like other transcription factors, remain difficult therapeutic targets, and the prognostic and biologic significance of MYB fusion genes are uncertain [6]. Other driver mutations in ACC involve genes that encode epigenetic regulators (e.g., MLL2, MLL3, EP300, SMARCA2, SMARCC1, CREBBP, and KDM6A), pro-growth factors (e.g., FGFR2, PIK3CA, MYC, KRAS, BRAF), and DNA damage/checkpoint regulators (ATM, CDKN2A, TP53) [2,3,4,5, 7]. Recurrent copy number losses (e.g., of chromosome 1p36) and amplifications (e.g., of chromosome 7p14.1 and 14q11.2) have also been noted [2,3,4,5]. However, the fraction of ACC with mutations in these genes that present opportunities for targeted therapy is small.
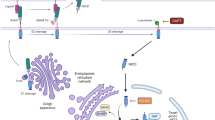
By contrast, recently emerging data suggest that the NOTCH pathway is a tractable, rational therapeutic target in ACC [7, 8]. The NOTCH gene family contains four paralogs encoding large type 1 transmembrane receptor signaling proteins, NOTCH1, 2, 3, and 4. Normal NOTCH signaling is initiated by binding the receptor to a ligand (JAG1, JAG2, DLL1, or DLL4) on an adjacent cell. This event elicits conformational changes in the extracellular domain of NOTCH that makes it susceptible to successive cleavages by ADAM10 and γ-secretase. The latter cleavage releases the NOTCH intracellular domain (NICD), which migrates to the nucleus and forms a transcription complex that upregulates the expression of downstream target genes. These upregulated genes, in turn, regulate diverse cellular functions in a context-specific fashion, many of which have the potential to influence the behavior of cancers through both cell autonomous and non-autonomous mechanisms [9,10,11,12,13].
Particular cancers are marked by the presence of NOTCH-activating mutations, which either disrupt the extracellular NOTCH negative regulatory region (NRR), leading to ligand-independent proteolysis and NICD generation, or a C-terminal PEST domain, leading to stabilization of NICD [9]. Among these cancers is ACC, in which activating NOTCH mutations are found in ~20% of tumors [7, 8, 14, 15]. According to the largest analysis (n = 1045) conducted across several retrospective sequencing datasets [8], these mutations predominantly occur in NOTCH1, although mutations in NOTCH2, NOTCH3, and NOTCH4 have also been identified [2, 5, 7, 8]. A retrospective analysis of three independent cohorts demonstrated that ACCs with NOTCH1 activating mutations are associated with a poorer median overall survival and progress four times faster than those patients without activating mutations [8, 15, 16]. The first retrospective study of 102 ACCs revealed that tumors with NOTCH1 mutations are associated with solid histology, advanced-stage disease at presentation, liver and bone metastases, and shorter relapse-free (12.5 vs 33.9 months) and median overall survival (29.6 vs 121.9 months) compared to tumors with wild-type (WT) NOTCH1 [15]. The second retrospective study in 84 patients with recurrent/metastatic ACC found that median overall survival was significantly shorter in those with NOTCH1-mutated tumors compared to NOTCH1 WT malignancies (55.1 vs 204.5 months) [8]. Furthermore, among NOTCH1 mutant tumors, activating mutations were associated with significantly poorer survival (31.1 vs 73.8 months) [8]. These findings were recently supported in an independent cohort in which activating NOTCH1 mutations were again associated with poorer overall survival (48 vs 195.6 months) [16]. Recurrent mutations in genes encoding proteins that regulate the activity of the NOTCH transcription complex, such as SPEN and FBXW7, have also been identified in ACC genomes, further implicating the NOTCH pathway in ACC tumorigenesis [2, 7, 15].
Based on the proposed multifaceted pro-oncogenic roles of NOTCH signaling in cancer [17,18,19,20], several γ-secretase inhibitors (GSIs) have been developed [15, 21,22,23,24,25,26] and tested in preclinical studies [27,28,29,30,31,32] and Phase I/II trials in patients with advanced solid tumors, including ACC [15, 33], either as a single agent or in combination with targeted therapeutics or chemotherapy [33]. However, the overall response rates to NOTCH-targeted therapy have been suboptimal. Factors limiting success to date include use of GSIs with suboptimal pharmacokinetic (PK)/pharmacodynamic properties, dose-limiting toxicity, and the failure to use biomarkers of NOTCH activation as selection criteria for clinical trial entry [33,34,35,36]. Thus, the potential of NOTCH inhibitors such as GSIs has yet to be fully explored.
AL101 is an investigational small molecule GSI that potently inhibits all four NOTCH paralogs and prevents the upregulation of NOTCH target genes [22]. While a number of recent preclinical studies have shown robust AL101 antitumor activity in several in vivo cancer models [22, 37], a comprehensive evaluation of its effect in ACC is lacking, in part due to the scarcity of experimental model systems for ACC.
Here we demonstrate that AL101 monotherapy inhibits the proliferation of NOTCH-activated ACC organoids at nanomolar concentrations. Furthermore, AL101 is well tolerated in vivo and has significant antitumor effects in ACC patient-derived xenograft (PDX) models with NOTCH1 activating mutations, but not in PDX models with WT NOTCH genes. Taken together, our results provide a strong foundation for the clinical development of AL101 as a targeted monotherapy in patients with NOTCH-activated ACC.
Methods
Cell lines and reagents
The human ACC cell line HACC2A was received from Dr Jacques Nȍr (University of Michigan), the UFH2 cell line was received from Dr Frederic Kaye (University of Florida), and the ACC52 cell line was received from Dr Lurdes Quiemado (University of Oklahoma). Cells were monitored for mycoplasma using the MycoDetect kit (Greiner Bio-One). HACC2A cells were cultured in DMEM medium (Gibco) supplemented with 10% FBS, 200 mM L-glutamine, antibiotic-antimycotic (100X) (Gibco), 400 ng/ml hydrocortisone, 20 ng/ml epidermal growth factor, 5 µg/ml insulin and bovine brain extract (Lonza). UFH2 cells were cultured in DMEM+GlutaMAX medium (Gibco) supplemented with 10% FBS and 5000 U/ml penicillin-streptomycin (Gibco 15070063). ACC52 cells were cultured in RPMI 1640 medium (Corning) supplemented with 10% FBS, antibiotic-antimycotic (100X), 20 ng/ml epidermal growth factor, 400 ng/ml hydrocortisone, and 5 μg/ml insulin. AL101 (batch: 3H66027) was obtained from Ayala Pharmaceuticals. All other chemicals used in this study were purchased from Sigma and prepared according to the manufacturer’s recommendations.
Organoid preparation
Freshly obtained surgical samples were digested first with a mixture of collagenase and dispase, followed by TrypLE (ThermoFisher). After passing through a mesh, cells were embedded in Matrigel (Corning) and cultured in four different organoid media formulations in parallel. These formulations have the same base media containing the growth factors EGF, Noggin (Sigma), R-spondin (Sigma), and FGF10 (ThermoFisher) as well as N-acetylcysteine (ThermoFisher), nicotinamide, Y-27632 (Rho kinase inhibitor), A83-01 (TGF-β signaling inhibitor), N2, and B27 (all from Sigma). Additional components of these media include forskolin (adenylyl cyclase activator), CHIR99021 (Wnt activator), gastrin I, prostaglandin E2, FGF2, hydrocortisone, and heregulin β1 (all from Sigma). The cells that demonstrated the most robust growth were further expanded, and fractions of cells were harvested for histology, DNA and RNA isolation, or cryopreserved at low passage numbers.
IHC staining
Sections (4-micron) prepared from FFPE PDX tumors were stained for NOTCH1 intracellular domain (NICD1) (Cell Signaling, 4147S, 1:200), MYC (Abcam, clone Y69, catalog ab32072; 0.56 μg/ml), or Ki67 (BioCare, clone SP6, catalog CRM326, 1:100) using a Bond III automated immunostainer (Leica). Staining was carried out in the Dana Farber/Harvard Cancer Center Specialized Histopathology Core Laboratory, which is certified by the College of American Pathologists and meets Clinical Laboratory Improvement Amendments standards. Staining was developed using the Bond Polymer Refine Detection Kit (Leica). Slides were counterstained with hematoxylin and reviewed by a board-certified anatomic pathologist (JCA).
Cell viability assays
For cell lines, relative viability was determined using an Alamar Blue assay as outlined by the manufacturer (AbDSerotec). New media containing 1/10 volume of Alamar Blue reagent was added to the wells and cells were incubated at 37 °C for 1 h. Fluorescence (560 nm excitation, 590 nm emission wavelengths) was measured using a SpectraMax-Plus384 fluorometer (Sunnyvale). Percent viability was determined by comparing DMSO treatment to inhibitor treatment. For organoids, ATP measurements (CellTiter-Glo Luminescent Cell Viability Assays, Promega) were used to assess proliferation and viability. Medium was discarded and CellTiter-Glo 3D reagent was added to each well. After incubating 30 min at room temperature on a rotary shaker, bioluminescence activity was assessed using a plate luminometer.
Reverse transcription and real-time PCR
RNA was reverse transcribed to cDNA using Superscript III (Invitrogen) and then used as a template for real-time PCR. Gene amplification was carried out on a StepOnePlus system (Applied Biosystems) using TaqMan Gene Expression Assays (Applied Biosystems). Assay IDs were: ACTB-Hs01060665_g1, HEY1-Hs00232618_m1. HEY2-Hs01012057_m1, NRARP-Hs04183811_s1, MYC-Hs00153408_m1, and HES5-Hs01387463_g1. All reactions were performed in triplicate and relative RNA quantity was calculated after normalizing to ACTB expression by the 2−ΔΔCT method.
Xenograft models
Early passage PDX tissues were obtained through the Adenoid Cystic Carcinoma Research Foundation. All animal procedures were performed at XenoSTART (South Texas Accelerated Research Therapeutics, San Antonio) following Institutional Animal Care and Use Committee protocols. Fragments of tumor (~70 mm3) were implanted subcutaneously into the flanks of 6–12-week-old female nu/nu athymic nude mice (The Jackson Laboratories or Charles River Laboratories). Upon reaching 150–300 mm3 tumor volume, mice were randomized to either treatment (n = 5) or vehicle (n = 5–10) groups using blinded block randomization and therapeutic dosing was implemented. Tumor dimensions were measured using digital calipers blinded to the treatment group and tumor volume was calculated using the formula: TV = width2 × length × 0.52. Tumors were harvested at termination, weighted, and used for histology, immunohistochemical staining, and RNA sequencing (RNA-seq) analysis (n = 3–5 per arm). Percent mean tumor growth inhibition (%TGI) induced by AL101 was calculated relative to the untreated control group using the formula: %TGI = 1 – (AL101 final – AL101 baseline) / (Control final – Control baseline).
NOTCH luciferase reporter assay
Full-length (FL) human NOTCH1 (FL-NOTCH1), ΔECD-NOTCH1, ACCx9-I1680N, and ACCx11-S1723ins28 transcripts were synthesized by GenScript and ligated into the pcDNA3.1 (+FLAG) expression vector. Constructs were transiently transfected into U2OS cells using Lipofectamine 2000 reagent (Invitrogen, #11668019) and assessed for their ability to activate a NOTCH-sensitive luciferase reporter gene [38, 39]. U2OS cells were used because of their transfectability and low basal NOTCH activity. Briefly, cells were co-transfected (in five biological replicates) with 10 ng of pcDNA3.1 expression construct, a NOTCH-sensitive firefly luciferase reporter gene (TP1-CSLx12-FF), and an internal control Renilla luciferase plasmid (pRL-TK, Promega). Normalized firefly luciferase activities were measured in whole cell extracts prepared 48 h after transfection using the Dual-Luciferase kit (Promega #E1960) and an Infinite M200 luminometer (Tecan).
RNA sequencing
RNA-seq was performed on an Illumina NovaSeq-6000 instrument. Adapters were trimmed with Cutadapt [40]. For PDXs, mouse reads were filtered out using an approach described by Callari et al. [41]. Human reads were aligned to human reference genome GRCh37/hg19 using STAR and STAR-fusion [42, 43]. Gene expression levels were calculated using featureCounts [44] and gene expression levels were normalized using DESeq2 [45]. Differentially expressed (DE) genes were detected using DESeq2 according to the following parameters: (i) average gene expression >50 normalized reads, (ii) log2(fold-change) >1 or log2(fold-change) <−1, (iii) false discovery rate (FDR) < 0.05.
Pathway enrichment analysis
Pathway enrichment analysis was performed using minimal hyper-geometric statistics [46], based on an approach similar to that described by Eden et al. [47]. MSigDB c2 (curated gene sets: Kyoto Encyclopedia of Genes and Genomes (KEGG)/Reactome pathways) [48] were used as pathway references. Multiple hypothesis correction of the p values was performed by controlling for FDR [49].
NOTCH target gene signature and gene expression clustering analysis
Known NOTCH target genes were curated from the literature and used to create a 21-gene signature that co-clustered ACC and triple-negative breast cancer (TNBC) tumors [50] that were responsive to GSIs, including specimens harboring NOTCH gain of function alterations such as single-nucleotide variants, indels or fusions [51, 52]. Hierarchical clustering analysis was done based on the Euclidean Distance method (Ward.D2 linkage) and cluster selection was done using SigClust2 [53]. A signature of 480 NOTCH-related genes was collected from (1) PathwaysCommons (the gene-gene interaction table was downloaded, filtered out, and only cases where NOTCH genes regulate the expression of the second gene, were retained (interaction type: “controls-expression-of”)); (2) KEGG [54]; (3) PID [55]; (4) MSigDB (https://www.gsea-msigdb.org/gsea/msigdb/collections.jsp); and (5) the 21 direct NOTCH target genes described above.
NOTCH activation caller
The NOTCH activation caller is a method relying on two algorithms: single sample Gene Set Enrichment Analysis (ssGSEA) [48, 56]; and k–Top Scoring Pairs (kTSP), a ranking-based classification algorithm that selects gene pairs whose expression levels switch their ranking between the two classes of interest [57,58,59]. For ssGSEA, a GSVA R package [60] was used with the set of 21 NOTCH-related genes described above. For each sample, a higher score indicates greater enrichment for NOTCH target genes. For kTSP, the in-house scripts were used on a set of 21 NOTCH-related genes, whose expression is expected to be higher in NOTCH-activated tumors, and a pre-defined set of four genes (LXN, RAPGEF3, TMEM154, and LGR6) whose expression is expected to be lower in NOTCH-activated tumors (derived from analyzing downregulated genes in TNBC and ACC PDX models as well as TNBC [50] and chronic lymphocytic leukemia [61] cell lines with activating NOTCH mutations curated from the literature). Briefly, the kTSP algorithm scores each sample based on the proportion of how many gene pairs comply with the expected rule of: “down” < “up”. Tumors were judged as being “NOTCH activated” based on the combination of ssGSEA and kTSP scores.
Statistical analysis
Student t-tests were used for statistical analysis between two groups in in vitro experiments. For in vivo studies, natural logarithm (ln) transformation was performed on tumor volume and regression models were created for tumor volume by study day per animal. Analysis of variance was run on the slopes obtained from the regression analysis, and the Tukey–Cramer method was used to compare treatment groups. Statistical analyses were performed using GraphPad Prism software. Statistical analysis of the RNA-seq data was performed using DESeq2 package in R. DE genes were identified using the Wald test [45], with three mice per condition/treatment. Multiple hypothesis correction was performed using FDR [49], where significance was defined as FDR <0.05.
Results
AL101 inhibits cell viability in NOTCH1-mutated ACC organoid but not in NOTCH wild-type ACC cell lines
Due to the rarity of ACC, its slow growth rate, and the limited number of passages that primary ACC cells typically grow in vitro, ACC cell lines have rarely been established. Although approximately 10 ACC cell lines have been reported to date and some were experimentally used, the origin of most of these cell lines has been questioned [62,63,64]. The naturally immortalized HACC2A and UFH2 cell lines [65, 66], as well as hTERT-transformed ACC52 cells [67], are among the few available authenticated salivary ACC cell lines. As whole-exome sequencing (WES) of these cell lines did not reveal any activating mutations in NOTCH genes (not shown), we established an organoid model (Supplementary Fig. 1A) using surgical resection material from a patient with an ACC tumor carrying two activating mutations in NOTCH1, as detected by the OncoPlus cancer mutation panel [68]. Specifically, a missense mutation (c.4787T>C, p.L1586P) that disrupts the structure of the NRR, leading to ligand-independent production of NICD1; and a complex change in exon 34 of NOTCH1 (c.7283_7286delinsCAG) that produces a coding frameshift starting at amino acid 2428 and creates a premature stop codon after 7 new amino acids, thereby leading to loss of a C-terminal PEST degron domain and stabilization of NICD1 [69]. Organoid histology and NICD1 expression were similar to those seen in the parental tumor (Supplementary Fig. 1B) and persistence of both NOTCH1 mutations in the organoid model was confirmed by WES.
RNA-seq revealed that expression of most of the 21 known NOTCH-related genes was substantially higher in the organoid model harboring activating NOTCH1 mutation compared to the three ACC cell lines bearing WT NOTCH genes (Fig. 1A). Reflecting the NOTCH signaling status, AL101 significantly inhibited the survival of the organoid model in a dose-dependent manner (Supplementary Fig. 2A), whereas only a minor response to AL101 was seen in the three WT ACC cell lines (Fig. 1B). Furthermore, treatment of the organoid model with AL101 resulted in downregulation of NICD1 (Supplementary Fig. 2B) and multiple NOTCH downstream target genes, such as HES1, HES2, HEY2, HEYL, and NRARP; NOTCH-dependent regulators of cell-cycle progression such as CDK6 and CCND1; and pro-survival oncogenes such as KIT and MYC (Fig. 1C), confirming that NOTCH signaling was functionally inhibited. In parallel, tumor suppressor genes such as MVP and cyclin-dependent kinase inhibitors CDKN1B and CDKN2D were upregulated following AL101 treatment (Fig. 1C). When using the NOTCH caller to visualize expression levels of the NOTCH target genes, a striking reduction in NOTCH signaling was observed in the organoid model harboring NOTCH1 activating mutations and not in NOTCH1 WT ACC cell lines (Fig. 1D).

A Hierarchical clustering of normalized baseline expression of 21 NOTCH-related genes in an organoid bearing NOTCH1 activating mutations and three ACC cell lines with wild-type NOTCH genes displayed as a heatmap. Higher or lower expression of genes is indicated with shades of red or blue cells, respectively. B In vitro ACC cell models were treated with increasing concentrations of AL101 and relative cell viability was determined on day 14. *p < 0.05, **p < 0.01, and ***p < 0.001. C Normalized expression of 21 NOTCH-related genes in the ACC in vitro cell models treated with increasing concentrations of AL101 displayed as a heatmap. Higher or lower expression of genes is indicated with shades of red or blue cells, respectively. D NOTCH activation caller plot scores the level of NOTCH signaling activity in the indicated samples by using ssGSEA (axis Y) and kTSP (axis X) algorithms on a set of NOTCH-related genes, where a shift down and to the left indicates reduced NOTCH signaling activity.
Characterization of ACC patient-derived xenograft (PDX) models
To evaluate the effect of AL101 in vivo, we selected two ACC PDX models reported to harbor NOTCH1 activating alterations: ACCx9, which carries an activating I1680N mutation; and ACCx11, which harbors a tandem duplication 3′ of NOTCH1 and has high levels of NICD1 [15]. We also selected two models without activating aberrations in NOTCH genes, ACCx6, and ACCx5M1. The NOTCH1 signaling activation status of these four ACC tumor models was confirmed by IHC analysis (Fig. 2A). As expected, strong diffuse nuclear staining for NICD1, a hallmark of NOTCH1 signaling activation, was observed in ACCx9 and ACCx11 tumors, whereas staining for NICD1 in ACCx6 and ACCx5M1 models, which have biphasic growth patterns, was weaker and confined to a subset of cells. Expression of MYC, a well-known oncogene and NOTCH target gene [70], and Ki67 (a marker of cell proliferation) were also significantly elevated in ACCx9 and ACCx11 PDX models (Fig. 2A).

A Tumor sections from four PDX models (ACCx9, ACCx11, ACCx5M1, and ACC6) collected at baseline (before implantation) were stained for NICD1, MYC, and Ki67 using an IHC. Representative images are shown. B Transactivation activity of wild-type full-length NOTCH1 (FL-NOTCH1), constitutively active γ-secretase dependent form of the NOTCH1 receptor (ΔECD-NOTCH1), and alleles containing mutations carried by ACCx9 and ACCx11 PDX tumors (ACC9-I1680N and ACC11-S1723ins28, respectively) was assessed using a dual-luciferase reporter assay. Bars represent the average of five independent experiments performed in triplicates and indicate fold-change relative to FL-NOTCH1 (red line). Percent inhibition induced by AL101 (gray bars) relative to DMSO (white bars) is indicated in blue. ***p < 0.001.
Detection of NOTCH1 intronic retention in ACCx11
The tandem repeat that lies 3’ of NOTCH1 in ACCx11 does not disrupt the NRR or PEST coding region, raising the possibility that some other genomic abnormality might explain the high levels of NICD1 production in this model. Since sequencing of exonic DNA failed to detect activating mutations in NOTCH1, we turned to RNA-seq, which revealed a tandem duplication involving a sequence normally encoded by exon 27. This tandem duplication results in the insertion of 28 amino acids between the structured part of the NRR and the transmembrane domain, creating a new “unprotected” ADAM10-cleavage site (Supplementary Fig. 3). Similar in-frame insertions of 11–36 amino acids stemming from internal tandem duplications in the 3’ end of intron 27 and/or the proximal region of exon 28 (NOTCH1 extracellular juxtamembrane region), called juxtamembrane expansion (JME) mutations, have been reported in T-ALL and ACC and are known to result in a strong constitutive production of NICD1 [71, 72]. Since the NOTCH1 exon 27 was wildtype, we reasoned that the additional sequence must be encoded by an aberrant “new” exon in intron 27. Indeed, DNA sequencing of intron 27 revealed an insertion, dubbed exon 27a, flanked by splice donor and acceptor sites encoding the duplicated sequence (Supplementary Fig. 3). This type of mutation, which to our knowledge has not been reported previously, is consistent with the idea that tumors with high constitutive levels of NICD1 harbor mutations that disrupt the NOTCH NRR and/or PEST domain.
NOTCH1 mutations in ACCx9 and ACCx11 tumors are activating and sensitive to AL101
To confirm the functional relevance of NOTCH1 variants expressed by ACCx9 and ACCx11 tumors and their sensitivity to AL101 inhibition, U2OS cells were transfected with a NOTCH reporter gene and cDNAs encoding full-length WT NOTCH1 (FL-NOTCH1), a strong gain-of-function form of NOTCH1 with a truncated extracellular domain (ΔECD-NOTCH1), the ACCx9 NRR variant (ACCx9-I1680N), or the ACCx11 NRR variant (ACCx11-S1723ins27a), in the presence and absence of 10 nM AL101. Importantly, like ΔECD-NOTCH1, the ACCx9-I1680N and ACCx11-S1723ins27a mutant alleles produced >100-fold higher activation of the NOTCH1 reporter gene than the FL-NOTCH1 allele (Fig. 2B). Furthermore, AL101 resulted in a marked reduction of NOTCH1 signaling in cells expressing the ACCx9-I1680N and ACCx11-S1723ins27a variants (99.2 and 99.4% inhibition, respectively) (Fig. 2B), confirming the dependency of these polypeptides on γ-secretase cleavage for NICD1 production and validating the use of these models in subsequent in vivo studies.
Tolerability of AL101 in vivo
Intravenous and oral administration of AL101 at a minimum effective dose (1 mg/kg) [22] had very similar PK profiles in mice (Supplementary Fig. 4A), and we therefore used oral administration in all in vivo experiments. To confirm AL101 tolerability in tumor-free mice, animals were treated with 3 escalating doses of AL101 (3, 5, or 7.5 mg/kg; 4 days on/3 days off) for 2 weeks. We observed that AL101 was well tolerated, with no overt toxicity (as assessed by altered behavior/appearance, or significant weight loss) (Supplementary Fig. 4B), consistent with prior reports showing that AL101 is well tolerated in vivo [22].
AL101 inhibits tumor growth in NOTCH1-activated ACC PDX models
To determine the impact of AL101 on tumor growth in vivo, animals were subcutaneously implanted with each of the 4 ACC tumor models and treated with either 7.5 mg/kg of AL101 or vehicle on a 4 day on/3 day off schedule. Tumor volumes and body weight were assessed twice a week, and percentage TGI was calculated as described in the Methods section. Although a long-term 7.5 mg/kg dosing schedule was tolerated in the tumor-bearing animals, with an observed weight loss of <10% (Supplementary Fig. 5), a slight decrease in body weight occurred at three consecutive timepoints (days 22, 25, and 28) in ACCx5M1 model (Supplementary Fig. 5). Thus, the dosage in this model was reduced to 5 mg/kg and maintained at this level until the end of the experiment. Importantly, while AL101 therapy induced potent TGI in both models with NOTCH1 gain-of-function mutations (110 and 74% for ACCx9 and ACCx11, respectively; p < 0.0001) (Fig. 3A and Supplementary Fig. 6), no significant antitumor effect was seen in models lacking NOTCH-activating aberrations (Fig. 3B and Supplementary Fig. 6). Notably, treatment of animals with 3 mg/kg of AL101, the lowest dose evaluated for tolerability in vivo (Supplementary Fig. 4), produced TGIs (Supplementary Fig. 7) and had effects on body weights (Supplementary Fig. 8) that were similar to those induced by treatment with a 7.5 mg/kg dose, suggesting that AL101 has a fairly broad therapeutic window for treating ACC cancers driven by activated NOTCH1.

Two ACC PDX models harboring tumors with NOTCH1 activating alterations (NOTCH-Act) (A) and two PDX models without NOTCH-activating mutations (NOTCH WT) (B) were treated with either AL101 or vehicle. Graphs show the average tumor volume for five to ten animals ±SD. Red arrow: dosage reduced on day 28 and administered at 5 mg/kg. **p < 0.01, ****p < 0.001. Act activated NOTCH1, WT wild-type NOTCH1.
AL101-induced tumor growth inhibition is associated with a decrease in NOTCH-mediated tumorigenic signaling
Consistent with TGI, NICD1, MYC, and Ki67 protein levels were sharply reduced by AL101 treatment in tumors with constitutively active NOTCH1 alleles (ACCx9 and ACCx11), but not in tumors with WT NOTCH1 (ACCx5M1 and ACCx6) (Fig. 4). Moreover, RNA-seq revealed that expression of 21 NOTCH-related genes was substantially altered by AL101 in NOTCH1-mutated models as compared to NOTCH WT counterparts (Fig. 5A). These results were further validated by RT-PCR analysis of five selected NOTCH-related genes (Fig. 5B). As NOTCH signaling involves a highly interconnected and complex network of protein modifiers that can affect its activity [73], we generated a set of 478 NOTCH-related genes (taken from KEGG, PID, MSigDB, and PathwayCommons databases) that may be affected by γ-secretase inhibition (Supplementary Table 1). Notably, in response to AL101 treatment, a substantially higher number of NOTCH-related genes, as well as a total number of genes, were DE in NOTCH1-mutated tumors compared to NOTCH WT specimens (Fig. 5E). Furthermore, downregulated genes (colored blue in Supplementary Fig. 9A, B) in AL101-treated ACCx11 and ACCx9 tumors were significantly enriched (p value < 10−16, exact binomial test) in “pathways in cancer”, a KEGG pathway (hsa05200) that contains 325 known tumor driving genes, suggesting that inhibition of NOTCH signaling has broad anti-oncogenic effects in ACC.

Tumor sections derived from ACCx9 and ACCx11 (A) or ACCx5M1 and ACCx9 B xenograft model treated with either AL101 or vehicle were stained for NICD1, MYC, and Ki67 using an IHC. Representative images are shown. Act activated NOTCH1, WT wild-type NOTCH1.

A Fold-change in expression of 21 NOTCH-related genes (from normalized RNA-seq data) in animals treated with AL101 relative to vehicle-treated animals, displayed as a hierarchical clustering heatmap. Increased and reduced expression of genes is indicated with shades of red or blue cells, respectively. B Confirmation of RNA-seq data with RT-PCR analysis of selected NOTCH target genes in AL101-treated relative to vehicle-treated animals (red line). Gray bars: animals carrying wild-type NOTCH1 alleles; white bars: animals harboring NOTCH1 activating mutations. *p < 0.05, and ***p < 0.01. C, D Volcano plots depicting gene expression changes in AL101 versus vehicle-treated animals. The red circles represent genes included in a 478 NOTCH-related geneset curated from KEGG, PID, MSigDB, and PathwayCommons databases (see Supplementary Table 1 for details). E The table summarizes the total number of differentially expressed (DE) genes and NOTCH-related DE genes in AL101-treated animals relative to vehicle-treated counterparts.
Discussion
Despite a relatively indolent growth rate, late relapse and distant metastasis occur in approximately 50% of patients with ACC [1]. To date, there are no FDA-approved targeted agents for patients with ACC, and responses to conventional chemotherapeutic agents are limited.
Cumulative evidence suggests that ~20% of ACC tumors carry mutations in NOTCH1, a critical regulator of cell proliferation, differentiation, and survival [7, 8, 14, 15], and that ACCs with alterations involving portions of NOTCH1 encoding the NRR and PEST domains are associated with worse prognosis [7, 8, 15, 72, 74, 75]. Our studies here indicate that ACCs with aberrant NOTCH signaling driven by activating genetic alterations are sensitive to AL101, a GSI that potently and specifically inhibits all 4 NOTCH receptors at nanomolar concentrations [22, 37, 76, 77], providing strong preclinical evidence for the ongoing Phase II open-label, single-arm, multicenter study (ACCURACY; NCT03691207) in relapsed/refractory ACC patients harboring activating NOTCH alterations.
A major challenge in the development of new therapeutic approaches for ACC has been the limited number of appropriate cellular and animal models. Developing ACC cell lines has been challenging, and to date, there are only five cell lines authenticated as ACC [63, 64, 66, 78, 79]. None of the three ACC cell lines that were available for this study (HACC2A, UFH2, and ACC52) harbored a NOTCH-activating mutation and consequently have low endogenous NOTCH signaling activity. Identification of the population of cancer stem cells in ACC and their ability to form cellular aggregates [74, 80, 81] makes this tumor an excellent candidate for establishing three-dimensional organoid models. While organoids remain genetically and phenotypically stable and maintain predictive value for drug responses of individual patients [82, 83], due to the rarity of the disease, the use of ACC organoids remains extremely limited [84], and no cellular model bearing a NOTCH-activating alteration is currently available. To study the effect of AL101 ex vivo, we established a human organoid model from a surgically excised ACC bearing a confirmed NOTCH1 activating mutation. We observed that AL101 had potent dose-dependent inhibitory effects on growth and NOTCH target gene expression in this model, whereas limited effects were seen in three ACC cell lines with WT NOTCH1. This suggests that the antitumor activity of AL101 may be restricted to ACC tumors with a constitutively activated NOTCH pathway.
To further test this idea, we studied the activity of AL101 in ACC PDX models. It is established that PDX models typically maintain the histology, gene expression patterns, and drug response of the tumors from which they were derived [85]. We selected 4 PDX models of known NOTCH1 mutation/activation status that were previously used to assess the efficacy of brontictuzumab, a monoclonal antibody that inhibits NOTCH1 by binding to its extracellular domain [15]. Although brontictuzumab had an inhibitory effect against ACCx9 and ACCx11, it was less impressive than the activity of AL101, possibly because mutations that altered the structure of the NOTCH1 NRR, particularly the type of JME mutation found in ACCx11, may abrogate the ability of blocking antibodies to prevent ADAM10 cleavage [86], the first step in NICD1 generation. This limitation argues that GSIs may be more generally reliable in targeting mutated NOTCH receptors than blocking antibodies. A second practical concern raised by the unusual mutation found in ACCx11 is that such mutations will be missed with standard exome sequencing, suggesting that RNA-seq platforms such as the Tempus xT genomic profiling test (https://www.ncbi.nlm.nih.gov/gtr/tests/558436) may have greater sensitivity in identifying NOTCH gain of function mutations. Our experience with ACCx11 also highlights the utility of immunohistochemical analysis for NICD1, as diffuse NICD1 staining in ACC is strongly correlated with the presence of NOTCH1 activating aberrations [72].
Treatment of tumor-bearing mice with AL101 led to significant responses in PDX models with activating NOTCH1 mutations but not in models with WT NOTCH genes. Responses were paralleled by a reduction in activated NOTCH1 and expression of NOTCH downstream target genes, as demonstrated by IHC, RNA-seq and RT-PCR analyses, with no significant toxicity observed in AL101-treated mice. These results further support the use of biomarkers that correlate with constitutive NOTCH activation to identify tumors that are likely to respond to AL101 and other NOTCH inhibitors [15, 50, 87]. While further studies in additional ACC in vitro models (once established) and PDXs are required to determine whether mutation-independent NOTCH pathway activation may also help predict response to NOTCH inhibitors, particularly when used in combination with other agents [37, 63, 77], our data emphasize the need to carefully assess ACCs for NOTCH1 activating mutations when selecting patients for treatment with GSIs.
In conclusion, our work adds to the growing body of evidence underscoring the critical role of NOTCH pathway activation in aggressive subtypes of ACC [15] and provides further support for the clinical development of GSIs for this indication. An ongoing Phase II ACCURACY clinical trial [88], which is assessing the effect of AL101 in patients with ACCs bearing NOTCH-activating mutations, is showing promising clinical activity and a favorable safety profile. Future extensions of the studies described here and ongoing clinical trials are likely to revolve around the identification of rational combinations of GSIs and other agents that increase tumor response and diminish the emergence of resistance to GSIs [89].
Data availability
The data supporting the findings of this study are available within the article and its Supplementary information files.
References
Dillon PM, Chakraborty S, Moskaluk CA, Joshi PJ, Thomas CY. Adenoid cystic carcinoma: a review of recent advances, molecular targets, and clinical trials. Head Neck. 2016;38:620–7.
Rettig EM, Talbot CC Jr, Sausen M, Jones S, Bishop JA, Wood LD, et al. Whole-genome sequencing of salivary gland adenoid cystic carcinoma. Cancer Prev Res (Phila). 2016;9:265–74.
Ho AS, Kannan K, Roy DM, Morris LG, Ganly I, Katabi N, et al. The mutational landscape of adenoid cystic carcinoma. Nat Genet. 2013;45:791–8.
Chahal M, Pleasance E, Grewal J, Zhao E, Ng T, Chapman E, et al. Personalized oncogenomic analysis of metastatic adenoid cystic carcinoma: using whole-genome sequencing to inform clinical decision-making. Cold Spring Harb Mol Case Stud. 2018;4:a002626.
Stephens PJ, Davies HR, Mitani Y, Van Loo P, Shlien A, Tarpey PS, et al. Whole exome sequencing of adenoid cystic carcinoma. J Clin Invest. 2013;123:2965–8.
Wysocki PT, Izumchenko E, Meir J, Ha PK, Sidransky D, Brait M. Adenoid cystic carcinoma: emerging role of translocations and gene fusions. Oncotarget. 2016;7:66239–54.
Ferrarotto R, Mitani Y, McGrail DJ, Li K, Karpinets TV, Bell D, et al. Proteogenomic analysis of salivary adenoid cystic carcinomas defines molecular subtypes and identifies therapeutic targets. Clin Cancer Res. 2021;27:852–64.
Ho AS, Ochoa A, Jayakumaran G, Zehir A, Valero Mayor C, Tepe J, et al. Genetic hallmarks of recurrent/metastatic adenoid cystic carcinoma. J Clin Invest. 2019;129:4276–89.
Aster JC, Pear WS, Blacklow SC. The varied roles of Notch in cancer. Annu Rev Pathol. 2017;12:245–75.
Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–6.
Hori K, Sen A, Artavanis-Tsakonas S. Notch signaling at a glance. J Cell Sci. 2013;126(Pt 10):2135–40.
Izumchenko E, Sun K, Jones S, Brait M, Agrawal N, Koch W, et al. Notch1 mutations are drivers of oral tumorigenesis. Cancer Prev Res (Phila). 2015;8:277–86.
Schubert AD, Zamuner FT, London NR, Zhavoronkov A, Mehra R, Hoque MO, et al. Role of the NOTCH signaling pathway in head and neck cancer. In: Burtness B, Golemis EA, editors. Molecular Determinants of Head and Neck Cancer. Cham: Current Cancer Research. Humana Press; 2018, p 229–48. https://link.springer.com/chapter/10.1007/978-3-319-78762-6_8.
Ferrarotto R, Heymach JV. Taking it up a NOTCH: a novel subgroup of ACC is identified. Oncotarget. 2017;8:81725–6.
Ferrarotto R, Mitani Y, Diao L, Guijarro I, Wang J, Zweidler-McKay P, et al. Activating NOTCH1 mutations define a distinct subgroup of patients with adenoid cystic carcinoma who have poor prognosis, propensity to bone and liver metastasis, and potential responsiveness to Notch1 inhibitors. J Clin Oncol. 2017;35:352–60.
Hapuarachi BS, Feeney L, Rack S, Adderley H, Morgan D, Betts G, et al. Clinical disease course and survival outcomes following disease recurrence in adenoid cystic carcinoma (ACC) with NOTCH signaling pathway activation. J Clin Oncol. 2021;39(15_suppl):6072.
Capaccione KM, Pine SR. The Notch signaling pathway as a mediator of tumor survival. Carcinogenesis. 2013;34:1420–30.
Wang Z, Li Y, Banerjee S, Sarkar FH. Emerging role of Notch in stem cells and cancer. Cancer Lett. 2009;279:8–12.
Zeng Q, Li S, Chepeha DB, Giordano TJ, Li J, Zhang H, et al. Crosstalk between tumor and endothelial cells promotes tumor angiogenesis by MAPK activation of Notch signaling. Cancer Cell. 2005;8:13–23.
Gu F, Ma Y, Zhang Z, Zhao J, Kobayashi H, Zhang L, et al. Expression of Stat3 and Notch1 is associated with cisplatin resistance in head and neck squamous cell carcinoma. Oncol Rep. 2010;23:671–6.
Wu CX, Xu A, Zhang CC, Olson P, Chen L, Lee TK, et al. Notch inhibitor PF-03084014 inhibits hepatocellular carcinoma growth and metastasis via suppression of cancer stemness due to reduced activation of Notch1-Stat3. Mol Cancer Ther. 2017;16:1531–43.
Gavai AV, Quesnelle C, Norris D, Han WC, Gill P, Shan W, et al. Discovery of clinical candidate BMS-906024: a potent Pan-Notch inhibitor for the treatment of leukemia and solid tumors. ACS Med Chem Lett. 2015;6:523–7.
Mohamed AA, Tan S-H, Xavier CP, Katta S, Huang W, Ravindranath L, et al. Synergistic activity with NOTCH inhibition and androgen ablation in ERG-positive prostate cancer cells. Mol Cancer Res. 2017;15:1308–17.
Barat S, Chen X, Cuong Bui K, Bozko P, Gotze J, Christgen M, et al. Gamma-secretase inhibitor IX (GSI) impairs concomitant activation of Notch and Wnt-beta-catenin pathways in CD44+ gastric cancer stem cells. Stem Cells Transl Med. 2017;6:819–29.
De Kloe GE, De Strooper B. Small molecules that inhibit Notch signaling. Methods Mol Biol. 2014;1187:311–22.
Pant S, Jones SF, Kurkjian CD, Infante JR, Moore KN, Burris HA, et al. A first-in-human phase I study of the oral Notch inhibitor, LY900009, in patients with advanced cancer. Eur J Cancer. 2016;56:1–9.
Yao J, Duan L, Fan M, Wu X. Gamma-secretase inhibitors exerts antitumor activity via down-regulation of Notch and Nuclear factor kappa B in human tongue carcinoma cells. Oral Dis. 2007;13:555–63.
Hijioka H, Setoguchi T, Miyawaki A, Gao H, Ishida T, Komiya S, et al. Upregulation of Notch pathway molecules in oral squamous cell carcinoma. Int J Oncol. 2010;36:817–22.
Lee SH, Hong HS, Liu ZX, Kim RH, Kang MK, Park NH, et al. TNFalpha enhances cancer stem cell-like phenotype via Notch-Hes1 activation in oral squamous cell carcinoma cells. Biochem Biophys Res Commun. 2012;424:58–64.
Yoshida R, Nagata M, Nakayama H, Niimori-Kita K, Hassan W, Tanaka T, et al. The pathological significance of Notch1 in oral squamous cell carcinoma. Lab Invest. 2013;93:1068–81.
Inamura N, Kimura T, Wang L, Yanagi H, Tsuda M, Tanino M, et al. Notch1 regulates invasion and metastasis of head and neck squamous cell carcinoma by inducing EMT through c-Myc. Auris Nasus Larynx. 2017;44:447–57.
Zhao ZL, Zhang L, Huang CF, Ma SR, Bu LL, Liu JF, et al. NOTCH1 inhibition enhances the efficacy of conventional chemotherapeutic agents by targeting head neck cancer stem cell. Sci Rep. 2016;6:24704.
Yuan X, Wu H, Xu H, Xiong H, Chu Q, Yu S, et al. Notch signaling: an emerging therapeutic target for cancer treatment. Cancer Lett. 2015;369:20–7.
Andersson ER, Lendahl U. Therapeutic modulation of Notch signalling-are we there yet? Nat Rev Drug Discov. 2014;13:357–78.
Diaz-Padilla I, Wilson MK, Clarke BA, Hirte HW, Welch SA, Mackay HJ, et al. A phase II study of single-agent RO4929097, a gamma-secretase inhibitor of Notch signaling, in patients with recurrent platinum-resistant epithelial ovarian cancer: a study of the Princess Margaret, Chicago and California phase II consortia. Gynecol Oncol. 2015;137:216–22.
Piha-Paul SA, Munster PN, Hollebecque A, Argiles G, Dajani O, Cheng JD, et al. Results of a phase 1 trial combining ridaforolimus and MK-0752 in patients with advanced solid tumours. Eur J Cancer. 2015;51:1865–73.
Morgan KM, Fischer BS, Lee FY, Shah JJ, Bertino JR, Rosenfeld J, et al. Gamma secretase inhibition by BMS-906024 enhances efficacy of paclitaxel in lung adenocarcinoma. Mol Cancer Ther. 2017;16:2759–69.
Blain J, Bédard J, Thompson M, Boisvert F-M, Boucher M-J. C-terminal deletion of NOTCH1 intracellular domain (N1ICD) increases its stability but does not amplify and recapitulate N1ICD-dependent signalling. Sci Rep. 2017;7:5034.
Pajcini KV, Xu L, Shao L, Petrovic J, Palasiewicz K, Ohtani Y, et al. MAFB enhances oncogenic Notch signaling in T cell acute lymphoblastic leukemia. Sci Signal. 2017;10:eaam6846.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet j. 2011;17:10–2.
Callari M, Batra AS, Batra RN, Sammut S-J, Greenwood W, Clifford H, et al. Computational approach to discriminate human and mouse sequences in patient-derived tumour xenografts. BMC Genomics. 2018;19:19.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2012;29:15–21.
Haas BJ, Dobin A, Li B, Stransky N, Pochet N, Regev A. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 2019;20:213.
Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2013;30:923–30.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Eden E, Lipson D, Yogev S, Yakhini Z. Discovering motifs in ranked lists of DNA sequences. PLoS Comput Biol. 2007;3:e39.
Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinforma. 2009;10:48.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B (Methodol). 1995;57:289–300.
Stoeck A, Lejnine S, Truong A, Pan L, Wang H, Zang C, et al. Discovery of biomarkers predictive of GSI response in triple-negative breast cancer and adenoid cystic carcinoma. Cancer Discov. 2014;4:1154–67.
Broner EC, Alpert G, Gluschnaider U, Mondshine A, Solomon O, Sloma I, et al. AL101 mediated tumor inhibition in notch-altered TNBC PDX models. J Clin Oncol. 2019;37(15_suppl):1064.
Ferrarotto R, Alpert G, Gluschnaider U, Rauch R, Mondshine A, Solomon O, et al. Abstract 4885: AL101 mediated tumor inhibition in Notch mutated ACC PDX models. Cancer Res. 2019;79(13 Supplement):4885.
Kimes PK, Liu Y, Neil Hayes D, Marron JS. Statistical significance for hierarchical clustering. Biometrics. 2017;73:811–21.
Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2016;45:D353–61.
Schaefer CF, Anthony K, Krupa S, Buchoff J, Day M, Hannay T, et al. PID: the Pathway Interaction Database. Nucleic Acids Res. 2008;37(suppl_1):D674–9.
Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–12.
Geman D, d’Avignon C, Naiman DQ, Winslow RL. Classifying gene expression profiles from pairwise mRNA comparisons. Stat Appl Genet Mol Biol. 2004;3:Article19.
Tan AC, Naiman DQ, Xu L, Winslow RL, Geman D. Simple decision rules for classifying human cancers from gene expression profiles. Bioinformatics. 2005;21:3896–904.
Tan AC, Naiman DQ, Xu L, Winslow RL, Geman D. Simple decision rules for classifying human cancers from gene expression profiles. Bioinforma (Oxf, Engl). 2005;21:3896–904.
Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinforma. 2013;14:7.
Pozzo F, Bittolo T, Vendramini E, Bomben R, Bulian P, Rossi FM, et al. NOTCH1-mutated chronic lymphocytic leukemia cells are characterized by a MYC-related overexpression of nucleophosmin 1 and ribosome-associated components. Leukemia. 2017;31:2407–15.
Phuchareon J, Ohta Y, Woo JM, Eisele DW, Tetsu O. Genetic profiling reveals cross-contamination and misidentification of 6 adenoid cystic carcinoma cell lines: ACC2, ACC3, ACCM, ACCNS, ACCS and CAC2. PLoS One. 2009;4:e6040.
Liu N-N, Zhang S-L, Chen W. The cross-contaminated cell lines of adenoid cystic carcinoma: a crucial concern. Transl Surg. 2017;2:10–3.
Li J, Perlaky L, Rao P, Weber RS, El-Naggar AK. Development and characterization of salivary adenoid cystic carcinoma cell line. Oral Oncol. 2014;50:991–9.
Jiang Y, Gao R, Cao C, Forbes L, Li J, Freeberg S, et al. MYB-activated models for testing therapeutic agents in adenoid cystic carcinoma. Oral Oncol. 2019;98:147–55.
Warner KA, Oklejas AE, Pearson AT, Zhang Z, Wu W, Divi V, et al. UM-HACC-2A: MYB-NFIB fusion-positive human adenoid cystic carcinoma cell line. Oral Oncol. 2018;87:21–8.
Queimado L, Lopes C, Du F, Martins C, Fonseca I, Bowcock AM, et al. In vitro transformation of cell lines from human salivary gland tumors. Int J Cancer. 1999;81:793–8.
Kadri S, Long BC, Mujacic I, Zhen CJ, Wurst MN, Sharma S, et al. Clinical validation of a next-generation sequencing genomic oncology panel via cross-platform benchmarking against established amplicon sequencing assays. J Mol Diagn. 2017;19:43–56.
Wang K, Zhang Q, Li D, Ching K, Zhang C, Zheng X, et al. PEST domain mutations in Notch receptors comprise an oncogenic driver segment in triple-negative breast cancer sensitive to a γ-secretase inhibitor. Clin Cancer Res. 2015;21:1487–96.
Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci USA. 2006;103:18261–6.
Sulis ML, Williams O, Palomero T, Tosello V, Pallikuppam S, Real PJ, et al. NOTCH1 extracellular juxtamembrane expansion mutations in T-ALL. Blood. 2008;112:733–40.
Sajed DP, Faquin WC, Carey C, Severson EA, A HA, C AJ, et al. Diffuse staining for activated NOTCH1 correlates with NOTCH1 mutation status and is associated with worse outcome in adenoid cystic carcinoma. Am J Surg Pathol. 2017;41:1473–82.
Schubert AD, Zamuner FT, London NR, Zhavoronkov A, Mehra R, Hoque MO, et al. Role of the NOTCH signaling pathway in head and neck cancer. In: Burtness B, Golemis EA, editors. Molecular determinants of head and neck cancer. Cham: Springer International Publishing; 2018. p. 229–48.
Panaccione A, Chang MT, Carbone BE, Guo Y, Moskaluk CA, Virk RK, et al. NOTCH1 and SOX10 are essential for proliferation and radiation resistance of cancer stem-like cells in adenoid cystic carcinoma. Clin Cancer Res. 2016;22:2083–95.
Bell D, Hanna EY, Miele L, Roberts D, Weber RS, El-Naggar AK. Expression and significance of notch signaling pathway in salivary adenoid cystic carcinoma. Ann Diagn Pathol. 2014;18:10–3.
Ran Y, Hossain F, Pannuti A, Lessard CB, Ladd GZ, Jung JI, et al. gamma-Secretase inhibitors in cancer clinical trials are pharmacologically and functionally distinct. EMBO Mol Med. 2017;9:950–66.
Sosa Iglesias V, Theys J, Groot AJ, Barbeau LMO, Lemmens A, Yaromina A, et al. Synergistic effects of NOTCH/gamma-secretase inhibition and standard of care treatment modalities in non-small cell lung cancer cells. Front Oncol. 2018;8:460.
Dong L, Wang YX, Li SL, Yu GY, Gan YH, Li D, et al. TGF-beta1 promotes migration and invasion of salivary adenoid cystic carcinoma. J Dent Res. 2011;90:804–9.
Drier Y, Cotton MJ, Williamson KE, Gillespie SM, Ryan RJ, Kluk MJ, et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat Genet. 2016;48:265–72.
Panaccione A, Zhang Y, Ryan M, Moskaluk CA, Anderson KS, Yarbrough WG, et al. MYB fusions and CD markers as tools for authentication and purification of cancer stem cells from salivary adenoid cystic carcinoma. Stem Cell Res. 2017;21:160–6.
Fujita S, Ikeda T. Cancer stem-like cells in adenoid cystic carcinoma of salivary glands: relationship with morphogenesis of histological variants. J Oral Pathol Med. 2012;41:207–13.
Drost J, Clevers H. Organoids in cancer research. Nat Rev Cancer. 2018;18:407–18.
Kondo J, Inoue M. Application of cancer organoid model for drug screening and personalized therapy. Cells. 2019;8:470.
Gao XL, Wu JS, Cao MX, Gao SY, Cen X, Jiang YP, et al. Cytokeratin-14 contributes to collective invasion of salivary adenoid cystic carcinoma. PLoS One. 2017;12:e0171341.
Izumchenko E, Meir J, Bedi A, Wysocki PT, Hoque MO, Sidransky D. Patient-derived xenografts as tools in pharmaceutical development. Clin Pharmacol Ther. 2016;99:612–21.
Aste-Amézaga M, Zhang N, Lineberger JE, Arnold BA, Toner TJ, Gu M, et al. Characterization of Notch1 antibodies that inhibit signaling of both normal and mutated Notch1 receptors. PLoS One. 2010;5:e9094.
Rettig EM, Talbot CC Jr, Sausen M, Jones S, Bishop JA, Wood LD, et al. Whole-genome sequencing of salivary gland adenoid cystic carcinoma. Cancer Prev Res (Philos). 2016;9:265–74.
Ferrarotto R, Wirth LJ, Muzaffar J, Rodriguez CP, Xia B, Perez CA, et al. 919MO ACCURACY a phase II trial of AL101, a selective gamma secretase inhibitor, in subjects with recurrent/metastatic (R/M) adenoid cystic carcinoma (ACC) harboring Notch activating mutations (Notchmut). Ann Oncol. 2020;31:S663.
Ferrarotto R, Rauch R, Leibovich T, Shitrit A, Solomon O, Herz E, et al. 1789P The gamma secretase inhibitor AL101 combined with other drugs for dual targeting of Notch dysregulated tumors. Ann Oncol. 2021;32:S1222.
Acknowledgements
We would like to thank the ACC Research Foundation (ACCRF) for funding this study. Specifically, we acknowledge Jeff Kaufmann and Dr Nicole Spardy-Burr for their contributions to study planning. We also thank The University of Chicago Organoid and Primary Culture Research Core, especially Ye Li, for their assistance with generating organoids. This work was supported in part by the National Institutes of Health grants R01DE027809 and R01DE028674.
Author information
Authors and Affiliations
Contributions
Conceptualization: RF, JK, JCA, NA, and EI. Methodology: EI, JK, TL-R, VM, NA, EI, RR, AY, and AM. Validation: EH, AY, OS, RR, AM, XC, MM, TL-R, and AS. Investigation: EI, RF, JK, EH, AY, OS, RR, AM, MM, TL-R, MD, JCA, VM, AS, and XC Resources: RF, JK, and EI. Data curation: EH, AY, OS, RR, AM, MM, and MD. Writing—original draft preparation: EI, JK, VM, TL-R, NA, and AS. Writing—review and editing: EI, JK, TL-R, RF, RR, JCA, AS, AJR, ATP, CRW, LS, and VM. Visualization: EI, AY, RR, and EH. Supervision: EI, JK, NA, and RF. Funding acquisition: JK, EI, and RF. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
EH, AY, OS, RR, AM, MM, TL-R, MD, and JK are affiliated with Ayala Pharmaceuticals. RF, JCA, and EI are consultants to Ayala Pharmaceuticals. The remaining authors declare no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Stephen Tait
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ferrarotto, R., Mishra, V., Herz, E. et al. AL101, a gamma-secretase inhibitor, has potent antitumor activity against adenoid cystic carcinoma with activated NOTCH signaling. Cell Death Dis 13, 678 (2022). https://doi.org/10.1038/s41419-022-05133-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-022-05133-9
This article is cited by
-
Molecular Aspects of Mucoepidermoid Carcinoma and Adenoid Cystic Carcinoma of the Salivary Gland
Head and Neck Pathology (2024)
-
Molecularly defined sinonasal malignancies: an overview with focus on the current WHO classification and recently described provisional entities
Virchows Archiv (2024)
-
Targeted therapy for head and neck cancer: signaling pathways and clinical studies
Signal Transduction and Targeted Therapy (2023)
-
Current understanding of adenoid cystic carcinoma in the gene expression and targeted therapy
Holistic Integrative Oncology (2023)
