
Abstract
Since the discovery of cell apoptosis, other gene-regulated cell deaths are gradually appreciated, including pyroptosis, ferroptosis, and necroptosis. Necroptosis is, so far, one of the best-characterized regulated necrosis. In response to diverse stimuli (death receptor or toll-like receptor stimulation, pathogenic infection, or other factors), necroptosis is initiated and precisely regulated by the receptor-interacting protein kinase 3 (RIPK3) with the involvement of its partners (RIPK1, TRIF, DAI, or others), ultimately leading to the activation of its downstream substrate, mixed lineage kinase domain-like (MLKL). Necroptosis plays a significant role in the host’s defense against pathogenic infections. Although much has been recognized regarding modulatory mechanisms of necroptosis during pathogenic infection, the exact role of necroptosis at different stages of infectious diseases is still being unveiled, e.g., how and when pathogens utilize or evade necroptosis to facilitate their invasion and how hosts manipulate necroptosis to counteract these detrimental effects brought by pathogenic infections and further eliminate the encroaching pathogens. In this review, we summarize and discuss the recent progress in the role of necroptosis during a series of viral, bacterial, and parasitic infections with zoonotic potentials, aiming to provide references and directions for the prevention and control of infectious diseases of both human and animals.
Similar content being viewed by others
Facts
-
Regulated necrosis, including necroptosis, pyroptosis, and ferroptosis, can release damage-associated molecular patterns (DAMPs) which promote inflammation, a process termed necroinflammation.
-
Necroptosis can be initiated by death receptor or toll-like receptor stimulation, pathogenic infection, or other stimuli.
-
Pathogens like viruses, bacteria, and parasites evolved exquisite strategies to either inhibit or promote necroptosis for higher replication and persistent infection.
Open questions
-
What are the exact roles and detailed mechanisms of regulated necrosis during pathogenic infections of animals and humans? How these pathogens interact with the host immune system for better survival in hosts.
-
Regulated necrosis is a double-edged sword, and can we precisely manipulate regulated necrosis as a potential therapeutic method in pathogenic infections, sepsis, Alzheimer’s disease, diverse stresses, and other relevant diseases and disorders?
Introduction
Delicate control of cell proliferation and cell death plays a critical role during the development and homeostasis of multicellular organisms and during host defense against invading pathogens. There is no doubt that the breakdown of this delicate balance between cell proliferation and cell death can lead to physiological malfunction, organ damage, or even a variety of diseases in the body. In the past several decades, apoptosis has traditionally been considered the only form of programmed cell death, a gene-regulated active cell death. However, necrosis was considered an accidental form of cell death, an uncontrolled passive cell death [1]. However, increasing evidence shows that cell apoptosis is not the unique form of programmed cell death, and regulated necrosis (also termed necroptosis or caspase-independent cell death), pyroptosis, and ferroptosis have been successively identified by several groups [2,3,4]. Currently, necroptosis is one of the best-characterized regulated necrosis. Necroptosis is generally initiated through RIP3/RIP1 or RIP3/ZBP1 and subsequently executed by MLKL. Necroptosis requires the kinase activity of RIP3, a member of the family of the death-domain-containing kinase so-called “RIP1” [3, 5], and the downstream “executioner” protein MLKL [6]. RIP3 can be activated by one of the upstream proteins that contain the RIP homotypic interaction motif domain-like RIP1, TRIF (Toll/IL-1R domain-containing adapter-inducing IFN-β, also known as TICAM1), or DAI (also known as ZBP1) [7,8,9,10]. The role of necroptosis in a wild-type setting is not completely clear, but its activation pathways have been suggested as part of inflammation-mediated immunity during infection [11]. In this review, we summarize and discuss the recent research progress on the mechanism of necroptosis and its modulatory role in diverse zoonotic pathogen infections.
Regulation of pyroptosis
Pyroptosis is a form of lytic-regulated cell death (RCD) first described in macrophages infected with the Gram-negative bacteria Shigella fexneri by Zychlinsky and colleagues in 1992 as apoptosis [12] but later renamed pyroptosis in 2001 by Cookson and Brennan [13] to reflect its inflammatory nature. Literately, pyroptosis is an inflammatory cell death caused by various stimuli like microbial infection and cancer, accompanied by activation of inflammasomes and maturation of proinflammatory cytokines interleukin-1β and interleukin-18 [14]. Pyroptosis plays a protective role in the host’s response to microbial infection [14,15,16] but also drives pathogenic inflammation [15, 16]. Distinct from apoptosis and necroptosis, pyroptosis is, in fact, the most immunogenic of all the cell death mechanisms. It has distinct morphological features such as cellular swelling, chromatin condensation, and plasma membrane permeabilization. Inflammasome activation was first coined in 2002 by Dr. Jurg Tshopp [14] and is a hallmark of pyroptosis. Inflammasomes are cytoplasmic multimeric protein complexes initiated by detecting PAMPs and DAMPs like DNA, bacterial flagellin, type 3 secretion system (T3SS) needle and rod subunits, and toxins which are detected by cytosolic sensors (NOD-like and RIG-I like receptor). The inflammasome formation leads to oligomerization of pro-Caspase-1 and pro-Caspase-11 (Caspase-4 and -5 in humans), resulting in the induced generation of their active form. Active Caspase-1 mediates the cleavage of gasdermin D (GSDMD), causing the release of the active form of N-terminal, which can induce cell death by pore formation through directly incorporating into the cellular membrane from inside [17] and outside of bacteria [18]. Proinflammatory cytokines IL-1β and IL-18 were cleaved by active caspase-1 during inflammasome activation. Distinct from caspase-1 mediated canonical inflammasome activation, Caspase-11 mediated noncanonical inflammasome activation is directly triggered by recognition of intracellular LPS and subsequently induces lytic cell death through cleaved gasdermin D [19, 20]. The executor of pyroptosis is mainly focused on gasdermin D, one of the Gasdermin family (GSDMs). Up to now, six genes have been classified as members of GSDMs based on their conserved N-terminal and C-terminal regions in humans, including gasdermin A (GSDMA), B (GSDMB), C (GSDMC), D (GSDMD), E (GSDME)/DFNA5, and DFNB59 [21]. All the members of the Gasdermin family, except DFNB59, are cleaved into two fragments and form pores on the cell membrane by the released N-terminal domain ensued by cell lysis [22, 23] (Fig. 1). In the context of pyroptosis, upon inflammasome activation, both GSDMD and GSDMB can be cleaved by caspase-1 [17, 24] and caspase-4 (in humans) [25, 26]. Of note, caspase-3, an important mediator of apoptosis, has been shown to cleave both GSDMB in human immune-related diseases [25, 27] and GSDME in antitumor studies, respectively [28,29,30]. While caspase-8, another important mediator of apoptosis, has been shown to cleave GSDMD [31] and GSDMB [31, 32] during Yersinia infection. These results further expand the understanding of the connection between pyroptosis and apoptosis through these functional proteins.

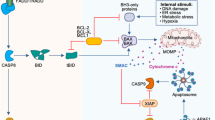
A MLKL stimulation by RIPK3 is necessary for the execution of necroptosis. The initiators of necroptosis include death receptors (FAS and TNF), TLRs, viruses, bacteria, others (e.g., STING, RIG-1), and they activate RIPK1, TICAM1, DAI/ZBP1, or others downstream factors, leading to the RIPK3/MLKL activation, membrane rupture, and necroptosis. DAMP (e.g., HMGB1, IL-1α, ATP…) can be released during this process. B Cleavage of GSDMD mediated by active caspase-1 or caspase-11 (mouse) or caspase-4, 5 (human) is the executor for pyroptosis. Proinflammatory cytokines IL-1β and IL-18 were cleaved by active caspase-1. Caspase-1 or caspase-4, 5, 11 can be induced by upstream initiators, DAMPS, PAMPs, LPS, and others, leading to GSDMD cleavage, membrane rupture, and finally pyroptosis. C Ferroptosis, an iron-dependent regulated necrosis, is generally induced by lipid peroxidation. Ferroptosis can be triggered by extrinsic pathway (e.g., GPX4, NOS, ALOX) or intrinsic pathway (e.g., inhibition system XC-, prevention cystine import). The membrane is damaged, followed by DAMP release. TLRs Toll-like receptors, RIPK3 receptor-interacting protein kinase 3, MLKL mixed lineage kinase domain-like, DAI IFN-regulatory factors, also known as ZBP1, GSDMD gasdermin D, DAMPs damage-associated molecular patterns, PAMPs pathogen-associated molecular patterns.
Regulation of ferroptosis
Ferroptosis is a newly identified type of regulated cell death originally proposed by Dixon in 2012, and it is characterized by iron accumulation and lipid peroxidation. Ferroptosis is distinct from apoptosis, pyroptosis, and necroptosis in terms of morphological, biochemical, and genetic features [33,34,35]. Ferroptosis can be induced mainly by two pathways, either an intrinsic pathway or an extrinsic pathway. The intrinsic pathway is normally initiated by regulation of transporters (e.g., inhibiting system Xc-, preventing cystine import, activation of the iron transporters transferrin), and the extrinsic pathway is generally mediated by suppressing the intracellular antioxidant enzymes expression or activities (like GPX4, NOS, and ALOX) [36, 37] (Fig. 1). Some new regulators of ferroptosis are constantly reported, like FSP1-CoQ10-NADPH and BH4 [37]. The regulation of ferroptosis is quite a complex process. Numerous studies have shown that ferroptosis is closely associated with many diseases or disorders, including cancers, kidney, liver, gastrointestinal, and neurological disorders [33, 36, 37]. It is anticipated that manipulating the ferroptosis process, either activating or inhibiting, can be served as an effective therapeutic strategy to relieve the diseases described above. The regulatory mechanisms of ferroptosis are quite complex, and more gaps or hypotheses still need to be filled or confirmed.
Mechanism of necroptosis
Unlike apoptosis featured by cell shrinkage, DNA fragmentation, and membrane blebbing, necroptosis exhibits cell swelling and rupture of the plasma membrane [38]. Necroptosis is a form of nonapoptotic cell death and can be initiated by a series of factors, including death ligands, microbial infection, Toll-like receptor (TLR), interferon (IFN), and sterile cell injury [2, 11, 39,40,41,42]. Given the importance of cell death during the host hemostasis, disease development, and host defense, our review will focus on the current progress illustrating the intricate role of necroptosis during host–pathogen (virus, bacteria, and parasites) interplay.
TNF is the best-studied extracellular death ligand among the diverse triggers for necroptosis. In 1988, Laster and colleagues showed that TNF induces target cells to undergo apoptosis and necrosis [43]. After TNF binding with its receptor TNFR1, multiple proteins are sequentially recruited to the cytosolic portion of TNFR1, thereby constituting a platform named “complex I” [44, 45]. Complex I can further activate the NF-kB and mitogen-activated protein kinases (MAPK), and the former is generally considered to initiate a survival signal [46]. Inhibition of cIAP or deubiquitination of RIP1 by cylindromatosis (CYLD) impedes the NF-κB pathway and hence induces cell apoptosis mediated by another platform, ‘complex II’ as named by some investigators [45, 47]. Genetic ablation or functional inhibition of caspase-8 or FADD can sensitize the cell to die in necroptosis instead of apoptosis, indicating that caspase-8 and FADD are crucial for guarding cells away from diverse cell death [48,49,50] (Fig. 2). In addition to necroptosis and apoptosis, caspase-8 also plays a role in inflammation-induced pyroptotic cell death, as shown by a recent study in mice expressing an enzymatically dead caspase-8 [51].

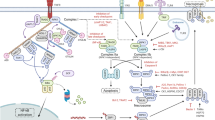
Various stimuli, including the TLRs, IFN, death ligands, and pathogenic infections (Viruses, Bacteria, and Parasites), can induce cell death in necroptosis. TNF engages with its receptor and induces cell apoptosis by inhibiting cIAP or deubiquitination of RIP1 by cylindromatosis (CYLD). Instead, inhibition of FADD or caspase-8 can sensitize the cells to undergo necroptosis. Microbial infections can also initiate necroptosis via activation of the receptor-interacting protein kinase 1 (RIP1) and RIP3 complex. Further phosphorylation of MLKL by RIP3 can induce the oligomerization and translocation of MLKL to the membrane, eventually causing cell death. Besides the RIP1-RIP3-MLKL dependent-necroptosis, some RHIM-containing factors like TRIF and IFN-regulatory factors (DAI, also known as ZBP1) mediate the noncanonical necroptosis without the involvement of RIP1. TLRs Toll-like receptors, IFN interferon, FADD Fas-associated protein with death-domain, MLKL mixed lineage kinase domain-like, RHIM RIP homology interaction motifs (RHIM), ZBP1 Z-DNA-binding protein 1.
Cellular FLICE-like inhibitory protein (cFLIP), an antiapoptotic protein, is another important factor regulating the apoptosis pathway and necroptosis. cFLIP is mainly expressed as three isoforms in humans (cFLIPL, cFLIPS, and cFLIPR) and two isoforms in mice (cFLIPL and cFLIPR) [52,53,54]. As described above, complex I normally activate the cellular survival pathway by activating NF-kB and cFLIP production. Generally, a sufficient cFLIP can prevent caspase-8 activation followed by inhibition of apoptosis, and insufficient cFLIP can fully activate caspase-8 leading to apoptosis activation. Insufficient cFLIP can prevent necroptosis via cleaving RIPK1 and RIPK3 [55]. Moreover, during the microbial infection, some viruses evolve a series of strategies to counteract the host protective machinery like apoptosis, and the data showed that several herpesviruses or poxviruses also express FLIP, sometimes termed vFLIP [56, 57]. vFLIP encoded by Kaposi’s sarcoma-associated herpesvirus can inhibit caspase-8 activation, thus preventing apoptosis and activating NF-kB [58, 59]. A recent report showed that mice constitutively expressing vFLIP showed excessive necroptosis in hepatocytes [60]. The data above indicates the critical functions of cFLIP in apoptosis and necroptosis, especially in the case of the TNF-mediated signaling pathway. Also, during the normal development of mice, cFLIPL is extensively involved in inhibiting apoptosis and necroptosis from maintaining homeostasis. The precise roles of these isoforms of cFLIP during the regulation of cell survival, cell death, and normal mammalian development are still under investigation.
Upon the inhibition of apoptosis by pharmacological inhibitors or during microbial infection, necroptosis can be initiated through activation of the receptor-interacting protein kinase 1 (RIP1) and RIP3 complex, named “necrosome” (several researchers also call it “complex IIb”) [45, 61]. The main components of the necrosome complex are FADD, caspase-8, RIP1, RIP3, and TRADD. The presence of caspase-8 in the necrosome can prevent the interaction of RIP1 and RIP3 via the direct cleavage of these two kinases [50, 62, 63]. RIP1 and RIP3 interact through their respective C-terminal RIP homology interaction motifs (RHIM) [6]. Overexpression of RIP3 can also lead to necroptosis [64]. Several groups demonstrated that pseudokinase MLKL, a substrate of RIP3, is a downstream mediator for the execution of necroptosis. It is shown that phosphorylation of MLKL by RIP3 can further induce the oligomerization and translocation of MLKL to the inner leaflet of the membrane, subsequently causing a cellular rupture and final cell death (Fig. 2) [6, 65, 66]. Besides the RIP1-RIP3-MLKL dependent canonical necroptosis, some RHIM-containing factors like TRIF and IFN-regulatory factors (DAI, also known as ZBP1/DLM-1) mediate the noncanonical necroptosis without the involvement of RIP1 [10, 40, 67]. DAI can recognize the viral double-strand DNA or viral RNA, recruit RIPK3, and induce RIPK1-independent/RIPK3-dependent-necroptosis [9, 68, 69]. Tanshinol A, a natural compound, can induce reactive oxygen species production in lung cancer cells, which further activates necroptosis mediated by MLKL, independent of RIPK1 and RIPK3 [67]. Besides DAI, TRIF, another RHIM domain-containing protein, is also capable of activating RIPK3 independent of RIPK1 [10]. Under certain circumstances, as the inhibition of caspase, TLR3 or TLR4 stimulation induce necroptosis with the contribution of TRIF and RIP3 [10, 40, 70]. In addition to TRIF, studies from several groups demonstrated that RIP3, in combination with direct interaction with DAI plays a critical role in initiating RIP3-dependent-necroptosis during MCMV infection [71, 72]. Despite some initiators for necroptosis, most researchers agree that the eventual execution of necroptosis is universally driven by RIP3 and its substrate MLKL [11, 40, 65], although RIP3 was shown to induce apoptosis through its adapter function [73].
zVAD-fmk, a pharmacological inhibitor of caspase-8, has been extensively used to inhibit apoptosis from inducing necroptosis of cells [74]. Using a cell-based assay, Degterev and colleagues have successfully identified a small molecular necrostatin-1 (Nec-1) as an effective RIP1 inhibitor, and Nec-1 is capable of suppressing necroptosis [7, 75]. Nec-1 and genetic deficiency of RIP3 or MLKL have been widely applied to block the necroptosis pathway either in the cell model in vitro or in the knockout mouse model in vivo to decipher the mechanism of necroptosis.
Immunological potentials of regulated necrosis-necroinflammation
Different from apoptosis without causing obvious inflammation, regulated necrosis, including necroptosis, pyroptosis, and ferroptosis, can release damage-associated molecular patterns (DAMPs), including HMGB1, IL-1alpha, DNA fragments, mitochondrial content, and ATP. Immune cells in an organism can respond to the exposed DAMPs, and this process can be generally defined as necroinflammation [76, 77]. In brief, DAMPs can further engage with the pattern recognition receptors (PRRs), like TLRs, and NODs, leading to the production of inflammatory cytokines via diverse inflammatory transcriptional pathways, including NF-κB signaling [78]. It is anticipated that certain diseases, especially those with overt inflammatory features (e.g., Crohn’s disease, multiple sclerosis, ischemia-reperfusion injury (IRI) in liver and kidney), may be relieved via inhibition of regulated necrosis. Research showed that inhibition of necroptosis via blocking RIPK1 kinase activity can protect against TNF-induced systemic inflammatory response syndrome in vivo [79, 80]. Notably, inhibiting regulated necrosis, including necroptosis and ferroptosis, showed an improved phenotype in a kidney IRI [81]. Regulated necrosis can be blocked and induced in some diseases, like cancers. It is well known that cancer cells exhibit resistance to cell apoptosis, necroptosis, and ferroptosis, showing promising performance in eliminating certain kinds of therapy-resistant cancer cells [80, 82, 83].
Moreover, in testing with various tumor types, RIPK3 activation can induce the expression of immunostimulatory cytokines, leading to an increased antitumor immunity [83]. Data showed that loss of GPX4 function either by genetic inactivation or pharmacological treatment could induce selective ferroptosis in cancer cells in vitro and also guard against tumor recrudesce in mice [84, 85], indicating that rationally manipulating ferroptosis or necroptosis or both may be an attractive way to treat certain kinds of cancers in clinic. However, we can not rule out the possibility that regulated necrosis can exhibit anti-inflammatory effects under some conditions. Although the exact links between regulated necrosis and clinical diseases are still unclear, some evidence from clinical settings and animal model research emphasizes the relevance of regulated necrosis in the diseases.
In response to certain stresses, like infection or tissue damage, hosts immediately take action to eliminate the foreign materials or restore tissue integrity via various defense mechanisms, including cell death, aiming to maintain body homeostasis [86, 87]. Dexamethasone, often utilized as an anti-inflammatory agent, is also one kind of stress. Several studies showed the potential of dexamethasone in modulating cell death, like regulated necrosis. Very recently, it has been proved that dexamethasone sensitizes erastin-induced ferroptosis via glutathione depletion [88]. Dexamethasone can also alleviate corneal alkali injuries by inhibiting the caspase-1/GSDMD-mediated pyroptotic signaling pathway [89]. Ballegeer et al. demonstrated that dexamethasone could inhibit TNF-induced-necroptosis in intestinal epithelial cells [90], indicating the potential application of dexamethasone in treating clinical diseases with the extensive involvement of ferroptosis and pyroptosis, like acute kidney injury, stroke, alkali injuries or others. In addition, Dexamethasone can induce cell apoptosis, necroptosis, and pyroptosis under certain pathophysiologic conditions in hosts [91,92,93,94]. The report showed that dexamethasone and SMAC mimetic cooperate to induce necroptosis dependent on RIPK3 and MLKL in lymphoblastic leukemia cells without caspase activation [94]. Dieckol can relieve dexamethasone-induced muscle atrophy via inhibiting NLRP3/GSDMD-mediated pyroptosis [92]. Thus, precisely manipulating regulated cell death can be served as a potential therapeutic method in diverse stresses.
Necroptosis and microbial infection
During the past several decades, countless discoveries have been obtained through studying the host–pathogen interactions, like diverse evasion strategies utilized by crafty pathogens. Some discoveries have been applied to protect the host from infection. Traditionally, as non-inflammatory but programmed cell death, apoptosis is crucial in protecting the host from microbial infection without eliciting an excess inflammatory response. However, the recent progress of necroptosis has greatly drawn the researchers’ attention to study host–pathogen interplay.
It is known that dying or stressed cells can continuously release immunogenic endogenous molecular contents, DAMPs [78]. It is generally considered that apoptosis release no or very limited DAMPs. However, most researchers agree that necroptosis can release massive amounts of DAMPs, a strong inflammatory trigger to activate immune responses against pathogenic infections, as discussed above [77, 78].
A variety of pathogens, including viruses and bacteria, have evolved exquisite strategies to modulate cell apoptosis of host cell, including inhibiting apoptosis at the early stage of infection and promoting apoptosis at the late stage to subvert the host immune response for higher replication and persistent infection [95, 96]. What is the role of necroptosis during pathogenic infection? Is it beneficial or detrimental to the host? The following sections discuss the role of necroptosis during microbial infection and emphasize pathogenic infections.
Viral infection
The host processes a variety of mechanisms to sense the invading virus to activate proinflammatory responses and induces cell death of infected cells for further clearance of viral pathogens. Thus programmed cell death, including apoptosis, necroptosis, and other types of cell death, is a vital component of host defense, although many viruses have evolved diverse strategies to subvert this host response.
Murine cytomegalovirus (MCMV)
Both MCMV and human cytomegalovirus (HCMV) belong to herpesvirus, one kind of large and complex DNA virus. It is known that HCMV infection is very prevalent worldwide and can cause serious diseases, especially in the population with depressed or naïve immune responses [97]. Most members of cytomegalovirus have very strong host species specificity. HCMV can mainly infect humans or human cells, and MCMV often serves as an invaluable model for studying the pathogenesis of HCMV infection in humans [97, 98]. MCMV can trigger apoptosis, necroptosis, or both pathways to control the viral spread of even cross-species infection if certain cell death inhibitors are absent [39, 99,100,101]. In the meantime, MCMV has evolved many genes that exhibit immune-modulatory functions to manipulate the timing and ways of cell death. This sophisticated pathogen can express both the suppressors for apoptosis and inhibitors to prevent necroptosis [102, 103].
It was shown that MCMV could encode some viral inhibitors to prevent cell death, including cell apoptosis and necroptosis, by targeting diverse apoptotic and necroptotic pathways [72, 100, 104]. It is generally agreed that caspase-8 and Bak/Bax mediate the initiation and execution of cell apoptosis. One of the MCMV protein products, vICA, can bind to the pro-domain of caspase-8 and prevent its activation [105]; another viral protein, M36, can also target and block caspase-8 [100]. Similarly, HCMV also encodes several apoptosis inhibitors to modulate the host responses. HCMV UL36 (vICA, a homolog of M36) can restrain cell apoptosis via inhibiting caspase-8 activation [106]. Besides caspase-8 inhibition, HCMV pUL37x1 (vMIA) can also block the mitochondrial release of cytochrome C, resulting in the inhibition of caspase9-mediated cell apoptosis [107, 108]. Other viral proteins like vIBO and vMIA can function at the mitochondrial level and inhibit Bak and Bax, respectively [103]. Viral inhibitor of RIP activation (vIRA) encoded by the viral M45 gene can disrupt RHIM-dependent RIP3-RIP1 interaction, suppressing necroptosis [39]. It has recently been shown that mice deficient in Caspase-8 and RIPK3 mount elevated levels of CD8 T cells in response to MCMV infection, suggesting the cell death-independent role of Caspase-8 during restricting antiviral CD8 T cell hyperaccumulation [109].
Nevertheless, the role of RIPK3 and MLKL played during the restriction of antiviral CD8 T cell by Caspase-8 awaits further investigation. Besides the inhibitory effects of MCMV on the execution of necroptosis, MCMV infection can also indirectly trigger necroptosis of retinal neurons via inflammatory response mediated by ocular immune cells [110], clearly showing that the complex modulation of MCMV infection on host cell death. Lane et al. utilized an antiviral necroptosis-based CRISPR knockout screen to analyze this complex virus-host interaction and identified a critical host factor mediating early viral infection [111]. Similarly, HCMV can also block the necroptosis through the degradation of MLKL by UL36 [106, 112]. Therefore, HCMV UL36 is a dual cell death inhibitor for apoptosis and necroptosis. A recent study demonstrated that HCMV-induced autophagy could prevent cells from undergoing necroptosis [113], facilitating the viral spread in hosts. All of these studies with MCMV infection, either in cells or host body, unambiguously demonstrated that this virus could utilize diverse strategies to manipulate or subvert the immune response effectively, either leashing or delaying the proinflammatory reactions of cell death signaling to favor viral persistence in the host.
Influenza virus
Influenza virus, an enveloped negative-sense and single-stranded RNA virus consists of types A, B, and C [114]. Influenza A virus (IAV) infection can cause severe diseases in mammals and birds through seasonal epidemics and occasional pandemics and pose a significant threat to human health, especially for populations with relatively compromised immune responses like young children and the elderly, and pregnant women [115].
Several studies showed that IAV infection could trigger programmed cell death, including apoptosis, necroptosis, or pyroptosis in various cell types, like macrophages, fibroblast, alveolar epithelial cells, and monocytes as in vivo animal models [69, 116,117,118,119,120]. Thapa et al. identified a host protein DAI which is originally considered to be a dsDNA sensor [71], and discovered that DAI could also sense genomic RNA of IAV and further induced RIPK3 independent apoptosis and parallel RIPK3-dependent-necroptosis in murine fibroblasts to eliminate the infected cells or tissue [69]. This group further found that compared to wild-type mice, RIPK3 knockout mice are more susceptible to IAV infection. This phenomenon is not observed in MLKL knockout mice. They demonstrated that apoptosis and inflammasome might also contribute to effectively controlling IAV infection besides necroptosis. A recent study showed that RNA from IAV can be recognized by ZBP1, leading to the activation of RIP3 and MLKL and following necroptosis [121,122,123], although the exact mechanism of how ZBP1 senses RNA is still under investigation. A study by Kuriakose showed that IAV infection in primary murine BMDMs could trigger multiple and parallel cell death pathways via this innate sensor, ZBP1/DAI [119]. The same group further identified a transcriptional regulator (IRF1) of ZBP1 promoting necroptosis during IAV infection [124].
Interestingly, Downey and colleagues identified a novel role of RIPK3 in regulating type I IFN antiviral immunity at both transcriptional and posttranscriptional levels during IAV infection, differing from its traditional role in necroptosis [125]. Recently, using a transgenic mouse model, Shubina et al. reported that necroptosis also plays a vital role in the antiviral host defense in the absence of apoptosis [126]. Conversely, RIPK3 deficiency can protect against Influenza H7N9 virus infection [127]. Thus, the exact role of necroptosis during the IAV infection is complex and dynamic, depending on infection stages, infection conditions, and others.
Few studies are available regarding the viral factors mediating necroptosis during viral infection. A study from Gaba et al. identified NS1 protein of IAV as a viral factor to interact with MLKL, leading to its oligomerization and eventually execution of necroptosis [128]. Besides the role of RIPK3 in regulating cell death and IFN secretion, RIPK3 also modulates the immune response of monocytes and DCs by promoting the expression of co-stimulatory molecules and T cell proliferation abilities [129]. This indirect modulation is mediated through the supernatant of necroptotic cells. These studies above provide concrete evidence that RIPK3 can exhibit distinct roles in different cell types, and the exact contribution of these pathways or viral factor(s) during IAV infection in vivo demands further investigation.
Besides IAV, animal-source influenza viruses emerged in the last decade and caused diseases in poultry, domestic animals, and human beings. These viruses include the best-known avian influenza virus (AIV) and swine-origin pandemic influenza virus [130, 131]. Taking AIV as an example, AIV mainly circulates in wild aquatic birds and domestic poultry but occasionally infects humans through direct and indirect contact with farmed poultry or migratory birds [132]. Both High pathogenic AIV H5N1 and low pathogenic AIV H7N9 can cause severe influenza diseases, even death in humans. Notably, H5N1 infection in humans generally causes a >50% fatality rate [133, 134], clearly manifesting their epidemic and pandemic potential and threat to public health. Little information is available regarding the role of necroptosis during AIV infection in animals and humans. Thus, understanding cell death, this key defense mechanism for the host during AIV infection in poultry or humans, await investigation and may promote early and quick diagnosis method and treatment.
Sendai virus
Sendai virus (SeV) is an enveloped negative-strand RNA virus, and it mainly infects mice and rats, causing acute respiratory diseases resembling the human parainfluenza infection [135]. It was shown that SeV could induce cell apoptosis via activation of Caspase-8 and Caspase-3 [135]. The MVAS/MAPK kinase7/c-Jun N-terminal kinase 2 signaling pathway plays a role somehow [136]. SeV infection can also trigger dramatic induction of necroptosis in a RIG-1 (RNA sensor) dependent manner, and viral proteins Y1 and Y2 can sensitize the cells towards necroptotic death through degradation of cIAPs [137]. Interestingly, the viral protein Y2 has also been involved in antiapoptotic activities [138], showing that this versatile virus has adapted to counteract the host defense. Although SeV infection causes an augmented inflammation in RIP3 deficient mice, the exact role of necroptosis during different stages of Sev infection in vivo is not yet clear.
Bacterial infection
Salmonella
Gastroenteritis is the main cause of morbidity and mortality in humans, especially young children. Salmonella species are a leading cause of gastroenteritis [139]. Worldwide, typhoidal and nontyphoidal Salmonella infections have resulted in at least 118.3 million reported human cases with 355,000 deaths each year and posed a significant threat to human health and life [139,140,141].
Salmonella Typhimurium (S. Typhimurium), a typical Gram-negative intracellular enteric Salmonella species, is the etiologic agent of salmonellosis, and S. Typhimurium infection can cause gastroenteritis, septicemia, hepatitis, and even rapid death in humans [142]. Mouse inoculated with a very low dose (as low as 100 bacteria) of S. Typhimurium can die within a short period (as short as 7 days) [143]. More than 20 years ago, Salmonella infection could cause cell death, mainly apoptosis and accidental necrosis [144,145,146]. Recently, researchers realized that S. Typhimurium infection could induce necroptosis and pyroptosis in macrophages, the most crucial innate immune cells for Salmonella control [143, 147, 148]. Early efficient control of S. Typhimurium depends on the innate immune cells, mainly macrophages, and subsequent T cell response will be engaged late in the infection [142, 149]. Jorgensen et al. showed that S. Typhimurium infection in macrophages can induce the release of type I interferon, which further drives the necroptosis of macrophages in a RIP3/RIP1-dependent manner, contributing to evasion of host immune response [148], and another group discovered that intestinal epithelial Caspase-8 is essential to prevent S. Typhimurium infection induced-necroptosis [150]. Jorgensen’s group further demonstrated the link between type I interferon production and augmentation of necroptosis in S. Typhimurium infected macrophages are antioxidative stress response impairment via RIP3 [151]. At the same time, data from another group indicated that K45A medicates the kinase activities of RIP1 in the initiation of necroptosis in macrophages, and mice with K45A mutation in RIP1 exhibited attenuated inflammatory response and are highly susceptible to S. Typhimurium challenge [152]. It is well known that Salmonella pathogenicity island 2 (SPI-2)-encoded type III secretion system (T3SS), the most extensively studied and vital virulence factor of Salmonella, mediated the translocation of effector proteins into host cells, triggering proinflammatory responses or cell death [153, 154]. Several effector proteins, including SseK1, SseK3, and SpvB, inhibited cell necroptosis in infected macrophages and intestinal epithelial cells [155,156,157].
Moreover, TLRs, the widely studied PRRs to sensor the pathogen-associated molecular patterns (PAMPs), play a vital role in the pathogen recognition and induction of immune response during infections [158]. Ligands for TLR2, TLR4, TLR5, TLR9, and TLR11 are all present in Salmonella, which are lipoproteins, lipopolysaccharide (PLS), flagellin, CpG DNA, and flagellin, respectively [159,160,161]. Very limited reports illustrate the role of specific TLRs in regulating the necroptosis of host/cells during Salmonella infection, and Zhan et al. were the first to show TLR9 protects the host against necroptosis, preventing or delaying the systematic spend of the bacteria in the host [162]. Therefore, more research awaits further investigation, e.g., do other TLRs or NOD-like receptors play a role during necroptosis regulation in the cells/host and their contributions to disease development. Indeed, when caspases are inactivated in mouse macrophages, following recognition of poly(I:C) and LPS by TLR3 and TLR4, respectively, necroptosis can be induced with the mediation by TRIF [163]. Besides macrophages, neutrophils are another important cell type involved in bacterial control by the host [164]. Do neutrophils regulate necroptosis in collaboration with macrophages or independently? Intriguingly, premature cell death of neutrophils infected by Brucella abortus can promote phagocytosis by macrophages, favoring further bacterial replication in macrophages and in hosts [165], indicating the potential “cooperative work” between neutrophils and macrophages manipulated by evading pathogens.
Moreover, NleB, the effector protein of Escherichia (E.) coli, is capable of blocking cell death receptor signaling, including necroptosis, as discussed below [61], and NleB homologs are proved to present in Salmonella [166]. It needs to be determined whether Salmonella NleB is capable of modulating necroptosis. Further investigation of the Salmonella virulence factors during modulation of cell death would allow a better understanding of how hosts counteract bacterial infection. Recently, Doerflinger and colleagues showed that necroptosis coordinated with pyroptosis and apoptosis protects the hosts from Salmonella infection [167], indicating a complex defense mechanism hosts utilize in response to intracellular infections.
Salmonella causes persistent infection in humans and various animals, including poultry, pigs, cattle, and some pet animals (pigeons, fish, reptiles) [168,169,170,171]. Related production, carcass, or excretion from these animals with Salmonella infection or water/food contamination can serve as a potential bacterial reservoir for spread, and humans with direct or indirect contact with them could get infected. Cell death can cause bacterial spread into the environment leading to a public health concern. However, little information is available regarding the possible contribution of cell necroptosis to the pathology, bacterial shedding, and disease development during Salmonella infection in animals, especially farm and pet animals, and the responsible virulence factors during this process remain much to be uncovered.
Mycobacterium
Tuberculosis (TB) is the ninth leading cause of death worldwide caused by Mycobacterium tuberculosis (M.tb). According to a WHO report, roughly 10.4 million people fell ill with TB globally, and 1.67 million deaths were estimated in 2016 (WHO, 2017). M.tb has co-evolved with human beings for tens of thousands of years, and several strategies have been evolved to efficiently manipulate host immune responses for better survival [172]. M.tb is an intracellular pathogen that generally infects and survives within the host macrophages and dendritic cells, neutrophils, and other non-professional phagocytes to a lesser extent [173,174,175]. Virulent M.tb can induce or inhibit cell apoptosis and can trigger necrosis of host macrophages to evade innate immunity and modulate the adaptive immune response [176, 177]. Currently, apoptosis and necrosis are the research focus for studying the host–pathogen interaction, and very limited studies are available about the modulation of necroptosis by this pathogen.
One report from 2013 shows that the TNF-RIP1-RIP3 axis mediates necroptosis of infected macrophages via mitochondrial reactive oxygen species (ROS) production [178]. Very recently, Butler and colleagues showed that M.tb infection and TNF-α synergize to induce necroptosis of murine fibroblasts via a RIP1-dependent manner in vitro and also the occurrence of necroptosis in granulomas in mice, possibly contributing to bacteria dissemination and transmission [179]. Another group confirmed that M.tb infection establishes a pro-necroptotic environment by up-regulating MLKL and ZBP1 and further demonstrated that macrophage necroptosis is eventually restricted to mitigate the disease outcome [180]. Several models of how M.tb induces necroptosis in macrophages have been established. Tuberculosis necrotizing toxin (TNT) can induce necroptosis in M.tb-infected macrophages through the release of ROS [181], and ROS could further promote necroptotic cell death in M.tb-infected macrophages. Although a subsequent study showed a contradictory conclusion, the discrepancy is possibly due to the difference in the cell line used or other unidentified reasons [182]. M.tb can also trigger macrophages necroptosis by consuming NAD by bacterial factor TNT [183]. Interestingly, the same group further discovered that the supply of NAD+ could reduce ROS levels in Mtb-infected macrophages and prevent cell death, contributing to the restriction of bacterial replication [184]. This study provides direction for better treatment of TB patients in the future.
It is unclear which other virulence factor(s) of M.tb modulate(s) the execution of necroptosis during the late stage of infection in vivo. The main genomic difference between the attenuated Bacillus Calmette-Guerin (BCG) and M.tb is the so-called region of difference 1 (RD1), which contributes to the full virulence of M.tb [185]. An intriguing report showed that the ESX-1 system and its substrate EAST-6, encoded by the RD1 region of M.tb, can induce necroptosis in macrophages dependent on inflammasome activation during bacterial infection [186, 187]. Thus it is worthy to further clarify the regulatory role of RD1 during the necroptosis of host cells during M.tb infection.
Besides human TB, zoonotic TB is another form of TB caused by M. bovis in humans. In 2016, an estimated 147,000 new human zoonotic TB cases and 125000 deaths were reported worldwide (WHO, 2017). Bovine TB can be transmitted from animals to humans through food consumption, usually raw or improperly cooked meat or dairy product from diseased animals and from humans to humans [188]. Currently, no report is available about the induction of necroptosis of host cells by M. bovis or its virulence factors. Quite early analysis using genomic subtraction showed that RD1 is present in all tested virulent M.tb strain and M. bovine strains [189], and it is reasonable to speculate some similarities between M. bovine infection and M.tb infection in the host, which needs to be further clarified.
Helicobacter
Helicobacter (H.) pylori is a microaerophilic Gram-negative bacillus that infects more than half of the world’s population and is the best-studied Helicobacter species that can cause severe gastric diseases in human beings [190]. H. pylori has evolved diverse strategies and successfully adapted to human after colonizing the human stomach for at least 50,000 years [191]. Several virulence factors from H. pylori have been shown to cause cell apoptosis and necrosis, including Vacuolating cytotoxin A (VacA) and gamma-glutamyl transpeptidase (GGT) [192,193,194]. Persistent colonization of H. pylori in the host stomach leads to chronic inflammation, which further drives the occurrence of peptic ulceration and even gastric cancer. Radin reported that VacA might augment mucosal inflammation via inducing necroptosis of gastric epithelial cells and contribute to the pathogenesis of gastric diseases [195]. Due to the limited information, the exact role and contribution of necroptosis during H. pylori infection in vivo is still unclear, e.g., what is the disease outcome if RIP3 or MLKL is knockout in the mouse? Does necroptosis of gastric epithelial cells in the stomach play a protective or detrimental role during bacterial infection in the host? H. pylori infection in hosts is a complex and dynamic process. The effect of gastric epithelial cell necroptosis on disease outcome should be considered from different infection stages. Before the bacterial infection in the mucosa, necroptosis of gastric epithelial cells can trigger the immune response and expose the bacteria to the host immune environment. However, uncontrolled necroptosis may lead to excess inflammation and chronic infection as the disease continues.
Besides H. pylori, non-H. pylori helicobacters have also been detected in the human stomach; these bacteria can cause gastric diseases [196]. H. suis is the most prevalent gastric non-H. pylori helicobacters in humans and pigs are the natural host for this bacterium. Pigs and pork are most likely the main sources of human H. suis infection [196,197,198]. It was shown that H. suis could induce cell apoptosis and necrosis in the gastric epithelial cells and lymphocytes [194, 199]. However, no data is available about necroptosis’s involvement and possible contribution during H. suis infection in vitro and in vivo. More studies need to be done considering its zoonotic potential.
Pathogenic Escherichia (E.) coli
Pathogenic E. coli, typically enteropathogenic E. coli (EPEC), uropathogenic E. coli (UPEC), Enteroaggregative E. coli (EATC), and Enterotoxigenic E. coli (ETTC), has been widely studied in humans, animals and food due to their ability to cause significant morbidity and mortality worldwide [200, 201]. It was reported that UPEC infection could induce RIP1/PIP3 dependent-necroptosis of macrophages via bacterial pore-forming toxin, and under the current experimental setup, necroptosis is proved to be the principal model of cell death. Intriguingly, they also demonstrated that other pore-forming toxin-producing bacteria exhibit similar effects on macrophages [202], highlighting the feasibility of counteracting these toxin-producing-bacterial infections and controlling disease outcomes via these pathways.
EPEC, another pathogenic E. coli, are important diarrheal pathogens for young children. Li and colleagues showed that EPEC NleB, a type III secretion system effector (T3SS), targets several death-domain-containing proteins in TNFR, FAS, and TRAIL death receptors complexes to block several host cell deaths, including necroptosis, making it the first identified bacterial virulence factor capable of hijacking the death receptor complexes to modulate host defense [61]. During EPEC infection, inhibition of proinflammatory reaction and apoptosis by diverseT3SS effector factors is one of the major mechanisms utilized by the bacteria to block antimicrobial host defense [203]. Recently, one group discovered that EspL, another cysteine protease effector of T3SS, can degrade the PHIM-containing proteins RIP1, RIP3, TRIF, and ZBP1/DAI to block necroptosis [204]. Although several T3SS effectors are shown to target the necroptosis machinery, they may function in different manners, clearly showing the complex features of host response modulation by pathogens. It is still unclear whether other bacteria, especially gastrointestinal bacteria, also possess NleB or EspL effector protein homologs and whether these effectors exhibit similar or different effects on host cell death signaling. Programmed cell death, including necroptosis and pathogens like EPEC, is always in an evolutionary race [205]. Thus exploring cell death mechanisms mediated by emerging or evolved bacterial virulence factors will be continually in progress. In addition, to our knowledge, no data is available about the involvement of necroptosis during EATC infection or ETTC infection.
Klebsiella pneumoniae
Klebsiella pneumoniae (K. pneumoniae), a Gram-negative bacterium, is an important pathogen that can cause infection and severe disease in pulmonary and urinary tracts of humans globally, especially nosocomial pneumoniae and sepsis. Notably, K. pneumonae infection can be fatal in some patients with compromised immune conditions [206]. This bacterium has evolved several mechanisms to evade the attack of antibiotics or antimicrobial material. Due to its profound significance in clinical diseases, K. pneumonae has drawn the great attention of researchers and doctors.
K. pneumonae infection can cause host cells, especially macrophages and neutrophils, to die in diverse forms such as apoptosis, necroptosis, and pyroptosis [95, 207, 208]. To some extent, K. pneumoniae infection can inhibit or delay cell apoptosis or pyroptosis, leading to bacterial survival and further bacterial dissemination in the host, and this mechanism is utilized by some other pathogens as well. However, the mechanism or bacterial factor(s) that mediate cell death modulation is still unknown. In addition, one group showed that carbapenem-resistant K. pneumoniae could activate necroptosis in THP-1 cell lines, and further study using a transgenic mouse model indicates that necroptosis may contribute to the substantial immune dysregulation during K. pneumoniae infection [208]. Recently another group showed that K. pneumoniae could wisely subvert efferocytotic clearance of neutrophils by depressing cell apoptotic signatures and induction of necroptosis, demonstrating the ability of K. pneumoniae to modulate cell death to more inflammatory cell death [209]. The mechanism by which K. pneumoniae manipulates efferocytosis through cell apoptosis and necroptosis remains unidentified.
Other bacteria
Other bacteria are also implicated in the induction and regulation of necroptosis in cell or animal models. Clostridium perfringens (C. perfringens) infection can cause histotoxic, enteric, or enterotoxemic diseases in animals and humans by producing many toxins [210]. β-toxin from C. perfringens type C strain can induce cell death of primary porcine endothelial cells. This observed cell death is inhibited by Nec-1, indicating regulated necrosis is most likely involved [211]. However, other experiments must be further performed to tamp this conclusion, e.g., involvement of RIP3, MLKL, and other necroptosis markers. A recent study demonstrated that a high level of enterotoxin from C. perfringens type C strain could induce necroptosis in Caco-2, Vero cells, and human enterocyte-like T84 cell lines [212]. These data showed that necroptosis is involved in cell death mediated by virulence factors, like diverse toxins, from C. perfringens. It is, however, necessary to elucidate the potential role, either beneficial or detrimental, of necroptosis during C. perfringens infection in vivo. Whether other toxins from C. perfringens also mediate the necroptosis needs to be explored, which can help determine if necroptosis induced in patients or animals can be targeted for clinical therapeutics.
Parasitic infection
Some parasites can cause human diseases, like leishmaniasis, toxoplasmosis, malaria, and Chagas disease [213]. Leishmaniasis is often found in tropics and sub-tropics, and the etiologic agent is Leishmania, intracellular protozoa. A higher level of heme, an inducer for RIP1/RIP3/MLKL dependent-necroptosis, was observed in the leishmaniasis patients. However, RIP1 kinase regulates leishmania replication-independent heme-induced-necroptosis [214]. Angiostrongylus cantonensis infection can cause major nerve diseases, and both necroptosis and apoptosis occur in the brain tissue of mice infected by Angiostrongylus cantonensis [215]. Toxoplasma gondii (T. gondii), a typical, prevalent, and also the world’s most successful intracellular parasite, can asymptomatically persist in the central nervous system, causing devastating diseases in animals and humans. It was shown that CD8+ T cell-mediated immune response plays a vital role during T. gondii infection [216]. An early study showed that T cell-specific FADD−/− mice succumb to T. gondii infection more easily than wild-type mice, underscoring the importance of keeping necroptosis in check-in T cells during parasite infection [49]. Interestingly, Ripk3−/−mouse exhibits a similar survival rate in response to T. gondii. However, Ripk3−/− Casp8−/− mice succumbed to T. gondii infection, indicating the vital role of Caspase-8 in controlling T. gondii infection in vivo [217]. Although studies with T. gondii show that necroptosis may not be crucial for the egress of Toxoplasma gondii [218], we still can not rule out the possible role of necroptosis during Toxoplasma gondii infection solely based on this cell model and experimental condition. Brief summary of necroptosis induced by common pathogens is described in Table 1.
Role of cell death in sepsis
Dysregulated host responses to microbial infections can cause sepsis by hyper inflammation, life-threatening organ dysfunction, multiple organ failure, or even death if not treated properly [219]. World Health Organization’s first global report on sepsis showed that sepsis is the 3rd cause of neonatal death, causing more than 11 million death each year. Diverse cell deaths, including apoptosis, necrosis, necroptosis, pyroptosis, and autophagy, can be induced during sepsis in infections. Comprehensive reports showed that these cell death pathways are frequently aberrantly-regulated, playing a vital role during the occurrence and progression of sepsis [220]. Altered apoptosis and necrosis are observed in sepsis and are closely associated with sepsis-induced organ dysfunction and increased inflammation [221, 222]. In certain cases, uncontrolled apoptosis sometimes can lead to a compromised immune response, which is related to a higher risk of secondary infection and the immunosuppressive pathophysiology of sepsis [223, 224]. Necroptosis is also involved in the sepsis progression. Inhibition of necroptosis via RIPK3 knockdown or Nec-1 treatment can relieve the inflammation and attenuate the symptoms of organ dysfunction in septic mouse, zebrafish, and piglet models [225,226,227,228]. Evidence demonstrated that excessive pyroptosis also contributes to sepsis progression. Inhibition or disruption of pyroptosis can protect hosts against sepsis [229, 230].
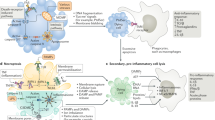
Interestingly, Chen et al. discovered that the synergetic effects of RIPK3-mediated necroptosis and GSDMD-mediated pyroptosis contribute to the multiple organ injury of sepsis in mice, and blockage of both pathways can protect mice from lethal sepsis [33, 231]. Ferroptosis is another form of cell death discovered in sepsis, and evidence shows that treatment with a ferroptotic inhibitor can mitigate the LPS-induced sepsis [227, 232], indicating that targeting ferroptotic cell death is another way to counteract the sepsis (Fig. 3). Overall, regulated necrosis is a double-edged sword, it can protect hosts from pathogenic infections, but on the other hand, excessive execution is also inevitably capable of leading to sepsis. Therefore, modulating these cell death signaling pathways in the future may provide new therapeutic directions for sepsis treatment.

Severe infections can cause sepsis, and apoptosis, necrosis, necroptosis, pyroptosis, and autophagy, can be induced during sepsis. Altered regulation of cell death can lead to local inflammation, tissue injury, organ dysfunction, immunosuppressive pathophysiology, and multiple organ failure. Uncontrolled cell death can be manipulated to restore homeostasis.
Concluding remarks
Necrosis is not accidental anymore; it can be highly regulated and genetically controlled. Although regulated necrosis has been studied for more than 20 years, the term necroptosis was coined by the group of Prof. Junying Yuan in 2005. The pathways of necroptosis are well characterized, and the regulatory role of necroptosis in microbial infection has also been partially revealed. Necroptosis is implicated not only during microbial infection but also in various diseases, such as Alzheimer’s disease, neurodegenerative disorders, acute kidney injury, atherosclerosis, severe inflammatory response syndrome, and cancer. The precise mechanism and pathophysiological significance of necroptosis in diseases of humans and animals still need further in-depth investigation.
Data availability
All data in this study are included in this published article, and additional information are available from the corresponding authors upon reasonable request.
References
Lockshin RA, Zakeri Z. Programmed cell death and apoptosis: origins of the theory. Nat Rev Mol Cell Biol. 2001;2:545–50.
Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–95.
Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–23.
Christofferson DE, Yuan JY. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol. 2010;22:263–8.
He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–11.
Sun LM, Wang HY, Wang ZG, He SD, Chen S, Liao DH, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–27.
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–9.
Rebsamen M, Heinz LX, Meylan E, Michallet MC, Schroder K, Hofmann K, et al. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. Embo Rep. 2009;10:916–22.
Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11:290–7.
Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288:31268–79.
Chan FK, Luz NF, Moriwaki K. Programmed necrosis in the cross talk of cell death and inflammation. Annu Rev Immunol. 2015;33:79–106.
Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature. 1992;358:167–9.
Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9:113–4.
Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–26.
Ceballos-Olvera I, Sahoo M, Miller MA, Del Barrio L, Re F. Inflammasome-dependent pyroptosis and IL-18 protect against Burkholderia pseudomallei lung infection while IL-1beta is deleterious. Plos Pathog. 2011;7:e1002452.
Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109.
Shi JJ, Zhao Y, Wang K, Shi XY, Wang Y, Huang HW, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5.
Wang JY, Deobald K, Re F. Gasdermin D protects from melioidosis through pyroptosis and direct killing of bacteria. J Immunol. 2019;202:3468–73.
Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, et al. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature. 2012;490:288–+.
Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–71.
Tamura M, Tanaka S, Fujii T, Aoki A, Komiyama H, Ezawa K, et al. Members of a novel gene family, Gsdm, are expressed exclusively in the epithelium of the skin and gastrointestinal tract in a highly tissue-specific manner. Genomics. 2007;89:618–29.
Shi JJ, Gao WQ, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42:245–54.
Chao KL, Kulakova L, Herzberg O. Gene polymorphism linked to increased asthma and IBD risk alters gasdermin-B structure, a sulfatide and phosphoinositide binding protein. Proc Natl Acad Sci USA. 2017;114:E1128–E1137.
Ding JJ, Wang K, Liu W, She Y, Sun Q, Shi JJ, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–6.
Chen Q, Shi PL, Wang YF, Zou DY, Wu XW, Wang DY, et al. GSDMB promotes non-canonical pyroptosis by enhancing caspase-4 activity. J Mol Cell Biol. 2019;11:496–508.
Shi PL, Tang A, Xian L, Hou SY, Zou DY, Lv YS, et al. Loss of conserved Gsdma3 self-regulation causes autophagy and cell death. Biochem J. 2015;468:325–36.
Panganiban RA, Sun MY, Dahlin A, Park HR, Kan MY, Himes BE, et al. A functional splice variant associated with decreased asthma risk abolishes the ability of gasdermin B to induce epithelial cell pyroptosis. J Allergy Clin Immun. 2018;142:1469–+.
Wang YP, Gao WQ, Shi XY, Ding JJ, Liu W, He HB, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103.
Yu JH, Li S, Qi J, Chen ZL, Wu YH, Guo J, et al. Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis. 2019;10:193.
Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128.
Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci USA. 2018;115:E10888–E10897.
Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science. 2018;362:1064–9.
Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11:88.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Tang DL, Kang R, Vanden Berghe T, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29:347–64.
Tang D, Kroemer G. Ferroptosis. Curr Biol. 2020;30:R1292–7.
Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31:107–25.
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–14.
Sridharan H, Upton JW. Programmed necrosis in microbial pathogenesis. Trends Microbiol. 2014;22:199–207.
Grootjans S, Vanden Berghe T, Vandenabeele P. Initiation and execution mechanisms of necroptosis: an overview. Cell Death Differ. 2017;24:1184–95.
Huang Z, Wu SQ, Liang Y, Zhou X, Chen W, Li L, et al. RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe. 2015;17:229–42.
Yu X, Li Y, Chen Q, Su C, Zhang Z, Yang C, et al. Herpes simplex virus 1 (HSV-1) and HSV-2 mediate species-specific modulations of programmed necrosis through the viral ribonucleotide reductase large subunit R1. J Virol. 2016;90:1088–95.
Laster SM, Wood JG, Gooding LR. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol. 1988;141:2629–34.
Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–90.
Zhang DW, Zheng M, Zhao J, Li YY, Huang Z, Li Z, et al. Multiple death pathways in TNF-treated fibroblasts: RIP3- and RIP1-dependent and independent routes. Cell Res. 2011;21:368–71.
Chen LW, Egan L, Li ZW, Greten FR, Kagnoff MF, Karin M. The two faces of IKK and NF-kappaB inhibition: prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat Med. 2003;9:575–81.
Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Bio. 2017;18:127–36.
Bell BD, Leverrier S, Weist BM, Newton RH, Arechiga AF, Luhrs KA, et al. FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proc Natl Acad Sci USA. 2008;105:16677–82.
Osborn SL, Diehl G, Han SJ, Xue L, Kurd N, Hsieh K, et al. Fas-associated death domain (FADD) is a negative regulator of T-cell receptor-mediated necroptosis. Proc Natl Acad Sci USA. 2010;107:13034–9.
Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–72.
Fritsch M, Gunther SD, Schwarzer R, Albert MC, Schorn F, Werthenbach JP, et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature. 2019;575:683–7.
Han DK, Chaudhary PM, Wright ME, Friedman C, Trask BJ, Riedel RT, et al. MRIT, a novel death-effector domain-containing protein, interacts with caspases and BclXL and initiates cell death. Proc Natl Acad Sci USA. 1997;94:11333–8.
Rasper DM, Vaillancourt JP, Hadano S, Houtzager VM, Seiden I, Keen SLC, et al. Cell death attenuation by ‘Usurpin’, a mammalian DED-caspase homologue that precludes caspase-8 recruitment and activation by the CD-95 (Fas, APO-1) receptor complex. Cell Death Differ. 1998;5:271–88.
Silke J, Strasser A. The FLIP side of life. Sci Signal. 2013;6:pe2.
He MX, He YW. A role for c-FLIP(L) in the regulation of apoptosis, autophagy, and necroptosis in T lymphocytes. Cell Death Differ. 2013;20:188–97.
Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997;386:517–21.
Bittel M, Kremer AE, Sturzl M, Wirtz S, Stolzer I, Neurath MF, et al. Modulation of the extrinsic cell death signaling pathway by viral Flip induces acute-death mediated liver failure. Cell Death Dis. 2019;10:878.
Belanger C, Gravel A, Tomoiu A, Janelle ME, Gosselin J, Tremblay MJ, et al. Human herpesvirus 8 viral FLICE-inhibitory protein inhibits Fas-mediated apoptosis through binding and prevention of procaspase-8 maturation. J Hum Virol. 2001;4:62–73.
Matta H, Chaudhary PM. Activation of alternative NF-kappa B pathway by human herpes virus 8-encoded Fas-associated death domain-like IL-1 beta-converting enzyme inhibitory protein (vFLIP). Proc Natl Acad Sci USA. 2004;101:9399–404.
Bittel M, Kremer AE, Sturzl M, Wirtz S, Stolzer I, Neurath MF, et al. Modulation of the extrinsic cell death signaling pathway by viral Flip induces acute-death mediated liver failure. Cell Death Dis. 2019;10:878.
Li S, Zhang L, Yao Q, Li L, Dong N, Rong J, et al. Pathogen blocks host death receptor signalling by arginine GlcNAcylation of death domains. Nature. 2013;501:242–6.
Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, et al. Catalytic activity of the caspase-8-FLIPL complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–7.
Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez-Majada V, Ermolaeva M, et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011;477:330–4.
Moriwaki K, Chan FK. RIP3: a molecular switch for necrosis and inflammation. Genes Dev. 2013;27:1640–9.
Wu JF, Huang Z, Ren JM, Zhang ZR, He P, Li YX, et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 2013;23:994–1006.
Cai ZY, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55.
Liu X, Zhang YY, Gao HW, Hou Y, Lu JJ, Feng YL, et al. Induction of an MLKL mediated non-canonical necroptosis through reactive oxygen species by tanshinol A in lung cancer cells. Biochem Pharmacol. 2020;171:113684.
Maelfait J, Liverpool L, Bridgeman A, Ragan KB, Upton JW, Rehwinkel J. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. Embo J. 2017;36:2529–43.
Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH, Ingram JP, et al. RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza A virus. Cell Host Microbe. 2016;20:13–24.
Kim SJ, Li J. Caspase blockade induces RIP3-mediated programmed necrosis in Toll-like receptor-activated microglia. Cell Death Dis. 2013;4:e716.
Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2019;26:564–564.
Sridharan H, Ragan KB, Guo HY, Gilley RP, Landsteiner VJ, Kaiser WJ, et al. Murine cytomegalovirus IE3-dependent transcription is required for DAI/ZBP1-mediated necroptosis. Embo Rep. 2017;18:1429–41.
Vanden Berghe T, Kaiser WJ, Bertrand MJ, Vandenabeele P. Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol Cell Oncol. 2015;2:e975093.
Feoktistova M, Leverkus M. Programmed necrosis and necroptosis signalling. FEBS J. 2015;282:19–31.
Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–21.
Jost PJ, Hockendorf U. Necroinflammation emerges as a key regulator of hematopoiesis in health and disease. Cell Death Differ. 2019;26:53–67.
Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–20.
Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–23.
Najjar M, Suebsuwong C, Ray SS, Thapa RJ, Maki JL, Nogusa S, et al. Structure guided design of potent and selective ponatinib-based hybrid inhibitors for RIPK1. Cell Rep. 2015;10:1850–60.
Conrad M, Angeli JP, Vandenabeele P, Stockwell BR. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Disco. 2016;15:348–66.
Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci USA. 2014;111:16836–41.
Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell. 2019;35:830–49.
Park HH, Kim HR, Park SY, Hwang SM, Hong SM, Park S, et al. RIPK3 activation induces TRIM28 derepression in cancer cells and enhances the anti-tumor microenvironment. Mol Cancer. 2021;20:107.
Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551:247–50.
Chen GQ, Benthani FA, Wu J, Liang D, Bian ZX, Jiang X. Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death Differ. 2020;27:242–54.
Shimba A, Ejima A, Ikuta K. Pleiotropic effects of glucocorticoids on the immune system in circadian rhythm and stress. Front Immunol. 2021;12:706951.
Chovatiya R, Medzhitov R. Stress, inflammation, and defense of homeostasis. Mol Cell. 2014;54:281–8.
von Massenhausen A, Zamora Gonzalez N, Maremonti F, Belavgeni A, Tonnus W, Meyer C, et al. Dexamethasone sensitizes to ferroptosis by glucocorticoid receptor-induced dipeptidase-1 expression and glutathione depletion. Sci Adv. 2022;8:eabl8920.
Tan Y, Zhang M, Pan Y, Feng H, Xie L. Suppression of the caspase-1/GSDMD-mediated pyroptotic signaling pathway through dexamethasone alleviates corneal alkali injuries. Exp Eye Res. 2022;214:108858.
Ballegeer M, Van Looveren K, Timmermans S, Eggermont M, Vandevyver S, Thery F, et al. Glucocorticoid receptor dimers control intestinal STAT1 and TNF-induced inflammation in mice. J Clin Invest. 2018;128:3265–79.
Zerlotin R, Oranger A, Pignataro P, Dicarlo M, Maselli F, Mori G, et al. Irisin and secondary osteoporosis in humans. Int J Mol Sci. 2022;23:690.
Oh S, Yang J, Park C, Son K, Byun K. Dieckol attenuated glucocorticoid-induced muscle atrophy by decreasing NLRP3 inflammasome and pyroptosis. Int J Mol Sci. 2021;22:8057.
Wang L, Jiao XF, Wu C, Li XQ, Sun HX, Shen XY, et al. Trimetazidine attenuates dexamethasone-induced muscle atrophy via inhibiting NLRP3/GSDMD pathway-mediated pyroptosis. Cell Death Disco. 2021;7:251.
Rohde K, Kleinesudeik L, Roesler S, Lowe O, Heidler J, Schroder K, et al. A Bak-dependent mitochondrial amplification step contributes to Smac mimetic/glucocorticoid-induced necroptosis. Cell Death Differ. 2017;24:83–97.
Lee CH, Chuah SK, Tai WC, Chang CC, Chen FJ. Delay in human neutrophil constitutive apoptosis after infection with Klebsiella pneumoniae serotype K1. Front Cell Infect Microbiol. 2017;7:87.
Lamkanfi M, Dixit VM. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe. 2010;8:44–54.
Cannon MJ, Schmid DS, Hyde TB. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev Med Virol. 2010;20:202–13.
Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. 2010;7:302–13.
Ohmer M, Weber A, Sutter G, Ehrhardt K, Zimmermann A, Hacker G. Anti-apoptotic Bcl-XL but not Mcl-1 contributes to protection against virus-induced apoptosis. Cell Death Dis. 2016;7:e2340.
Daley-Bauer LP, Roback L, Crosby LN, McCormick AL, Feng Y, Kaiser WJ, et al. Mouse cytomegalovirus M36 and M45 death suppressors cooperate to prevent inflammation resulting from antiviral programmed cell death pathways. Proc Natl Acad Sci USA. 2017;114:E2786–95.
Garcia LR, Tenev T, Newman R, Haich RO, Liccardi G, John SW, et al. Ubiquitylation of MLKL at lysine 219 positively regulates necroptosis-induced tissue injury and pathogen clearance. Nat Commun. 2021,12:3364.
McCormick AL, Skaletskaya A, Barry PA, Mocarski ES, Goldmacher VS. Differential function and expression of the viral inhibitor of caspase 8-induced apoptosis (vICA) and the viral mitochondria-localized inhibitor of apoptosis (vMIA) cell death suppressors conserved in primate and rodent cytomegaloviruses. Virology. 2003;316:221–33.
Cam M, Handke W, Picard-Maureau M, Brune W. Cytomegaloviruses inhibit Bak- and Bax-mediated apoptosis with two separate viral proteins. Cell Death Differ. 2010;17:655–65.
Jurak I, Brune W. Induction of apoptosis limits cytomegalovirus cross-species infection. Embo J. 2006;25:2634–42.
Skaletskaya A, Bartle LM, Chittenden T, McCormick AL, Mocarski ES, Goldmacher VS. A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc Natl Acad Sci USA. 2001;98:7829–34.
Muscolino E, Castiglioni C, Brixel R, Frascaroli G, Brune W. Species-specific inhibition of necroptosis by HCMV UL36. Viruses 2021,13:2134.
Arnoult D, Bartle LM, Skaletskaya A, Poncet D, Zamzami N, Park PU, et al. Cytomegalovirus cell death suppressor vMIA blocks Bax- but not Bak-mediated apoptosis by binding and sequestering Bax at mitochondria. Proc Natl Acad Sci USA. 2004;101:7988–93.
Boya P, Cohen I, Zamzami N, Vieira HL, Kroemer G. Endoplasmic reticulum stress-induced cell death requires mitochondrial membrane permeabilization. Cell Death Differ. 2002;9:465–7.
Feng Y, Daley-Bauer LP, Roback L, Guo H, Koehler HS, Potempa M, et al. Caspase-8 restricts antiviral CD8 T cell hyperaccumulation. Proc Natl Acad Sci USA. 2019;116:15170–7.
Xu J, Mo J, Liu X, Marshall B, Atherton SS, Dong Z, et al. Depletion of the receptor-interacting protein kinase 3 (RIP3) decreases photoreceptor cell death during the early stages of ocular murine cytomegalovirus infection. Invest Ophthalmol Vis Sci. 2018;59:2445–58.
Lane RK, Guo HY, Fisher AD, Diep J, Lai Z, Chen YD, et al. Necroptosis-based CRISPR knockout screen reveals Neuropilin-1 as a critical host factor for early stages of murine cytomegalovirus infection. Proc Natl Acad Sci USA. 2020;117:20109–16.
Fletcher-Etherington A, Nobre L, Nightingale K, Antrobus R, Nichols J, Davison AJ, et al. Human cytomegalovirus protein pUL36: a dual cell death pathway inhibitor. Proc Natl Acad Sci USA. 2020;117:18771–9.
Altman AM, Miller MJ, Mahmud J, Smith NA, Chan GC. Human cytomegalovirus-induced autophagy prevents necroptosis of infected monocytes. J Virol. 2020;94:e01022–20.
Palese P. The genes of influenza virus. Cell. 1977;10:1–10.
Kumar D, Michaels MG, Morris MI, Green M, Avery RK, Liu C, et al. Outcomes from pandemic influenza A H1N1 infection in recipients of solid-organ transplants: a multicentre cohort study. Lancet Infect Dis. 2010;10:521–6.
Stray SJ, Air GM. Apoptosis by influenza viruses correlates with efficiency of viral mRNA synthesis. Virus Res. 2001;77:3–17.
Herold S, Ludwig S, Pleschka S, Wolff T. Apoptosis signaling in influenza virus propagation, innate host defense, and lung injury. J Leukoc Biol. 2012;92:75–82.
Arora S, Lim W, Bist P, Perumalsamy R, Lukman HM, Li F, et al. Influenza A virus enhances its propagation through the modulation of Annexin-A1 dependent endosomal trafficking and apoptosis. Cell Death Differ. 2016;23:1243–56.
Kuriakose T, Man SM, Malireddi RKS, Karki R, Kesavardhana S, Place DE, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol. 2016;1:aag2045.
Oltean T, Van San E, Divert T, Vanden Berghe T, Saelens X, Maelfait J, et al. Viral dosing of influenza A infection reveals involvement of RIPK3 and FADD, but not MLKL. Cell Death Dis. 2021;12:471.
Zhang T, Yin C, Boyd DF, Quarato G, Ingram JP, Shubina M, et al. Influenza virus Z-RNAs induce ZBP1-mediated necroptosis. Cell. 2020;180:1115–29 e1113.
Jiao H, Wachsmuth L, Kumari S, Schwarzer R, Lin J, Eren RO, et al. Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature. 2020;580:391–5.
Zheng M, Kanneganti TD. The regulation of the ZBP1-NLRP3 inflammasome and its implications in pyroptosis, apoptosis, and necroptosis (PANoptosis). Immunol Rev. 2020;297:26–38.
Kuriakose T, Zheng M, Neale G, Kanneganti TD. IRF1 is a transcriptional regulator of ZBP1 promoting NLRP3 inflammasome activation and cell death during influenza virus infection. J Immunol. 2018;200:1489–95.
Downey J, Pernet E, Coulombe F, Allard B, Meunier I, Jaworska J, et al. RIPK3 interacts with MAVS to regulate type I IFN-mediated immunity to Influenza A virus infection. PLos Pathog. 2017;13:e1006326.
Shubina M, Tummers B, Boyd DF, Zhang T, Yin CR, Gautam A, et al. Necroptosis restricts influenza A virus as a stand-alone cell death mechanism. J Exp Med. 2020;217:e20191259.
Xu YL, Tang HL, Peng HR, Zhao P, Qi ZT, Wang W. RIP3 deficiency ameliorates inflammatory response in mice infected with influenza H7N9 virus infection. Oncotarget. 2017;8:27715–24.
Gaba A, Xu F, Lu Y, Park HS, Liu G, Zhou Y. The NS1 protein of influenza A virus participates in necroptosis by interacting with MLKL and increasing its oligomerization and membrane translocation. J Virol. 2019;93:e01835-18.
Lee ACY, Zhang AJX, Chu H, Li C, Zhu H, Mak WWN, et al. H7N9 influenza A virus activation of necroptosis in human monocytes links innate and adaptive immune responses. Cell Death Dis. 2019;10:442.
Peiris JSM, de Jong MD, Guan Y. Avian influenza virus (H5N1): a threat to human health. Clin Microbiol Rev. 2007;20:243–67.
Vidana B, Martinez J, Martinez-Orellana P, Garcia-Migura L, Montoya M, Martorell J, et al. Heterogeneous pathological outcomes after experimental pH1N1 influenza infection in ferrets correlate with viral replication and host immune responses in the lung. Vet Res. 2014;45:85.
Wang ZF, Loh L, Kedzierski L, Kedzierska K. Avian influenza viruses, inflammation, and CD8(+) T cell immunity. Front Immunol. 2016;7:1–10.
Ungchusak K, Auewarakul P, Dowell SF, Kitphati R, Auwanit W, Puthavathana P, et al. Probable person-to-person transmission of avian influenza A (H5N1). N. Engl J Med. 2005;352:333–40.
Li FC, Choi BC, Sly T, Pak AW. Finding the real case-fatality rate of H5N1 avian influenza. J Epidemiol Community Health. 2008;62:555–9.
Bitzer M, Prinz F, Bauer M, Spiegel M, Neubert WJ, Gregor M, et al. Sendai virus infection induces apoptosis through activation of caspase-8 (FLICE) and caspase-3 (CPP32). J Virol. 1999;73:702–8.
Huang YF, Liu H, Li SL, Tang YJ, Wei B, Yu HS, et al. MAVS-MKK7-JNK2 defines a novel apoptotic signaling pathway during viral infection. PLos Pathog. 2014,10:e1004020.
Schock SN, Chandra NV, Sun Y, Irie T, Kitagawa Y, Gotoh B, et al. Induction of necroptotic cell death by viral activation of the RIG-I or STING pathway. Cell Death Differ. 2017;24:615–25.
Koyama AH, Irie H, Kato A, Nagai Y, Adachi A. Virus multiplication and induction of apoptosis by Sendai virus: role of the C proteins. Microbes Infect. 2003;5:373–8.
Majowicz SE, Musto J, Scallan E, Angulo FJ, Kirk M, O’Brien SJ, et al. The global burden of nontyphoidal Salmonella gastroenteritis. Clin Infect Dis. 2010;50:882–9.
Crump JA, Luby SP, Mintz ED. The global burden of typhoid fever. B World Health Organ. 2004;82:346–53.
Verbrugghe E, Dhaenens M, Leyman B, Boyen F, Shearer N, Van Parys A, et al. Host stress drives Salmonella recrudescence. Sci Rep. 2016;6:20849.
Jones BD, Falkow S. Salmonellosis: host immune responses and bacterial virulence determinants. Annu Rev Immunol. 1996;14:533–61.
Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol. 2012;13:954–62.
Monack DM, Raupach B, Hromockyj AE, Falkow S. Salmonella typhimurium invasion induces apoptosis in infected macrophages. Proc Natl Acad Sci USA. 1996;93:9833–8.
Boise LH, Collins CM. Salmonella-induced cell death: apoptosis, necrosis or programmed cell death? Trends Microbiol. 2001;9:64–67.
Zeng H, Wu HX, Sloane V, Jones R, Yu YM, Lin P, et al. Flagellin/TLR5 responses in epithelia reveal intertwined activation of inflammatory and apoptotic pathways. Am J Physiol Gastr L. 2006;290:G96–108.
Bleriot C, Lecuit M. The interplay between regulated necrosis and bacterial infection. Cell Mol Life Sci. 2016;73:2369–78.
Jorgensen I, Zhang Y, Krantz BA, Miao EA. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med. 2016;213:2113–28.
Albaghdadi H, Robinson N, Finlay B, Krishnan L, Sad S. Selectively reduced intracellular proliferation of Salmonella enterica Serovar Typhimurium within APCs limits antigen presentation and development of a rapid CD8 T cell response. J Immunol. 2009;183:3778–87.
Hefele M, Stolzer I, Ruder B, He GW, Mahapatro M, Wirtz S, et al. Intestinal epithelial Caspase-8 signaling is essential to prevent necroptosis during Salmonella Typhimurium induced enteritis. Mucosal Immunol. 2018;11:1191–202.
Hos NJ, Ganesan R, Gutierrez S, Hos D, Klimek J, Abdullah Z, et al. Type I interferon enhances necroptosis of Salmonella Typhimurium-infected macrophages by impairing antioxidative stress responses. J Cell Biol. 2017;216:4107–21.
Shutinoski B, Alturki NA, Rijal D, Bertin J, Gough PJ, Schlossmacher MG, et al. K45Amutation of RIPK1 results in poor necroptosis and cytokine signaling in macrophages, which impacts inflammatory responses in vivo. Cell Death Differ. 2016;23:1628–37.
Wemyss MA, Pearson JS. Host cell death responses to non-typhoidal Salmonella Infection. Front Immunol. 2019;10:1758.
Figueira R, Holden DW. Functions of the Salmonella pathogenicity island 2 (SPI-2) type III secretion system effectors. Microbiology. 2012;158:1147–61.
Gunster RA, Matthews SA, Holden DW, Thurston TLM. SseK1 and SseK3 type III secretion system effectors inhibit NF-kappa B signaling and necroptotic cell death in Salmonella-infected macrophages. Infect Immun. 2017;85:e00010–17.
Dong K, Zhu Y, Deng Q, Sun L, Yang S, Huang K, et al. Salmonella pSLT-encoded effector SpvB promotes RIPK3-dependent necroptosis in intestinal epithelial cells. Cell Death Disco. 2022;8:44.
Wemyss MA, Pearson JS. Host cell death responses to non-typhoidal Salmonella infection. Front Immunol. 2019;10:1758.
Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–95.
Arpaia N, Godec J, Lau L, Sivick KE, McLaughlin LM, Jones MB, et al. TLR signaling is required for Salmonella typhimurium virulence. Cell. 2011;144:675–88.
Broz P, Ohlson MB, Monack DM. Innate immune response to Salmonella typhimurium, a model enteric pathogen. Gut Microbes. 2012;3:62–70.
Hatai H, Lepelley A, Zeng WY, Hayden MS, Ghosh S. Toll-like receptor 11 (TLR11) interacts with flagellin and profilin through disparate mechanisms. PLos ONE 2016;11:e0148987.
Zhan RH, Han QJ, Zhang C, Tian ZG, Zhang J. Toll-like receptor 2 (TLR2) and TLR9 play opposing roles in host innate immunity against Salmonella enterica Serovar Typhimurium infection. Infect Immun. 2015;83:1641–9.
He SD, Liang YQ, Shao F, Wang XD. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci USA. 2011;108:20054–9.
de Jong HK, Parry CM, van der Poll T, Wiersinga WJ. Host-pathogen interaction in invasive Salmonellosis. PLos Pathog. 2012;8:e1002933.
Gutierrez-Jimenez C, Mora-Cartin R, Altamirano-Silva P, Chacon-Diaz C, Chaves-Olarte E, Moreno E, et al. Neutrophils as Trojan horse vehicles for Brucella abortus macrophage infection. Front Immunol. 2019;10:1012.
Choy SLK, Boyle EC, Gal-Mor O, Goode DL, Valdez Y, Vallance BA, et al. SseK1 and SseK2 are novel translocated proteins of Salmonella enterica serovar typhimurium. Infect Immun. 2004;72:5115–25.
Doerflinger M, Deng Y, Whitney P, Salvamoser R, Engel S, Kueh AJ, et al. Flexible usage and interconnectivity of diverse cell death pathways protect against intracellular infection. Immunity. 2020;53:533–47 e537.
Hoelzer K, Switt AIM, Wiedmann M. Animal contact as a source of human non-typhoidal salmonellosis. Vet Res. 2011;42:34.
Haesendonck R, Rasschaert G, Martel A, Verbrugghe E, Heyndrickx M, Haesebrouck F, et al. Feral pigeons: a reservoir of zoonotic Salmonella Enteritidis strains? Vet Microbiol. 2016;195:101–3.
Yue M, Han X, De Masi L, Zhu C, Ma X, Zhang J, et al. Allelic variation contributes to bacterial host specificity. Nat Commun. 2015;6:8754.
Verbrugghe E, Dhaenens M, Leyman B, Boyen F, Shearer N, Van Parys A, et al. Host stress drives Salmonella recrudescence. Sci Rep. 2016;6:20849.
Srinivasan L, Ahlbrand S, Briken V. Interaction of Mycobacterium tuberculosis with host cell death pathways. Cold Spring Harb Perspect Med. 2014;4:a022459.
Houben EN, Nguyen L, Pieters J. Interaction of pathogenic mycobacteria with the host immune system. Curr Opin Microbiol. 2006;9:76–85.
Moraco AH, Kornfeld H. Cell death and autophagy in tuberculosis. Semin Immunol. 2014;26:497–511.
Etna MP, Giacomini E, Pardini M, Severa M, Bottai D, Cruciani M, et al. Impact of Mycobacterium tuberculosis RD1-locus on human primary dendritic cell immune functions. Sci Rep. 2015;5:17078.
Behar SM, Divangahi M, Remold HG. Evasion of innate immunity by Mycobacterium tuberculosis: is death an exit strategy? Nat Rev Microbiol. 2010;8:668–74.
Afriyie-Asante A, Dabla A, Dagenais A, Berton S, Smyth R, Sun J. Mycobacterium tuberculosis exploits focal adhesion kinase to induce necrotic cell death and inhibit reactive oxygen species production. Front Immunol. 2021;12:742370.
Roca FJ, Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell. 2013;153:521–34.
Butler RE, Krishnan N, Garcia-Jimenez W, Francis R, Martyn A, Mendum T, et al. Susceptibility of Mycobacterium tuberculosis-infected host cells to phospho-MLKL driven necroptosis is dependent on cell type and presence of TNFalpha. Virulence. 2017;8:1820–32.
Stutz MD, Ojaimi S, Allison C, Preston S, Arandjelovic P, Hildebrand JM, et al. Necroptotic signaling is primed in Mycobacterium tuberculosis-infected macrophages, but its pathophysiological consequence in disease is restricted. Cell Death Differ. 2018;25:951–65.
Zhang YY, Su SS, Zhao SB, Yang ZT, Zhong CQ, Chen X, et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun. 2017;8:1–14.
Grab J, Suarez I, van Gumpel E, Winter S, Schreiber F, Esser A, et al. Corticosteroids inhibit Mycobacterium tuberculosis-induced necrotic host cell death by abrogating mitochondrial membrane permeability transition. Nat Commun. 2019;10:688.
Pajuelo D, Gonzalez-Juarbe N, Tak U, Sun J, Orihuela CJ, Niederweis M. NAD(+) depletion triggers macrophage necroptosis, a cell death pathway exploited by Mycobacterium tuberculosis. Cell Rep. 2018;24:429–40.
Pajuelo D, Gonzalez-Juarbe N, Niederweis M. NAD hydrolysis by the tuberculosis necrotizing toxin induces lethal oxidative stress in macrophages. Cell Microbiol. 2020;22:e13115.
Brosch R, Gordon SV, Marmiesse M, Brodin P, Buchrieser C, Eiglmeier K, et al. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci USA. 2002;99:3684–9.
Abdallah AM, Bestebroer J, Savage ND, de Punder K, van Zon M, Wilson L, et al. Mycobacterial secretion systems ESX-1 and ESX-5 play distinct roles in host cell death and inflammasome activation. J Immunol. 2011;187:4744–53.
Wong KW, Jacobs WR Jr. Critical role for NLRP3 in necrotic death triggered by Mycobacterium tuberculosis. Cell Microbiol. 2011;13:1371–84.
Chen Y, Chao Y, Deng Q, Liu T, Xiang J, Chen J, et al. Potential challenges to the Stop TB Plan for humans in China; cattle maintain M. bovis and M. tuberculosis. Tuberculosis. 2009;89:95–100.
Mahairas GG, Sabo PJ, Hickey MJ, Singh DC, Stover CK. Molecular analysis of genetic differences between Mycobacterium bovis BCG and virulent M. bovis. J Bacteriol. 1996;178:1274–82.
Ernst PB, Gold BD. The disease spectrum of Helicobacter pylori: the immunopathogenesis of gastroduodenal ulcer and gastric cancer. Annu Rev Microbiol. 2000;54:615–40.
Atherton JC, Blaser MJ. Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J Clin Invest. 2009;119:2475–87.
Cover TL, Krishna US, Israel DA, Peek RM Jr. Induction of gastric epithelial cell apoptosis by Helicobacter pylori vacuolating cytotoxin. Cancer Res. 2003;63:951–7.
Gong M, Ling SS, Lui SY, Yeoh KG, Ho B. Helicobacter pylori gamma-glutamyl transpeptidase is a pathogenic factor in the development of peptic ulcer disease. Gastroenterology. 2010;139:564–73.
Flahou B, Haesebrouck F, Chiers K, Van Deun K, De Smet L, Devreese B, et al. Gastric epithelial cell death caused by Helicobacter suis and Helicobacter pylori gamma-glutamyl transpeptidase is mainly glutathione degradation-dependent. Cell Microbiol. 2011;13:1933–55.
Radin JN, Gonzalez-Rivera C, Ivie SE, McClain MS, Cover TL. Helicobacter pylori VacA induces programmed necrosis in gastric epithelial cells. Infect Immun. 2011;79:2535–43.
Haesebrouck F, Pasmans F, Flahou B, Chiers K, Baele M, Meyns T, et al. Gastric helicobacters in domestic animals and nonhuman primates and their significance for human health. Clin Microbiol Rev. 2009;22:202–23.
Hellemans A, Chiers K, De Bock M, Decostere A, Haesebrouck F, Ducatelle R, et al. Prevalence of ‘Candidatus Helicobacter suis’ in pigs of different ages. Vet Rec. 2007;161:189–92.
De Cooman L, Houf K, Smet A, Flahou B, Ducatelle R, De Bruyne E, et al. Presence of Helicobacter suis on pork carcasses. Int J Food Microbiol. 2014;187:73–76.
Zhang G, Ducatelle R, Pasmans F, D’Herde K, Huang L, Smet A, et al. Effects of Helicobacter suis gamma-glutamyl transpeptidase on lymphocytes: modulation by glutamine and glutathione supplementation and outer membrane vesicles as a putative delivery route of the enzyme. PLoS ONE. 2013;8:e77966.
Ochoa TJ, Contreras CA. Enteropathogenic escherichia coli infection in children. Curr Opin Infect Dis. 2011;24:478–83.
Croxen MA, Law RJ, Scholz R, Keeney KM, Wlodarska M, Finlay BB. Recent advances in understanding enteric pathogenic Escherichia coli. Clin Microbiol Rev. 2013;26:822–80.
Gonzalez-Juarbe N, Gilley RP, Hinojosa CA, Bradley KM, Kamei A, Gao G, et al. Pore-forming toxins induce macrophage necroptosis during acute bacterial pneumonia. PLos Pathog. 2015;11:e1005337.
Pearson JS, Giogha C, Ong SY, Kennedy CL, Kelly M, Robinson KS, et al. A type III effector antagonizes death receptor signalling during bacterial gut infection. Nature. 2013;501:247–51.
Pearson JS, Giogha C, Muhlen S, Nachbur U, Pham CL, Zhang Y, et al. EspL is a bacterial cysteine protease effector that cleaves RHIM proteins to block necroptosis and inflammation. Nat Microbiol. 2017;2:16258.
Lacey CA, Miao EA. Programmed cell death in the evolutionary race against bacterial virulence factors. Cold Spring Harb Perspect Biol. 2019;12:a036459.
Russo TA, Marr CM. Hypervirulent Klebsiella pneumoniae. Clin Microbiol Rev. 2019;32:e00001-19. .
Codo AC, Saraiva AC, Dos Santos LL, Visconde MF, Gales AC, Zamboni DS, et al. Inhibition of inflammasome activation by a clinical strain of Klebsiella pneumoniae impairs efferocytosis and leads to bacterial dissemination. Cell Death Dis. 2018;9:1182.
Ahn D, Prince A. Participation of necroptosis in the host response to acute bacterial pneumonia. J Innate Immun. 2017;9:262–70.
Jondle CN, Gupta K, Mishra BB, Sharma J. Klebsiella pneumoniae infection of murine neutrophils impairs their efferocytic clearance by modulating cell death machinery. PLos Pathog. 2018;14:e1007338.
Kiu R, Hall LJ. An update on the human and animal enteric pathogen Clostridium perfringens. Emerg Microbes Infect. 2018;7:141.
Autheman D, Wyder M, Popoff M, D’Herde K, Christen S, Posthaus H. Clostridium perfringens beta-toxin induces necrostatin-inhibitable, calpain-dependent necrosis in primary porcine endothelial cells. PLoS ONE. 2013;8:e64644.
Shrestha A, Mehdizadeh Gohari I, McClane BA. RIP1, RIP3, and MLKL contribute to cell death caused by Clostridium perfringens Enterotoxin. mBio 2019;10:e02985-19.
Walker DM, Oghumu S, Gupta G, McGwire BS, Drew ME, Satoskar AR. Mechanisms of cellular invasion by intracellular parasites. Cell Mol Life Sci. 2014;71:1245–63.
Farias Luz N, Balaji S, Okuda K, Barreto AS, Bertin J, Gough PJ, et al. RIPK1 and PGAM5 control Leishmania replication through distinct mechanisms. J Immunol. 2016;196:5056–63.
Mengying Z, Yiyue X, Tong P, Yue H, Limpanont Y, Ping H, et al. Apoptosis and necroptosis of mouse hippocampal and parenchymal astrocytes, microglia and neurons caused by Angiostrongylus cantonensis infection. Parasit Vectors. 2017;10:611.
Mendez OA, Koshy AA. Toxoplasma gondii: Entry, association, and physiological influence on the central nervous system. PLos Pathog. 2017;13:e1006351.
DeLaney AA, Berry CT, Christian DA, Hart A, Bjanes E, Wynosky-Dolfi MA, et al. Caspase-8 promotes c-Rel-dependent inflammatory cytokine expression and resistance against Toxoplasma gondii. Proc Natl Acad Sci USA. 2019;116:11926–35.
Yao Y, Liu M, Ren C, Shen J, Ji Y. Exogenous tumor necrosis factor-alpha could induce egress of Toxoplasma gondii from human foreskin fibroblast cells. Parasite. 2017;24:45.
Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315:801–10.
Lelubre C, Vincent JL. Mechanisms and treatment of organ failure in sepsis. Nat Rev Nephrol. 2018;14:417–27.
Bahar I, Elay G, Baskol G, Sungur M, Donmez-Altuntas H. Increased DNA damage and increased apoptosis and necrosis in patients with severe sepsis and septic shock. J Crit Care. 2018;43:271–5.
Pinheiro da Silva F, Nizet V. Cell death during sepsis: integration of disintegration in the inflammatory response to overwhelming infection. Apoptosis. 2009;14:509–21.
Girardot T, Rimmele T, Venet F, Monneret G. Apoptosis-induced lymphopenia in sepsis and other severe injuries. Apoptosis. 2017;22:295–305.
Cao C, Yu M, Chai Y. Pathological alteration and therapeutic implications of sepsis-induced immune cell apoptosis. Cell Death Dis. 2019;10:782.
Sharma A, Matsuo S, Yang WL, Wang ZM, Wang P. Receptor-interacting protein kinase 3 deficiency inhibits immune cell infiltration and attenuates organ injury in sepsis. Crit. Care 2014;18:R142.
Bolognese AC, Yang WL, Hansen LW, Denning NL, Nicastro JM, Coppa GF, et al. Inhibition of necroptosis attenuates lung injury and improves survival in neonatal sepsis. Surgery. 2018;S0039-6060:30096–5.
Li JY, Ren C, Wang LX, Yao RQ, Dong N, Wu Y, et al. Sestrin2 protects dendrite cells against ferroptosis induced by sepsis. Cell Death Dis. 2021;12:834.
Wang Z, Gu ZY, Hou Q, Chen WJ, Mu D, Zhang YX, et al. Zebrafish GSDMEb cleavage-gated pyroptosis drives septic acute kidney injury in vivo. J Immunol. 2020;204:1929–42.
Liu L, Sun BW. Neutrophil pyroptosis: new perspectives on sepsis. Cell Mol Life Sci. 2019;76:2031–42.
Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol. 2018;3:eaat2738.
Chen H, Li YS, Wu JF, Li GP, Tao X, Lai KM, et al. RIPK3 collaborates with GSDMD to drive tissue injury in lethal polymicrobial sepsis. Cell Death Differ. 2020;27:2568–85.
He RY, Liu BH, Xiong R, Geng BX, Meng H, Lin WC, et al. Itaconate inhibits ferroptosis of macrophage via Nrf2 pathways against sepsis-induced acute lung injury. Cell Death Discov. 2022;8:43.
Acknowledgements
The authors are grateful to Prof. Astar Winoto from the University of California, Berkeley, for his valuable comments and discussion.
Funding
This study was supported by the Agricultural Science and Technology Innovation Program (ASTIP-IAS15), Central Public-interest Scientific Institution Basal Research Fund (2021-YWF-ZYSQ-10), National Natural Science Foundation of Guangdong, China (2021A1515010478), and the National Natural Science Foundation of China (32100734).
Author information
Authors and Affiliations
Contributions
GZ, JW, and ZZ designed the study and drafted the manuscript. TX, XF, QS, AR, and CRL assisted in manuscript preparation and designed the figures. HJ and JD designed and supervised the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Hans-Uwe Simon
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, G., Wang, J., Zhao, Z. et al. Regulated necrosis, a proinflammatory cell death, potentially counteracts pathogenic infections. Cell Death Dis 13, 637 (2022). https://doi.org/10.1038/s41419-022-05066-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-022-05066-3
This article is cited by
-
Erythronecroptosis: an overview of necroptosis or programmed necrosis in red blood cells
Molecular and Cellular Biochemistry (2024)
-
Macrophage PTEN controls STING-induced inflammation and necroptosis through NICD/NRF2 signaling in APAP-induced liver injury
Cell Communication and Signaling (2023)
-
A positive feedback cycle between the alarmin S100A8/A9 and NLRP3 inflammasome-GSDMD signalling reinforces the innate immune response in Candida albicans keratitis
Inflammation Research (2023)
