
Abstract
Cannabinoids are known to modulate oligodendrogenesis and developmental CNS myelination. However, the cell-autonomous action of these compounds on oligodendroglial cells in vivo, and the molecular mechanisms underlying these effects have not yet been studied. Here, by using oligodendroglial precursor cell (OPC)-targeted genetic mouse models, we show that cannabinoid CB1 receptors exert an essential role in modulating OPC differentiation at the critical periods of postnatal myelination. We found that selective genetic inactivation of CB1 receptors in OPCs in vivo perturbs oligodendrogenesis and postnatal myelination by altering the RhoA/ROCK signaling pathway, leading to hypomyelination, and motor and cognitive alterations in young adult mice. Conversely, pharmacological CB1 receptor activation, by inducing E3 ubiquitin ligase-dependent RhoA proteasomal degradation, promotes oligodendrocyte development and CNS myelination in OPCs, an effect that was not evident in OPC-specific CB1 receptor-deficient mice. Moreover, pharmacological inactivation of ROCK in vivo overcomes the defects in oligodendrogenesis and CNS myelination, and behavioral alterations found in OPC-specific CB1 receptor-deficient mice. Overall, this study supports a cell-autonomous role for CB1 receptors in modulating oligodendrogenesis in vivo, which may have a profound impact on the scientific knowledge and therapeutic manipulation of CNS myelination by cannabinoids.
Similar content being viewed by others


Introduction
During developmental CNS myelination, oligodendrocyte progenitor cells (OPCs) proliferate, migrate, and differentiate into mature myelinating oligodendrocytes (OLs), which generate the myelin sheath internode and, thereby, interact with axons to organize the nodal, paranodal, and juxtaparanodal regions [1, 2]. Thus, the OL developmental program is temporally and spatially controlled by a high number of extracellular signals that coordinately regulate essential intracellular signaling pathways and their downstream transcriptional programs. Alterations in essential genes modulating OPC differentiation and/or OL maturation cause myelination defects, which is translated into neuronal dysfunction and, eventually, behavioral alterations in mice [2,3,4,5]. Despite the identification of a high number of these regulatory signals, the whole molecular network that controls developmental oligodendrogenesis and CNS myelination has remained incomplete.
Almost two decades after the first evidence supporting that cannabinoid compounds modulate OL development and CNS myelination [6, 7], several pharmacological studies have shown that synthetic cannabinoids [8], phytocannabinoids [9], and endocannabinoids (eCBs) [10] modulate oligodendrogenesis and CNS myelination. Elevated levels of the endocannabinoid 2-arachidonoylglycerol (2-AG) upon pharmacological inhibition of the 2-AG-degrading enzyme monoacylglycerol lipase (MAGL) enhances OL development in cultured OPCs [11], both at embryonic stages in mice [10] and in the Theiler’s murine encephalomyelitis virus (TMEV) progressive mouse model of multiple sclerosis (MS) [12, 13]. Moreover, administration of the phytocannabinoid Δ9-tetrahydrocannabinol (THC) [9] or of synthetic cannabinoids [8] promotes OPC differentiation and developmental CNS myelination, as well as OL regeneration and functional CNS remyelination upon cuprizone-induced demyelination [14]. Moreover, the phytocannabinoid cannabidiol (CBD) prevents hypoxia/ischemia-induced hypomyelination in newborn rats [15]. Of note, collectively, these studies have only been based on systemic pharmacological approaches. Owing to the abundant expression of CB1 receptors in various neuronal and glial cell populations [16], the cell-autonomous action of cannabinoid compounds in OPCs to modulate OL development in vivo has remained unexplored. Thus, deciphering the cellular neurobiology of the eCB system may help to identify the cellular targets of cannabinoids under physiological or pathophysiological settings.
Here, by using genetic mouse models aimed to inactivate CB1 receptor gene expression selectively in OPCs, we show that CB1 receptors exert an essential function in modulating OPC differentiation and oligodendrogenesis during postnatal myelination in vivo. We found that selective depletion of CB1 receptor signaling in OPCs, by altering the RhoA/ROCK signaling pathway, prevents cell differentiation, perturbs oligodendrogenesis and postnatal myelination, and causes hypomyelination as well as motor and cognitive defects at young adult ages. Moreover, pharmacological inactivation of ROCK in vivo overcomes the defects in oligodendrogenesis and functional CNS myelination of CB1 receptor deficient OPCs. This study supports an essential role for CB1 receptors in modulating OPC functions and CNS myelination, which may contribute to understanding the complex molecular network that controls CNS myelination.
Results
Selective CB1 receptor gene inactivation in OPCs in vivo
To interrogate a cell-autonomous role for CB1 receptors in the modulation of oligodendrogenesis and CNS myelination in vivo, we generated a new mouse line by crossing the CB1f/f [17] mouse line with the Ng2-CreERT2 [18] and Rosa-stop-Ai6 [19] mouse lines. This would conceivably deplete CB1 receptors gene (Cnr1) expression selectively in OPCs upon tamoxifen (TAM) administration, and ZsGreen1 fluorescently label OPCs to track OL differentiation along the process of CNS myelination (Fig. S1A). We found approximately 77.7% of recombination efficiency in Ng2/Ai6-CB1KO and Ng2/Ai6-CB1HET mice by quantifying the percentage of oligodendroglial-lineage Olig2+ cells that expressed the Ai6 recombinant transgene by immunofluorescence analysis in the developing corpus callosum (CC) (Fig. S1B). We verified the recombination in the CB1 receptor locus by genomic DNA analysis of Ng2/Ai6-CB1KO and Ng2/Ai6-CB1HET CC FAC-sorted cells at postnatal day 10 (P10) (Fig. S1C, D) [20]. We also confirmed CB1 protein depletion in Ng2/Ai6-CB1KO CC isolated cells at P10 by immunofluorescence (Fig. S1E).
CB1 receptor gene inactivation in OPCs disrupts postnatal oligodendrogenesis
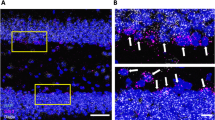
To study the role for CB1 receptors in oligodendrogenesis, we induced TAM-driven recombination in Ng2/Ai6-CB1KO and Ng2/Ai6-CB1HET mice at P6 and P7 and performed a differentiation-state analysis of the OL-lineage populations by immunofluorescence in the CC at P15 and at a young adult age (P60) (Fig. 1A). Analysis revealed a higher proportion of NG2+ OPCs and a reduced proportion of CC1+ OLs within the recombinant Ai6+ population in the CC of Ng2/Ai6-CB1KO mice compared to their controls (Ng2/Ai6-CB1HET), thus pointing to a blockade of OPC differentiation (Fig. 1B and Fig. S2A). Similar data were obtained when analyzing the Olig2+ cell population in the Ng2-CB1KO mouse line at P15 (Fig. 1C and Fig. S2B). Ng2-CB1KO mice also showed a reduced density of CC1+ OLs compared to their respective controls (Ng2-CB1WT mice) at both ages (Fig. 1C). We did not find differences in Olig2+ cell densities between Ng2-CB1KO mice and their control Ng2-CB1WT littermates at both ages (Fig. 1C), indicating that CB1 receptor gene inactivation in OPCs disrupts cell differentiation without affecting OPC cell survival. We provided further support of these data by generating another OPC-specific mouse line upon crossing Pdgfrα-Cre [21] and CB1f/f animals. Pdgfrα-CB1KO mice also showed a reduced CC1+ OL cell density as well as a reduced proportion of CC1+ cells among Olig2+ cells in the developing CC compared to their control Pdgfrα-CB1WT mice at P15 (Fig. 1D and Fig. S2C). Taken together, these results indicate that CB1 receptors regulate OPC differentiation during postnatal development in a cell autonomous manner.

A Timeline of tamoxifen administrations and time points of analysis. Tamoxifen-driven recombination was induced at postnatal day 6 (P6-P7) in B Ng2/Ai6-CB1KO, C, E Ng2-CB1KO, D Pdgfrα-CB1KO and control mice, and oligodendrogenesis was analyzed in the corpus callosum at P15 and P60. Immunofluorescence analysis and quantification of B the percentage of NG2+ OPCs, CC1+ OLs and GSTπ+CC1+ mature OLs among the recombinant Rosa-Ai6+ population at P15, (C, left panel, D) the percentage of NG2+ OPCs, CC1+ OLs among Olig2+ cells at P15, (C, D) Olig2+ and CC1+ OL cell densities at P15 C, D and P60 C, and E the percentage of mature GSTπ+ OLs among CC1+ OLs at P15 and P60. Arrows in B indicate Olig2+CC1neg cells. Data are shown as mean + /− SEM. n = 4 for B, n = 5–9 for C, n = 3 for D, n = 3–5 for E independent data points used per experimental group. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 vs Ng2/Ai6-CB1HET, Ng2-CB1WT or Pdgfrα-CB1WT mice, by two-tailed unpaired Student’s t-test for B, D, E, and by two-tailed unpaired Student’s t-test or Mann Whitney test for C. Scale bars, 80 µm for B, and 30 µm for C and E’.
Next, we studied the maturation state of the OL population in the CC of OPC-CB1 receptor-deficient mice. Immunofluorescence analysis of OL (CC1) and mature myelinating OL markers (glutathione S-transferase P, GSTπ; myelin-associated glycoprotein MAG) showed a reduced density of GSTπ+CC1+ or MAG+ mature myelinating OLs (Fig. S3A, B), together with a reduced proportion of myelinating GSTπ+ OLs within CC1+ OLs cells, in Ng2-CB1KO mice compared to their Ng2-CB1WT control mice at P15 and P60 (Fig. 1E). We also found a reduced proportion of CC1+GSTπ+ cells within the Ai6+ population in the CC of Ng2/Ai6-CB1KO mice compared to their Ng2/Ai6-CB1HET controls (Fig. 1B and Fig. S3C), thus suggesting that CB1 receptors modulate not only OPC differentiation but also OL maturation. Altogether, these findings indicate that CB1 receptor ablation in OPCs prevents cell differentiation and disrupts oligodendrogenesis during the critical period of postnatal myelination.
CB1 receptor gene inactivation in OPCs disrupts postnatal CNS myelination
To address whether the observed defects in oligodendrogenesis shown by Ng2-CB1KO and Pdgfrα-CB1KO mice impact the myelination process, we analyzed myelination in the CC of these mice and their respective controls at P15. Western blot analysis of dissected CC extracts revealed reduced levels of myelin-associated proteins, such as MAG, myelin oligodendrocyte glycoprotein (MOG), and myelin basic protein (MBP) from Ng2-CB1KO and Pdgfrα-CB1KO mice compared to their controls (Ng2-CB1WT and Pdgfrα-CB1WT mice respectively) (Fig. 2A). Fluoromyelin staining, as well as MBP and proteolipid protein (PLP) immunofluorescence analysis in the developing CC, supported the hypomyelinated phenotype of Ng2-CB1KO mice compared to their respective controls (Fig. 2B, C). Moreover, ultrastructural analysis by electron microscopy showed a reduced density of myelinated axons in CC of Ng2-CB1KO mice compared to their controls (Fig. 2D). These results indicate that CB1 receptor deficiency in OPCs at early postnatal ages disrupts developmental CNS myelination.

Tamoxifen-driven recombination was induced in Ng2-CB1KO, Pdgfrα-CB1KO and control mice at postnatal day 6 (P6-P7) and tissue was analyzed in the corpus callosum (CC) at P15 and P60. A and F Western blot analysis of myelin protein levels, such as myelin-associated glycoprotein (MAG), myelin oligodendrocyte glycoprotein (MOG), and myelin basic protein (MBP) from CC extracts at P15 (A) or P60 (F), or cerebellar (Crb), hippocampal (Hipp), and medial prefrontal cortex (mPFC) extracts at P60 (F). Quantification of optical density (O.D.). B Immunofluorescence analysis of proteolipid protein (PLP) and MBP in Ng2-CB1KO and Ng2-CB1WT mice at P15. C Fluoromyelin staining in the CC at P15. D Electron microscopy analysis and quantification of myelinated axon density in Ng2-CB1KO and Ng2-CB1WT mice at P15. E Representative images of Sudan black staining in the CC of Ng2-CB1KO and Ng2-CB1WT mice at P60. G Immunofluorescence analysis and quantification of node density and paranode (Caspr) and node (Nav1.6) length in the CC of Ng2-CB1KO and Ng2-CB1WT at P60. Data are shown as mean ± SEM. n = 3–5 for A, n = 3 for D, E, n = 3–4 for E, F, and n = 4 for G, independent data points used per each experimental group. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 vs Ng2-CB1WT mice, by two-tailed unpaired Student’s t-test for A, D, E, F and G. Scale bars, 80 µm for B, 100 µm for C, 4 µm for D, 600 µm for F, and 3 µm for G.
CB1 receptor gene inactivation in OPCs causes hypomyelination in young adult mice
We next studied whether CB1 receptor deficiency in OPCs at early postnatal ages leads to alterations of myelin levels at P60. Tamoxifen-driven recombination was induced at P6 in Ng2-CB1KO and their controls, and myelination was evaluated in the CC at P60. Sudan black staining unveiled a hypomyelinated CC in adult Ng2-CB1KO mice compared to controls (Fig. 2E and Fig. S4B). Western blot and real time-PCR analyses revealed reduced protein and mRNA levels of myelin-associated proteins, such as MAG, MOG and MBP, in CC extracts from Ng2-CB1KO mice compared to their controls (Fig. 2F and Fig. S4A). Furthermore, we also found reduced myelin-related protein levels in other CNS areas of Ng2-CB1KO mice, such as the cerebellum, the hippocampus, and the medial prefrontal cortex, compared to Ng2-CB1WT mice (Fig. 2F). Then, we analyzed whether the hypomyelinated phenotype observed upon OPC-CB1 receptor depletion impacts node/paranode density or structure. Immunofluorescence analysis of nodal (Nav1.6) and paranodal (Caspr) markers in the CC at P60 revealed an altered nodal and paranodal length, with equal node density in Ng2-CB1KO mice compared to their controls (Fig. 2G), confirming a defect in myelination in Ng2-CB1KO mice.
CB1 receptor gene inactivation in OPCs leads to motor and cognitive defects in young adult mice
To address whether the hypomyelinated phenotype observed in Ng2-CB1KO mice is associated with behavioral alterations, we performed a battery of tests at P60. Ng2-CB1KO mice showed reduced motor activity and coordination in the open field (Fig. 3A), beam walking (Fig. 3B), and Actitrack (Fig. 3C) tests compared to Ng2-CB1WT mice. Ng2-CB1KO also showed an anxiety-like behavior in the open field and elevated plus-maze (EPM) (Fig. 3A, D) tests, and memory deficits in the novel object recognition (NOR, Fig. 3E) and y-maze (Fig. 3F) tests. Altogether, these observations show that CB1 receptor deficiency in OPCs at early postnatal ages causes hypomyelination at young adult ages, which is translated into impaired motor and memory functions, and anxiety-like behaviors.

Tamoxifen-driven recombination was induced in Ng2-CB1KO, and control mice at postnatal day 6 (P6-P7) and a battery of behavioural tests was performed at P60. A Open field test. Representative mouse trajectories, quantification of total distance travelled and the number of entries into the center of the arena. B Beam walking test. Quantification of the number of missteps and the time spent to cross the beam. C Actitrack test. Quantification of locomotor activity, mean velocity, and the total distance travelled. D Elevated plus-maze (EPM) test. Quantification of the time spent in open arms and of the number of entries into open arms. E Novel object recognition (NOR) test. Quantification of the discrimination index and the preference index. F Y-maze test. Quantification of the time spent in the new arm. Data are shown as mean ± SEM. n = 8–25 independent data points used per each experimental group. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 vs Ng2-CB1WT mice, by two-tailed unpaired Student’s t-test, two-tailed unpaired Student’s t-test with Welch’s correction or Man Whitney test.
CB1 receptors modulate OPC differentiation through the RhoA/ROCK signaling axis
The Ras homolog family member A (RhoA) is a multifunctional small GTPase protein that has recently emerged as a central control point of OPC differentiation and OL maturation [22, 23]. RhoA, by stabilizing actin fibers, regulates cytoskeletal reorganization, thereby modulating the morphological changes necessary for OPC differentiation and OL maturation. Thus, activation of the RhoA/Rho-associated protein kinase (ROCK) signaling axis impedes OPC differentiation, and RhoA/ROCK inactivation promotes OPC differentiation in vitro [22, 24, 25], or under inhibitory conditions, such as in the presence of myelin [26], or following spinal cord injury [27] or hypoxia [28] in mice. Thus, we studied whether the aforementioned deficits in cell differentiation of CB1 receptor deficiency in OPCs may be caused by an altered RhoA signaling. Western blot analysis revealed increased RhoA protein levels and increased activation of the RhoA downstream target ROCK, paralleled by a reduced activation of cofilin, in CC extracts from Ng2-CB1KO mice (Fig. 4A). Immunofluorescence analysis in the developing CC corroborated the increased RhoA protein levels in Ng2/Ai6-CB1KO OPCs compared to their Ng2/Ai6-CB1HET controls (Fig. 4B). In line with these observations, acute THC administration (3 mg/kg, i.p.) to Ng2-CB1WT mice reduced RhoA protein and ROCK activation levels, and increased cofilin activation levels, in CC extracts (Fig. 4C). Immunofluorescence analysis in the developing CC of Ng2-dsRed mice confirmed the reduced RhoA protein levels in THC-treated Ng2-dsRed+ OPCs compared to Veh-treated mice (Fig. 4D).

Tamoxifen-driven recombination was induced at postnatal day 6 (P6-P7) in Ng2-CB1KO, Ng2-CB1WT, Ng2/Ai6-CB1KO, or Ng2/Ai6-CB1HET mice, followed by Δ9-Tetrahydrocannabinol (THC, 3 mg/kg) administrations the following day, for 1 time C, D or for 2 E, or 4 F, G consecutive days. A Western blot analysis for Ras homolog gene family member A (RhoA), phosphorylated RhoA/Rho-associated protein kinase 2 (pROCK2), and pCofilin of dissected corpus callosum (CC) extracts from Ng2-CB1KO and Ng2-CB1WT at P15. Quantification of optical density (O.D.) B Immunofluorescence analysis of RhoA expression in the recombinant Ai6+ population in Ng2/Ai6-CB1KO, or Ng2/Ai6-CB1HET mice at P15. C Western blot analysis of RhoA, pROCK2 and pCofilin protein levels of dissected CC extracts from THC or Veh-treated Ng2-CB1KO or Ng2-CB1WT mice, at 6 hours after THC administration. D Immunofluorescence analysis of RhoA in Ng2-dsRed+ OPCs at 6 h after THC or Veh administration. E-G Ng2-CB1KO, Ng2-CB1WT, Ng2/Ai6-CB1KO, or Ng2/Ai6-CB1HET mice were administered with THC or Veh for 2 E or 4 F, G days. E Immunofluorescence analysis and quantification of the percentage of CC1+ cells among the recombinant Ai6+ population. Western blot (F) or immunofluorescence (G) analysis of myelin-related proteins such as myelin-associated glycoprotein (MAG), myelin oligodendrocyte glycoprotein (MOG), and myelin basic protein (MBP) or proteolipid protein (PLP). Data are shown as mean ± SEM. n = 3 for E, n = 3–4 for A, C, n = 4 for F, independent data points were used for each experimental group. *p ≤ 0.05, **p ≤ 0.01 and ***p ≤ 0.001 vs Ng2-CB1WT or vehicle-treated groups, by two-tailed unpaired Student’s t-test for A, two-tailed unpaired Student’s or Mann Whitney test for C and F, and two-way ANOVA followed by Turkey´s multiple comparison for E. Scale bars, 20 µm for B, D and E, and 100 µm for G.
To address the cell-autonomous action of THC in targeting RhoA selectively in OPCs through CB1 receptors activation to drive oligodendrogenesis, we administered THC or Veh to Ng2-CB1KO mice. THC failed to alter RhoA protein levels and activation of its downstream targets in CC extracts from Ng2-CB1KO mice compared to Veh-treated animals (Fig. 4C). Similarly, immunofluorescence (Fig. 4E, G) and western blot (Fig. 4F) analyses revealed that THC enhanced OL differentiation and increased myelin-associated protein levels in the CC of Ng2-CB1WT or Ng2/Ai6-CB1HET control mice, but not in their Ng2-CB1KO or Ng2/Ai6-CB1KO littermates. These results indicate a cell-autonomous action of THC in inducing OPC differentiation and myelination in vivo through CB1 receptors activation, at least in part, by targeting RhoA protein levels.
Modulation of protein stability by proteasomal degradation plays an important role in RhoA biological functions [29]. To explore the possibility that CB1 receptors target proteasomal degradation to modulate RhoA protein levels we used a reversible proteasome inhibitor, MG-132. Thus, pretreatment with MG-132 prevented the THC-induced reduction in RhoA protein levels in CC extracts (Fig. 5A), as well as the THC-induced OL differentiation determined by immunofluorescence analysis in the CC of Ng2/Ai6-CB1WT mice (Fig. 5B). We excluded the transcriptional modulation of RhoA by CB1 receptors in OPCs, as we did not find differences in RhoA mRNA levels in CC extracts from acutely THC-treated mice compared to Veh-treated controls (Fig. 5C). THC-treated mice also exhibited a reduction in RhoA activity in CC extracts compared to Veh-treated controls (Fig. 5C), in line with the differences observed at RhoA protein levels. These results indicate that CB1 receptors modulate OPC differentiation, at least in part, by regulating RhoA proteasomal degradation.

A, B Ng2/Ai6-CB1WT mice were administered with the proteasomal inhibitor MG-132 (5 mg/kg) at 30 min before THC. A Western blot analysis for RhoA in dissected CC extracts at 6 hours after THC. Quantification of optical density (O.D.). B Immunofluorescence analysis and quantification of the percentage of Ai6+ cells that expressed the OL marker CC1 at 48 hours after THC administrations. C Relative levels of RhoA protein levels, activity, or mRNA levels in CC extracts at 6 hours after THC. D Maternal Tetrahydrocannabinol (THC) administration induces oligodendrocyte development in postnatal pups. THC (3 mg/kg) or Veh were administered to CD1 mouse mothers the day their pups were 1 day old, once a day for 8 consecutive days, and oligodendrogenesis was analyzed in corpus callosum at postnatal days 15. Immunofluorescence analysis and quantification of the percentage of CC1+ OLs among the Olig2+ population. E Electroporation experiments with siRNAs against Cullin-3 (CUL-3), Smurf-1 or SCFFBXL19, and with a GFP reporter plasmid control in Pups from Veh or THC-treated mothers. Quantification of CC1+ OLs among GFP+Olig2+ cells in the subcortical white matter at P30. Data are shown as mean ± SEM. n = 3 for A and D, n = 3–4 for B, n = 3–5 for C, and n = 4–9 for E, independent data points were used for each experimental group. *p ≤ 0.05, **p ≤ 0.01 and ***p ≤ 0.001 vs vehicle-treated groups; #p < 0.05 and ###p < 0.001 vs THC-treated group, by one-way ANOVA followed by Tukey´s multiple comparisons for A and B, two-tailed unpaired Student’s t-test for C and D, and by one-way ANOVA Kruskal-Wallis followed by Dunn´s test for E. Scale bar, 20 µm for B, D, E.
To further study the molecular mechanism of CB1 receptor-mediated RhoA proteasomal degradation, we performed electroporation experiments in P2 pups from THC or Veh-treated mothers by using siRNAs against the three main E3 ubiquitin-protein ligases reported so far to target RhoA for proteasomal degradation, namely Smurf-1, Cullin-3 (CUL-3), and SCFFBXL19 [30,31,32,33]. First, we observed that maternal THC administration during the early postnatal period indirectly induced OPC differentiation in pups´ CC (Fig. 5D). Then, we electroporated a control GFP reporter plasmid and quantified by immunofluorescence the percentage of CC1+ OLs within GFP+Olig2+ cells. This analysis revealed an enhanced oligodendrogenesis in siC-electroporated pups of THC-treated mothers compared to SiC-electroporated pups from Veh-treated mothers (Fig. 5E), confirming that maternal THC administration induced OPC differentiation in control pups. Moreover, we found that Smurf-1 and CUL-3 silencing prevented the THC-induced OPC differentiation in the developing CC (Fig. 5E), indicating that CB1 receptor-mediated modulation of OPC differentiation requires E3 ubiquitin ligase-mediated RhoA proteasomal degradation.
ROCK inactivation overcomes the defects in oligodendrogenesis and functional myelination of CB1 receptor deficient OPCs
To confirm the involvement of the RhoA/ROCK axis in the CB1 receptor-mediated modulation of OPC differentiation, we analyzed the effect of inhibiting ROCK pharmacologically in vivo by administering the selective inhibitor Y-27632. CB1 receptor deficiency was induced in Ng2-CB1KO and Ng2/Ai6-CB1KO and control mice, followed by Y-27632 or Veh administration (Fig. 6A). Immunofluorescence analysis in the developing CC confirmed a reduced proportion of CC1+ OLs within the Ai6+ population in Veh-treated Ng2/Ai6-CB1KO mice compared to Veh-treated Ng2/Ai6-CB1HET animals (Fig. 6B). Importantly, ROCK inhibition induced OPC differentiation in Ng2/Ai6-CB1KO mice, rescuing CB1 receptor-null OPC deficiency up to the levels found in Veh or Y-27632-treated Ng2/Ai6-CB1HET littermates (Fig. 6B). Immunofluorescence (Fig. S5) and western blot (Fig. 6C) analysis of myelin-associated proteins evidenced that ROCK blockade also restored Ng2-CB1KO myelin protein levels up to the levels of Ng2-CB1WT-Veh or Y-27632-treated mice at both, postnatal and adult ages. Notably, we also found that ROCK pharmacological blockade overcame the motor and cognitive deficits observed in Ng2-CB1KO mice at adult ages in the open field (Fig. 6D), beam walking (Fig. 6E), elevated plus-maze (EPM, Fig. 6F) and novel object recognition (NOR, Fig. 6G) tests. Altogether, these findings show that ROCK pharmacological inactivation overcomes the defects in oligodendrogenesis and functional CNS myelination of CB1 receptor deficient OPCs during postnatal development in mice.

A Tamoxifen-driven recombination was induced at postnatal day 6 (P6-P7) in Ng2-CB1KO, Ng2-CB1WT, Ng2/Ai6-CB1KO and Ng2/Ai6-CB1HET mice followed by administrations of the ROCK inhibitor Y-27632 (10 mg/kg), and tissue was analyzed at P11 or P60. B Immunofluorescence analysis and quantification of the percentage of CC1+ oligodendrocytes among the recombinant Ai6+Olig2+ population at P11. C Western blot analysis of the myelin associated proteins, myelin-associated glycoprotein (MAG), myelin oligodendrocyte glycoprotein (MOG), and myelin basic protein (MBP) in corpus callosum extracts at P60. Quantification of optical density (O.D.). D–G Behavioral analysis of Ng2-CB1KO and Ng2-CB1WT mice at P60. D Open field test. Representative images of mouse trajectories. Quantification of total distance travelled. E Beam walking test. Quantification of missteps. F Elevated plus maze (EPM) test. Quantification of the time spent in open arms. G Novel object recognition (NOR) test. Quantification of the discrimination index. Data are shown as mean ± SEM. n = 6–9 for B, n = 3 for C, 16–24 for D–G, independent data points were used for each experimental group. *p ≤ 0.05, **p ≤ 0.01 vs Veh-treated Ng2/Ai6-CB1HET or Ng2-CB1WT mice; #p ≤ 0.05, ##p ≤ 0.01 vs Veh-treated Ng2/Ai6-CB1KO mice or Ng2-CB1KO mice by two-way ANOVA followed by Tukey´s multiple comparisons. Scale bar, 20 µm in B.
Discussion
CB1 receptors modulate OPC differentiation cell-autonomously
Here, by using new conditional mouse models, we provide the first evidence for a cell-autonomous role of CB1 receptors in modulating OPC differentiation and OL development during postnatal myelination in mice. Our observations support the relevance of eCB signaling in the control of postnatal myelination through the activation of CB1 receptors located on OPCs. However, mouse OPCs express CB1 and CB2 receptors at developmental stages [9], respond to THC administration in vivo (present study), and pharmacologically blocking either of the two receptors prevents THC effects [9]. Moreover, we show that THC administration to Ng2-CB1KO mice fails to modulate oligodendrogenesis and postnatal myelination in the CC, addressing that CB1 receptor expression in OPCs is strictly required for cannabinoid modulation of developmental oligodendrogenesis. Thus, the involvement of CB2 receptors may be substantiated by the existence of a functional interaction between both receptors in OPCs, such as the CB1-CB2 heteromers observed in neuronal cells [34], or the non-cell autonomous modulation of oligodendrogenesis by CB2 receptors. Therefore, our study also provides evidence for a cell-autonomous action of THC administration in modulating OPC differentiation and OL development in the CC during postnatal myelination in mice, and suggests that previous pharmacological studies based on the use of synthetic cannabinoids [8], phytocannabinoids [14] or eCBs [10] may have also involved the targeting of OPCs cell-autonomously to enhance OL development during postnatal myelination.
Our results show that genetic inactivation of CB1 receptors in OPCs not only blocks cell differentiation, but also affects OL maturation and CNS myelination in the CC. These data are in line with previous findings in which administration of cannabinoid agonists induce OPC differentiation and enhance OL maturation and CNS myelination [8, 10, 15]. As premature and mature OLs derived from CB1 receptor deficient OPCs shall inherit genetic depletion of CB1 receptor, we cannot exclude the possibility that some of the observed maturation defects arise from an altered OPC differentiation. Thus, assessing the selective action of CB1 receptors in modulating OL maturation and myelin formation would require the generation of selective premature/mature OL Cre-dependent mouse models.
CB1 receptor-mediated RhoA proteasomal degradation in OPCs
The CB1 receptor is one of the most abundant G protein-coupled receptors (GPCRs) in the mammalian brain. To date, other GPCRs, such as GPR17 [35], GPR37 [36], GPR56 [37], and GPR126 [38] have been shown to modulate OL development and/or CNS myelination. Thus, our present study adds evidence for GPCRs in modulating oligodendrogenesis and myelination. Mechanistically, GPR37 regulates OPC differentiation and CNS myelination via cAMP-dependent Ras-ERK1/2 activation [39], while GPR56 modulates OPC proliferation via Gα12/13-RhoA, without affecting OL differentiation. Here, we observed that the CB1 receptor-mediated modulation of OPC differentiation occurs by inducing Smurf-1/CUL-3-mediated RhoA proteasomal degradation. Thus, CB1 receptors may modulate RhoA proteasomal degradation by targeting both, the inactive (GDP-bound) and the active (GTP-bound) forms of RhoA by CUL3 and Smurf-1 respectively [29]. Indeed, pharmacological activation of CB1 receptor has been shown to induce RhoA proteasomal degradation in migrating pyramidal neurons during mouse corticogenesis [40]. Therefore, it is plausible to speculate that CB1 receptor-mediated RhoA degradation under those settings may involve Smurf-1 or CUL-3 actions (Fig. S6). Moreover, CB1 receptors may exert opposing effects regarding RhoA activation. CB1 receptors activation promote bone marrow-derived macrophage migration or phagocytosis [41,42,43] by increasing RhoA activity and subsequent ROCK activation via Gαi/o, but also via Gα12/13, in neurons [44] or platelets [45]. Conversely, CB1 receptors also inhibit carcinoma cell migration [46] by reducing RhoA activity, indicating that the differential CB1 receptor-modulation of RhoA signaling may depend on factors such as the cell type and the pathophysiological context.
CB1 receptors in OPCs and motor and cognitive development
Here, we also reveal that CB1 receptor deficiency in OPCs at early postnatal ages affects the number and maturation state of the OL population in the CC in young adult mice, which is also associated with hypomyelination in several CNS regions and important behavioral deficits. In line with these results, increasing evidence has linked deregulated developmental myelination to impaired functional performance or neuropsychiatric alterations in adult mice [3, 47], such as those related with motor function [48, 49], memory [50], and anxiety-like behaviors [51,52,53]. Thus, it is plausible that the altered behavioral traits found in Ng2-CB1KO mice is due to a reduced number of myelinating OLs, and, thereby, to reduced myelin levels in CNS regions responsible for motor, memory, anxiety-like behaviors, which, in turn, would impact neuronal and whole-body functionality. In fact, alterations of the eCNB system during embryonic, postnatal, or adolescent ages evokes multiple long-lasting behavioral alterations in mice that persist in adulthood [54, 55]. Moreover, THC exposure during embryonic or postnatal development has been linked to psychiatric disorders, such as depression and anxiety [56, 57], spontaneous behavior, or habituation [58, 59], thus suggesting that, in addition to the neuronal component, at least part of these behavioral defects are mediated by the restricted OPC CB1 receptor population.
Conversely, on top of the observed behavioral alterations in complete CB1 receptor-deficient mice [54], the development of mouse models with CB1 receptors selectively deleted in dorsal telencephalic glutamatergic neurons or forebrain GABAergic neurons have contributed to dissect the populations of neuronal CB1 receptors responsible for the modulation of motor, memory, anxiety-like behaviors [60,61,62]. To sum up, this is the first study that supports the necessary role for CB1 receptors located on OPCs in the modulation of motor and cognitive development in mice, therefore contributing to understand the complex control of motor and cognitive development exerted by the eCB system.
The therapeutic potential of targeting OPC CB1 receptors in demyelinating disorders
A number of studies have pointed out important similarities in the effects of molecules that modulate OPC differentiation during postnatal myelination and during remyelination [63, 64]. Specifically, cannabinoids modulate oligodendrogenesis under demyelinating conditions, enhancing OL regeneration, and functional CNS remyelination following cuprizone-induced demyelination [14], or in the TMEV animal model of progressive MS [12, 13], thus suggesting that CB1 receptors also modulate OPC differentiation under demyelinating conditions. In MS, there is a failure in OPC differentiation, which limits their remyelinating potential. Although the cause of this failure is not completely known, several studies have denoted the presence of inhibitory signals in the demyelinated CNS that prevent OPC to become mature myelinating cells. Indeed, inhibition of OPC differentiation or OL maturation by myelin debris [26], or by astrocytic chondroitin sulfate proteoglycans [27, 65], are mediated by RhoA/ROCK axis modulation [23]. Moreover, elevated levels of the eCB 2-AG upon pharmacological inhibition of MAGL reduces astrocytic chondroitin sulfate proteoglycan production and enhances OL differentiation under inhibitory conditions [12]. Thus, it is plausible that, following demyelination, CB1 receptor-mediated modulation of RhoA signaling controls OPC differentiation and functional remyelination in a cell-autonomous manner though OPC CB1 receptors, but also non-cell-autonomously through astroglial CB1 receptors. In any event, the CB1 receptor-evoked control of RhoA signaling could be potentially targeted to promote functional recovery, which would open new avenues to the therapeutic manipulation of currently intractable demyelinating diseases.
Materials and methods
Animal procedures
Experimental designs and procedures were approved by the Complutense University Animal Research Committee and Comunidad de Madrid, in accordance with the European Commission regulations (Directive 2010/63/EU), and addressing the issues raised in ‘Implementing guidelines on reporting research using animals (ARRIVE) guidelines. All animals used were bred into C57BL/6 J background. Animals were housed, three in a cage, in temperature-controlled rooms on an artificial 12-h light/dark cycle at 22–24 °C and relative humidity of 50–60%, with water, food availability ad libitum and sterile cardboard tubes as a housing refinement. All mice were pathogen free. Jackson Laboratories (Bar Harbor, ME, USA) mouse lines used: Ng2-dsRed (Cspg4-DsRed.T1 1Akik/J Cat# RRID:IMSR_JAX:008241) [18], Ng2-CreERT2 [18], Pdgfrα-creERT2 (RRID:IMSR_JAX:018280 [21], Rosa-Stop-Ai6, RRID:IMSR_JAX:007906) [19], CB1ff (IMSR Cat# JAX:036107, RRID:IMSR_JAX:036107 [17] and CD1 mice. Mice were bred to finally generate Ng2-CB1KO mice (containing homozygous CB1-floxed/floxed alleles and heterozygous Ng2-CreERT2 allele), Ng2-CB1WT (containing homozygous CB1-floxed/floxed alleles and negative for Ng2-CreERT2 allele), Pdgfrα-CB1KO mice (containing homozygous CB1-floxed/floxed alleles and heterozygous Pdgfrα-CreERT2 allele), Pdgfrα-CB1WT mice (containing homozygous CB1-floxed/floxed alleles and negative for Pdgfrα-CreERT2 allele), Ng2/Ai6-CB1KO mice (containing homozygous CB1-floxed/floxed alleles, homozygous ROSA-stop-Ai6 alleles and heterozygous Ng2-CreERT2 allele), Ng2/Ai6-CB1HET mice (containing heterozygous CB1-floxed alleles, homozygous ROSA-stop-Ai6 alleles and heterozygous Ng2-CreERT2 allele), and Ng2/Ai6-CB1WT mice (containing homozygous CB1-WT alleles, homozygous ROSA-stop-Ai6 alleles and heterozygous Ng2-CreERT2 allele) (in C57BL/6 N background). Of note, Ng2/Ai6-CB1HET mice were used as controls for Ng2/Ai6-CB1KO mice, by crossing CB1HET x CB1KO mice, to obtain a higher number of littermates of the same litter, as the dynamics of oligodendrogenesis in Ng2/Ai6-CB1HET mice were similar to Ng2/Ai6-CB1WT mice in immunofluorescence studies (not shown). Delta-9-tetrahydrocannabinol (THC, 3 mg/kg), MG-132 (5 mg/kg, Medchem, NJ, USA), or vehicle, were dissolved in 100 μL Tween-80/NaCl (1:18, v/v) and 1% (v/v) of dimethyl sulfoxide [9]. MG-132 was administered intraperitoneally 30 min before THC. In maternal THC administration experiments, we confirmed CB1 receptors activation in the CNS of pups at 4 h after administering THC or Veh to mothers by analyzing pS6 and cFos levels (not shown). Y27632 (10 mg/kg, Medchem, Monmouth Junction, NJ, USA) was dissolved in saline and administered intraperitoneally. Recombination was induced by tamoxifen (Tam; 24 h apart; 37.5 mg/kg i.p. dissolved in Ethanol-sunflower oil 1:9). Experiments included male and female mice.
Fluorescent-activated cell sorting
Rosa-Ai6+ cells were isolated from Ng2/Ai6-CB1KO and Ng2/Ai6-CB1HET at postnatal day 10, after two tamoxifen injections (37.5 mg/kg), at P6 and p7, as previously described [20]. Briefly, CC was dissected following a second step of dissociation using papain (30 μg/ml in DMEM-Glutamax, with 0.24 μg/ml L-cystein and 40 μg/ml DNase I) and cells were put on a preformed 30% Percoll density gradient before centrifugation for 15 min. Rosa-Ai6+ cells were FACS sorted on an FACS Aria II (BD Biosciences). Propidium iodide (PI) was used to exclude dead cells. For genomic analysis, cells were washed twice in PBS 1×, then the dry cell pellets were frozen at −80 °C.
Electroporation experiments
siRNAs purchased from Santa Cruz (Dallas, TX, USA), siC (SC-37007) siSmurf-1 (ref: sc-41674), siCUL-3 (ref: sc-35131), SCFFBXL19 (39393) were electroporated into postnatal day 2 (P2) CD1 pups. Vectors were diluted at 1 μg/μl in PBS with 2.5 mg/ml fast green (Sigma, St Louis, MO, USA) and injected, together with a constitutive GFP overexpression plasmid (pCAG-GFP), in the lumen of the diencephalic ventricle. Electroporation was performed with a BTX electroporator (Holliston, MA, USA) with these parameters: 5 pulses, intensity = 95 V, pulse length = 50 ms, inter-pulse interval = 50 ms. The same day of electroporation, mothers received THC (3 mg/kg) or Veh administrations, once a day for 14 days. Efficient silencing of ubiquitin ligases was confirmed by immunofluorescence analysis at 4 days after electroporation, by using anti-Smurf-1, anti-CUL-3 and anti-SCFFBXL19 antibodies for each experimental condition (not shown).
Immunofluorescence
Brain tissue was processed as previously described [66]. Briefly, mice were perfused transcardially with 4% paraformaldehyde (PFA), and brains postfixed overnight in 4% PFA and treated with 30% sucrose before freezing. 30-μm-thick coronal free-floating brain cryosections were washed in PBS, blocked with 10% goat serum, and incubated with the indicated primary antibodies (overnight at 4 °C). When needed, antigen retrieval immunostaining was performed with citric acid (10 mM, pH 6, 65 °C for 30 min) or with retrieve-all antigen unmasking system 3. Acidic (Biolegend) at 92 °C for 10 min. For immunofluorescence analysis in vitro, CC extracts from P10 Ng2/Ai6-CB1KO and Ng2/Ai6-CB1WT mice administered with tamoxifen (37.5 mg/kg) at P6 and P7, was dissociated, and individual cells were attached to glass slides by cytospin. After air-drying, slides were treated with 4% PFA for 10 minutes. The primary antibodies used are as follows, anti-CB1R (1:500, Frontier Institute, CB1-GP-Af530), anti-CC1 (1:400, Millipore, Cat#OP80, RRID:AB_2057371), anti‐Olig2 (1:250, Millipore Burlington, MA, USA) Cat# AB9610, RRID:AB_570666), anti‐GSTπ (1:200, MBL International Cat# 312, RRID:AB_591792, Wobrun, MA, USA), anti-MBP (1:200, BioLegend, (San Diego, CA, USA) Cat# 836504, RRID:AB_2616694), anti-NG2, (1:200, MBL International, Cat# AB5320, RRID:AB_11213678), anti‐PLP (1:500, Abcam (Waltham, MA, USA), Cat#ab28486, RRID:AB_776593), RhoA (1:500, Cytosqueleton (Denver, CO, USA) Cat# ARH03, RRID:AB_10708069), Nav1.6 (1:300, Alobome labs (Jerusalem, Israel), Cat# ASC-009, RRID:AB_2040202), Caspr (1:300, Millipore, Cat# MABN69, RRID:AB_10806491) FluoroMyelin (1:300, Thermo Fisher Scientific (Waltham, MA, USA) Cat# F34652, RRID:AB_2572213). Nuclei were visualized with DAPI. The appropriate mouse, rabbit or guinea pig highly cross-adsorbed Alexa Fluor 488, 594, and 647 secondary antibodies (1:1000, Invitrogen, Waltham, MA, USA) were used.
Confocal microscopy
Optical sections (z = 0.5 µm) of confocal epifluorescence images were sequentially acquired using a confocal laser-scanning microscope TCS-SP8 (20×, 40× and 63×, Leica DMI6000 B instrument) and Leica Application Suite X (LAS X) software. Images were acquired in the CC of each animal, a minimum of six correlative slices from a 1-in-10 series located between +0.7 and −0.5 mm from bregma were analysed. Cell counts were performed blindly using ImageJ software (NIH) in the corpus callosum (CC) and data are presented as the mean cell number per mm2 or as a percentage of positive cells. For CB1 inmunostaining of cytospined cells, the microscope was configured to capture 16 images following the z axis, which resulted in 8 µm thick stacks.
Electron microscopy
Mice were transcardially perfused with PBS followed by a fixative solution containing 4% PFA, 2.5% glutaraldehyde, and 0.5% NaCl in 0.1 M PB, as described [67]. Brains were postfixed overnight at 4 °C and stored in 1% paraformaldehyde. Vibratome sections (100 µm) containing the CC were cut in the coronal plane on a vibratome (VT1000S, Leica (Wetzlar, Germany) and incubated in 1% OsO4, then embedded in epoxy resin overnight to polymerize at 60˚C, and then trimmed and glued onto epoxy resin capsules. Semi‐thin sections (500 nm) were cut using a Power Tome ultramicrotome (RMC Boeckeler) and stained with 1% toluidine blue. Ultrathin (60 nm) sections were cut with a diamond knife (Diatome), collected on nickel mesh grids, and stained with 4% uranyl acetate and 2.5% lead citrate for electron microscope visualization. Electron microscopy images of the rostral CC were taken from randomly selected fields with a Jeol JEM Plus electron microscope at the Service of Analytical and High-Resolution Microscopy in Biomedicine of University of the Basque Country UPV/EHU. The mean number of myelinated axons was analyzed in 10 non-serial electron micrographs per animal taken systematically at a magnification of 5,000×.
Sudan black
Floating sections were mounted on to TESPA-coated glass slides, dehydrated in 70% ethanol and stained with 0.5% Sudan Black in 70% ethanol for 20 min. Excess staining was removed by washing the slides in 70% ethanol and finally rinsed with water. Samples were observed under light microscopy in a Zeiss Axioplan2 microscope.
Western blot
CC tissue was microdissected from 500-μm-thick coronal sections and proteins were extracted using RIPA buffer (SDS 0.1%, Sodium deoxycholate 0.5%, NP40 1%, NaCl 150 mM, Tris-HCl 50 mM pH8 in PBS) containing PMSF, protease inhibitors, and sodium orthovanadate. Protein samples were separated on 12% acrylamide (Bio-Rad) gels and transferred to polyvinylidene difluoride (PVDF membranes (Millipore). Membranes were placed in blocking buffer (5% w/v BSA in TBS-T + Azida 0.02%) and probed with primary antibodies overnight at 4 °C. The primary antibodies used are as follows: anti-MAG (1:1000, Abcam, Cat#ab89780, RRID:AB_2042411), anti-MOG (1:2000, Abcam Cat#ab32760, RRID:AB_2145529), anti-MBP (1:1000, BioLegend, Cat#836504, RRID:AB_2616694), RhoA (1:500, Cytosqueleton Cat# ARH03, RRID:AB_10708069), pROCK2 (1:750, Genetex, Hsinchu City, Taiwan Cat# GTX122651, RRID:AB_2560946), pCofilin (1:500, Cell Signaling Technology Cat# 3313, RRID:AB_2080597) and anti-α-tubulin (1:5000, Sigma-Aldrich, Cat#T9026, RRID:AB_477593). After incubation with corresponding HRP-conjugated secondary antibody proteins were visualized using an enhanced chemiluminescence substrate mixture (ECL Plus; GE Healthcare; Santa Cruz Biotechnology; 1:5000). Band intensity of films was quantified using Adobe Photoshop software. Protein levels were normalized to the internal control α-tubulin. See full length uncropped original western blots in supplemental material.
Quantitative PCR
RNA was isolated using RNeasy Plus kit (Quiagen). cDNA was obtained with Transcriptor (Roche). Real-time quantitative PCR (qPCR) assays were performed using the FastStart Master Mix with Rox (Roche, Basel, Switzerland) and probes were obtained from the Universal Probe Library Set (Roche). Amplifications were run in a 7900 HT-Fast Real-Time PCR System (Applied Biosystems). The sequence of primers used are as follows: F-β-ACTIN: AAGGCCAACCGTGAAAAGAT; R-β-ACTIN: GTGGTACGACCAGAGGCATAC; F-MAG: GGTGTTGAGGGAGGCAGTTG; R-MAG: CGTTGTCTGCTAGGCAAGCA; F-MBP: GGAAGGCAGGTGATGGTTGA; R-MBP: ACACTGGAGGGCAAACACTC; F-MOG: TCCATCGGACTTTTGATCCTCA; R-MOG: GCTCCAGGAAGACACAACCA; F-RhoA: GAATGACGAGCACACGAGAC; R-RhoA: AAAAGCGCCAATCCTGTTT; F-TBP: GGGGAGCTGTGATGTGAAGT; R-TBP: CCAGGAAATAATTCTGGCTCA Each value was adjusted by using β-actin and TBP mRNA levels as reference.
Genomic recombination analysis
Recombination was tested by PCR with genomic DNA isolated from sorted Rosa-Ai6 cells by the use of the GenElute mammalian genomic DNA Miniprep Kit (Sigma-Aldrich), as previously described [68]. PCR was performed using Taq DNA polymerase (Thermo Fisher Scientific, Inc.) For the recombined CB1 allele we used the forward primer 5′-GCTGTCTCTGGTCCTCTTAAA-3′ (G50) and the reverse primer 5′-CTCCTGTATGCCATAGCTCTT-3′ (G53) resulting in a 600 bp fragment.
RhoA activity essay
Dissected corpus callosum extracts were processed for active RhoA quantification with the G-LISA kit (Cytoskeleton, Cat#BK124) following manufacturer’s guide.
Behavioral assessments
All tests were conducted during the light cycle, with uniform lighting conditions and white noise in an isolated room. Animals were acclimated to the room for 45–60 min before testing. Beam walking test: To evaluate fine motor coordination, mice were trained to cross a narrow wood beam (100 cm length, 10 and 7 mm width) [69]. Open-field test: Mice were placed in the center of an open-field arena (70 × 70 × 40 cm) and allowed to freely explore it for 5 min. Behavior was recorded with a video camera placed above and the video tracking software SMART 3.0 (Panlab, Spain) was used for analysis [70]. Novel Object Recognition test (NOR): The day before the test, mice were habituated to an empty open-field arena for 10 min. The day of the test, mice were allowed to freely explore the arena with two identical objects for 10 min and, after 2 hours, mice could explore the arena containing a familiar object and a novel object. Discrimination index was calculated as the difference in exploration time between the novel and familiar object divided from the total exploration time and expressed as a percentage. Preference Index was calculated as the percentage of exploration time spent examining the novel object over the total exploration time. Modified Y-Maze test: Mice were allowed to explore the maze with one of the three arms closed for 3 min. After an inter-trial interval of 5 min, mice could freely explore all three arms of the maze for 3 min, and the time spent in each arm was registered. Actimeter/ Locomotor activity test: Spontaneous motor activity was evaluated using an automated actimeter (Acti-Track; Panlab, Barcelona, Spain). This consisted of a 22.5 × 22.5 cm area with 16 surrounding infrared beams coupled to a computerized control unit. After 1 min of habituation, activity was recorded for a period of 5 min and data were collected with Acti-Track v2.7 software (Panlab, Barcelona, Spain). Elevated plus-maze test (EPM): Mice were placed in the center of the maze (two open arms and two closed arms of 30 ×7 cm arranged orthogonally 60 cm above the floor) and allowed to explore it for 5 min. The test was conducted under red light conditions.
Statistics
The n number of animals per group or experiments per condition is indicated in every case. The declared group size is the number of independent data points, and that statistical analysis was done using these independent data points. Studies were designed to generate groups of equal size, using randomisation and blinded analysis. The numbers illustrated represent the animals used in each of the experiments, after considering any unexpected loss of data or exclusion criterion. In some cases, experimental losses may be determined by animals receiving the wrong treatment, infections unrelated to the experiment, sampling errors (e.g., inadequate calibration of equipment, software error during acquisition), or other human error (e.g., forgetting to switch on equipment). Power analysis was conducted with IBM SPSS software (IBM France, Bois-Colombes, France). Sample sizes were based on our prior studies where similar sample sizes were adequately powered to detect significant differences. All variables were first tested for both, normality (Shapiro test or D’Agostino & Pearson normality test with p > 0.05) and homogeneity of variances (Brown-Forsythe test with p > 0.05). When comparing two groups we use unpaired two-tailed t-test for normal distribution (unpaired t-tests were Welch-corrected if needed) or Mann-Whitney test when they did not distribute normally. For comparisons of more than two groups, if data were found to be normally distributed, one-way ANOVA followed by Tukey’s post hoc test was carried out. The post hoc tests were conducted only if F in ANOVA achieved P < 0.05 and there was no significant variance in homogeneity. If data were found to not be normally distributed, then a Kruskal–Wallis one-way ANOVA with uncorrected Dunn’s post hoc test was carried out. For data from Figs. 4E and 6, two-way ANOVA followed by Tukey’s post hoc test was carried out. Differences with p < 0.05 between group means were considered statistically significant. All data analyses were done using GraphPad Prism 7.00.
Data availability
All data generated or analyzed during this study are available in the supporting information.
References
Bercury KK, Macklin WB. Dynamics and mechanisms of CNS myelination. Dev Cell. 2015;32:447–58.
Fields RD. Oligodendrocytes changing the rules: action potentials in glia and oligodendrocytes controlling action potentials. Neuroscientist 2008;14:540–3.
Nave KA, Ehrenreich H. Myelination and oligodendrocyte functions in psychiatric diseases. JAMA Psychiatry. 2014;71:582–4.
Haroutunian V, Katsel P, Roussos P, Davis KL, Altshuler LL, Bartzokis G. Myelination, oligodendrocytes, and serious mental illness. Glia 2014;62:1856–77.
Elbaz B, Popko B. Molecular control of oligodendrocyte development. Trends Neurosci. 2019;42:263–77.
Molina-Holgado E, Vela JM, Arevalo-Martin A, Almazan G, Molina-Holgado F, Borrell J, et al. Cannabinoids promote oligodendrocyte progenitor survival: involvement of cannabinoid receptors and phosphatidylinositol-3 kinase/Akt signaling. J Neurosci. 2002;22:9742–53.
Ilyasov AA, Milligan CE, Pharr EP, Howlett AC. The endocannabinoid system and oligodendrocytes in health and disease. Front Neurosci. 2018;12:733.
Arevalo-Martin A, Garcia-Ovejero D, Rubio-Araiz A, Gomez O, Molina-Holgado F, Molina-Holgado E. Cannabinoids modulate Olig2 and polysialylated neural cell adhesion molecule expression in the subventricular zone of post-natal rats through cannabinoid receptor 1 and cannabinoid receptor 2. Eur J Neurosci. 2007;26:1548–59.
Huerga-Gomez A, Aguado T, Sanchez-de la Torre A, Bernal-Chico A, Matute C, Mato S, et al. Delta(9) -Tetrahydrocannabinol promotes oligodendrocyte development and CNS myelination in vivo. Glia. 2020.
Alpar A, Tortoriello G, Calvigioni D, Niphakis MJ, Milenkovic I, Bakker J, et al. Endocannabinoids modulate cortical development by configuring Slit2/Robo1 signalling. Nat Commun. 2014;5:4421.
Gomez O, Arevalo-Martin A, Garcia-Ovejero D, Ortega-Gutierrez S, Cisneros JA, Almazan G, et al. The constitutive production of the endocannabinoid 2-arachidonoylglycerol participates in oligodendrocyte differentiation. Glia 2010;58:1913–27.
Feliu A, Mestre L, Carrillo-Salinas FJ, Yong VW, Mecha M, Guaza C. 2-arachidonoylglycerol reduces chondroitin sulphate proteoglycan production by astrocytes and enhances oligodendrocyte differentiation under inhibitory conditions. Glia 2020;68:1255–73.
Feliu A, Bonilla Del Rio I, Carrillo-Salinas FJ, Hernandez-Torres G, Mestre L, Puente N, et al. 2-Arachidonoylglycerol reduces proteoglycans and enhances remyelination in a progressive model of demyelination. J Neurosci. 2017;37:8385–98.
Aguado T, Huerga-Gomez A, Sanchez-de la Torre A, Resel E, Chara JC, Matute C, et al. Delta(9) -Tetrahydrocannabinol promotes functional remyelination in the mouse brain. Br J Pharm. 2021;178:4176–92.
Ceprian M, Vargas C, Garcia-Toscano L, Penna F, Jimenez-Sanchez L, Achicallende S, et al. Cannabidiol administration prevents hypoxia-ischemia-induced hypomyelination in newborn rats. Front Pharm. 2019;10:1131.
Di Marzo V, Stella N, Zimmer A. Endocannabinoid signalling and the deteriorating brain. Nat Rev Neurosci. 2015;16:30–42.
Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, et al. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 2003;302:84–8.
Zhu X, Bergles DE, Nishiyama A. NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development 2008;135:145–57.
Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–40.
Zimmermann T, Maroso M, Beer A, Baddenhausen S, Ludewig S, Fan W, et al. Neural stem cell lineage-specific cannabinoid type-1 receptor regulates neurogenesis and plasticity in the adult mouse hippocampus. Cereb Cortex. 2018;28:4454–71.
Rivers LE, Young KM, Rizzi M, Jamen F, Psachoulia K, Wade A, et al. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci. 2008;11:1392–401.
Liang X, Draghi NA, Resh MD. Signaling from integrins to Fyn to Rho family GTPases regulates morphologic differentiation of oligodendrocytes. J Neurosci. 2004;24:7140–9.
Harlow DE, Macklin WB. Inhibitors of myelination: ECM changes, CSPGs and PTPs. Exp Neurol. 2014;251:39–46.
Zhao CF, Liu Y, Que HP, Yang SG, Liu T, Liu ZQ, et al. Rnh1 promotes differentiation and myelination via RhoA in oligodendrocytes. Cell Tissue Res. 2013;353:381–9.
Pedraza CE, Taylor C, Pereira A, Seng M, Tham CS, Izrael M, et al. Induction of oligodendrocyte differentiation and in vitro myelination by inhibition of rho-associated kinase. ASN Neuro. 2014;6.
Baer AS, Syed YA, Kang SU, Mitteregger D, Vig R, Ffrench-Constant C, et al. Myelin-mediated inhibition of oligodendrocyte precursor differentiation can be overcome by pharmacological modulation of Fyn-RhoA and protein kinase C signalling. Brain 2009;132:465–81.
Dyck SM, Alizadeh A, Santhosh KT, Proulx EH, Wu CL, Karimi-Abdolrezaee S. Chondroitin sulfate proteoglycans negatively modulate spinal cord neural precursor cells by signaling through LAR and RPTPsigma and modulation of the Rho/ROCK pathway. Stem Cells. 2015;33:2550–63.
Wang F, Yang YJ, Yang N, Chen XJ, Huang NX, Zhang J, et al. Enhancing oligodendrocyte myelination rescues synaptic loss and improves functional recovery after chronic hypoxia. Neuron 2018;99:689–701 e5.
Nethe M, Hordijk PL. The role of ubiquitylation and degradation in RhoGTPase signalling. J Cell Sci. 2010;123:4011–8.
Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20.
Ding F, Yin Z, Wang HR. Ubiquitination in Rho signaling. Curr Top Med Chem. 2011;11:2879–87.
Wei J, Mialki RK, Dong S, Khoo A, Mallampalli RK, Zhao Y, et al. A new mechanism of RhoA ubiquitination and degradation: roles of SCF(FBXL19) E3 ligase and Erk2. Biochim Biophys Acta. 2013;1833:2757–64.
Wang HR, Ogunjimi AA, Zhang Y, Ozdamar B, Bose R, Wrana JL. Degradation of RhoA by Smurf1 ubiquitin ligase. Methods Enzymol. 2006;406:437–47.
Callen L, Moreno E, Barroso-Chinea P, Moreno-Delgado D, Cortes A, Mallol J, et al. Cannabinoid receptors CB1 and CB2 form functional heteromers in brain. J Biol Chem. 2012;287:20851–65.
Lecca D, Raffaele S, Abbracchio MP, Fumagalli M. Regulation and signaling of the GPR17 receptor in oligodendroglial cells. Glia. 2020;68:1957–67.
Smith BM, Giddens MM, Neil J, Owino S, Nguyen TT, Duong D, et al. Mice lacking Gpr37 exhibit decreased expression of the myelin-associated glycoprotein MAG and increased susceptibility to demyelination. Neuroscience 2017;358:49–57.
Ackerman SD, Garcia C, Piao X, Gutmann DH, Monk KR. The adhesion GPCR Gpr56 regulates oligodendrocyte development via interactions with Galpha12/13 and RhoA. Nat Commun. 2015;6:6122.
Lyons DA, Talbot WS. Glial cell development and function in zebrafish. Cold Spring Harb Perspect Biol. 2014;7:a020586.
Yang HJ, Vainshtein A, Maik-Rachline G, Peles E. G protein-coupled receptor 37 is a negative regulator of oligodendrocyte differentiation and myelination. Nat Commun. 2016;7:10884.
Diaz-Alonso J, de Salas-Quiroga A, Paraiso-Luna J, Garcia-Rincon D, Garcez PP, Parsons M, et al. Loss of cannabinoid CB1 receptors induces cortical migration malformations and increases seizure susceptibility. Cereb Cortex. 2017;27:5303–17.
Mai P, Tian L, Yang L, Wang L, Yang L, Li L. Cannabinoid receptor 1 but not 2 mediates macrophage phagocytosis by G(alpha)i/o /RhoA/ROCK signaling pathway. J Cell Physiol. 2015;230:1640–50.
Mai P, Yang L, Tian L, Wang L, Jia S, Zhang Y, et al. Endocannabinoid system contributes to liver injury and inflammation by activation of bone marrow-derived monocytes/macrophages in a CB1-dependent manner. J Immunol. 2015;195:3390–401.
Wang L, Yang L, Tian L, Mai P, Jia S, Yang L, et al. Cannabinoid receptor 1 mediates homing of bone marrow-derived mesenchymal stem cells triggered by chronic liver injury. J Cell Physiol. 2017;232:110–21.
Dalton GD, Peterson LJ, Howlett AC. CB(1) cannabinoid receptors promote maximal FAK catalytic activity by stimulating cooperative signaling between receptor tyrosine kinases and integrins in neuronal cells. Cell Signal. 2013;25:1665–77.
Signorello MG, Giacobbe E, Passalacqua M, Leoncini G. The 2-arachidonoylglycerol effect on myosin light chain phosphorylation in human platelets. Biochimie 2013;95:1620–8.
Nithipatikom K, Gomez-Granados AD, Tang AT, Pfeiffer AW, Williams CL, Campbell WB. Cannabinoid receptor type 1 (CB1) activation inhibits small GTPase RhoA activity and regulates motility of prostate carcinoma cells. Endocrinology 2012;153:29–41.
Fields RD. White matter in learning, cognition and psychiatric disorders. Trends Neurosci. 2008;31:361–70.
Ishii A, Furusho M, Dupree JL, Bansal R. Role of ERK1/2 MAPK signaling in the maintenance of myelin and axonal integrity in the adult CNS. J Neurosci. 2014;34:16031–45.
Schneider S, Gruart A, Grade S, Zhang Y, Kroger S, Kirchhoff F, et al. Decrease in newly generated oligodendrocytes leads to motor dysfunctions and changed myelin structures that can be rescued by transplanted cells. Glia 2016;64:2201–18.
Poggi G, Boretius S, Mobius W, Moschny N, Baudewig J, Ruhwedel T, et al. Cortical network dysfunction caused by a subtle defect of myelination. Glia 2016;64:2025–40.
Carson RP, Kelm ND, West KL, Does MD, Fu C, Weaver G, et al. Hypomyelination following deletion of Tsc2 in oligodendrocyte precursors. Ann Clin Transl Neurol. 2015;2:1041–54.
Chen X, Zhang W, Li T, Guo Y, Tian Y, Wang F, et al. Impairment of oligodendroglia maturation leads to aberrantly increased cortical glutamate and anxiety-like behaviors in juvenile mice. Front Cell Neurosci. 2015;9:467.
Roy K, Murtie JC, El-Khodor BF, Edgar N, Sardi SP, Hooks BM, et al. Loss of erbB signaling in oligodendrocytes alters myelin and dopaminergic function, a potential mechanism for neuropsychiatric disorders. Proc Natl Acad Sci USA. 2007;104:8131–6.
Martinez LR, Black KC, Webb BT, Bell A, Baygani SK, Mier TJ, et al. Components of endocannabinoid signaling system are expressed in the perinatal mouse cerebellum and required for its normal development. eNeuro. 2020;7.
Diaz-Alonso J, Aguado T, Wu CS, Palazuelos J, Hofmann C, Garcez P, et al. The CB(1) cannabinoid receptor drives corticospinal motor neuron differentiation through the Ctip2/Satb2 transcriptional regulation axis. J Neurosci. 2012;32:16651–65.
Volkow ND, Baler RD, Compton WM, Weiss SR. Adverse health effects of marijuana use. N Engl J Med. 2014;370:2219–27.
Fried PA, Smith AM. A literature review of the consequences of prenatal marihuana exposure. An emerging theme of a deficiency in aspects of executive function. Neurotoxicol Teratol. 2001;23:1–11.
Trezza V, Campolongo P, Cassano T, Macheda T, Dipasquale P, Carratu MR, et al. Effects of perinatal exposure to delta-9-tetrahydrocannabinol on the emotional reactivity of the offspring: a longitudinal behavioral study in Wistar rats. Psychopharmacol (Berl). 2008;198:529–37.
Philippot G, Nyberg F, Gordh T, Fredriksson A, Viberg H. Short-term exposure and long-term consequences of neonatal exposure to Delta(9)-tetrahydrocannabinol (THC) and ibuprofen in mice. Behav Brain Res. 2016;307:137–44.
Busquets-Garcia A, Desprez T, Metna-Laurent M, Bellocchio L, Marsicano G, Soria-Gomez E. Dissecting the cannabinergic control of behavior: The where matters. Bioessays 2015;37:1215–25.
Ruehle S, Rey AA, Remmers F, Lutz B. The endocannabinoid system in anxiety, fear memory and habituation. J Psychopharmacol. 2012;26:23–39.
de Salas-Quiroga A, Diaz-Alonso J, Garcia-Rincon D, Remmers F, Vega D, Gomez-Canas M, et al. Prenatal exposure to cannabinoids evokes long-lasting functional alterations by targeting CB1 receptors on developing cortical neurons. Proc Natl Acad Sci USA. 2015;112:13693–8.
Patel JR, Klein RS. Mediators of oligodendrocyte differentiation during remyelination. FEBS Lett. 2011;585:3730–7.
Itoh K, Maki T, Lok J, Arai K. Mechanisms of cell-cell interaction in oligodendrogenesis and remyelination after stroke. Brain Res. 2015;1623:135–49.
Pendleton JC, Shamblott MJ, Gary DS, Belegu V, Hurtado A, Malone ML, et al. Chondroitin sulfate proteoglycans inhibit oligodendrocyte myelination through PTPsigma. Exp Neurol. 2013;247:113–21.
Palazuelos J, Klingener M, Raines EW, Crawford HC, Aguirre A. Oligodendrocyte Regeneration and CNS Remyelination Require TACE/ADAM17. J Neurosci. 2015;35:12241–7.
Mobius W, Cooper B, Kaufmann WA, Imig C, Ruhwedel T, Snaidero N, et al. Electron microscopy of the mouse central nervous system. Methods Cell Biol. 2010;96:475–512.
Moyon S, Dubessy AL, Aigrot MS, Trotter M, Huang JK, Dauphinot L, et al. Demyelination causes adult CNS progenitors to revert to an immature state and express immune cues that support their migration. J Neurosci. 2015;35:4–20.
Skripuletz T, Miller E, Moharregh-Khiabani D, Blank A, Pul R, Gudi V, et al. Beneficial effects of minocycline on cuprizone induced cortical demyelination. Neurochem Res. 2010;35:1422–33.
Prut L, Belzung C. The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: a review. Eur J Pharm. 2003;463:3–33.
Acknowledgements
We acknowledge the animal and microscopy facility cores at Complutense University.
Funding
This work was supported by the MINECO grants SAF2017–83516 and PID2020–112640RB-I00, and the Comunidad de Madrid grants 2016-T1/BMD-1060 and 2020–5 A/BMD-19728, Atracción del Talento Investigador Program, to JP. AHG and TA. AHG was also supported by the Comunidad de Madrid contract PEJD-2017-PRE/BMD-3703, and A.S.T by Fundación Tatiana Pérez de Guzmán el Bueno. Support was also provided by MINECO (grants RTI2018–095311-B-I00 to MG. and SAF2016–75292-R to CM), CIBERNED (grants CB06/05/0005 to M.G. and CB06/0005/0076 to C.M.), FEDER and ISCIII (AES 2018 grants PI18–00941 to IG-R and PI18/00513 to SM), Basque Government (grants IT1203–19 to C.M. and PIBA19–0059 to SM), and ARSEP Foundation (grant to SM).
Author information
Authors and Affiliations
Contributions
TA and JP designed research, analyzed data, and wrote the manuscript. AST, TA, AH, SS, AG and JCC performed research. MG, IGR, SM, CM, KM and BL contributed to write the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics statement
All the experimental procedures used were ethically reviewed and approved and were performed under the guidelines and with the approval of the Animal Welfare Committee of Universidad Complutense de Madrid and Comunidad de Madrid and under the directives of the European Commission (Directive 2010/63/EU).
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Alexei Verkhratsky
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sánchez-de la Torre, A., Aguado, T., Huerga-Gómez, A. et al. Cannabinoid CB1 receptor gene inactivation in oligodendrocyte precursors disrupts oligodendrogenesis and myelination in mice. Cell Death Dis 13, 585 (2022). https://doi.org/10.1038/s41419-022-05032-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-022-05032-z
