
Abstract
Genomic instability plays a key role in the initiation and progression of colorectal cancer (CRC). Although cancer driver genes in CRC have been well characterized, identifying novel genes associated with carcinogenesis and treatment remains challenging because of tumor heterogeneity. Here, we analyzed the genomic alterations of 45 samples from CRC patients in northern China by whole-exome sequencing. In addition to the identification of six well-known CRC driver genes (APC, TP53, KRAS, FBXW7, PIK3CA, and PABPC), two tumor-related genes (MTCH2 and HSPA6) were detected, along with RRP7A and GXYLT1, which have not been previously linked to cancer. GXYLT1 was mutated in 40% (18/45) of the samples in our cohort. Functionally, GXYLT1 promoted migration and invasion in vitro and metastasis in vivo, while the GXYLT1S212* mutant induced significantly greater effect. Furthermore, both GXYLT1 and GXYLT1S212* interacted with ERK2. GXYLT1 induced metastasis via a mechanism involving the Notch and MAPK pathways, whereas the GXYLT1S212* mutant mainly promoted metastasis by activating the MAPK pathway. We propose that GXYLT1 acts as a novel metastasis-associated driver gene and GXYLT1S212* might serve as a potential indicator for therapies targeting the MAPK pathway in CRC.
Similar content being viewed by others


Introduction
Colorectal cancer (CRC) is the second leading cause of cancer-related death worldwide [1, 2]. Although most CRCs are regarded as sporadic diseases, the development and progression of CRC is also associated with the accumulation of genetic aberrations and mutations in tumor-suppressor genes and oncogenes [3].
Many recent studies illustrate the genomic landscape underlying the carcinogenesis of CRC [4,5,6,7,8,9]. A genetic study by The Cancer Genome Atlas (TCGA) profiled genomic changes in CRC and identified common somatic mutations in several genes critical in CRC development [6]. These common mutations include mutations in APC, TP53, SMAD4, PIK3CA, and KRAS, which are commonly observed in various cancers, as well as frequent mutations in ARID1A, SOX9, and FAM123B/WTX. Moreover, the components of several pathways were found to have hypermutation status and recurrent alterations in CRC [10, 11].
Recent studies have indicated that the frequency of gene mutation and altered genetic events are different in CRC patients from different populations. Whole-exome sequencing (WES) on African-American CRC patients identified significant distinct mutational landscapes, which suggests different disease mechanisms in patients from diverse ethnic backgrounds [12]. Furthermore, analysis of the genomic landscape in Japanese CRC patients revealed different somatic gene mutations and mutation frequencies compared with those in Caucasian patients [13]. Consistently, the frequency and sequence of gene mutations are different between Chinese CRC patients and those in other countries [14]. Moreover, genomic alterations of Chinese CRC patients show considerable heterogeneity across different regions [14,15,16,17]. Therefore, exploring the genetic events in CRC Chinese patients from different regions may improve our understanding of the somatic mutations involved in CRC progression.
In this study, we aimed to investigate the genomic landscape of patients with CRC in northern China to identify novel mutations and corresponding driver genes associated with CRC development. WES was performed in 45 patients with CRC to determine genomic alterations. Our results identified GXYLT1 as a novel cancer-related gene with the ability to promote metastasis in CRC cells, and the stop-gain mutant GXYLT1S212* mainly enhanced metastasis through MAPK pathway activation. These findings suggest that the GXYLT1S212* mutant may serve as a biomarker for MAPK pathway-targeting treatments in CRC patients.
Materials and methods
Patients
This study included 45 CRC patients who underwent CRC resection from 2015 to 2016 at Peking University Cancer Hospital & Institute (Beijing, China) and were pathologically confirmed by two pathologists. Clinical stage was determined according to the eighth staging system of the American Joint Committee on Cancer. Patient clinicopathological characteristics are listed in Supplementary Tables 1 and 2. Written informed consent was obtained from each patient before fresh cancer tissues and peripheral blood were obtained for WES. This study was approved and supervised by the Research Ethics Committee of Peking University Cancer Hospital & Institute (No. 2014KT97).
Whole-exome sequencing
For blood samples and cancer samples with sufficient high-quality DNA, we carried out molecular biological tests to determine whether the two DNAs were matched and from the same patient. After bacterial and viral contamination had been removed, DNA samples were sheared using a Covaris S220 sonicator and libraries were prepared using an Agilent SureSelect Human All Exon v7 kit (cat #5991–9039, Agilent, Wilmington, DE, USA), according to the manufacturer’s protocols. DNA fragments of 200 bp were sequenced using 150bp paired-end reads with a NovaSeq-6000 (Illumina), achieving a mean depth of ≥200× for tumor DNA and ≥100× for normal DNA.
Mutational signature analysis
To explore the overall mutational features in our dataset, we focused on single-nucleotide mutations and calculated the frequency of the six substitution patterns. Based on a comprehensive analysis of COSMIC data [18] and the COSMIC mutational signature analysis pipeline, each substitution pattern was further separated into 16 substitution categories based on the flanking nucleotides surrounding the mutated base. A data matrix was constructed to represent each of the 45 exome-sequenced samples (columns) and the proportion of the 96 substitution categories (rows). Using the R package “MutationalPatterns” [19], we applied the nonnegative matrix factorization (NMF) algorithm to our data matrix and identified four mutational signatures in our 45 samples. By overlapping the 30 previously identified mutational signatures from COSMIC, we evaluated the associations between our four mutational signatures and the 30 known signatures by calculating the cosine similarity.
Cell lines, cell culture, and reagents
Human CRC cell lines (RKO, LoVo, HCT116, and SW480) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA), recently been authenticated by STR, and tested mycoplasma negative. Cells were cultured in RPMI-1640 or DMEM medium (HyClone, Logan, UT, USA) supplemented with 10% FBS, penicillin (100 units/mL), and streptomycin (100 µg/mL) in a humidified incubator with 5% CO2 at 37 °C. The Notch inhibitor DAPT (Cat #S2215) and ERK inhibitor SCH772984 (Cat #S7101) were purchased from Selleck Chemicals (Houston, TX, USA).
In vivo metastasis assays
All animal experiments were approved by the Ethics Committee of the Peking University Cancer Hospital & Institute and performed in accordance with experimental animal management ordinance. Female BALB/c nude mice (6–8 weeks old) were purchased from the Hua-Fu-Kang Corporation (Beijing, China), randomly divided into three groups with comparable body weight (n = 5 per group), and housed under specific pathogen-free conditions. After anesthetizing the mice using 1.2% avertin, their abdominal cavities were opened and the spleens were injected with 5 × 106 HCT116 cells stably expressing wild-type (WT) GXYLT1 or the mutant GXYLT1S212*. Bioluminescence imaging was conducted using an IVIS (PerkinElmer, Hopkinton, MA, USA), and image radiance values were normalized using Living Image software (PerkinElmer). After four weeks, mice were sacrificed and the livers with metastasis were isolated, fixed with 4% paraformaldehyde, and metastatic nodules counted in a single-blinded manner. The tissues were then embedded in paraffin and stained with hematoxylin and eosin (H&E) for histological analysis.
Statistical analyses
The human CRC gene expression profiles used in this study were downloaded from the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) and The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/). The disease-free survival and overall survival of CRC patients were determined using Kaplan–Meier survival analysis and the log-rank test. GSEA was performed to evaluate the mRNA levels of GXYLT1 with associated pathways using GSEA software 4.1.0. Pearson correlation analysis was used to evaluate the association between mRNA expressions of EGFR, ERK2, and GXYLT1. Two-tailed Student’s t-tests were performed to compare two different groups, while one-way ANOVA was used to compare more than two groups. Data are presented as the mean ± standard deviation (SD) of at least three independent experiments. All statistical analyses were performed using SPSS 23.0 software (SPSS Inc., Chicago, IL) or GraphPad Prism 8 software (GraphPad Prism, La Jolla, CA, USA). Statistical significance was set at p-value <0.05.
Additional materials and methods are provided in Supplementary materials and methods.
Results
Mutation overview and tumor mutational burden (TMB) analysis in 45 CRC samples
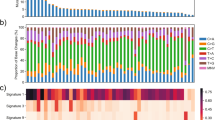
We first performed WES on tumors and paired blood samples from 45 Chinese patients with CRC and obtained approximately 460 Gb of read data with an average sequencing depth of 200-fold for tumors and 100-fold for blood samples. Further mapping to the human reference genome identified 22472 somatic single-nucleotide variants (SNVs), which were classified as missense, nonsense, frameshift, splice site, translation start site, and nonstop mutations (Supplementary Fig. 1a and Table 3). Moreover, 1063 short insertions and 4005 short deletions were detected in our cohort (Supplementary Fig. 1b and Table 3). Mutational signature analysis revealed that transition mutations were more common than transversion mutations (Supplementary Fig. 1c), and C > T substitutions were predominant in our samples (Fig. 1a and Supplementary Fig. 1d).

a Proportional contribution of six base-substitution patterns in each sample. b Relative contribution of the four mutational signatures in individual patients. c Patterns of the four mutational signatures A–D in 45 CRC samples.
To examine the overall genomic features of our data, we compared 33 publicly available TCGA cancer mutational datasets using TMB analysis. The TMB of our data was similar to that of TCGA COAD (CRC) dataset, suggesting that our data had a similar mutational burden to the well-established CRC cohort (Supplementary Fig. 2).
Identification of four mutational signatures in our cohort
A previous signature analysis of all cancer mutation data from the COSMIC database linked different cancer types to different contributions from each mutational spectrum [18]. To better understand the pathogenesis of CRC, we performed mutational signature analysis on all 22472 SNVs by analyzing the six mutation classes (C > T, C > A, T > C, T > G, C > G, and T > A). We identified four mutational signatures in our CRC samples (Signatures A–D) based on the calculated frequency of the six substitution patterns (Fig. 1b, c). Moreover, the relative contributions of these four signatures, which are the counterparts of the contributions observed for the 30 COSMIC signatures, varied among the 45 samples (Fig. 1b and Supplementary Fig. 3a).
Signature A was characterized by dominant C > A substitution with 92% similarity to COSMIC Signature 6 (Fig. 1c and Supplementary Fig. 3b), which is the most common signature in CRC, and this signature was detected in less than 3% of examined samples of other cancer types [18]. Samples with Signature 6 are generally microsatellite-unstable tumors associated with defective DNA mismatch repair.
Signature B in our cohort displayed a unique C > A feature in TpCpT and T > G in TpTpT (Fig. 1c), with 98% similarity to COSMIC Signature 10 (Supplementary Fig. 3b), which is characterized by large numbers of mutations in small subsets of samples, notably colorectal and uterine cancers [18]. Therefore, tumor samples exhibiting this mutational signature are often grouped as ultrahypermutators.
Signature C, which was characterized by mutations in all six substitution categories, was a predominant signature in our 45 CRC samples with 80% similarity to COSMIC Signature 1 (Fig. 1c and Supplementary Fig. 3b), which is the most commonly reported signature in all cancer types including CRC.
Signature D exhibited a uniquely high proportion of T > C substitution (Fig. 1c), similar to COSMIC Signature 6 (cosine similarity = 0.81) (Supplementary Fig. 3b). Together, our mutational analysis identified some well-known CRC signatures and confirmed the results of our TMB analysis, suggesting that our data have similar signature features to known CRC cohorts.
Driver mutation analysis revealed GXYLT1 as a potential driver gene in CRC
To identify novel genetic mutations in our dataset (Fig. 2a), we used the driver mutation identification pipeline IntOGen [20], which identified 495 driver mutations in 79 genes (Supplementary Table 4). Among the top-10 ranked genes in the IntOGen results from our dataset, we identified six well-studied driver genes that have been associated with cancer: APC, TP53, KRAS, PABPC1, FBXW7, and PIK3DA (Fig. 2b). We also identified four novel genes with significant OncodriveFM and OncodriveCLUST scores that have not yet been reported in CRC: MTCH2, GXYLT1, RRP7A, and HSPA6.

a Schematic of the workflow for the identification of novel driver genes. b Sample-based OncoPrint of the top- 10 driver genes identified using the IntOGen algorithm. Each column represents an individual sample, and each row represents a gene. Top: total number of somatic alterations (y axis) in each sample (x axis). Right: percentage of somatic alterations in 45 CRC samples. c Analysis of GXYLT1 mRNA expression levels in CRC tumors and matched normal tissues (GSE32323). d Analysis of GXYLT1 mRNA expression levels with respect to tumor stage (GSE17537). e Kaplan–Meier survival analysis of GXYLT1 levels and survival (GSE17537) in GEO datasets. *p < 0.05, ***p < 0.001.
MTCH2 and HSPA6 play various roles in different cancers but have not been associated with CRC [21, 22]. Among the two genes that have not been previously linked to cancer (GXYLT1 and RRP7A), GXYLT1 was recurrently mutated in 18 of 45 samples (40%) (Fig. 2b). We therefore focused on GXYLT1 as a novel gene potentially associated with CRC.
GXYLT1 encodes a glucoside xylosyltransferase that contributes to the first xylose elongation of O-glucose glycans on the extracellular domain of Notch1 and Notch2 [23], which is required for the trafficking of Notch proteins to the cell surface [24]. To explore the potential role of GXYLT1 in CRC, we first examined the mRNA levels of GXYLT1 in GEO datasets. GXYLT1 transcript expressions were significantly elevated in CRC tissues compared with levels in matched normal tissues from datasets GSE32323 (p < 0.001), GSE24550 (p < 0.001), and GSE9348 (p < 0.001) (Fig. 2c and Supplementary Fig. 4a). Moreover, GXYLT1 mRNA levels gradually increased with tumor progression and differed significantly between tumor stages (p = 0.023, p < 0.001, and p = 0.056 for GSE17537, GSE33193, and GSE28702, respectively) (Fig. 2d and Supplementary Fig. 4b).
We next performed Kaplan–Meier analysis to explore the prognostic significance of GXYLT1 expression in CRC. The results showed that patients with elevated GXYLT1 levels had a shorter disease-free survival or overall survival than those with low GXYLT1 levels (p = 0.044, p = 0.069, and p = 0.082 for GSE17537, GSE38832, and GSE17538, respectively) (Fig. 2e and Supplementary Fig. 4c). Taken together, these results suggest that elevated GXYLT1 mRNA expression indicates a poor prognosis in CRC patients.
GXYLT1 enhances CRC metastasis, while GXYLT1S212* induces greater metastatic ability in CRC
Our results identified GXYLT1 as a novel gene with a high mutation frequency in CRC. Twenty-two mutations were identified, most of which were located in the functional “GT8 like 2” domain of GXYLT1, including three stop-gain mutations (GXYLT1S212*, GXYLT1R224*, and GXYLT1Y264*) (Fig. 3a). One of the most frequently occurring mutations, GXYLT1S212*, was validated and recognized as a nonsense mutation in four independent samples (4/45, 8.9%) (Fig. 3a, Supplementary Fig. 5 and Table 2), compared with GXYLT1R224* (2/45, 4.4%) and GXYLT1Y264* (2/45, 4.4%). Thus, we focused on examining the role of GXYLT1 and mutant GXYLT1S212* in CRC.

a Somatic mutations in the GXYLT1 functional domain. Numbers represent amino acid residues. Each dot represents an individual mutated tumor sample. Blue dot: missense mutation. Purple dot: nonsense mutation. b, f Cell viability was assessed using CCK-8 assays in CRC cells transfected as indicated. c, g Representative images of colony formation in CRC cells transfected as indicated. Graphs show the numbers of colonies. d, h Transwell migration and invasion assays in CRC cells transfected as indicated. Graphs show quantification of migrated cells. e, i Wound-healing assays to evaluate cell migration in CRC cells transfected as indicated. Graphs show quantification of the wound-healing area. Data are presented as the mean ± SD of at least three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
To investigate the functions of GXYLT1 and GXYLT1S212* in CRC, we first examined the expression of GXYLT1 in CRC cells (Supplementary Fig. 6a, b). Since the GXYLT1 antibody is against the C-terminal 30 amino acids of the protein, GXYLT1S212* could not be detected by anti-GXYLT1 (Supplementary Fig. 6c). We used the FLAG antibody to determine the overexpression levels of GXYLT1 and GXYLT1S212* in CRC cells transiently transfected with GXYLT1 and GXYLT1S212* plasmids (Supplementary Fig. 6d). Ectopic WT GXYLT1 expression had no significant effects on the proliferation or colony formation of CRC cells (Fig. 3b, c), but increased cell migration and invasion in the Transwell and wound-healing assays (Fig. 3d, e). Similarly, we found that GXYLT1S212* had no effect on proliferation or colony-formation ability. However, mutant GXYLT1S212* showed a stronger capability of promoting migration and invasion compared with WT GXYLT1 (Fig. 3d, e). We also knocked down GXYLT1 using short interfering RNA in the SW480 and RKO cells (Supplementary Fig. 6e). Consistent with the overexpression results, downregulation of GXYLT1 had no effect on CRC cell growth but attenuated CRC cell migration and invasion (Fig. 3f–i). These results indicated that GXYLT1 contributes to the migration and invasion of CRC cells and that GXYLT1S212* exhibits enhanced migration and invasion-promoting ability in vitro.
To confirm the in vitro findings, we next established LV-GXYLT1 and LV-S212* cell models (Fig. 4a). We evaluated whether WT GXYLT1 and GXYLT1S212* affected CRC metastasis in vivo by injecting HCT116 cells stably overexpressing GXYLT1 or GXYLT1S212* into the spleen of nude mice. Two weeks after injection, bioluminescence imaging was performed to detect tumor signals in mouse liver. Although LV-GXYLT1 showed a little increase in the metastatic potential of HCT116 cells compared with control group, LV-S212* exhibited a significant stronger effect, which was demonstrated by enhanced luminescence of the mouse liver (Fig. 4b, c). After 4 weeks, mouse livers were collected for analysis. Increased numbers of liver metastatic nodules and increased incidence of liver metastasis were observed in the LV-GXYLT1 and LV-S212* groups compared with the control group (Fig. 4d–f and Supplementary Table 5). Furthermore, the LV-S212* group had more liver metastatic nodules than the LV-GXYLT1 group (Fig. 4d–f), suggesting that LV-S212* exhibits a gain-of-function effect. Taken together, these findings suggest that GXYLT1 promotes CRC metastasis and GXYLT1S212* induces a greater metastatic ability both in vitro and in vivo.

The spleens of mice were injected with 5 × 106 HCT116 cells infected with LV-control, LV-GXYLT1, or LV-S212* (n = 5 mice per group). a GXYLT1 and GXYLT1S212* expressions were confirmed in HCT116 cells stably infected with LV-GXYLT1 and LV-S212*, respectively. b Representative bioluminescence imaging of mice two weeks after implantation. c Quantification of bioluminescence imaging in the mouse liver from the indicated groups. d Representative images of liver metastatic nodules from the indicated groups. e Representative images of H&E staining in liver metastatic lesions from the indicated group. Scale bars: top, 300 μm; bottom, 60 μm. f Number of metastatic liver nodules from the indicated groups. Data are presented as the mean ± SD (n = 5). *p < 0.05.
WT GXYLT1 promotes CRC metastasis via the Notch pathway and GXYLT1S212* increases metastasis partially via the Notch pathway
To investigate whether GXYLT1 regulates the Notch pathway in CRC cells, we examined the protein levels of the intracellular domain of the Notch protein (NICD) and Hes1, which is encoded by one of the major Notch-targeted genes. Overexpression of WT GXYLT1 and GXYLT1S212* in CRC cells upregulated NICD and Hes1 levels (Fig. 5a), indicating activation of the Notch pathway. Notably, GXYLT1S212* induced NICD and Hes1 expression to lower levels than those induced by WT GXYLT1. Consistently, GXYLT1 knockdown decreased NICD and Hes1 expression in SW480 and RKO cells (Fig. 5b). These results indicated that GXYLT1 regulates the Notch pathway in CRC cells. Furthermore, GXYLT1S212* promoted the Notch pathway cascade to a lower level compared with WT GXYLT1.

a, b Western blot analysis of the protein levels of the Notch protein intracellular domain (NICD) and Hes1 in CRC cells transfected as indicated. Bar graphs show quantitative analysis of protein levels. c, d Transwell migration and invasion assays of transfected CRC cells treated with the Notch pathway inhibitor DAPT or the DMSO control for 48 h. Charts show quantification of migrated cells. e, f Western blot analysis of NICD and Hes1 in cell lysates. Bar graphs show quantification of NICD and Hes1 protein levels. Data are presented as the mean ± SD of at least three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
To determine whether WT GXYLT1 and GXYLT1S212* promote CRC cell metastasis via the Notch pathway, we abolished Notch pathway activity using the Notch inhibitor, DAPT. Transwell assays demonstrated that treatment of CRC cells with DAPT significantly eliminated the induction of cell migration and invasion by WT GXYLT1, but had a modest effect on cells expressing GXYLT1S212* (Fig. 5c and Supplementary Fig. 7a). DAPT treatment also inhibited cell migration and invasion in both GXYLT1-depleted and control cells (Fig. 5d and Supplementary Fig. 7b). Moreover, DAPT markedly decreased NICD and Hes1 protein levels in WT GXYLT1- and GXYLT1S212*-overexpressing cells, as well as in GXYLT1-knockdown cells (Fig. 5e, f and Supplementary Fig. 7c, d). These results suggest that Notch pathway inhibition prevents cell migration and invasion induced by WT GXYLT1 but not GXYLT1S212*. Therefore, these data raise the possibility that GXYLT1S212* promotes CRC metastasis only partially through the Notch pathway, and that alternative pathways are potentially involved in the induction of metastasis by GXYLT1S212*.
GXYLT1S212* enhances metastasis mainly via the MAPK pathway
Accumulating evidence has indicated that the interactions between the Notch and MAPK pathways are associated with the progression of CRC [25, 26]. Gene set enrichment analysis (GSEA) of the TCGA dataset suggested that GXYLT1 expression was positively associated with MAPK pathway (Fig. 6a). Furthermore, Pearson correlation analyses of the public CRC databases indicated that mRNA levels of GXYLT1 were positively correlated with EGFR and ERK2 (Supplementary Fig. 8). Thus, we next investigated whether WT GXYLT1 and GXYLT1S212* regulate the MAPK pathway in CRC cells by detecting the phosphorylation levels of EGFR and ERK1/2. Both WT GXYLT1 and GXYLT1S212* overexpression enhanced the phosphorylation of EGFR and ERK1/2 in LoVo cells (Fig. 6b). In addition, we observed that GXYLT1S212* enhanced EGFR and ERK1/2 phosphorylation to greater levels compared with those observed in cells expressing WT GXYLT1. Consistently, depletion of GXYLT1 resulted in decreased phosphorylation of EGFR and ERK1/2 in RKO cells (Fig. 6c). These results suggested that compared with WT GXYLT1, GXYLT1S212* strongly promoted the MAPK pathway.

a GSEA analysis of TCGA dataset revealed the correlation between GXYLT1 expression and the MAPK pathway. b, c Western blot analysis of protein levels and phosphorylation levels of EGFR and ERK1/2 in transfected CRC cells. Bar graphs show quantification of pEGFR and pERK1/2 levels. d–f Exogenous GXYLT1 and GXYLT1S212* interact with EKR2. Co-immunoprecipitation was performed in cells transfected as indicated using Flag antibody to pull down Flag-GXYLT1 and Flag-GXYLT1S212* (d) or Myc antibody against Myc-ERK2 (e, f). Western blot was performed using the indicated antibodies. g, h Transwell migration and invasion assays of CRC cells expressing WT GXYLT1 or GXYLT1S212* and treated with the ERK1/2 inhibitor SCH772984 or the DMSO control for 48 h. Graphs show quantification of migrated cells. i, j Western blot analysis of cell lysates with the indicated antibodies. Bar graphs show quantification of pERK1/2 levels. Data are presented as the mean ± SD of at least three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
To further explore the relationship between GXYLT1 or GXYLT1S212* and ERK1/2, we performed co-immunoprecipitation. Myc-ERK2 was found in Flag-GXYLT1 and Flag-GXYLT1S212* immune complexes (Fig. 6d), while Flag-GXYLT1 and Flag-GXYLT1S212* were detected in Myc-ERK2 immunoprecipitates through reciprocal immunoprecipitation (Fig. 6e, f). These findings suggest that GXYLT1 and GXYLT1S212* interact with ERK2 in vivo.
To evaluate whether WT GXYLT1 and GXYLT1S212* enhanced cell metastasis via the MAPK pathway, we inhibited MAPK pathway activity using the ERK kinase inhibitor SCH772984 and analyzed the migration and invasion of CRC cells expressing WT GXYLT1 or GXYLT1S212*. SCH772984 significantly blocked migration and invasion (Fig. 6g, h) and suppressed ERK1/2 phosphorylation (Fig. 6i, j) induced by WT GXYLT1 and GXYLT1S212* in CRC cells.
Taken together, our results demonstrated that both WT GXYLT1 and GXYLT1S212* enhance CRC metastasis via the MAPK pathway by interacting with ERK, and GXYLT1S212* had stronger promoting effects compared with WT GXYLT1.
GXYLT1 and GXYLT1S212* are required for the activities of Notch and MAPK pathways
To further clarify whether the activities of Notch and MAPK pathways are dependent on GXYLT1 and GXYLT1S212*, LoVo cells were transfected with the GXYLT1 and GXYLT1S212* plasmid in a dose-dependent manner, and the endogenous expression of NICD, Hes1, and pERK1/2 was determined. The gradual increase in NICD, Hes1, and pERK1/2 levels was observed post GXYLT1 or GXYLT1S212* dose-dependent transfection (Fig. 7a, b).

a, b Western blot analysis of cell lysates with the indicated antibodies. LoVo cells were transfected with different amounts of GXYLT1 and GXYLT1S212* expression plasmid and harvested for western blot after 48 h. Bar graphs show quantification of NICD, Hes1, and pERK1/2 levels. c RKO cells with indicated transfection were harvested for RT-PCR analysis and Western blot analysis to determine the expression of GXYLT1 and GXYLT1S212*. d Transwell migration and invasion assays of RKO cells with indicated transfection. Graphs show quantification of migrated cells. e Western blot analysis of cell lysates with the indicated antibodies. RKO cells with indicated transfection were harvested for Western blot after 48 h. Bar graphs show quantification of NICD, Hes1, and pERK1/2 levels. Data are presented as the mean ± SD of at least three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
Furthermore, rescue experiments were performed in GXYLT1-depleted RKO cells transfected with Flag-GXYLT1 and Flag-S212* (Fig. 7c). Transwell assays showed that WT GXYLT1 and GXYLT1S212* significantly rescued the migration and invasion deficit induced by GXYLT1 knockdown in RKO cells, and GXYLT1S212* had a stronger effect (Fig. 7d). Moreover, restoring the expression of WT GXYLT1 and GXYLT1S212* rescued the GXYLT1 depletion-induced downregulation of NICD, Hes1, and pERK1/2 (Fig. 7e), which are effectors of the Notch and MAPK pathway. These results suggested that Notch and MAPK signaling are downstream of GXYLT1 and GXYLT1S212*.
Discussion
In this study, we performed WES on samples from 45 patients with CRC in northern China to identify genomic alterations and potential genomic targets for clinical diagnosis and treatment. We identified four mutation signatures in our cohort with a high prevalence of signature C. Our combined computational prediction-based prioritization and functional analysis identified that GXYLT1 is a potentially novel oncogene that promotes the metastasis of CRC via the Notch and MAPK pathways, and the stop-gain mutant GXYLT1S212* promoted a stronger malignant phenotype in CRC by activating MAPK signaling.
Increasing evidence has shown a high rate of C > T transitions in diverse types of cancers including CRC [27,28,29,30]. Consistent with previous studies, C > T substitutions were predominant in our cohort. In addition, the TMB in our cohort was similar to that of TCGA dataset, indicating a comparable mutational frequency in the protein-coding regions. Moreover, mutational signature analysis revealed four prominent mutational patterns in our CRC cohort. Signatures A and D were similar to the COSMIC Signature 6, while Signature B and C corresponded to COSMIC Signature 10 and 1, respectively.
Gene mutations, such as those in APC, KRAS, and TP53, have been established as core elements that facilitate the malignant transformation of CRC cells [6, 8]. Similarly, we found that APC (82%), TP53 (47%), KRAS (33%), PABPC1 (31%), FBXW7 (22%), and PIK3CA (18%) were mutated at a high frequency in our cohort. In addition, we identified previously uncharacterized mutated genes in CRC, such as MTCH2 (42%), GXYLT1 (40%), RRP7A (16%), and HSPA6 (11%).
In addition to APC, TP53, and MTCH2, GXYLT1 displayed a high frequency of somatic mutations (40%) in our cohort. However, mutations in GXYLT1 have only been detected in 0.9‒2% of CRC samples from TCGA and other published cohorts [6, 8, 27]. The differences between our study and previous studies may be from substantial differences in the epidemiological characteristics of the patient cohorts.
Gene mutations play critical roles in the tumorigenesis and metastasis of CRC [31]. Previous studies showed that stop-gain mutations of APC deactivate the tumor-suppressor functions of the encoded proteins and confer oncogenic gain-of-function activity, resulting in the rapid development of aggressive carcinoma [32,33,34]. Moreover, the truncated PPM1D induced by exon mutation impairs the activation of p53 pathway, and promotes tumor growth in Apcmin mice, compared with the wild-type PPM1D [35]. Our study identified new recurrent alterations in GXYLT1, three of which were stop-gain mutations (GXYLT1S212*, GXYLT1R224*, and GXYLT1Y264*). Thus, GXYLT1S212* and GXYLT1Y264* with relatively short and long amino acid sequences, respectively, were selected to explore their roles in CRC. The results showed that the GXYLT1S212* exhibited significantly increased migration and invasion ability compared with WT GXYLT1. However, no significant effects were observed for the GXYLT1Y264* (Supplementary Fig. 9). Therefore, GXYLT1S212* was chosen for mechanistic studies.
Although the Notch pathway has been associated with CRC development and progression [36,37,38], the role of xylosylation in the Notch ECD remains controversial. Xylose modification of Notch by GXYLT1 and GXYLT2 suppresses Notch activity in Drosophila [39, 40], whereas xylosylation by GXYLT2 upregulates the Notch pathway in human cancers [41]. Since there is no significant association between the expression of GXYLT1 and GXYLT2 in available databases (Supplementary Fig. 10), we investigated the functions of WT GXYLT1 and GXYLT1S212*. Consistent with a previous study on GXYLT2, our results showed that the Notch signaling pathway is required for WT GXYLT1-regulated metastasis. However, GXYLT1S212* increased the metastatic ability of CRC cells with relatively lower Notch pathway activation compared with WT GXYLT1. Despite the stronger metastatic ability induced by GXYLT1S212*, these effects were not completely blocked by the Notch inhibitor DAPT, indicating that other mechanisms may be involved in the effects of GXYLT1S212* on CRC metastasis.
Research has demonstrated that the crosstalk between the MAPK and Notch pathways is involved in cancer progression [25, 26]. MAPK pathway activation promotes Notch signaling [42, 43], whereas its inhibition suppresses Notch signaling by decreasing NICD [44] and Hes1 [45] expression. Therefore, we evaluated the effects of WT GXYLT1 and GXYLT1S212* on the MAPK signaling pathway. Our results showed that both WT GXYLT1 and GXYLT1S212* interact with ERK2, and GXYLT1S212* induced a greater effect on the MAPK cascade than WT GXYLT1 (Fig. 8). Inhibiting the MAPK pathway completely abrogated the promotion of CRC cell metastasis by WT GXYLT1 and GXYLT1S212*. Since the Notch pathway inhibitor only partially repressed the metastasis induced by GXYLT1S212*, we hypothesize that GXYLT1S212* mainly enhances metastasis via the MAPK signaling pathway. Together, these data suggest that ERK1/2 inhibitors could potentiate CRC therapies targeting GXYLT1 in the presence of mutant GXYLT1.

GXYLT1 promoted CRC metastasis via the Notch and MAPK pathways, whereas GXYLT1S212* exhibited stronger metastasis-promoting ability mainly through activating the MAPK pathway.
The roles of WT GXYLT1 and GXYLT1S212* in CRC metastasis of our study should be interpreted with some considerations. First, our patients are from northern China, and this mutation pattern may be unique to CRC patients from northern China. Second, a larger cohort of patients with CRC, including detailed clinicopathological information, is required to validate the clinical significance of WT GXYLT1 and GXYLT1S212* in patients with CRC. Third, although the ERK1/2 inhibitor completely abolished GXYLT1S212*-induced metastasis and the mutation site may serve as a therapy indicator, further studies are required to justify the potential of ERK1/2 inhibitors to treat GXYLT1-mutant CRC.
In conclusion, this study provides insights into the genomic landscape of Chinese patients with CRC and identified new recurrent alterations in GXYLT1 not previously described in CRC. In addition, we found that the gain-of-function GXYLT1S212* mutation showed a stronger capability to promote CRC metastasis than WT GXYLT1 by activating MAPK signaling. Furthermore, these findings suggest that GXYLT1S212* could be used as a potential indicator for therapies targeting the MAPK pathway.
Data availability
WES raw sequencing data generated during the current study have been submitted to the Sequence Read Archive (SRA) with BioProject ID: PRJNA745011 (http://submit.ncbi.nlm.nih.gov/). All other data generated during this study are included in this published article (and its supplementary information files).
References
Sung H, Ferlay J, Siegel R, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Siegel R, Miller K, Goding Sauer A, Fedewa S, Butterly L, Anderson J, et al. Colorectal cancer statistics, 2020. CA Cancer J Clin. 2020;70:145–64.
Dekker E, Tanis P, Vleugels J, Kasi P, Wallace M. Colorectal cancer. Lancet. 2019;394:1467–80.
Brannon A, Vakiani E, Sylvester B, Scott S, McDermott G, Shah R, et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol. 2014;15:454.
Bian S, Hou Y, Zhou X, Li X, Yong J, Wang Y, et al. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science. 2018;362:1060–3.
Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7.
Strickler J, Loree J, Ahronian L, Parikh A, Niedzwiecki D, Pereira A, et al. Genomic landscape of cell-free DNA in patients with colorectal cancer. Cancer Disco. 2018;8:164–73.
Yaeger R, Chatila W, Lipsyc M, Hechtman J, Cercek A, Sanchez-Vega F, et al. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell. 2018;33:125–36.
Pleguezuelos-Manzano C, Puschhof J, Rosendahl Huber A, van Hoeck A, Wood H, Nomburg J, et al. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature. 2020;580:269–73.
Koveitypour Z, Panahi F, Vakilian M, Peymani M, Seyed Forootan F, Nasr, et al. Signaling pathways involved in colorectal cancer progression. Cell Biosci. 2019;9:97.
Nguyen L, Goel A, Chung D. Pathways of colorectal carcinogenesis. Gastroenterology. 2020;158:291–302.
Guda K, Veigl M, Varadan V, Nosrati A, Ravi L, Lutterbaugh J, et al. Novel recurrently mutated genes in African American colon cancers. Proc Natl Acad Sci USA. 2015;112:1149–54.
Nagahashi M, Wakai T, Shimada Y, Ichikawa H, Kameyama H, Kobayashi T, et al. Genomic landscape of colorectal cancer in Japan: clinical implications of comprehensive genomic sequencing for precision medicine. Genome Med. 2016;8:136.
Ma R, Jing C, Zhang Y, Cao H, Liu S, Wang Z, et al. The somatic mutation landscape of Chinese colorectal cancer. J Cancer. 2020;11:1038–46.
Liu Z, Yang C, Li X, Luo W, Roy B, Xiong T, et al. The landscape of somatic mutation in sporadic Chinese colorectal cancer. Oncotarget. 2018;9:27412–22.
Teng H, Gao R, Qin N, Jiang X, Ren M, Wang Y, et al. Identification of recurrent and novel mutations by whole genome sequencing of colorectal tumors from the Han population in Shanghai, eastern China. Mol Med Rep. 2018;18:5361–70.
Ye W, Ling S, Liu R, Pan Z, Wang G, Gao S, et al. Exome sequencing reveals the genetic landscape and frequent inactivation of PCDHB3 in Chinese rectal cancers. J Pathol. 2018;245:222–34.
Alexandrov L, Kim J, Haradhvala N, Huang M, Tian Ng A, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101.
Blokzijl F, Janssen R, van Boxtel R, Cuppen E. MutationalPatterns: comprehensive genome-wide analysis of mutational processes. Genome Med. 2018;10:33.
Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Tamborero D, Schroeder M, Jene-Sanz A, et al. IntOGen-mutations identifies cancer drivers across tumor types. Nat Methods. 2013;10:1081–2.
Fan Q, Liu B. Identification of the anticancer effects of a novel proteasome inhibitor, ixazomib, on colorectal cancer using a combined method of microarray and bioinformatics analysis. Onco Targets Ther. 2017;10:3591–606.
Zhang T, Hu Y, Wang T, Cai P. Dihydroartemisinin inhibits the viability of cervical cancer cells by upregulating caveolin 1 and mitochondrial carrier homolog 2: Involvement of p53 activation and NAD(P)H:quinone oxidoreductase 1 downregulation. Int J Mol Med. 2017;40:21–30.
Sethi M, Buettner F, Krylov V, Takeuchi H, Nifantiev N, Haltiwanger R, et al. Identification of glycosyltransferase 8 family members as xylosyltransferases acting on O-glucosylated notch epidermal growth factor repeats. J Biol Chem. 2010;285:1582–6.
Urata Y, Saiki W, Tsukamoto Y, Sago H, Hibi H, Okajima T, et al. Xylosyl extension of O-glucose glycans on the extracellular domain of NOTCH1 and NOTCH2 regulates notch cell surface trafficking. Cells. 2020;9:1220.
Schmidt E, Lamprecht S, Blaj C, Schaaf C, Krebs S, Blum H, et al. Targeting tumor cell plasticity by combined inhibition of NOTCH and MAPK signaling in colon cancer. J Exp Med. 2018;215:1693–708.
Neradugomma N, Subramaniam D, Tawfik O, Goffin V, Kumar T, Jensen R, et al. Prolactin signaling enhances colon cancer stemness by modulating Notch signaling in a Jak2-STAT3/ERK manner. Carcinogenesis. 2014;35:795–806.
Seshagiri S, Stawiski E, Durinck S, Modrusan Z, Storm E, Conboy C, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488:660–4.
Li M, Liu F, Zhang F, Zhou W, Jiang X, Yang Y, et al. Genomic ERBB2/ERBB3 mutations promote PD-L1-mediated immune escape in gallbladder cancer: a whole-exome sequencing analysis. Gut. 2019;68:1024–33.
Sawada G, Niida A, Uchi R, Hirata H, Shimamura T, Suzuki Y, et al. Genomic landscape of esophageal squamous cell carcinoma in a Japanese population. Gastroenterology. 2016;150:1171–82.
Yachida S, Wood L, Suzuki M, Takai E, Totoki Y, Kato M, et al. Genomic sequencing identifies ELF3 as a driver of ampullary carcinoma. Cancer Cell. 2016;29:229–40.
Huang D, Sun W, Zhou Y, Li P, Chen F, Chen H, et al. Mutations of key driver genes in colorectal cancer progression and metastasis. Cancer Metastasis Rev. 2018;37:173–87.
Nelson S, Li Z, Newton I, Fraser D, Milne R, Martin D, et al. Tumorigenic fragments of APC cause dominant defects in directional cell migration in multiple model systems. Dis Model Mech. 2012;5:940–7.
Kim S, Zhang L, Yoon J, Lee J, Min J, Li W, et al. Truncated adenomatous polyposis coli mutation induces Asef-activated Golgi fragmentation. Mol Cell Biol. 2018;38:e00135–18.
Aghabozorgi A, Bahreyni A, Soleimani A, Bahrami A, Khazaei M, Ferns G, et al. Role of adenomatous polyposis coli (APC) gene mutations in the pathogenesis of colorectal cancer; current status and perspectives. Biochimie. 2019;157:64–71.
Burocziova M, Burdova K, Martinikova A, Kasparek P, Kleiblova P, Danielsen S, et al. Truncated PPM1D impairs stem cell response to genotoxic stress and promotes growth of APC-deficient tumors in the mouse colon. Cell Death Dis. 2019;10:818.
Aster J, Pear W, Blacklow S. The varied roles of notch in cancer. Annu Rev Pathol. 2017;12:245–75.
Jackstadt R, van Hooff S, Leach J, Cortes-Lavaud X, Lohuis J, Ridgway R, et al. Epithelial NOTCH signaling rewires the tumor microenvironment of colorectal cancer to drive poor-prognosis subtypes and metastasis. Cancer Cell. 2019;36:319–36.
Tyagi A, Sharma A, Damodaran C. A review on notch signaling and colorectal cancer. Cells. 2020;9:1549.
Lee T, Sethi M, Leonardi J, Rana N, Buettner F, Haltiwanger R, et al. Negative regulation of notch signaling by xylose. PLoS Genet. 2013;9:e1003547.
Lee T, Pandey A, Jafar-Nejad H. Xylosylation of the notch receptor preserves the balance between its activation by trans-Delta and inhibition by cis-ligands in Drosophila. PLoS Genet. 2017;13:e1006723.
Cui Q, Xing J, Gu Y, Nan X, Ma W, Chen Y, et al. GXYLT2 accelerates cell growth and migration by regulating the notch pathway in human cancer cells. Exp Cell Res. 2019;376:1–10.
Tremblay I, Pare E, Arsenault D, Douziech M, Boucher M. The MEK/ERK pathway promotes NOTCH signalling in pancreatic cancer cells. PLoS ONE. 2013;8:e85502.
Yamashita A, Geraldo M, Fuziwara C, Kulcsar M, Friguglietti C, da Costa R, et al. Notch pathway is activated by MAPK signaling and influences papillary thyroid cancer proliferation. Transl Oncol. 2013;6:197–205.
Pope J, Bhat A, Sharma A, Ahmad R, Krishnan M, Washington M, et al. Claudin-1 regulates intestinal epithelial homeostasis through the modulation of Notch-signalling. Gut. 2014;63:622–34.
Traversi F, Stooss A, Dettmer M, Charles R. BRAF(V600E) overrides NOTCH signaling in thyroid cancer. Thyroid. 2021;31:787–99.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 81872022, 82173218, 82171720, 31671375, 31871339, and 81672439), Beijing Natural Science Foundation (No. 5202008, 7162039), Capital’s Funds for Health Improvement and Research (CFH 2018-2-2153), The National Key Research and Development Program of China (No. 2017YFC1201200), and Beijing Hospitals Authority Clinical Medicine Development of Special Funding Support (ZYLX202116).
Author information
Authors and Affiliations
Contributions
HQ, BJ, and XS conceived and designed the study. LP, TL, JC, PG, and PX performed the experiments. MZ performed the bioinformatics data analysis. LC, ZW, JD, and QX contributed samples, reagents, materials, and comments on the paper. LP, MZ, and BJ wrote the paper. All authors read and approved the final paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was approved and supervised by the Research Ethics Committee of Peking University Cancer Hospital & Institute (No. 2014KT97).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Massimiliano Agostini
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peng, L., Zhao, M., Liu, T. et al. A stop-gain mutation in GXYLT1 promotes metastasis of colorectal cancer via the MAPK pathway. Cell Death Dis 13, 395 (2022). https://doi.org/10.1038/s41419-022-04844-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-022-04844-3
