
Abstract
Autosomal-dominant polycystic kidney disease (ADPKD) is the most common inherited kidney disease and is characterized by progressive growth of fluid-filled cysts. Growth factors binding to receptor tyrosine kinases (RTKs) stimulate cell proliferation and cyst growth in PKD. Nintedanib, a triple RTK inhibitor, targets the vascular endothelial growth-factor receptor (VEGFR), platelet-derived growth-factor receptor (PDGFR), and fibroblast growth-factor receptor (FGFR), and is an approved drug for the treatment of non-small-cell lung carcinoma and idiopathic lung fibrosis. To determine if RTK inhibition using nintedanib can slow ADPKD progression, we tested its effect on human ADPKD renal cyst epithelial cells and myofibroblasts in vitro, and on Pkd1f/fPkhd1Cre and Pkd1RC/RC, orthologous mouse models of ADPKD. Nintedanib significantly inhibited cell proliferation and in vitro cyst growth of human ADPKD renal cyst epithelial cells, and cell viability and migration of human ADPKD renal myofibroblasts. Consistently, nintedanib treatment significantly reduced kidney-to-body-weight ratio, renal cystic index, cystic epithelial cell proliferation, and blood-urea nitrogen levels in both the Pkd1f/fPkhd1Cre and Pkd1RC/RC mice. There was a corresponding reduction in ERK, AKT, STAT3, and mTOR activity and expression of proproliferative factors, including Yes-associated protein (YAP), c-Myc, and Cyclin D1. Nintedanib treatment significantly reduced fibrosis in Pkd1RC/RC mice, but did not affect renal fibrosis in Pkd1f/fPkhd1Cre mice. Overall, these results suggest that nintedanib may be repurposed to effectively slow cyst growth in ADPKD.
Similar content being viewed by others
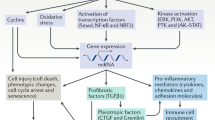


Introduction
In PKD kidneys, cysts develop from the renal tubules and progressively enlarge due to cell proliferation and fluid secretion by tubular epithelial cells [1]. Progressive renal fibrosis accompanies cyst expansion in all forms of PKD, irrespective of the underlying gene mutation, and along with inflammation, could be a final common pathway to end-stage renal disease (ESRD) [2, 3].
The RTK family consists of 58 known transmembrane receptors that regulate cell proliferation, differentiation, migration, metabolism, fibrosis, and angiogenesis. High-affinity binding to ligands such as growth factors, cytokines, or hormones leads to activation and consequent autophosphorylation of RTKs. This, in turn, leads to the recruitment of a wide range of proteins that propagate cell-signaling pathways, including JAK2/STAT, RAS/RAF/MEK/ERK (MAPK), PI3 kinase, AKT, or mTOR signaling.
Under normal physiologic conditions, the activity of RTKs is tightly regulated. However, in various human diseases, aberrant RTK activity occurs due to gain-of-function mutations in their ligand-binding or kinase domains, overexpression of RTKs by genomic amplification, overproduction of ligands, or by autocrine activation of RTKs [4]. Multiple small-molecule inhibitors of RTKs have been developed to specifically target the ATP-binding site of the intracellular tyrosine kinase domain. Moreover, FDA-approved small-molecule inhibitors and monoclonal antibodies that interfere with RTK activation are currently used for the treatment of cancer and lung disease [5, 6].
In PKD, two decades of research have shown that RTKs play an important role in disease progression. The main RTKs that were studied in PKD are EGFR, HER2, and VEGFR. Inhibition of EGFR and VEGFR was shown to slow cyst growth in various PKD rodent models [7,8,9,10,11,12,13]. Moreover, tesevatinib, an inhibitor of EGFR, HER2, c-SRC, and VEGFR, which reduced cyst growth in autosomal-recessive PKD (ARPKD) mice [10], was recently tested for efficacy and safety in a clinical phase-2 study in ADPKD patients (ClinicalTrials.gov Identifier: NCT03203642).
Nintedanib (BIBF 1120, Vargatef®, Ofev®), is a potent small-molecule RTK inhibitor that targets PDGFRα and β, FGFR1,2 and 3, and VEGFR1,2 and 3 [14]. Competitive binding of nintedanib to the ATP-binding pocket of RTKs interferes with cross-autophosphorylation of the receptor homodimers, thereby blocking the signaling cascade. Importantly, nintedanib has undergone extensive human trials [5, 15, 16] and is currently approved by the US FDA for therapy for idiopathic pulmonary fibrosis, interstitial lung disease associated with systemic sclerosis, and chronic fibrosing interstitial lung diseases, and by the European Medicines Agency for non-small-cell lung carcinoma. Preclinical studies have demonstrated nintedanib’s antiproliferative and antitumor effects in various cancers, including kidney, ovarian, pancreatic, colorectal, prostate, and non-small-cell lung cancers [14, 17]. In addition, Nintedanib has been shown to be a potent antifibrotic agent that stimulates promatrix metalloproteinases-2 expression and inhibits tissue inhibitor of metalloproteinases-2 (TIMP2) expression, a combination that promotes extracellular matrix (ECM) degradation in the lungs [18]. Nintedanib treatment also inhibited fibrosis in various cancers and in chronic disease of the lungs and liver [14, 19,20,21]. Importantly, nintedanib treatment significantly reduced renal fibrosis in unilateral ureteric obstruction and folic acid induced chronic kidney disease in mice [22]. However, the effect of nintedanib has not been tested in PKD. Here we examined the effects of nintedanib on cyst growth and fibrosis in two rapid and slow progressive ADPKD mouse models. The results of these studies are presented.
Results
Nintedanib suppressed ADPKD cyst epithelial cell proliferation and cyst growth in vitro
To test the effect of nintedanib on epithelial cell proliferation, primary culture human ADPKD renal cyst epithelial cells were treated with nintedanib for 24 h and cell proliferation was determined by BrdU incorporation. Nintedanib treatment at 1.5 µM dose reduced cell proliferation by over 40% compared with vehicle treatment (Fig. 1A). However in primary culture normal human kidney (NHK) epithelial cells or M1 mouse-collecting duct cells, at 1.5 µM dose, nintedanib did not significantly affect cell proliferation and even at 3 µM, nintedanib reduced proliferation by only 22 and 15%, respectively (Supplemental 1A, B). Nintedanib also reduced cyst growth by human ADPKD renal epithelial cells seeded within a 3D collagen matrix (Fig. 1B) by 5-fold when compared with vehicle-treatment group (Fig. 1C).

A BrdU-incorporation assay in human ADPKD renal cystic epithelial cells treated with nintedanib (1.5 µM for 24 h). n = 4 biological replicates (patient samples) and seven technical replicates each. B Images of microcyst growth in 3D collagen matrix by human ADPKD renal cystic epithelial cells treated with vehicle or nintedanib (1.5 µM) for 12 days. 1X magnification, and (C) quantitation of cyst diameter. n = 4 biological replicates and 5 or 6 technical replicates each. D Human ADPKD renal myofibroblasts treated with nintedanib for 48 h. MTT assay and E migration (% wound closure in a scratch assay) in the presence of 0 or 1 µM nintedanib. n = 4 biological replicates and three technical replicates each for D and E. F Human renal ADPDK myofibroblasts immunostained for αSMA, FSP1, vimentin, or pan-cytokeratin. G NRK-49F rat renal fibroblasts treated with nintedanib for 48 h. MTT assay. n = 7. H Migration of NRK-49F cells treated with nintedanib n = 4. I Immunoblot of NRK-49F cells incubated with TGFβ (2 ng/ml) and nintedanib (1.5 μM) for 48 h and (J) quantitation of band density. *P < 0.05 **P < 0.01, ***P < 0.001, vs vehicle or 0 µM nintedanib.
Nintedanib reduced myofibroblast-cell viability and migration in vitro
Nintedanib significantly reduced cell viability (Fig. 1D) and migration (Fig. 1E) of primary-culture myofibroblasts isolated and cultured from human ADPKD kidneys, compared with vehicle. These cells expressed αSMA, vimentin, and fibroblast-specific protein-1 (FSP-1), markers for differentiated myofibroblasts, mesangial cell, and fibroblast, respectively, but not pan-cytokeratin, an epithelial cell marker (Fig. 1F). The αSMA expression in human ADPKD myofibroblasts was not affected by nintedanib at doses that reduced their migration and cell proliferation (Supplemental 1C, D). However, in NRK-49F rat renal fibroblasts, nintedanib treatment not only dose-dependently reduced cell viability (Fig. 1G) and migration (Fig. 1H), but also reduced TGFβ-induced fibroblast-to-myofibroblast differentiation indicated by reduced αSMA levels (Fig. 1I, J).
Nintedanib treatment reduced renal cystic epithelial cell proliferation and cyst growth in Pkd1 RC/RC mice
In five-month old Pkd1RC/RC mouse kidneys, significant increase in protein levels of PDGFRβ and FGFR1, but not VEGFR2, was observed when compared with WT kidneys (Supplemental 2A, B). Both PDGFRα and PDGFRβ mRNA levels were increased in Pkd1RC/RC kidneys, when compared with WT kidneys (Supplemental 2C). In Pkd1RC/RC mice, nintedanib treatment (20 mg/kg body weight) on alternate days from three months to five months of age (Fig. 2A) significantly reduced kidney size (Fig. 2B), kidney to body-weight ratio (Fig. 2C), blood urea nitrogen (BUN) levels (Fig. 2D), and cystic index (percent cystic area) (Fig. 2E), but not cyst number when compared with vehicle treatment (Fig. 2F). Cystic epithelial cell division indicated by Ki-67 staining was significantly reduced by 42% in nintedanib-treated Pkd1RC/RC kidneys, when compared with vehicle treatment (Fig. 2G, H). In WT mice, nintedanib treatment did not significantly affect renal morphology, kidney to body-weight ratio, or cell proliferation when compared with vehicle treatment (Supplemental 3A–D).

A Scheme of treatment. WT and Pkd1RC/RC mice were treated with vehicle or Nintedanib (20 mg/Kg BWt) by IP injection on alternate days from 3 to 5 months of age. B H&E staining of kidney sections. Scale bar = 1 mm. C Two kidney to body-weight ratios (%). D Blood-urea nitrogen (BUN) levels. E Cystic index (%), (F) Cyst number. G Ki-67 immunostaining (scale bar = 50 µM), and (H) quantitation of Ki-67-stained nuclei in kidney sections, n = 5. I Immunoblot of kidney tissue and (J) quantitation of band density. *P < 0.05, **P < 0.01, ***P < 0.001 by T-test.
Nintedanib reduced RTK cell signaling in Pkd1 RC/RC mouse kidneys
We examined the effect of nintedanib on intracellular RTK cell-signaling components, including ERK, AKT, and STAT3 activity, which also regulate cell proliferation, inflammation, and fibrosis in ADPKD kidneys [9, 11, 23]. When compared with WT kidneys, Pkd1RC/RC kidneys showed significant increases in pERK1/2 /ERK1/2, pAKT/AKT, and pSTAT3/STAT3 ratios (Fig. 2I, J), suggesting increased activity, which was significantly reduced by nintedanib. YAP, an important Hippo signaling component and mediator of RTK signaling in cancer cells [24], promotes cyst growth [25, 26] and fibrosis [26] in ADPKD. In Pkd1RC/RC kidneys, we found significantly increased YAP levels when compared with WT kidneys, and nintedanib treatment significantly reduced YAP levels in Pkd1RC/RC kidneys (Fig. 2I, J). However, similarly increased cyclin D1 and c-myc expression in Pkd1RC/RC kidneys were not significantly altered by nintedanib treatment (Supplemental 4A, B).
Nintedanib treatment reduced fibrosis and myofibroblast population in Pkd1 RC/RC mice
Renal fibrosis in Pkd1RC/RC kidneys indicated by Sirius Red staining and quantitation was significantly reduced in the nintedanib-treated Pkd1RC/RC kidneys (Fig. 3A, B). Nintedanib also reduced mRNA levels of ECM proteins such as collagen-1a and fibronectin (FN1), but not collagen-IIIa (Fig. 3C). High αSMA mRNA and protein levels (Fig. 3D–G) in Pkd1RC/RC kidneys were also significantly reduced by nintedanib treatment. However, in Pkd1RC/RC kidneys, nintedanib treatment did not change SMAD3 activity, an important component of TGFβ-signaling pathway and a contributor to renal fibrosis, as suggested by no change in pSMAD3/SMAD3 ratio, when compared with vehicle treatment (Supplemental 4A, B).

A Sirius red-stained tissue images under polarized light (scale bar = 1 mm) and (B) quantitation of fibrosis based on Sirius red staining. C Renal mRNA levels for ECM proteins. D Renal αSMA mRNA levels. E Immunostaining for αSMA (green) in mouse kidney tissue (scale bar = 50 µM). F Immunoblot for αSMA in kidney tissue and (G) quantitation of band density. *P < 0.05, **P < 0. 01, ***P < 0.001 by T-test.
In Pkd1KO mice, nintedanib treatment reduced renal cyst growth, but not fibrosis
We also tested the effect of nintedanib in Pkd1KO (Pkd1f/fPkhd1Cre) mice, an ADPKD model with rapid disease progression and a high rate of cystic epithelial cell proliferation [26, 27].
Pkd1KO and WT littermates (Pkd1f/f with no cre) were treated with vehicle or nintedanib (20 mg/kg BWt) on postnatal days P10, P12, P14, and P16, and sacrificed on P18 (Fig. 4A). Nintedanib treated Pkd1KO mice had smaller kidneys (Fig. 4B), 50% reduction in kidney to body-weight ratio (Fig. 4C), 28% reduction in BUN (Fig. 4D), 40% fewer cysts (Fig. 4E) and 25% smaller percent cyst area in the kidneys (Fig. 4F) when compared with vehicle-treated Pkd1KO mice. In WT mice, nintedanib treatment did not alter renal morphology or kidney to body-weight ratio when compared with vehicle treatment (Supplemental 5A–C).

A Scheme of treatment. WT or Pkd1KO mice were treated with vehicle or Nintedanib (20 mg/Kg BWt, IP) on P10, P12, P14, and P16, and sacrificed on P18. B H&E staining of kidney sections of WT or Pkd1KO mice treated with vehicle or Nin sacrificed at P18. Scale bar = 1 mm. C Two kidney to body-weight ratios (%). D Plasma blood-urea nitrogen (BUN) levels. E Cyst number/kidney, (F) Cystic index (%). *P < 0.05, **P < 0.01, ***P < 0.001 by T-test.
Pkd1KO kidneys showed no significant change in FGFR1, PDGFRβ, or VEGFR2 expression compared with WT kidneys (Supplemental 6A, B). However, downstream effectors of RTK cell signaling including ERK, AKT, and mTOR [4] activity, and proproliferative factors such as c-Myc [28] and Cyclin D1 were increased in Pkd1KO kidneys and nintedanib treatment significantly reduced their activity or expression compared with vehicle treatment (Fig. 5A–D). Nintedanib treatment did not significantly change YAP, or pSTAT3/STAT3 ratio or αSMA protein levels in Pkd1KO kidneys (Supplemental 7A–D). No significant difference was observed in mRNA levels of ECM proteins (data not shown) or pSMAD3/SMAD3 ratio between nintedanib and vehicle treatment in Pkd1KO mice (Supplemental 7E, F).

A Immunoblot of kidney tissue lysate of mice treated with vehicle (Veh.) or Nin., and (B) quantitation of band density. C Immunoblot of kidney tissue lysate for proproliferative factors and (D) quantitation of band density. *P < 0.05, **P < 0. 01, ***P < 0.001 by T-test for Nin vs vehicle-treated Pkd1KO mice.
Discussion
In this study, we show for the first time that nintedanib, an FDA-approved triple RTK inhibitor, significantly reduces ADPKD disease progression. We demonstrated that in vitro, nintedanib reduced human ADPKD renal cystic epithelial cell proliferation and cyst growth, as well as reduced ADPKD renal myofibroblast migration and cell viability. In Pkd1RC/RC and Pkd1KO mice, nintedanib treatment significantly inhibited renal RTK cell signaling, reduced cystic epithelial cell proliferation and cyst growth, and overall disease progression. Nintedanib treatment also significantly inhibited fibrosis in Pkd1RC/RC kidneys.
Aberrant expression or activity of RTKs results in altered intracellular signaling and contributes to the progression of chronic diseases of the lungs, kidneys and liver, and in various cancers [4]. The roles of EGF/EGFR and VEGF/VEGFR in PKD are complicated. EGFR inhibitors, such as EKI-785, EKB-569, and Tesevatinib, and the Src inhibitor SKI-606, which also inhibits the EGFR signaling, have been shown to slow cyst growth in rodent models of ARPKD, ADPKD, and other forms of PKD [7, 9,10,11, 13]. The role of EGF/EGFR appears to also depend on the postnatal developmental stage because in neonatal ARPKD mice, EGF treatment did not reduce cyst expansion and also caused early death [7]. Increased expression of VEGF in renal cystic epithelial cells and VEGFR2 in endothelial cells has been detected in patients with ADPKD [29]. Moreover, a VEFGA gene polymorphism has been positively correlated with ADPKD progression [30, 31]. Expression of VEGF, VEGFR1, and VEGFR2 has also been shown in Cy/+ rats, an ADPKD rodent model characterized by proximal tubule-derived cysts [12]. Consistently, ribozymes that specifically inhibit VEGFR1 and VEGFR2 mRNA expression significantly reduced renal cyst growth in Cy/+ rats [12], while SU5416, a small-molecule inhibitor of VEGFR2, reduced hepatic cyst growth in Pkd2 KO mice [8]. However, targeting the ligand using anti-VEGF antibody treatment induced severe renal failure and glomerular damage, and increased proximal tubular cyst growth in Cy/+ rats [32]. By contrast, anti-VEGF antibody treatment in cancer patients did not change the size of simple renal cysts [33].
The role of PDGFR and FGFR is not well studied in PKD. PDGF immunoreactivity was detected in cyst-lining epithelium of human kidneys with acquired cystic disease [34, 35] and in ADPKD [35]. Moreover, PDGF treatment stimulated proliferation of ADPKD fibroblasts, but not ADPKD epithelial cells in vitro [35]. Similarly, FGF had a greater effect on the proliferation and intracellular RTK-mediated cell signaling in human ADPKD renal fibroblasts than normal renal fibroblasts [36]. In Pkd1RC/RC kidneys, we found significant increase in PDGFRβ and FGFR1 expression compared with WT kidneys, but VEGFR2 expression remained unchanged. In Pkd1KO kidneys, PDGFRβ, VEGFR2, and FGFR1 levels showed no difference compared to WT littermates. However, known cell signaling mediated by these RTKs was found to be upregulated. PDGFRs, VEGFRs, and FGFRs signal via the SRC kinase and regulate STAT3, AKT, ERK, MAPK, or JNK-mediated proliferative and migratory pathways in tumor cells [18]. These cell-signaling pathways also stimulate renal cyst expansion and inflammation in PKD [9, 11, 23], and could be regulated by multiple mechanisms in the PKD kidney [37]. Hence, our findings that ERK, AKT, mTOR and STAT3 activities are elevated in PKD and were significantly reduced by nintedanib treatment suggests that the activities of RTKs (PDGFR, VEGFR and FGFR) are important in the Pkd1RC/RC and Pkd1KO kidneys. Consistent with reduced intracellular RTK-mediated cell signaling, nintedanib-treatment reduced human ADPKD epithelial cell proliferation and cyst growth in vitro, and reduced cystic epithelial cell proliferation and renal cyst growth in both Pkd1RC/RC and Pkd1KO models in our study. While the percent cystic area in the nintedanib treatment group was significantly reduced in both Pkd1RC/RC and Pkd1KO kidneys, the cyst numbers were reduced only in Pkd1KO kidneys. This could be because at three months of age when nintedanib treatment was started in Pkd1RC/RC mice, their kidneys already had small cysts. By comparison, when nintedanib treatment was started at postnatal day 10 in the Pkd1KO mice, their renal cysts were very small or not detectable [38].
In PKD kidneys, increased ECM production, reduced degradation, and changes in its composition lead to fibrosis, and subsequent loss of renal function [3, 39,40,41,42]. Nintedanib has been demonstrated to be a very effective antifibrotic drug in preclinical and clinical studies [14,15,16, 19,20,21], including a CKD mouse model [22]. In our in vitro studies, nintedanib treatment significantly inhibited cell proliferation and migration of cultured human ADPKD renal myofibroblasts and significantly reduced renal fibrosis in the Pkd1RC/RC mouse. However, nintedanib had no effect in the Pkd1KO mouse. This could be because disease progression in the Pkd1RC/RC model is relatively slow and fibrosis becomes evident in the adult mice by 4–5 months of age. The Pkd1KO model, on the other hand, is a preweaning, rapidly progressing model, which may be suitable to study early increases in the myofibroblast population and ECM deposition, but perhaps not mature fibrosis. However, it should also be noted that in the Pkd1RC/RC mouse, all cells, including the myofibroblasts, are Pkd1 hypomorphs, while in the Pkd1KO mouse, Pkd1 deletion only occurs in the collecting ducts. Nintedanib is known to reduce TGFβ-induced fibroblast-to-myofibroblast differentiation, migration and proliferation of tumor stromal fibroblasts, and lung and renal fibroblasts [14, 43]. While nintedanib treatment in our study significantly reduced renal fibrosis in Pkd1RC/RC mice, it did not affect activity of SMAD3, an important component of TGFβ signaling.
In conclusion, these findings demonstrate that nintedanib, a triple RTK inhibitor, reduces proliferation and in vitro cyst formation of human ADPKD cystic cells. Nintedanib also inhibits epithelial cell proliferation and cyst growth in slowly progressing adult and rapidly progressing prenatal models of ADPKD. As such, we propose that nintedanib could be a useful pharmacological approach to slow ADPKD progression in patients.
Materials and methods
In vivo studies
Mouse models
(A) The Pkd1RC/RC mouse, a slowly progressive orthologous ADPKD model that contains a temperature-sensitive folding hypomorphic mutation (R3277C) in polycystin-1 [44, 45] and wild-type (WT, Pkd1+/+) mice, was maintained on BALB/c background. (B) The Pkd1f/fPkhd1Cre (Pkd1KO) mouse, a rapidly progressive ADPKD model caused by a mostly collecting duct-specific Pkd1 gene deletion [26, 27, 38, 46], and WT (Pkd1f/f) mouse littermates without Cre, was on C57Bl/6 J background.
Nintedanib study
Nintedanib (Selleck Chemicals, TX) was administered at 20 mg/Kg BWt dose on alternate days by intraperitoneal injections. This dose was well tolerated in adult WT C57/Bl6J mice in our pilot studies (data not shown). Pkd1RC/RC mice were treated from 3 to 5 months of age and sacrificed and Pkd1KO mice from postnatal day P10 to P16 and sacrificed on P18. Mouse sample size for the two studies was selected based on our preliminary data from pilot studies (data not shown). Mice were not randomized, instead, littermates were assigned either to vehicle or to nintedanib-treatment groups. For some analyses such as immunoblot and immunostaining, tissues of all mice were not analyzed, instead, few samples in each study group were randomly chosen. All animal studies were performed according to protocols approved by the University of Kansas Medical Center’s (KUMC) Institutional Animal Care and Use Committee.
Quantification of cysts and tissue fibrosis
Hematoxylin and eosin staining was performed on kidney tissue sections (5-µm thick) and imaged using Nikon 80i upright microscope (Tokyo, Japan). Quantification of cyst number, area, and total kidney area was performed using ImageJ (Fiji, Madison, WI, USA) by an observer blinded to sample identity. Picro sirius red staining (Polysciences, Warrington, PA, USA) was performed for kidney tissue sections following the manufacturer’s protocol. Stained tissue sections were imaged under polarized light and the amount of polarizable collagen per unit area was quantified.
Blood-urea nitrogen (BUN)
BUN levels were measured in serum as described previously [27] using the QuantiChrom Urea Assay Kit from BioAssay Systems (Hayward, CA, USA).
Western blot
Mouse kidneys were homogenized in SDS Laemmli buffer and loaded onto 10% SDS-polyacrylamide agarose electrophoresis gels essentially as described previously [27, 47]. Primary antibodies pERK1/2 (9101 S), pAKT (4060 S), pSTAT3 (9145 S), STAT3 (9139 S), cyclinD1 (2978 S), S6 (2317 S), pS6 (4858 S), PDGFRβ (3169), VEGFR2 (9698), and FGFR1 (9740) from Cell signaling (Danvers, MA, USA); YAP (SC-101199), GAPDH (SC-32233), ERK1/2 (SC-94), cMyc (SC-40) from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA), and α-SMA (ab5694; Abcam, Cambridge, MA, USA); and secondary antibodies, anti-mouse (P0447) and anti-rabbit (P0448) from Dako (Santa Clara, CA, USA) and ECL reagent from Amersham (GE Health care, Buckinghamshire, UK) were used.
Immunohistochemistry/immunofluorescence (IHC/IF)
Fixed and paraffin tissue sections were processed as described before [48]. Primary antibodies, α-SMA (ab5694) and FSP1 (ab27957) from Abcam (Cambridge, MA, USA), Ki67 (94495) from Cell Signaling, (Danvers, MA, USA), Vimentin (SC7557) from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA), and Pan-cytokeratin (F0397) from (MilliporeSigma, St. Louis, MO) were used. For IHC, secondary antibody application was followed by incubation with Streptavidin HRP conjugate (Invitrogen, Carlsbad, CA, USA), and DAB (Vector Laboratories, Burlingame, CA, USA), and counterstained with Harris Haematoxylin, dehydrated, and mounted with Permount (Fisher Scientific, Waltham, MA, USA). For IF, goat anti-Rabbit IgG fluor and Goat anti-mouse IgG Texas red (Invitrogen, Carlsbad, CA, USA), secondary antibodies were applied, incubated, washed and stained with DAPI, and mounted with Flour-G (Invitrogen, Carlsbad, CA, USA). Images were captured using a Nikon 80i upright microscope (Tokyo, Japan).
Quantitative real-time PCR
From whole kidney tissue lysate, RNA was isolated using the trizol method (Ambion, Austin, TX, USA) and cDNA was prepared using High capacity CDNA reverse transcription kit from Applied Biosystems (Foster City, CA, USA) and QRTPCR performed using power SYBR Green PCR master mix Applied Biosystems (Foster City, CA, USA) following the manufacturer’s protocol. Primer sequences are shown before [26].
In vitro studies
Primary culture human ADPKD cells
Primary culture human renal ADPKD cystic epithelial cells and normal human kidney (NHK) cells [26] were provided by the KUMC PKD Biomarkers and Biomaterials Core, a part of the national PKD Research Resource Consortium (PKD-RRC). Primary culture human myofibroblasts from ADPKD kidneys were isolated using a standard protocol described before [49] and cells were used in their first passage. ADPKD kidneys and NHK were obtained by this core from the surgery department at KUMC by KU Cancer Center’s Biospecimen Resource Core and hospitals participating in Tissue Donation Program of PKD Foundation (Kansas City, MO). These deidentified tissue samples have only alphanumeric codes and basic clinical information, and have IRB approval (IRB #5929; approval date 8/25/2020) to consent patients for “medical discard” nephrectomy (routine care) samples to use for research. Mycoplasma contamination was monitored by DAPI staining and confirmed using a mycoplasma testing kit (PCR method).
In vitro cystogenesis assay
In vitro 3D cyst-growth assay was performed as before [38] using ADPKD epithelial cells. On day 4, when cysts were evident, nintedanib (1.5 µM) or vehicle (DMSO 1 µl/ml) was added for 12 days. Cysts were photographed and cyst diameter (≥30 μM) measured using Image-Pro Premier software.
Fibroblast to myofibroblast differentiation
NRK-49F (ATCC® CRL-1570, Manassas, VA, USA) rat renal fibroblast cells grown in DMEM media, when 50% confluent, were treated with 2 ng/ml of TGFβ for 48 h, lysed, and αSMA levels measured.
Migration (wound-closure) assay
NRK-49F or human ADPKD renal myofibroblasts were seeded in 6-well plates, grown in DME/F-12 media, and wound closure assay performed as described earlier [26].
Cell proliferation and cell viability
To assess cell proliferation, M-1 mouse renal cortical collecting duct cell (#CRL-2038, American Type Culture Collection, Manassas, VA) and human primary culture NHK and ADPKD epithelial cells grown on glass coverslips were treated with vehicle or Nintedanib for 24 h and treated with BrdU (#10280879001, MilliporeSigma, Burlington, MA) (3 μg/mL) for 3 h and cell proliferation measured as described [50]. To measure cell viability, cells were seeded in 24-well plates and MTT assay performed as described [51].
Statistics
Values were expressed as mean ± standard error for in vivo, and mean ± standard deviation for in vitro studies. Data were analyzed by two-tailed unpaired T-test with Welch’s correction, or one-way ANOVA followed by Dunnett’s multiple-comparison test using GraphPad Prism software (Version 5.0d). P ≤ 0.05 was considered significant.
Data availability
The data and materials generated from this study are available upon reasonable request from the corresponding author.
References
Grantham JJ, Mulamalla S, Swenson-Fields KI. Why kidneys fail in autosomal dominant polycystic kidney disease. Nat Rev Nephrol. 2011;7:556–66.
Fragiadaki M, Macleod FM, Ong ACM. The controversial role of fibrosis in autosomal dominant polycystic kidney disease. Int J Mol Sci. 2020;21:8936.
Norman J. Fibrosis and progression of autosomal dominant polycystic kidney disease (ADPKD). Biochim Biophys Acta. 2011;1812:1327–36.
Yamaoka T, Kusumoto S, Ando K, Ohba M, Ohmori T. Receptor tyrosine kinase-targeted cancer therapy. Int J Mol Sci. 2018;19:3491.
Rivera-Ortega P, Hayton C, Blaikley J, Leonard C, Chaudhuri N. Nintedanib in the management of idiopathic pulmonary fibrosis: clinical trial evidence and real-world experience. Ther Adv Respir Dis. 2018;12:1753466618800618.
Shabani M, Hojjat-Farsangi M. Targeting receptor tyrosine kinases using monoclonal antibodies: the most specific tools for targeted-based cancer therapy. Curr Drug Targets. 2016;17:1687–703.
Nakanishi K, Gattone VH 2nd, Sweeney WE, Avner ED. Renal dysfunction but not cystic change is ameliorated by neonatal epidermal growth factor in bpk mice. Pediatr Nephrol. 2001;16:45–50.
Spirli C, Okolicsanyi S, Fiorotto R, Fabris L, Cadamuro M, Lecchi S, et al. ERK1/2-dependent vascular endothelial growth factor signaling sustains cyst growth in polycystin-2 defective mice. Gastroenterology. 2010;138:360–71. e367
Sweeney WE, Chen Y, Nakanishi K, Frost P, Avner ED. Treatment of polycystic kidney disease with a novel tyrosine kinase inhibitor. Kidney Int. 2000;57:33–40.
Sweeney WE, Frost P, Avner ED. Tesevatinib ameliorates progression of polycystic kidney disease in rodent models of autosomal recessive polycystic kidney disease. World J Nephrol. 2017;6:188–200.
Sweeney WE Jr., von Vigier RO, Frost P, Avner ED. Src inhibition ameliorates polycystic kidney disease. J Am Soc Nephrology: JASN. 2008;19:1331–41.
Tao Y, Kim J, Yin Y, Zafar I, Falk S, He Z, et al. VEGF receptor inhibition slows the progression of polycystic kidney disease. Kidney Int. 2007;72:1358–66.
Torres VE, Sweeney WE Jr., Wang X, Qian Q, Harris PC, Frost P, et al. EGF receptor tyrosine kinase inhibition attenuates the development of PKD in Han:SPRD rats. Kidney Int. 2003;64:1573–9.
Roth GJ, Binder R, Colbatzky F, Dallinger C, Schlenker-Herceg R, Hilberg F, et al. Nintedanib: from discovery to the clinic. J medicinal Chem. 2015;58:1053–63.
Dimitroulis IA. Nintedanib: a novel therapeutic approach for idiopathic pulmonary fibrosis. Respir Care. 2014;59:1450–5.
Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, Nicholson AG, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N. Engl J Med. 2011;365:1079–87.
Bill R, Fagiani E, Zumsteg A, Antoniadis H, Johansson D, Haefliger S, et al. Nintedanib is a highly effective therapeutic for neuroendocrine carcinoma of the pancreas (PNET) in the Rip1Tag2 transgenic mouse model. Clin Cancer Res. 2015;21:4856–67.
Liu F, Zhuang S. Role of receptor tyrosine kinase signaling in renal fibrosis. Int J Mol Sci. 2016;17:972.
Kato R, Haratani K, Hayashi H, Sakai K, Sakai H, Kawakami H, et al. Nintedanib promotes antitumour immunity and shows antitumour activity in combination with PD-1 blockade in mice: potential role of cancer-associated fibroblasts. Br J cancer. 2021;124:914–24.
Ruscitti F, Ravanetti F, Bertani V, Ragionieri L, Mecozzi L, Sverzellati N, et al. Quantification of lung fibrosis in ipf-like mouse model and pharmacological response to treatment by micro-computed tomography. Front Pharmacol. 2020;11:1117.
Wollin L, Togbe D, Ryffel B. Effects of nintedanib in an animal model of liver fibrosis. Biomed Res Int. 2020;2020:3867198.
Liu F, Wang L, Qi H, Wang J, Wang Y, Jiang W, et al. Nintedanib, a triple tyrosine kinase inhibitor, attenuates renal fibrosis in chronic kidney disease. Clin Sci. 2017;131:2125–43.
Wang J, Zhuang S. Src family kinases in chronic kidney disease. Am J Physiol Ren Physiol. 2017;313:F721–F728.
Azad T, Nouri K, Janse van Rensburg HJ, Maritan SM, Wu L, Hao Y, et al. A gain-of-functional screen identifies the Hippo pathway as a central mediator of receptor tyrosine kinases during tumorigenesis. Oncogene. 2020;39:334–55.
Cai J, Song X, Wang W, Watnick T, Pei Y, Qian F, et al. A RhoA-YAP-c-Myc signaling axis promotes the development of polycystic kidney disease. Genes Dev. 2018;32:781–93.
Dwivedi N, Tao S, Jamadar A, Sinha S, Howard C, Wallace DP, et al. Epithelial vasopressin type-2 receptors regulate myofibroblasts by a YAP-CCN2-dependent mechanism in polycystic kidney disease. J Am Soc Nephrology. 2020;31:1697–710.
Tao S, Kakade VR, Woodgett JR, Pandey P, Suderman ED, Rajagopal M, et al. Glycogen synthase kinase-3beta promotes cyst expansion in polycystic kidney disease. Kidney Int. 2015;87:1164–75.
Sun X, Song Q, He L, Yan L, Liu J, Zhang Q, et al. Receptor tyrosine kinase phosphorylation pattern-based multidrug combination is an effective approach for personalized cancer treatment. Mol Cancer Ther. 2016;15:2508–20.
Bello-Reuss E, Holubec K, Rajaraman S. Angiogenesis in autosomal-dominant polycystic kidney disease. Kidney Int. 2001;60:37–45.
Pandita S, Maurya D, Ramachandran V, Verma J, Kohli S, Saxena R, et al. Vascular endothelial growth factor (VEGF) gene promoter polymorphisms and disease progression in north indian cohort with autosomal dominant polycystic kidney disease. Int J Mol Cell Med. 2017;6:164–73.
Reiterova J, Obeidova H, Lenicek M, Stekrova J, Merta M, Maixnerova D, et al. Influence of VEGF polymorphism on progression of autosomal dominant polycystic kidney disease. Kidney blood Press Res. 2008;31:398–403.
Raina S, Honer M, Kramer SD, Liu Y, Wang X, Segerer S, et al. Anti-VEGF antibody treatment accelerates polycystic kidney disease. Am J Physiol Ren Physiol. 2011;301:F773–783.
Grenader T, Shavit L. Influence of vascular endothelial growth factor inhibition on simple renal cysts in patients receiving bevacizumab-based chemotherapy. Korean J Urol. 2015;56:791–5.
Herrera GA. C-erb B-2 amplification in cystic renal disease. Kidney Int. 1991;40:509–13.
Wilson PD, Du J, Norman JT. Autocrine, endocrine and paracrine regulation of growth abnormalities in autosomal dominant polycystic kidney disease. Eur J Cell Biol. 1993;61:131–8.
Kuo NT, Norman JT, Wilson PD. Acidic FGF regulation of hyperproliferation of fibroblasts in human autosomal dominant polycystic kidney disease. Biochem Mol Med. 1997;61:178–91.
Malekshahabi T, Khoshdel Rad N, Serra AL, Moghadasali R. Autosomal dominant polycystic kidney disease: disrupted pathways and potential therapeutic interventions. J Cell Physiol. 2019;234:12451–70.
Kakade VR, Tao S, Rajagopal M, Zhou X, Li X, Yu AS, et al. A cAMP and CREB-mediated feed-forward mechanism regulates GSK3beta in polycystic kidney disease. J Mol cell Biol. 2016;8:464–76.
Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. N. Engl J Med. 2008;359:1477–85.
Ibrahim S. Increased apoptosis and proliferative capacity are early events in cyst formation in autosomal-dominant, polycystic kidney disease. ScientificWorldJournal. 2007;7:1757–67.
Song CJ, Zimmerman KA, Henke SJ, Yoder BK. Inflammation and fibrosis in polycystic kidney disease. Results Probl Cell Differ. 2017;60:323–44.
Weimbs T. Polycystic kidney disease and renal injury repair: common pathways, fluid flow, and the function of polycystin-1. Am J Physiol Ren Physiol. 2007;293:F1423–1432.
Wollin L, Maillet I, Quesniaux V, Holweg A, Ryffel B. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J Pharmacol Exp therapeutics. 2014;349:209–20.
Hopp K, Ward CJ, Hommerding CJ, Nasr SH, Tuan HF, Gainullin VG, et al. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J Clin Investig. 2012;122:4257–73.
Kleczko EK, Marsh KH, Tyler LC, Furgeson SB, Bullock BL, Altmann CJ, et al. CD8(+) T cells modulate autosomal dominant polycystic kidney disease progression. Kidney Int. 2018;94:1127–40.
Karihaloo A, Koraishy F, Huen SC, Lee Y, Merrick D, Caplan MJ, et al. Macrophages promote cyst growth in polycystic kidney disease. J Am Soc Nephrology: JASN. 2011;22:1809–14.
Norregaard R, Tao S, Nilsson L, Woodgett JR, Kakade V, Yu AS, et al. Glycogen synthase kinase 3alpha regulates urine concentrating mechanism in mice. Am J Physiol Ren Physiol. 2015;308:F650–660.
Singh SP, Tao S, Fields TA, Webb S, Harris RC, Rao R. Glycogen synthase kinase-3 inhibition attenuates fibroblast activation and development of fibrosis following renal ischemia-reperfusion in mice. Dis Model Mech. 2015;8:931–40.
Grimwood L, Masterson R. Propagation and culture of renal fibroblasts. Methods Mol Biol. 2009;466:25–37.
Sinha S, Dwivedi N, Woodgett J, Tao S, Howard C, Fields TA, et al. Glycogen synthase kinase-3beta inhibits tubular regeneration in acute kidney injury by a FoxM1-dependent mechanism. FASEB J. 2020;34:13597–608.
Sinha S, Dwivedi N, Tao S, Jamadar A, Kakade VR, Neil MO, et al. Targeting the vasopressin type-2 receptor for renal cell carcinoma therapy. Oncogene. 2020;39:1231–45.
Acknowledgements
We thank KU PKD Biomarkers and Biomaterials Core for human tissue and primary culture cells, Dr. Igarashi for Pkhd1cre mouse, Dr. Somlo and Yale PKD Center (P30 DK090744) for Pkd1f/f mouse, and Dr. Peter Harris and Dr. Katharina Hopp for Pkd1RC/RC mouse.
Author information
Authors and Affiliations
Contributions
RR conceptualized, designed studies, analyzed the results, and wrote the paper. AJ performed experiments and analyzed the results. SMS bred mice and performed some mouse studies. AJ and SMS wrote parts of the paper. SM, TAF, DPW, and JPC provided reagents and/or quantitated tissue staining, reviewed, and edited the paper. All authors read and approved the paper. Studies were supported by NIH-NIDDK R01-DK083525 to RR and Postdoctoral grant to AJ from Kansas Institutional Development Award from NIH-NIGMS P20GM103418; KU Cancer Center Biospecimen Repository (P30CA168524) and Kansas PKD Research and Translational Core Center (U54DK126126 to JPC and DPW).
Corresponding author
Ethics declarations
Ethics statement
All animal study protocols were approved by the KUMC Institutional Animal Care and Use Committee.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Anastasis Stephanou
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jamadar, A., Suma, S.M., Mathew, S. et al. The tyrosine-kinase inhibitor Nintedanib ameliorates autosomal-dominant polycystic kidney disease. Cell Death Dis 12, 947 (2021). https://doi.org/10.1038/s41419-021-04248-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-021-04248-9
