
Abstract
Axonal degeneration of retinal ganglion cells (RGCs) causes blindness in glaucoma. Currently, there are no therapies that target axons to prevent them from degenerating. Activation of the BAX protein has been shown to be the determining step in the intrinsic apoptotic pathway that causes RGCs to die in glaucoma. A putative role for BAX in axonal degeneration is less well elucidated. BCLXL (BCL2L1) is the primary antagonist of BAX in RGCs. We developed a mCherry-BCLXL fusion protein, which prevented BAX recruitment and activation to the mitochondria in tissue culture cells exposed to staurosporine. This fusion protein was then packaged into adeno-associated virus serotype 2, which was used to transduce RGCs after intravitreal injection and force its overexpression. Transduced RGCs express mCherry-BCLXL throughout their somas and axons along the entire optic tract. In a model of acute optic nerve crush, the transgene prevented the recruitment of a GFP-BAX fusion protein to mitochondria and provided long-term somal protection up to 12 weeks post injury. To test the efficacy in glaucoma, DBA/2J mice were transduced at 5 months of age, just prior to the time they begin to exhibit ocular hypertension. Gene therapy with mCherry-BCLXL did not affect the longitudinal history of intraocular pressure elevation compared to naive mice but did robustly attenuate both RGC soma pathology and axonal degeneration in the optic nerve at both 10.5 and 12 months of age. BCLXL gene therapy is a promising candidate for glaucoma therapy.
Similar content being viewed by others


Introduction
In glaucoma, blindness is caused by degeneration of retinal ganglion cells (RGCs), neurons that project axons from the retina to the brain. The most prominent risk factor for glaucoma is elevated intraocular pressure (IOP) [1], and currently, lowering IOP is the only viable treatment for this disease. While this treatment is often able to slow the progression of neurodegeneration [2], there is wide-spread acknowledgement that a therapeutic that directly targets the RGCs and their axons would significantly augment IOP lowering therapies.
Evidence suggests that RGC axons are injured early in glaucoma and can degenerate independently of the soma [3]. Mechanisms of axon degeneration have been an area of intense study for the past two decades. There is evidence for axon degeneration pathways mediated independently by SARM1 [3,4,5] and BAX [6,7,8,9]. In RGCs, BAX is essential for the execution of the caspase-dependent, intrinsic apoptotic program [6, 10, 11], but its role in glaucomatous axon degeneration is controversial [3, 5].
Bcl2l1 (hereafter designated as BclXL) is an anti-apoptotic member of the Bcl2 gene family, and in central nervous system neurons is the primary antagonist of BAX [12]. BCLXL exists primarily at the mitochondria and inhibits BAX activation by preventing BAX from accumulating on the mitochondrial outer membrane [13]. Overexpression of BCLXL protects neurons from death following trophic factor withdrawal and ischemia–reperfusion [14,15,16]. In RGCs, increasing the intracellular concentration of BCLXL prevents the degeneration of somas and proximal axon segments after axotomy [17, 18].
The DBA/2J mouse is a widely used model of glaucoma that develops spontaneous, asynchronous elevation of IOP around 6 months of age that persists until the mice are nearly a year old [19,20,21]. This causes degeneration of RGCs and makes the DBA/2J mouse a useful model for studying glaucomatous neurodegeneration. Experimentally, the ages of 10.5 and 12 months are frequently used for assessing degeneration. Some therapeutic approaches are protective exclusively at 10.5 months of age [6, 22], whereas others are protective at both 10.5 and 12 months of age [23,24,25]. The differential effect of therapies is evidence that multiple axon degenerative pathways contribute to glaucomatous neurodegeneration. Intriguingly, deletion of the Bax gene conferred protection only at 10.5 months of age but deletion of Bim, a protein that is important for activating BAX in RGCs, protected optic nerves (ONs) at both 10.5 and 12 months of age, suggesting that the BCL2 family may mediate axon degeneration independent of BAX activity in RGCs [6, 24]. Notably, both complete Bax and Bim deletion also affected the elevation of IOP, which may have contributed to their protective effect.
Gene therapy is a clinically relevant therapeutic paradigm for treating retinal disease [26]. The premise of gene therapy is to deliver a therapeutic gene to a susceptible population of cells, generally via a viral vector such as a recombinant adeno-associated virus (AAV). AAVs have low immunogenicity, are replication deficient, and are not known to cause any human disease [27]. In mice, intravitreal delivery of AAV serotype 2 (AAV2) of a sufficient titer can transduce around 85% of RGCs [28].
Gene therapy targeted directly to RGCs to prevent BAX activation in a glaucoma model has never been attempted. This approach is clinically relevant and can be performed in wild-type mice that do not have the developmental abnormalities associated with Bax deletion [29]. A previous study investigated BCLXL gene therapy in a model of ON axotomy and achieved significant albeit transient protection of RGCs [18]. The transient nature of the protection may have been due to loss of expression of the transgene, which was expressed using a neuron-specific promoter. Neuron-specific gene expression is rapidly silenced in RGCs following ON injury [30]. Loss of expression can be circumvented by using the Pgk promoter, whose expression is sustained in damaged RGCs [31].
We hypothesized that AAV2-mediated BCLXL gene therapy would prevent RGC degeneration in the DBA/2J mouse model of glaucoma. Overexpression of mCherry-BCLXL protected both somas and axons from glaucomatous neurodegeneration. This result demonstrates a role for the BCL2 family in glaucomatous RGC somal and axonal degeneration.
Methods
Cloning mCherry-BclXL
Moloney Murine Leukemia Virus Reverse Transcriptase (Promega, Madison, WI) and Oligo(dT)15 Primer (Promega) were used to make cDNA from total RNA isolated from BALB/c mouse brain tissue. Polymerase chain reaction (PCR) was used to amplify the coding region of the BclXL transcript from the cDNA using the following primers 5’-AAATGTCTCAGAGCAACCGGGAGCTG–3’ and 5’-CAGTGTCTGGTCACTTCCGACTGAAGAG-3’. This PCR product was ligated into the pGemT vector using T4 DNA Ligase (Promega). BclXL was amplified with XhoI and HindIII restriction sites using the following primers 5’-TGGCCGGCTCGAGAAATGTCTCAG-3’ and 5’-GATTCAGTAAGCTTTCACTTCCGACTGAAG-3’. This PCR product was digested with XhoI and HindIII (Promega) and ligated into a pmCherry-C1 plasmid (Clontech, Mountain View, CA), with BclXL placed downstream of mCherry. Sequencing was performed to verify that BclXL had the proper sequence and was in frame with mCherry.
Validation of mCherry-BCLXL in vitro
D407 cells were grown in Dulbecco’s modified Eagle media (DMEM) (Corning, Corning, NY) containing 3% (V/V) fetal bovine serum (Atlanta Biologicals, Norcross, GA) and 1% penicillin–streptomycin (V/V) (Thermo Fisher Scientific, Waltham, MA). The GFP-BAX and mitoBFP constructs have been previously described [32, 33].
D407 cells were nucleofected using a Lonza 4D nucleofector (Lonza, Basel, Switzerland) and treated 24 h later with 1 µM staurosporine (STS) (Alfa Aesar, Ward Hill, MA). Live cell imaging was performed using an Andor Revolution XD spinning disc confocal microscope (Andor, Belfast, UK) in a 37 °C imaging chamber. Images were taken every 3 min for 3.5 h during the experiment. Image analysis was performed using the Imaris 9.2.1 software (Oxford Instruments, Abingdon, UK). The significant differences in the distribution between groups were assessed using χ2 tests.
Viral packaging
The mCherry-BclXL construct was digested using NheI and ApaI and ligated into a bridge vector created from AAV-Pgk-Cre (a gift from Patrick Aebischer, Addgene plasmid #24593) where the Cre coding region was replaced with a multiple cloning site containing unique NheI and ApaI sites.
Viral packaging was performed by the University of North Carolina – Chapel Hill, Vector Core. AAV2-Pgk-mCherry-BclXL and AAV2-Pgk-GFP-Bax had titers of 1.7 × 1013 and 2.3 × 1012 viral genomes/mL, respectively.
Mouse housing and ethics
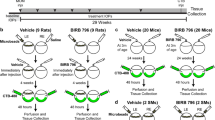
Mice were handled in accordance with the Association for Research in Vision and Ophthalmology’s Statement for the Use of Animals in Ophthalmic and Vision Research. The research protocol was approved by the Institutional Animal Care and Use Committee of the University of Wisconsin – Madison. Mice were kept in microisolator cages on a 12-h light/dark cycle and fed a 4% fat diet. Sample sizes were estimated using power calculations (0.8, 20% effective difference) with historical variance estimates for the type of assay used. All data shown used sample sizes that exceeded the “N” estimated by power calculations. Animals were randomly distributed into treatment groups with effort to balance male to female ratios. For DBA/2J mouse experiments, the male:female ratio is specifically indicated for each cohort in the figure legends. Any animals that exhibited distress after inclusion in a cohort were removed from the study.
Optic nerve crush (ONC) and intravitreal injections
For all surgeries, mice were anesthetized with 16 mg/mL ketamine and 1.5 mg/mL xylazine. Eyes were anesthetized with 0.5% proparacaine hydrochloride. Postoperative discomfort was alleviated with 0.03 mg/mL buprenorphine.
Intravitreal injection of virus or 1% cholera toxin subunit B (Alexa-488) (CTB-488; Thermo Fisher) was performed as previously described [30]. For experiments requiring injection of multiple viruses, titers were equalized by mixing the viruses followed by a single injection. Four weeks were allowed for viral genome replication and transgene expression. ONC was performed as previously described [34] using C57BL/6J mice.
DBA/2J experiments and IOP measurements using rebound tonometry
Ten-week-old DBA/2J mice were bilaterally injected with AAV2-Pgk-mCherry-BCLXL. An additional cohort of 10 uninjected DBA/2J mice was used as a naive control. Each cohort contained 5 male and 5 female mice. IOPs were measured every 2 weeks between 9 and 11 a.m. using a Tonolab rebound tonometer (Icare, Finland) as previously described [35]. Eyes that developed corneal abnormalities were excluded. Longitudinal IOP history was analyzed using Generalized Estimating Equation regression modeling (R, v4.0.5).
For experiments measuring ON degeneration, DBA/2J mice between 4 and 5 or 7 months of age were bilaterally transduced with AAV2-Pgk-mCherry-BclXL. Naive mice received no injection. Each cohort of glaucomatous mice contained at least 49 ONs and 29 retinas. We did not use a control viral injection because other studies have found no effect on IOP progression or protection of RGCs by control AAVs [35, 36]. Because glaucomatous disease progresses asymmetrically in the eyes of DBA/2J mice [21], each eye/retina/ON was considered as an independent variable.
Euthanasia, tissue removal, and fixation
Mice were euthanized with pentobarbital sodium and phenytoin sodium (Virbac, Westlake, TX) followed by cervical dislocation. Eyes were enucleated with a proximal piece of the ON attached and placed in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS, 100 mM phosphate buffer with 150 mM NaCl, pH 7.4) for 1 h. For ONC experiments, mice were euthanized 1 week after ONC. For glaucoma studies, mice were euthanized at 10.5 or 12 months of age. Young DBA/2J cohorts consisted of mice <4 months of age.
Frozen sectioning and staining of whole-eye sections
After fixation, a brief PBS washed was performed. Samples, including both retinas and ONs, were equilibrated in 30% sucrose in PBS overnight at 4 °C. Samples were then frozen on dry ice in plastic molds using O.C.T. Compound (Scigen, Paramount, CA). In all, 8 µm sections were obtained from the Translational Initiatives in Pathology (TRIP) lab at the University of Wisconsin – Madison.
Immunofluorescence was performed as previously described [30]. Mouse anti-BRN3A monoclonal antibody (MAB1585) (EMD Millipore, Temecula, CA) was used at a concentration of 1:50. Rabbit anti-mCherry polyclonal antibody (ab167453) (Abcam, Cambridge, UK) was used at a concentration of 1:500. The anti-mCherry antibody was only used to enhance the signal of the sections that were used to assess transduction efficiency. All other samples showed endogenous mCherry signal. Rabbit anti-calbindin antibody (SWANT, Marly, Switzerland) was used at a concentration of 1:1000. Goat anti-mouse immunoglobulin G (IgG) fluorescein isothiocyanate conjugated to FITC or Alexa488 and goat anti-rabbit IgG conjugated to Texas Red, Alexa594 or Alexa488 (Jackson Immunoresearch, West Grove, PA) were used at a concentration of 1:1000.
Transduction efficiency was calculated as the percentage of 4,6-diamidino-2-phenylindole (DAPI)-positive nuclei that had mCherry-BCLXL labeling in the adjacent cytosolic compartment and the percentage of BRN3A-positive nuclei that had adjacent mCherry-BCLXL labeling. One section from four transduced eyes was counted.
Imaging of the ON tract
Euthanized mice were decapitated and the heads were placed in 4% PFA for 24 h. The skullcap and brain were then dissected away, taking care to leave the optic tract intact. Imaging was performed on a Zeiss Discovery V8 Stereo fluorescent microscope (Zeiss, Oberkochen, Germany). Images were taken using the Zen blue software (Zeiss).
Whole mounting of retinas, imaging, and calculation of nuclear densities
Retinas were whole mounted as previously described [30] using VECTASHIELD Antifade Mounting Medium containing DAPI (Vector Laboratories, Burlingame, CA). Imaging and quantification of cellular density were performed as previously described [30]. Counted nuclei were restricted to cells with nuclear morphology typical of neurons (round euchromatic appearance with prominent nucleoli). At least 29 retinas were counted for each group. Statistical significance was calculated using a one-sided t test, which assumed equal variance between groups.
Quantitative PCR (qPCR)
Retinas that were used for qPCR were flash frozen in microcentrifuge tubes on dry ice. cDNA was created as previously described [30]. qPCR was performed using an ABI Quant Studio 7 RT PCR machine (Thermo Fisher Scientific). Primers for S16, BclXL, Nefl, Nrn1, Thy1, Sncg, Tubb3, Gfap, Hsp27, and Gap43 have been described previously [37]. Absolute abundances of each transcript were calculated using a standard curve of S16 ribosomal protein mRNA [37]. Four groups of three pooled eyes were used for each experimental cohort. Statistical significance between the transcript abundances of different groups were calculated using a one sided t test assuming equal variance.
ON fixation, sectioning, paraphenylenediamine (PPD) staining, and scoring
ONs were processed for PPD staining as previously described [38]. Sections were imaged on a Zeiss Axioimager Z2 upright microscope using a ×40 magnification oil objective. Images were scored using a semi-quantitative three-score system where each nerve was assigned a score of No or Early (NOE), Moderate (MOD), or Severe (SEV) glaucoma [20]. All ONs were scored by two masked observers. The statistical significance between the distributions of different groups was calculated using χ2 test.
Results
mCherry-BCLXL prevents GFP-BAX recruitment in vitro
The canonical function of mCherry-BCLXL of preventing BAX from accumulating on the mitochondria was verified in vitro. As expected, mCherry-BCLXL colocalized with a mitochondrially localized BFP (Fig. 1E, F, H). The ability of mCherry-BCLXL to prevent GFP-BAX translocation was assessed in cells after STS treatment. Over 95% of cells expressing mCherry and GFP-BAX exhibited GFP-BAX translocation to mitochondria within 3.5 h (Fig. 1A–D, I). Conversely, only 8% of cells expressing mCherry-BCLXL and GFP-BAX exhibited punctate GFP-BAX (Fig. 1E–I).

Representative images of D407 cells expressing mCherry (A–D), mCherry-BCLXL (E–H) along with mitoBFP and GFP-BAX 3.5 h after treatment with 1 µM staurosporine (STS). Note that mitoBFP fluorescence is rapidly lost when the mitochondrial outer membrane becomes permeabilized and the mitochondria become fragmented, which is why B has dramatically reduced fluorescence. Scale bar = 10 µm. Live cell imaging was used to track the localization of GFP-BAX for 3.5 h after STS treatment. The percentage of cells in each condition with punctate localization is quantified in I. N = 163 and 146 cells for the mCherry and mCherry-BCLXL expressing groups, respectively. χ2 test was used to assess the significance of the difference in percentage between each group at each time point. P < 0.0005 at 30 min, P < 0.00001 at 120 min, and P < 0.00001 at 210 min.
AAV2-Pgk-mCherry-BclX L efficiently transduces RGCs
Next, the transduction efficiency of AAV2-Pgk-mCherry-BclXL (Fig. 2A) was measured. Evaluation of retinal whole mounts 4 weeks after intravitreal injection showed wide-spread expression of the transgene in the ganglion cell layer (Fig. 2B). Histologic examination of transduced retinas showed that AAV2-Pgk-mCherry-BclXL transduced over 50% of the cells in the ganglion cell layer of the retina, which is consistent with the percentage of RGCs in this layer [39]. Colocalization with the RGC marker BRN3A demonstrated that nearly 80% of BRN3A+ cells were transduced (Fig. 2C–G). AAV2 also transduces other retinal cell types [31]. We observed transduction of both horizontal and amacrine cells based on colocalization with an antibody to Calbindin [40] (Fig. S1).

A A map showing the plasmid that was packaged into AAV2-Pgk-mCherry-BclXL. B A retinal whole-mount preparation, 4 weeks after intravitreal injection, showing wide-spread expression of the transgene across the retina. Scale bar = 450 µm. C–F Co-localization of BRN3A-positive cells that were transduced by AAV2-Pgk-mCherry-BclXL (arrows). An asterisk labels a transgene expressing cell that is BRN3A negative but appears to project a labeled axon into the nerve fiber layer. Only the ganglion cell layer (GCL) is shown. Scale bar = 20 µm. G Graph showing the percentages of total cells in the GCL and BRN3A-positive cells that expressed the mCherry-BCLXL transgene (mean ± standard deviation).
The mCherry-BCLXL fusion protein was also observed throughout the optic tract of transduced eyes, demonstrating its presence in RGC axons (Fig. 3). Unilateral injection of CTB-488, to label axons, in a mouse that was bilaterally injected with AAV2-Pgk-mCherry-BclXL showed colocalization with the fusion protein, confirming axonal labeling. CTB-488 also showed bilateral fluorescence in the suprachiasmatic nucleus (SCN; Fig. 3C arrows), the only region of the rodent brain that is innervated equally on the ipsilateral and contralateral sides by projections from one ON [41], principally from intrinsically photosensitive RGCs [42]. No mCherry-BCLXL fluorescence was detected in the SCN, suggesting that AAV2-Pgk-mCherry-BclXL did not transduce this population of RGCs. Transgenic mice expressing Thy1-mitoCFP to label RGC mitochondria were also injected. ON sections showed colocalization of mCherry-BCLXL and mitochondria in RGC axons (Fig. 3E–G).

A A brightfield image of the dorsal view of the optic nerves from a mouse transduced bilaterally with AAV2-Pgk-mCherry-BCLXL and later injected unilaterally (left eye only) with CTB-Alexa488. B The same field imaged for mCherry-BCLXL fluorescence. The fusion protein can be seen in the optic nerve distal to the optic chiasm (OC) and approaching the superior colliculus (SC). C The same field imaged for CTB-Alexa488 fluorescence. Arrows denote fluorescence in the suprachiasmatic nucleus (SCN). D A merged image of B, C showing bilateral mCherry-BCLXL expression and unilateral CTB-488. Scale bar = 1 mm. E–G Histologic sections of the optic nerve from a transduced eye of a transgenic mouse expressing Thy1-mitoCFP to label RGC mitochondria. Note that not all RGCs express Thy1-mitoCFP in this line. The mCherry-BCLXL transgene is present throughout individual axons but concentrated in regions that correspond to axonal mitochondria (arrows). Scale bar = 5 µm.
mCherry-BCLXL prevents cell loss in RGC layer following ONC
We tested the ability of mCherry-BCLXL to protect RGCs from apoptosis following ONC (Fig. 4). Mice were co-transduced with AAV2s expressing mCherry-BCLXL or GFP-BAX. Prevention of BAX translocation after ONC was assessed by counting the percentage of cells with punctate GFP-BAX in the presence or absence of mCherry-BCLXL (Fig. 4A, B). One week after ONC, mCherry-BCLXL overexpression prevented a significant change in the percentage of cells with punctate GFP-BAX (Fig. 4C).

A, B Panels showing mCherry-BCLXL and GFP-BAX expression, respectively, in a retinal whole mount from a mouse that that had undergone ONC to induce BAX activation. The cell expressing mCherry-BCLXL shows diffuse localization of GFP-BAX and the cell that is not expressing mCherry-BCLXL has punctate, translocated GFP-BAX. Scale bar = 10 µm. C A quantification of the phenomenon displayed in A, B. In this experiment, all mice were transduced with AAV2-Pgk-GFP-BAX. Half the mice also received an injection of AAV2-Pgk-mCherry-BCLXL. The percentage of GFP-BAX-labeled cells with punctate GFP-BAX was quantified 1 week after ONC in untransduced mice and mice that had been bilaterally transduced with AAV2-Pgk-mCherry-BclXL 1 month before ONC (mean ± standard deviation). Significance was determined using a one-sided t test. In all, 4–7 mice were used per group. D Cell loss was determined by quantifying the difference in nuclear density in the RGC layer of retinal wholemounts between the experimental and contralateral eye (mean ± standard deviation). In all, 6–10 mice were used per group. n.s. = not significant. *P < 0.05. **P < 0.01.
Next, we tested the ability of mCherry-BCLXL to provide sustained protection to RGCs by examining the pattern of cell loss in the RGC layer after ONC. Both 4 and 12 weeks post-ONC, AAV2-Pgk-mCherry-BclXL transduced retinas had significantly less cell loss than untransduced retinas analyzed 4 weeks post-ONC (Fig. 4D).
AAV2 transduction does not prevent IOP elevation in DBA/2J mice
Next, we examined whether AAV2-Pgk-mCherry-BclXL affected the progression of ocular hypertension in DBA/2J mice. Histological examination of transduced cells in the conventional aqueous outflow pathway showed that mCherry-BCLXL was expressed in the non-pigmented epithelial cells of the ciliary body but not in cells of the trabecular meshwork or Schlemm’s canal (Fig. 5A–D). Longitudinal measurements of IOP in AAV2-Pgk-mCherry-BclXL transduced mice, compared to untransduced mice, revealed four individual timepoints with statistically significant differences in IOP between groups (Fig. 5E). Regression modeling failed to detect a difference between the trends of the groups over the entire course of the experiment, indicating that mCherry-BCLXL did not significantly impact longitudinal IOP elevation.

A Brightfield image showing the relevant ocular anatomy for the outflow pathway of an eye that was transduced with AAV2-Pgk-mCherry-BclXL. B–D Fluorescent images showing the DAPI-positive nuclei (B) and the expression pattern of mCherry-BCLXL (C), as well as a combined view of B, C (D). Note that an mCherry antibody was used to amplify the mCherry-BCLXL signal. Expression is seen strongly in the retina and ciliary body but is not present in Schlemm’s canal, the iris, or the trabecular meshwork (TM). The exterior surface of the cornea also appears to be expressing mCherry-BCLXL, which may be the result of viral particles flowing onto the surface of the eye when the needle is removed from the eye during the intravitreal injection. E Intraocular pressures of AAV2-Pgk-mCherry-BclXL transduced and naive DBA/2J mice that were measured every 2 weeks from the age of 8 to 42 weeks. Transduction occurred the day after the second measurement of IOP when the mice were 10 weeks old. The data are represented as box and whisker plots with the mean shown as a bar in the center of the data, the 25th and 75th percentiles shown as the edges of the box, and the whiskers extending to show the minimum and maximum values for the data. For IOP measurements, N = an average of 18 eyes per group per time point. Generalized Estimating Equation regression modeling was used to assess the statistical difference between the IOPs of the groups at each timepoint and longitudinally. Individual timepoints that are statistically different from one another are shown. *P < 0.05, **P < 0.01, ***P < 0.001. Regression analysis performed on the data show that the longitudinal IOP progression of the naive and mCherry-BCLXL groups are similar.
mCherry-BCLXL prevents RGC degeneration in aged DBA/2J mice
Next, we tested whether mCherry-BCLXL would prevent RGC degeneration in aged DBA/2J mice. Retinal whole mounts showed robust expression of the transgene in transduced retinas at both 10.5 and 12 months of age (Fig. S2). The protective effect of mCherry-BCLXL was first measured by quantifying changes in the abundance of transcripts selectively expressed in RGCs using qPCR. At 10.5 months of age, BCLXL treated retinas exhibited a nearly 5-fold increase in BclXL transcript abundance (Fig. 6A). These retinas also had significantly higher abundances of the RGC-selective mRNAs Nefl, Nrn1, Thy1, Sncg, and Tubb3 transcripts than untreated retinas, which showed a nearly uniform decrease in these mRNAs (Fig. 6B–F), indicating that BCLXL treatment preserved RGC-specific gene expression. Both naive and treated retinas exhibited similar increases in Gfap and Hsp27 transcript abundance relative to young mice, indicating that BCLXL treatment did not prevent retinal stress (Fig. 6G, H). Interestingly, treated retinas expressed more Gap43 (Fig. 6I), a marker of axon regeneration [43].

Quantitative (real time) PCR was used to assess retinal gene expression in young mice and glaucomatous mice. A BclXL expression was five times higher in transduced, aged DBA/2J mice (grey bars) than either young or 10.5-month-old naive animals (black bars). B–F The transcript abundances of five RGC-specific/selective transcripts. RGC transcripts were consistently more abundant in aged mice that were BCLXL treated than age-matched mice that did not receive BCLXL. G, H The transcript abundance of Gfap and Hsp27, two transcripts produced in retinas experiencing glaucomatous stress. These transcripts were significantly more abundant in aged mice relative to the young mice that had not developed glaucoma. I The transcript abundance of Gap43, a gene that has been associated with neuroregeneration. Gap43 was more abundant in BCLXL-treated, 10.5-month-old mice than young or 10.5-month-old naive mice. Four groups of three pooled eyes were used for each experimental cohort. The graphs display the average abundance of the four groups and the error bars show the standard deviation from that average. All transcript levels are quantified as the number of target molecules present per molecule of S16 ribosomal protein mRNA. Statistical evaluation was made using unpaired t tests of the means (*P < 0.05, **P < 0.01).
BCLXL gene therapy prevented glaucomatous degeneration. BCLXL-treated mice had significantly fewer moderately and severely degenerated ONs compared to naive animals at 10.5 months of age (Fig. 7). BCLXL gene therapy was profoundly protective of axons in the ONs of 12-month-old animals, an age when Bax deficiency was not protective [6]. In fact, the distribution of ON scores for 12-month-old BCLXL-treated mice showed modestly, but significantly, less degeneration than the distribution of scores for 10.5-month-old BCLXL-treated mice. Stratification of data where we had both cell density counts and ON scores for the same eye showed that there was an association with reduced cell density and SEV ON score, while eyes exhibiting MOD ON damage typically showed no significant loss of cell density compared to eyes with NOE damage within a given cohort (Fig. 8). We interpret the presence of SEV damage and cell loss in some mice from treated cohorts as a consequence of less-than-optimal viral transduction, although other factors such as extreme ocular hypertension cannot be ruled out. Additionally, cell density in retinas from eyes with NOE ON damage, in most cohorts of aged mice, still exhibited an overall 10% decline in total cell density when compared with young mice, suggesting age-related effects not affected by the gene therapy.

A–C Examples of each potential optic nerve score. A The no or early (NOE) glaucoma example has uniform distribution of axons and the glial cells occupy minimal space on the image. B The moderate glaucoma (MOD) example has a large number of healthy axons but also displays increased dark spots (denoted by white arrows) where the myelin sheaths have collapsed around the axons. The glial cells may have become slightly enlarged but do not occupy much more space than the NOE example. C The severe glaucoma (SEV) example has very few healthy axons, most of what remains are dark spots. The glial cells in this image have expanded and occupy a larger percentage of the image than either the NOE or moderate glaucoma examples. Scale bar = 25 µm. D Distributions of each of the three scores for each experimental group. The inset number denotes the number of optic nerves scored for each group. The distribution of mice for each cohort (M/F) was: <4 (4/12); 10.5 Naive (21/21); 10.5 BCLXL (22/15); 12 Naive (13/12); 12 BCLXL (11/14). χ2 tests were performed to assess the statistical differences between the distributions of scores of different groups. *P < 0.01, **P < 0.00003 (young mice relative to all other cohorts in individual tests), ***P < 0.00001.

Eyes for which there was both an optic nerve score and analysis of the total neuronal cell density present in the retina were used to evaluate the symmetry of both RGC soma loss and axon loss in the same eye. Total cell counts reflect both RGC and amacrine neuronal populations in the ganglion cell layer. Combined scatter plot/bar graphs (mean ± standard deviation) are shown. In all cohorts (naive and BCLXL treated, aged 10.5 or 12 months), cell density was significantly lower in eyes with SEV optic nerves compared to eyes with NOE optic nerves in the same cohort (Student’s t test, ***P < 0.0001). Also, within cohorts, eyes exhibiting MOD optic nerves had no significant change in cell density, compared to eyes with NOE optic nerves, with the exception of naive mice aged to 10.5 months (**P = 0.0006). Compared to young naive mouse eyes with NOE optic nerves, most aged cohorts had modestly reduced cell densities in the NOE populations (on average a 10% reduction, ###P < 0.001), with the exception of BCLXL-treated eyes at 10.5 months, which showed no significant change. The distribution of mice for each cohort (M/F) was: <4 (6/10); 10.5 Naive (17/12); 10.5 BCLXL (10/5); 12 Naive (12/11); 12 BCLXL (8/12).
These data reflect the protective effect of BCLXL therapy if applied prophylactically (4–5 months). To test whether we can achieve similar protection during the period of first onset of elevated IOP, mice were transduced at 7 months and aged to 10.5 months. These mice exhibited a similar level of protection compared to mice transduced at an earlier age (Fig. S3).
Discussion
The mCherry-BCLXL fusion protein prevented BAX translocation both in vitro and in vivo (in RGCs) after exposure to acute apoptotic stimuli. In the DBA/2J mouse model of glaucoma, mCherry-BCLXL conferred extended protection to the retina and the ON that exceeded the reported protective effect of genetic deletion of Bax. Therefore, BCLXL must protect RGCs through more than simple inhibition of BAX.
There are several potential explanations for the additional protection conferred by mCherry-BCLXL. Recent literature has focused on the axodegenerative pathway catalyzed by SARM1 [5, 44, 45]. A combination of the Wlds allele, which prevents SARM1 activation, and depletion of BAX, via genetic deletion of one Bax allele, appeared to provide a greater protective effect to ONs of DBA/2J mice than either treatment alone [3]. Our results suggest that BCLXL is able to inhibit both the BAX and SARM1 pathways. SARM1 activation is the result of an increased ratio of nicotinamide mononucleotide (NMN) to NAD+ [46]. Future studies should examine whether BCLXL stabilizes the ratio of NMN:NAD+ or acts by some indirect mechanism, such as increasing the resiliency of the mitochondria, consistent with growing evidence that an age-related decline in mitochondrial performance is a contributing factor to glaucomatous neurodegeneration [25, 47]. Interestingly, BCLXL has been shown to improve the metabolic efficiency of mitochondria and increase overall mitochondrial biomass [48,49,50]. Future studies should explore how BCLXL augments mitochondrial function in RGCs.
A final possibility is that BCLXL helps the RGC maintain axonal transport. Axonal transport disruption is one of the earliest events in glaucoma [51]. The sustained protection in glaucomatous animals by our fusion protein suggests that mCherry-BCLXL was present in axons at high enough concentrations throughout the course of the disease to counteract degenerative signaling. A by-product of putative augmentation of mitochondrial physiology by BCLXL may be uninterrupted axonal transport.
BCLXL gene therapy yielded better preservation of ONs in 12-month-old DBA/2J mice than in 10.5-month-old mice. Since the 10.5- and 12-month groups were performed sequentially, this variation may be the result of a stochastic batch effect. However, the increased abundance of RGC-specific transcripts and the regeneration marker Gap43 suggests some level of spontaneous regeneration occurring in these mice after ocular hypertension subsides, a process which begins between 11 and 12 months of age in these mice [20]. The effect of BCLXL on the regenerative potential of RGCs is a promising, untested future direction.
These results suggest that BCLXL gene therapy preserves RGC anatomy and gene expression in a mouse model of glaucoma. To build on these findings, future studies should test BCLXL gene therapy in a larger animal model of glaucoma. Larger animals more accurately reflect the human anatomy and disease and the potential challenges with gene therapy in human retinas [52]. Additionally, our experiments did not examine the potential to preserve RGC function, which should be a priority in a model system better suited to clinical examination. Translational application of BCLXL gene therapy will also require evaluating the safety issues associated with long-term expression of this anti-apoptotic protein, including its possible effects on tumorigenesis [53, 54] and increased susceptibility to viral infections [55].
Data availability
Raw data files used for this study will promptly be made available upon reasonable request to the authors. The reagent “AAV2-Pgk-mCherry-BCLXL” is the subject of a provisional patent filed by the Wisconsin Alumni Research Foundation (WARF).
References
Quigley HA. Glaucoma. Lancet. 2011;377:1367–77.
Leske MC, Heijl A, Hussein M, Bengtsson B, Hyman L, Komaroff E, et al. Factors for glaucoma progression and the effect of treatment: the early manifest glaucoma trial. Arch Ophthalmol. 2003;121:48–56.
Howell GR, Libby RT, Jakobs TC, Smith RS, Phalan FC, Barter JW, et al. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J Cell Biol. 2007;179:1523–37.
Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 2012;337:481–4.
Fernandes KA, Mitchell KL, Patel A, Marola OJ, Shrager P, Zack DJ, et al. Role of SARM1 and DR6 in retinal ganglion cell axonal and somal degeneration following axonal injury. Exp Eye Res. 2018;171:54–61.
Libby RT, Li Y, Savinova OV, Barter J, Smith RS, Nickells RW, et al. Susceptibility to neurodegeneration in a glaucoma Is modified by Bax gene dosage. PLoS Genet. 2005. https://doi.org/10.1371/journal.pgen.0010004.
Nikolaev A, McLaughlin T, O’Leary DDM, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–9.
Simon DJ, Weimer RM, Mclaughlin T, Kallop D, Stanger K, Yang J, et al. A caspase cascade regulating developmental axon degeneration. J Neurosci. 2012;32:17540–53.
Simon DJ, Pitts J, Hertz NT, Tesic Mark M, Molina H, Tessier M, et al. Axon degeneration gated by retrograde activation of somatic pro-apoptotic signaling. Cell. 2016;164:1031–45.
Li Y, Schlamp CL, Poulsen KP, Nickells RW. Bax-dependent and independent pathways of retinal ganglion cell death induced by different damaging stimuli. Exp Eye Res. 2000;71:209–13.
Sun YF, Yu LY, Saarma M, Timmusk T, Arumae U. Neuron-specific Bcl-2 homology 3 domain-only splice variant of Bak is anti-apoptotic in neurons, but pro-apoptotic in non-neuronal cells. J Biol Chem. 2001;276:16240–7.
Levin LA, Schlamp CL, Spieldoch RL, Geszvain KM, Nickells RW. Identification of the bcl-2 family of genes in the rat retina. Investig Ophthalmol Vis Sci. 1997;38:2545–53.
Edlich F, Banerjee S, Suzuki M, Cleland MM, Arnoult D, Wang C, et al. Bcl-xL retrotranslocates Bax from the mitochondria into the cytosol. Cell. 2011;145:104–16.
González-García M, García I, Ding L, O’Shea S, Boise LH, Thompson CB, et al. bcl-x is expressed in embryonic and postnatal neural tissues and functions to prevent neuronal cell death. Proc Natl Acad Sci USA. 1995;92:4304–8.
Parsadanian AS, Cheng Y, Keller-Peck CR, Holtzman DM, Snider WD. Bcl-x(L) is an antiapoptotic regulator for postnatal CNS neurons. J Neurosci. 1998;18:1009–19.
Yin W, Cao G, Johnnides MJ, Signore AP, Luo Y, Hickey RW, et al. TAT-mediated delivery of Bcl-xL protein is neuroprotective against neonatal hypoxic-ischemic brain injury via inhibition of caspases and AIF. Neurobiol Dis. 2006;21:358–71.
Liu XH, Collier RJ, Youle RJ. Inhibition of axotomy-induced neuronal apoptosis by extracellular delivery of a Bcl-XL fusion protein. J Biol Chem. 2001;276:46326–32.
Malik JMI, Shevtsova Z, Bähr M, Kügler S, Shevstova Z, Bahr M, et al. Long-term in vivo inhibition of CNS neurodegeneration by Bcl-X L gene transfer. Mol Ther. 2005;11:373–81.
John SWM, Smith RS, Savinova OV, Hawes NL, Chang B, Turnbull D, et al. Essential iris atrophy, pigment dispersion, and glaucoma in DBA/2J mice. Investig Ophthalmol Vis Sci. 1998;39:951–62.
Libby RT, Anderson MG, Pang I-H, Robinson ZH, Savinova OV, Cosma IM, et al. Inherited glaucoma in DBA/2J mice: pertinent disease features for studying the neurodegeneration. Vis Neurosci. 2005;22:637–48.
Schlamp CL, Li Y, Dietz JA, Janssen KT, Nickells RW. Progressive ganglion cell loss and optic nerve degeneration in DBA/2J mice is variable and asymmetric. BMC Neurosci. 2006;7:1–14.
Harder JM, Williams PA, Braine C, Yang H, Thomas J, Foxworth N, et al. Anaphylatoxin receptor C3AR1 promotes optic nerve degeneration in DBA/2J mice. J Neuroinflammation. 2020. https://doi.org/10.21203/rs.3.rs-39367/v1.
Howell GR, Macalinao DG, Sousa GL, Walden M, Soto I, Kneeland SC, et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J Clin Investig. 2011;121:1429–44.
Harder JM, Fernandes KA, Libby RT. The Bcl-2 family member BIM has multiple glaucoma-relevant functions in DBA/2J mice. Sci Rep. 2012;2:530.
Williams PA, Harder JM, Foxworth NE, Cochran KE, Philip VM, Porciatti V, et al. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science. 2017;355:756–60.
Weng CY. Bilateral subretinal Voretigene Neparvovec-rzyl (Luxturna). Gene Ther Ophthalmol Retina. 2019;3:450.
Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. 2019;18:358–78.
Martin KRG, Quigley HA, Zack DJ, Levkovitch-Verbin H, Kielczewski J, Valenta D, et al. Gene therapy with brain-derived neurotrophic factor as a protection: retinal ganglion cells in a rat glaucoma model. Investig Opthalmol Vis Sci. 2003;44:4357–65.
Mosinger Ogilvie J, Deckwerth TL, Knudson CM, Korsmeyer SJ. Supression of developmental retinal cell death but not of photoreceptor degeneration in Bax-deficient mice. Invest Ophthalmol Vis Sci. 1998;39:1713–20.
Donahue RJ, Maes ME, Grosser JA, Nickells RW. BAX-depleted retinal ganglion cells survive and become quiescent following optic nerve damage. Mol Neurobiol. 2020;57:1070–84.
Nickells RW, Schmitt HM, Maes ME, Schlamp CL. AAV2-mediated transduction of the mouse retina after optic nerve injury. Investig Opthalmol Vis Sci. 2017;58:6091.
Semaan SJ, Nickells RW. The apoptotic response in HCT116 cancer cells becomes rapidly saturated with increasing expression of a GFP-BAX fusion protein. BMC Cancer. 2010;10:554.
Maes ME, Schlamp CL, Nickells RW. Live-cell imaging to measure BAX recruitment kinetics to mitochondria during apoptosis. PLoS ONE. 2017;12:e0184434.
Li Y, Schlamp CL, Nickells RW. Experimental induction of retinal ganglion cell death in adult mice. Investig Ophthalmol Vis Sci. 1999;40:1004–8.
Schmitt HM, Grosser JA, Schlamp CL, Nickells RW. Targeting HDAC3 in the DBA/2J spontaneous mouse model of glaucoma. Exp Eye Res. 2020;200:108244.
Bosco A, Anderson SR, Breen KT, Romero CO, Steele MR, Chiodo VA, et al. Complement C3-targeted gene therapy restricts onset and progression of neurodegeneration in chronic mouse glaucoma. Mol Ther. 2018;26:2379–96.
Nickells RW, Pelzel HR. Tools and resources for analyzing gene expression changes in glaucomatous neurodegeneration. Exp Eye Res. 2015;141:99–110.
Smith RS, John SWM, Nishina PM, Sundberg JP. Systematic evaluation of the mouse eye: anatomy, pathology, and biomethods. 1st ed. Boca Raton, FL: CRC Press; 2001.
Schlamp CL, Montgomery AD, MacNair CE, Schuart C, Willmer DJ, Nickells RW. Evaluation of the percentage of ganglion cells in the ganglion cell layer of the rodent retina. Mol Vis. 2013;19:1387–96.
Hendrickson A, Yan YH, Erickson A, Possin D, Pow D. Expression patterns of calretinin, calbindin and parvalbumin and their colocalization in neurons during development of Macaca monkey retina. Exp Eye Res. 2007;85:587–601.
Lawrence PM, Studholme KM. Retinofugal projections in the mouse. J Comp Neurol. 2014;522:3733–53.
Carrillo GL, Su J, Monavarfeshani A, Fox MA. F-spondin is essential for maintaining circadian rhythms. Front Neural Circuits. 2018. https://doi.org/10.3389/fncir.2018.00013.
Benowitz LI, Routtenberg A. GAP-43: an intrinsic determinant of neuronal development and plasticity. Trends Neurosci. 1997;20:84–91.
Goldner R, Yaron A. TIR axons apart: unpredicted NADase controls axonal degeneration. Neuron. 2017;93:1239–41.
Viar K, Njoku D, Secor McVoy J, Oh U. Sarm1 knockout protects against early but not late axonal degeneration in experimental allergic encephalomyelitis. PLoS ONE. 2020;15:e0235110.
Figley MD, Gu W, Nanson JD, Shi Y, Sasaki Y, Cunnea K, et al. SARM1 is a metabolic sensor activated by an increased NMN/NAD+ ratio to trigger axon degeneration. Neuron. 2021;109:1118–36. e11.
Harun-Or-Rashid M, Pappenhagen N, Zubricky R, Coughlin L, Jassim AH, Inman DM. MCT2 overexpression rescues metabolic vulnerability and protects retinal ganglion cells in two models of glaucoma. Neurobiol Dis. 2020;141:104944.
Alavian KN, Li H, Collis L, Bonanni L, Zeng L, Sacchetti S, et al. Bcl-x L regulates metabolic efficiency of neurons through interaction with the mitochondrial F1 FO ATP synthase. Nat Cell Biol. 2011;13:1224–33.
Chen YB, Aon MA, Hsu YT, Soane L, Teng X, McCaffery JM, et al. Bcl-x L regulates mitochondrial energetics by stabilizing the inner membrane potential. J Cell Biol. 2011;195:263–76.
Berman SB, Chen YB, Qi B, McCaffery JM, Rucker EB, Goebbels S, et al. Bcl-x L increases mitochondrial fission, fusion, and biomass in neurons. J Cell Biol. 2009;184:707–19.
Anderson DR, Hendrickson A. Effect of intraocular pressure on rapid axoplasmic transport in monkey optic nerve. Investig Ophthalmol Vis Sci. 1974;13:771–83.
Stieger K, Lhériteau E, Moullier P, Rolling F. AAV-mediated gene therapy for retinal disorders in large animal models. ILAR J. 2009;50:206–24.
Ramírez-Komo JA, Delaney MA, Straign D, Lukin K, Tsang M, Iritani BM, et al. Spontaneous loss of B lineage transcription factors leads to pre-B leukemia in Ebf1+/–Bcl-xLTg mice. Oncogenesis. 2017;6:e355.
Pena JC, Rudin CM, Thompson CB. A Bcl-x(L) transgene promotes malignant conversion of chemically initiated skin papillomas. Cancer Res. 1998;58:2111–6.
Wyżewski Z, Świtlik W, Mielcarska MB, Gregorczyk-Zboroch KP. The role of Bcl-xL protein in viral infections. Int J Mol Sci. 2021;22:1–16.
Acknowledgements
The authors would like to thank Mr. Joel Dietz for managing the mouse colony and for providing technical advice, Mr. Satoshi Kinoshita at the Translational Research Initiative in Pathology (TRIP) laboratory at the University of Wisconsin-Madison for cutting sections analyzed in this study, and Dr. Mark Banghart for providing statistical considerations of the longitudinal IOP data.
Funding
This work was supported by National Eye Institute grants R01 EY030123 (to RWN), T32 EY027721 (Department of Ophthalmology and Visual Sciences, University of Wisconsin-Madison), and a Vision Science Core grant P30 EY016665 (Department of Ophthalmology and Visual Sciences, University of Wisconsin-Madison), an unrestricted funding grant from Research to Prevent Blindness (Department of Ophthalmology and Visual Sciences, University of Wisconsin-Madison), and the Frederick A. Davis Endowment (to RWN).
Author information
Authors and Affiliations
Contributions
RJD and RWN conceived of the project. RJD performed experiments for every portion of the manuscript. RLF performed the in vitro validation of mCherry-BCLXL. JRG performed the analyses for the optic nerve crush experiments. RJD and RWN wrote the manuscript, which was approved by all the authors.
Corresponding author
Ethics declarations
Competing interests
The Wisconsin Alumni Research Foundation (WARF) has filed a provisional patent application around this research.
Ethics approval and consent to participate
No human subjects were used in this study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by N. Bazan
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Donahue, R.J., Fehrman, R.L., Gustafson, J.R. et al. BCLXL gene therapy moderates neuropathology in the DBA/2J mouse model of inherited glaucoma. Cell Death Dis 12, 781 (2021). https://doi.org/10.1038/s41419-021-04068-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-021-04068-x
This article is cited by
-
BAX activation in mouse retinal ganglion cells occurs in two temporally and mechanistically distinct steps
Molecular Neurodegeneration (2023)
-
Apoptotic cell death in disease—Current understanding of the NCCD 2023
Cell Death & Differentiation (2023)
-
BclXL (Bcl2l1) gene therapy lessens retinal ganglion cell soma loss but not axonal degeneration after acute axonal injury
Cell Death Discovery (2022)
