
Abstract
The prevalence of noise-induced hearing loss (NIHL) continues to increase, with limited therapies available for individuals with cochlear damage. We have previously established that the transcription factor FOXO3 is necessary to preserve outer hair cells (OHCs) and hearing thresholds up to two weeks following mild noise exposure in mice. The mechanisms by which FOXO3 preserves cochlear cells and function are unknown. In this study, we analyzed the immediate effects of mild noise exposure on wild-type, Foxo3 heterozygous (Foxo3+/−), and Foxo3 knock-out (Foxo3−/−) mice to better understand FOXO3’s role(s) in the mammalian cochlea. We used confocal and multiphoton microscopy to examine well-characterized components of noise-induced damage including calcium regulators, oxidative stress, necrosis, and caspase-dependent and caspase-independent apoptosis. Lower immunoreactivity of the calcium buffer Oncomodulin in Foxo3−/− OHCs correlated with cell loss beginning 4 h post-noise exposure. Using immunohistochemistry, we identified parthanatos as the cell death pathway for OHCs. Oxidative stress response pathways were not significantly altered in FOXO3’s absence. We used RNA sequencing to identify and RT-qPCR to confirm differentially expressed genes. We further investigated a gene downregulated in the unexposed Foxo3−/− mice that may contribute to OHC noise susceptibility. Glycerophosphodiester phosphodiesterase domain containing 3 (GDPD3), a possible endogenous source of lysophosphatidic acid (LPA), has not previously been described in the cochlea. As LPA reduces OHC loss after severe noise exposure, we treated noise-exposed Foxo3−/− mice with exogenous LPA. LPA treatment delayed immediate damage to OHCs but was insufficient to ultimately prevent their death or prevent hearing loss. These results suggest that FOXO3 acts prior to acoustic insult to maintain cochlear resilience, possibly through sustaining endogenous LPA levels.
Similar content being viewed by others
Introduction
Noise-induced hearing loss (NIHL) is a pervasive health threat. In 2017, the NIDCD estimated that despite increased awareness and access to protective equipment, nearly 40 million American adults had signs of NIHL [1]. Therapies for NIHL remain limited, in many cases due to the permanent loss of cochlear sensory cells. Individuals have varying susceptibilities to noise damage, partially due to differences in gene expression crucial for hearing recovery and cochlear preservation [2]. By studying the underlying genetic mechanisms of NIHL susceptibility, our goal is to better prevent or mitigate NIHL using biological approaches.
Transcriptional regulators comprise a class of NIHL susceptibility genes, including Forkhead Box O-3 (FOXO3) [3]. FOXO3 is a winged helix/forkhead class transcription factor involved in cellular processes including autophagy [4], survival [5], stress resistance [6,7,8], apoptosis [9,10,11], and longevity [12,13,14]. Individuals may harbor genetic variants that can lead to varying levels of FOXO3 transcription, with its spatiotemporal regulatory roles susceptible to manipulation in autoimmune diseases [15,16,17] and cancers [5, 10]. FOXO3 is expressed from birth into adulthood throughout the mammalian cochlea including inner and outer hair cells (IHCs and OHCs), supporting cells (SCs), and spiral ganglion neurons (SGNs) [18]. Within aged SGNs, FOXO3 is capable of nuclear translocation in response to non-traumatic noise [18]. It has previously been shown that 4 h following a stressor, evidence of the two-step induction of FOXO3 by AMPK used for transcription was present in glutamate-exposed neurons [19]. These neuronal observations suggest that FOXO3 is capable of driving gene transcription in stressed, damaged, or dysfunctional cells. We have previously determined that FOXO3 is required to preserve OHCs and hearing thresholds following mild noise exposure [20]. However, the mechanism by which FOXO3 confers protection has not been confirmed. In the hematopoietic system, loss of FOXO3 leads to the overaccumulation of reactive oxygen species (ROS), leading to the death of hematopoietic stem cells [21] and developing erythrocytes [22]. ROS accumulation in cochlear cells after noise exposure also drives cell death [23,24,25,26]. We hypothesize that in the absence of FOXO3 following noise exposure, OHCs would be vulnerable to apoptotic cascades resulting from oxidative stress induction and ROS accumulation. OHC loss is correlated with poorer hearing outcomes in humans [27].
To better understand FOXO3’s role in NIHL susceptibility, we analyzed the immediate effects of mild noise exposure on wild-type (WT, Foxo3+/+), Foxo3 heterozygous (Foxo3+/−), and Foxo3 knock-out (Foxo3−/−) mice. Previous studies have shown that after traumatic noise exposure, OHC damage drives apoptosis, necroptosis, and a third death pathway [28,29,30,31]. Depending on the severity of the traumatic exposure, OHC caspase-3 activation may be immediately evident [31], or it may develop over the following 24 h post noise (HPN) [32]. OHC loss continues over a period of days to weeks [32, 33]. The degree of OHC loss seen in the Foxo3−/− cochlea after mild noise exposure could reflect either change in cochlear homeostasis prior to damage or a compromised damage response. Determining the time course of OHC death could shed light on when FOXO3 acts to protect the cochlea.
To this end, we characterized the time course of OHC loss from noise in the Foxo3−/− mouse and identified a likely mode of cell death. We compared activation of known damage pathways in the Foxo3−/− cochlea to that of the WT cochlea to assess their involvement. We performed RNA-SEQ, identified a candidate downstream effector enzyme, and tested its involvement by exogenous supplementation of its product. Our goals were to determine what pathways were immediately active after the noise and when therapeutic intervention would be most efficacious to mitigate the effects of FOXO3’s absence.
Results
Establishing the time course of OHC loss in the Foxo3 −/− noise-exposed cochlea
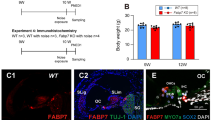
Foxo3−/− mice acquire severe permanent threshold shifts throughout the cochlea after mild noise exposure [20]. By two weeks, about one-third of Foxo3−/− OHCs were lost from the basal cochlea without any IHC loss [20]. WT littermates experienced only temporary threshold shifts and no cell loss from the same exposure [20]. These observations did not establish when or how Foxo3−/− OHCs are lost. Here we characterize cellular pathology in Foxo3−/− and WT cochleae immediately after noise exposure. We used cochlear cryosectioning to obtain cross-sections for detailed cellular analysis (Fig. S1A), and whole mounts preparations for cellular quantification according to tonotopic frequency (Fig. S1B). The latter is important as high frequency (basal) OHCs are more susceptible to trauma [34,35,36]. To test for haploinsufficiency, we analyzed the FOXO3 heterozygote (Foxo3+/−). We measured Foxo3+/− auditory brainstem responses (ABRs) and distortion product otoacoustic emissions (DPOAEs) at baseline, 1-day post-noise (1 DPN), and 14 DPN (Fig. S2). Since Foxo3+/− mice displayed temporary threshold shifts and recovery similar to WT mice [20], one Foxo3 allele appears sufficient for cochlear homeostasis.
To establish the time course of OHC loss in Foxo3−/− mice, we exposed adult WT, Foxo3+/−, and Foxo3−/− mice to mild noise (Fig. 1A). Cochleae were isolated at 0.5, 4, and 24 HPN, and fluorescent antibodies for Oncomodulin (OCM) and Myosin 7a (MYO7a) [37] were used to quantify OHCs. OCM is an EF-hand calcium-binding protein highly expressed in OHCs [38], and its deletion confers progressive hearing loss [39]. Representative 24 kHz (middle) regions of Foxo3−/− cochleae are presented in Fig. 1B–E′, with WT and Foxo3+/− cochleae in Fig. S3. Under baseline conditions, OCM and MYO7a were co-expressed within the OHCs (Fig. 1B–B′). OCM expression was reduced in some Foxo3−/− MYO7a+ OHCs after 4 HPN (Fig. 1D–D′, yellow arrows), becoming more apparent at 24 HPN in rows 2 and 3 (Fig. 1E–E′, yellow arrows). OCM/MYO7a+ OHCs were maintained in the WT and Foxo3+/− cochleae following noise (Fig. S3), indicating specific noise-induced modulations in FOXO3-absent OHCs.

A Noise exposure method for WT, Foxo3+/−, and Foxo3−/− mice. Cochleae were extracted for whole-mount immunohistochemistry immediately following the exposure (0.5 h post-noise exposure or HPN), at 4 HPN, or 24 HPN. Unexposed littermates served as baseline controls. B–E Expression of MYO7 (white) was maintained in the Foxo3−/− OHCs following noise exposure (yellow arrows). B′–E′ Over the same time course, OCM (pink) immunoreactivity decreased in OHCs, mainly in rows 2 and 3. n = 3, scale bar = 10 µm, ×20 magnification, ~24 kHz region. F–I Cochleograms of % mean OCM+ cell loss (y-axis) plotted against % distance from the cochlear apex (x-axis). Each dot represents the mean number of OCM+ cells counted in 100 µM intervals along the length of cochleae (4–86 kHz) pooled (n = 3–6 per genotype/condition). Cochlear tonotopic regions are presented by background color: Apex = light gray, Middle = gray, Base = dark gray. Non-parametric interpolation lines are presented for each group: WT (orange), Foxo3+/− (pink), and Foxo3−/− (green). G–I The noise band is presented as a red dotted outline. Kruskal–Wallis rank-sum test adjusted for multiple comparisons, alpha = 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To determine the overall number of OCM+ OHCs after noise exposure, cochleae were stained with DAPI and antibodies against OCM, MYO7a or Cytochrome-C (CytC), a mitochondria-associated hemeprotein strongly expressed in OHCs [40]. Cochleograms were generated for each genotype and time point. All three genotypes displayed slightly more baseline OCM-low OHCs in the basal cochlear third (Fig. 1F). The Foxo3−/− apex and Foxo3+/− base showed increased OCM loss (Fig. 1F; Apex p = 0.0042, Middle p = 0.3000, Base p = 0.0043, Kruskal–Wallis). Immediately the following the noise, loss of OCM+ OHCs increased in both Foxo3−/− and Foxo3+/− cochleae at the apical (p = 0.0002) and middle (p < 0.0001) regions. All three genotypes had similar losses in the base (p = 0.3000, Fig. 1G; Kruskal–Wallis). By 4 HPN, loss of OCM+ cells increased along the entire lengths of Foxo3−/− cochleae (all turns p < 0.0001) while Foxo3+/− and WT numbers recovered to baseline (Fig. 1H). Foxo3−/− cochleae had greater OCM+ cell loss by 24 HPN (all turns p < 0.0001) as the other two genotypes regained OCM immunoreactivity (Fig. 1I, Kruskal–Wallis). These data suggest that OCM dynamically modulates, and its normal response to noise exposure is exacerbated in the Foxo3−/− cochleae.
To assess cellular damage, we rendered the 12 (apical) and 24 (middle) kHz regions in Imaris (Figs. 2, S4, S5). We focus on 24 kHz WT and Foxo3−/− renderings (Fig. 2A–H″) and mean OHC counts of all three genotypes (Fig. 2I–K and Fig. S4). At baseline, CytC distribution in OHCs was similar to OCM (Fig. 2A″, E″). WT mice showed consistent OCM immunoreactivity at baseline and following noise exposure (Fig. 2A–D″). At baseline, some low OCM-expressing OHCs were detected in Foxo3+/− mice (Fig. S4N), and Foxo3−/− OHCs were comparable to WT cells (Fig. 2E–E″). Immediately following noise exposure, many Foxo3−/− OHCs lost OCM immunoreactivity (Fig. 2F–F″, yellow arrows). By 4 HPN, pyknotic nuclei were present in OHC regions lacking OCM (Fig. 2G–G″). By 24 HPN, many OCM+/CytC+ cells were lost and fewer OHC nuclei remained in the Foxo3−/− (Fig. 2H–H″, white arrows). Most WT OHCs expressed OCM by 24 HPN (Fig. 2D–D″, yellow arrows). Notably, their nuclei were neither fragmented nor expanded. Although WT and Foxo3+/− mice displayed fluctuations in OCM immunoreactivity, unlike the Foxo3−/− mice this did not resolve into OHC loss by 24 HPN (Fig. 2I, J, p < 0.0001, two-way ANOVA). The 12 kHz region exhibited similar but less drastic OCM changes than the 24 kHz region, and Foxo3−/− OHCs were better preserved (Fig. S5). In the absence of FOXO3, weak OCM immunoreactivity lasting beyond 4 HPN correlated with OHC apoptosis.

After immunostaining in whole-mount, the 24 kHz cochlear region was mapped, imaged using confocal microscopy at ×150 magnification, and rendered in Imaris (n = 3 cochleae per genotype/condition). A–H The rendered images of OHCs colored cyan for DAPI+ cell nuclei. A’–H’ Renderings of DAPI+/OCM + OHCs with OCM in magenta. A″–H″ Renderings of DAPI+/OCM+/CytC+ cells with CytC in gray. Scale bar = 10 µm. A–C″ Little to no change in OCM and CytC localization through 4 HPN in WT OHCs. D–D″ By 24 HPN, WT OHCs only expressing CytC+ (yellow arrows) are in the minority of cells likely damaged by the noise exposure. F′, F″ At 0.5 HPN, a large number of Foxo3−/− OHCs lose OCM immunoreactivity (yellow arrows). G, G″ By 4 HPN, several OHC regions with low OCM contain pyknotic or missing nuclei (yellow arrows). H′, H″ Only two OCM+/CytC+ OHCs remain in any of the standard three rows (white arrows); the other nuclei belong to SCs as they exist in a lower z-plane and have a larger mean diameter. I–K OHC counts for all three genotypes expressing specific fluorophores (y-axis) are graphed versus the noise exposure timeline (x-axis). I Numbers of DAPI + OHC nuclei (y-axis) present across the time course (x-axis); supporting cell nuclei unable to be excluded in the renderings due to thresholding limitations were omitted. J Counts of OHCs as calculated by totaling OCM+/CytC+ cells (y-axis). K Counts of OHCs only expressing CytC (y-axis). WT (black circles, orange mean bar), Foxo3+/− (gray squares, pink mean bar), and Foxo3−/− (light gray triangles, green mean bar) total cell counts per 1024 × 400 × 20-pixel selection. n = 3 per genotype/condition, two-way ANOVA with Tukey’s test for multiple comparisons, alpha = 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Caspase-independent parthanatos may drive OHC loss
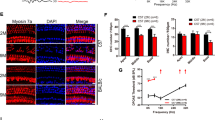
FOXO3 regulates caspase-dependent apoptosis in many tissues by promoting transcription of Bax and Bim [41, 42]. Nuclear fragmentation, indicating apoptosis, correlated with decreased OCM in Foxo3−/− OHCs following noise exposure. Activated CASP3 immunostaining was not observed (Fig. 3A–D′), suggesting a caspase-independent apoptotic pathway in noise-induced OHC death [28, 43,44,45,46]. Parthanatos is a rapid caspase-independent apoptotic mechanism seen in cells primed for death [40]. It is identified by the nuclear localization of the mitochondrial protein AIFM1, which initiates a cell death program [47]. No nuclear AIFM1 was seen in WT OHCs (Fig. 3). We saw low AIFM1 immunoreactivity in the Foxo3−/− at baseline, which became elevated in OCM-low OHC nuclei at 0.5 HPN (Fig. 3H-H’”, yellow arrows). These results are consistent with parthanatos as a pathway for OHC death in Foxo3−/− cochleae.

A–D′ Multiphoton images of the middle, ~24 kHz region, in whole-mounted cochleae over the established time course. Tissues were analyzed for OCM (magenta) and CASP3 (cyan). D′ Inset = positive control tissue: CASP3 + apoptotic SGNs after chronic cigarette smoke exposure. n = 3–5 per time point, ×20 magnification, scale bar = 50 μm. E–H‴ Single-matched optical sections from confocal images of 24 kHz OHCs under baseline conditions and 0.5 HPN. WT (E–F‴) and Foxo3−/− (G–H‴) cochleae were sectioned and immunostained for detection of DAPI (cyan, E–H), OCM (magenta, E′–H′), and AIFM1 (white, E″–H″). H–H‴ The mitochondrial protein AIFM1, localized to the nucleus in the Foxo3−/− OCM-low OHC (yellow arrow), an indication of parthanatos. n = 4 per genotype/condition, ×200 magnification, scale bar = 5 μm.
Differential expression analysis of genes following noise exposure shows enhancement in calcium signaling and oxidative stress response pathways
Absence of the transcription factor FOXO3 could alter gene expression, leaving OHCs primed for death. Foxo3−/− and WT cochleae were processed at 0, 4, and 24 HPN for mRNA extraction (Fig. 4A). Differentially expressed genes (DEGs) between genotypes and time points were identified with RNA-sequencing. Gene ontology was used to perform enrichment analysis on DEGs. Calcium signaling, the oxidative stress response, and some cytokine signaling pathways were enhanced by 4 HPN in the Foxo3−/− (Fig. 4B). Changes in calcium signaling genes correlated with the OCM modulation observed shortly after noise. Clustering analysis for the Foxo3−/− versus WT 4 HPN comparison found 8 distinct gene clusters (Figs. 4C, S6). Several genes matched with the canonical pathways assigned at 4 HPN and are listed in Supplementary Table 3. Volcano plots enable visualization of DEG distribution (Fig. 4D, E). Notably, the genes Gdpd3, Ypel3, and Gatsl2 were downregulated at baseline and following noise exposure (Fig. 4D–F). We include these genes in our analysis of OHC-associated factors (Table 1). The transcription factor JunB was up-regulated (Log FC = 1.73, Log CPM = 5.21, p = 8.52E−16) in the Foxo3−/− cochleae at 4 HPN. This was confirmed with RT-qPCR (fold gene expression = 4.35). Jun family members JunB, cJUN, and JNK are associated with the oxidative stress response [48, 49]. Provided these RNA sequencing results and known associations between FOXO3 and oxidative stress regulation in other tissues, we examined the oxidative stress response following noise.

A Foxo3−/− and WT mice were subjected to noise and had their cochleae removed for mRNA extraction at 0, 4, and 24 HPN (n = 6 pooled cochleae per genotype/condition). B Top canonical pathways in Foxo3−/− versus WT cochleae at 4 HPN using Ingenuity Gene Ontology. The p-value indicates the statistical significance of the overlapping genes within the canonical pathway. C Gene expression clustering was performed for the Foxo3−/− versus WT at 4 HPN to produce 8 clusters with similarly behaving genes. Clustering traces and the detailed gene table are available in Supplements (Fig. S6, Table S3). D–F Volcano plots of differentially expressed genes at baseline (D), 4 HPN (E), and 24 HPN (F). The y-axis represents −log10(p-value) and x-axis is the log2fold-change. Significantly differentiated genes (−log10(p-value) ≥ 2) are denoted by pink dots above the gray line. Non-significant genes are represented by black dots. Cyan lines divide genes that fall between a −1 to 1 log fold-change.
Loss of FOXO3 does not alter the cochlear oxidative stress response to mild noise
We utilized immunofluorescence to evaluate the oxidative stress pathway in Foxo3−/− and WT cochleae. No differences in HSP70 in OHCs or Deiters’ cells (DCs) were observed at baseline (Fig. S7). Immediately the following noise, both genotypes similarly expressed 4-hydroxy-2-nonenal (4-HNE), in middle OHC stereocilia, indicating similar levels of lipid peroxidation (Fig. 5C, F). At baseline, JNK phosphorylation (pJNK) was highly concentrated at both IHC and OHC synapses & neurites and has lower expression in the pillar and DCs (Fig. 5H, J). At 0.5 HPN, the DCs of Foxo3−/− and pillar cells of WT cochleae showed increased pJNK expression (Fig. 5I, K). Little to no pJNK was observed within the OHCs of either genotype. The baseline phosphorylation of cJUN (pcJUN) varied across animals but was mainly expressed in the DCs and IHC synapses (Fig. 5L, O). At 2 HPN, pcJUN was most pronounced in the DC nuclei of both genotypes (Fig. 5M, P). Low OCM+ Foxo3−/− OHCs contained pcJUN within their pyknotic nuclei, possibly reflecting cell stress [50] (Fig. 5P, Q′, yellow arrows). Surprisingly, p53, which directly interacts with FOXO3 [51], showed little difference in expression at the OHCs and DCs following noise exposure (Fig. S8). These data suggest that loss of FOXO3 does not alter the cochlear oxidative stress response following noise exposure. Given these data and the presence of parthanatos, we speculate that FOXO3’s importance to OHC survivability is due to its activity prior to insult.

A WT and Foxo3−/− mice were exposed to noise and their cochleae were extracted for cryosectioning and immunohistochemistry at designated time points. B–G Cochlear sections of the 24 kHz region immunostained for 4HNE (white) at baseline, 0.5 HPN, and 4 HPN. C, F At 0.5 HPN, 4HNE expression increased at the OHC stereocilia in both genotypes (yellow arrows). D, G 4HNE levels returned to baseline by 4 HPN. n = 3–4 per genotype/time point, 60x magnification, scale bar = 10 µm, TM = Tectorial membrane. H–Q′ Cochlear sections of the 24 kHz OHCs and SCs stained for DAPI (cyan) and with antibodies against OCM (magenta), pJnk (H–K′, white) and pcJun (L–Q′, white). n = 3–5 per genotype/condition, ×200 magnification, scale bar = 10 µm. Corresponding % fluorescent cellular expression graphs below each set of images: undetected = white, low = light gray, high = gray, and lost cells = black, DC Deiters’ cells, OHCs outer hair cells, OHC S&N: OHC synapses and neurites, PCs pillar cells, IHC S&N IHC synapses and neurites, and IHC inner hair cells. H–K′ Though strong at the hair cell synapses and highly variable across the organ of Corti, pJnk was absent from the OHCs in both genotypes after noise exposure. In both genotypes, pcJun expression was variable in DC nuclei at baseline (L–L′, O–O′) but strongly increased by 2 HPN (M–M′, P–P′). pcJun was present in damaged Foxo3−/− OHC nuclei at 2 HPN (P, P′), continuing through 4 HPN (Q–Q′) (yellow arrows).
LPA treatment delayed but did not prevent Foxo3 −/− OHC and hearing loss
RNA sequencing identified candidate DEGs that could contribute to increased noise sensitivity in the Foxo3−/− cochlea (Table 1). Actin dysregulation contributes to rapid cell death in noise damage [27]. Lysophosphatidic acid (LPA) administration can mitigate this effect by activating the ROCK2-RhoA pathway [52]. Gdpd3 encodes an enzyme that hydrolyzes lysoglycerophospholipids to generate LPA. Gdpd3 was downregulated at baseline in Foxo3−/− cochleae (Log FC = −1.36, Log CPM = 4.55, p = 9.2E−11). Decreased Gdpd3 transcription was confirmed with RT-qPCR (fold gene expression = 0.184). Immunofluorescence showed that GDPD3 protein was expressed in neurites and SCs (Fig. S9). GDPD3’s proximity to OHCs makes it a candidate for intrinsic cochlear LPA production, which has not previously been identified. Unlike the RNA transcripts, we observed no difference in immunofluorescence intensity between genotypes. GDPD3 has recently been postulated to regulate FOXO3a/β-catenin binding and inhibition of the AKT/mTORC1 pathway via the mediation of lysophospholipid metabolism [53]. It is possible that in the absence of FOXO3, residual GDPD3 may alter its LPA production.
We tested if cytoskeletal dysregulation contributes to rapid OHC loss in the Foxo3−/− by phenotypic rescue with LPA treatment. Both short and long-term analyses were performed (Fig. 6A, G). The 4 HPN cochleogram indicates that survival of OCM + OHCs improved with LPA versus saline treatment for Foxo3−/− mice basal to the noise band (Fig. 6B, p = 0.0020, Mann–Whitney U). Confocal images at 24 kHz showed more OCM-negative regions with pyknotic nuclei in the saline-treated versus LPA-treated Foxo3−/− cochlea (Fig. 6E, F, yellow arrows). However, by 14 DPN LPA-treated and saline-treated Foxo3−/− mice had similar levels of OHC loss, differing from WT littermates (Fig. 6H, p < 0.0001, Kruskal–Wallis). In both groups of Foxo3−/− mice, ABRs following noise damage revealed significant permanent threshold shifts (Fig. 6M, p < 0.0001, two-way ANOVA). LPA treatment improved WT hearing recovery, reducing all threshold shifts back to baseline (Fig. 6M, p = 0.0495, two-way ANOVA). Identically poor DPOAEs were present in both Foxo3−/− groups above 16 kHz at 14 DPN (Fig. 6N, 16 kHz p < 0.0001, 24 kHz p = 0.02, 32 kHz p = 0.03, two-way ANOVA). LPA treatment improved WT OHC function versus saline-treated mice at 32 kHz (Fig. 6N, p = 0.0004, Welch’s t-test). These results suggest that a brief LPA treatment delays rapid cell death in the Foxo3−/− cochlea, implicating cytoskeletal dysregulation in parthanatos. LPA treatment is insufficient, however, to overcome OHC fragility or prevent hearing threshold shifts. Altogether, these results implicate FOXO3 in actin stability rather than oxidative stress response for cochlear homeostasis.

A, G All mice were tested for baseline ABRs and DPOAEs the week prior to noise exposure. A One set of WT and Foxo3−/− mice (n = 3–5 per genotype) received three i.p. injections of LPA and were sacrificed at 4 HPN. Sham mice (n = 3–4 per genotype) received three 0.9% sterile saline injections at the same time points. Cochleae were extracted and stained for DAPI and with antibodies against OCM, and MYO7 for OHC quantification. B Cochleogram at 4 HPN of % mean OCM+ cell loss (y-axis) plotted against % distance from the cochlear apex (x-axis). Each dot represents the mean number of OCM+ cells counted in 100 µM intervals along the length of cochleae (4–86 kHz) pooled (n = 3 per genotype/condition). Non-parametric interpolation lines are presented with each group: WT Saline (orange squares), WT LPA (gold squares), Foxo3−/− Saline (green triangles), and Foxo3−/− LPA (purple triangles). Cochlear tonotopic regions are presented by background color: Apex = light gray, Middle = gray, Base = dark gray. The noise band is presented as a red dotted outline. Mann–Whitney U and Kruskal–Wallis rank-sum test adjusted for multiple comparisons, p < 0.05. C–F Confocal images of 24 kHz OHC regions of each genotype/condition at 4 HPN immunostained with DAPI (cyan) and OCM (magenta). E, F Foxo3−/− saline-treated OCM-low OHCs contained more pyknotic nuclei overall than the Foxo3−/− LPA-treated OHCs (yellow arrows). n = 3 per genotype/condition, ×20 magnification, scale bar = 10 µm. G A second set of mice (n = 3 per genotype) was injected four times with LPA and sacrificed at 336 h (14 DPN) following hearing tests. Sham mice (n = 3 per genotype) received four saline injections at the same time points and underwent the same tests. H Cochleogram at 14 DPN with the same parameters as in B. I–L Confocal images at 14 DPN with the same parameters as in C–F. K, L OCM + OHC immunoreactivity in both Foxo3−/− groups was similar. Mean ABR (M) and DPOAE (N) thresholds (y-axis) at baseline for WT (n = 16, orange circles) and Foxo3−/− (n = 14, green triangles) groups, and at 14 DPN for WT Saline (n = 3, orange open circles), WT LPA (n = 3, pink open squares), Foxo3−/− Saline (n = 3, green open triangles), and Foxo3−/− LPA (n = 3, purple open diamonds) groups are plotted along five frequencies tested (x-axis). ±s.e.m., two-way ANOVA with Tukey’s test for multiple comparisons, Welch’s t-test, alpha = 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Discussion
Irreversible loss and damage to sensory cells contribute to poor hearing outcomes in NIHL. Genetic variants in transcription factors necessary for survival and death can confer greater susceptibility to developing this disorder, including FOXO3 [54, 55]. To better understand how FOXO3 promotes cochlear homeostasis, we exposed Foxo3−/− mice, which lack FOXO3 in all cells throughout their lives, to mild noise. We determined the time course and cell death pathway for Foxo3−/− OHCs. We evaluated the effects of noise exposure on RNA expression and several damage response pathways. Finally, we tested if activation of the ROCK2-RhoA pathway could mitigate the effects of noise on Foxo3−/− OHCs [52]. FOXO3 is expressed in both OHCs and SCs and maybe playing different roles in each [18]. Due to the rapidity with which OHCs are lost basal to the noise band, FOXO3 could potentially mediate the expression of factors important to preserving OHCs, preventing parthanatos, and possibly enhancing cytoskeletal resilience. Contrary to our original hypothesis, we conclude that FOXO3 is not preserving the cochlea via oxidative stress regulation but rather via its transcriptional activity during development.
We show a correlation of early and prolonged OCM modulation with cell injury, possibly due to excessive calcium buffering experienced in noise exposure. OCM binds to calcium and is localized to the cuticular plate and cytoplasm of OHCs, likely to regulate their motility [38]. We found that OCM immunoreactivity normally fluctuates at baseline and in response to noise in viable OHCs (Figs. 1, S4, 2, Fig. S5). However, poor outcomes for OHCs harboring low OCM levels through 4 HPN were characteristic of the Foxo3−/− (Figs. 1, S4, 2, S5, 6). While not essential to cochlear development, Ocm−/− mice develop progressive hearing loss [39]. When modeled in vitro, OCM did not appear to have a direct impact on actin polymerization [56]. There have been no studies to confirm whether OCM protects against oxidative stress and whether it is responsible for multiple functions within OHCs [38]. The majority of OHCs containing fragmented nuclei expressed low OCM levels, making its loss a marker of permanent damage (Figs. 2, S5, 6).
Due to the rapidity of their demise, we consider Foxo3−/− OHCs primed to die. Several cell death pathways have been examined in NIHL models with apoptosis and necrosis the most prominent [45, 57]. Both intrinsic and extrinsic apoptotic pathways contain various players whose actions eventually converge with caspase activation [58]. These cells are characterized by shrunken volumes and pyknotic nuclei with surface blebs, in contrast to necrotic (oncotic) cells with swollen features [28]. We saw no evidence of caspase-dependent apoptosis nor necrosis post-noise exposure (Fig. 3). We did find similarities to the “3rd death pathway” as described by Bohne et al., with cells having a persistent apical region, debris arranged in a cylindrical cell-like shape with a deficient plasma membrane, and a normal-sized nucleus that may later enlarge and fragment [28] (Figs. 2, S5). Other causes of death for sensory cells have been proposed, including AIF-mediated parthanatos [47]. AIFM1 translocated to the nucleus of Foxo3−/− OHCs after noise exposure (Fig. 3), as previously described for guinea pig OHCs after severe, repeated acoustic insults [44]. Understanding the molecular basis for OHC fragility will lead to new models of cochlear homeostasis.
FOXO3 has previously been reported as a master regulator of oxidative stress elements in various tissues [19, 59,60,61,62]. Catalase and MnSOD are two examples of reactive oxygen species-detoxifying enzymes that are decreased in haematopoietic stem cells deficient for FOXO isoforms [63]. We analyzed control cochleae for initial signs of oxidative imbalance in the Foxo3−/−. There was no indication of elevated lipid peroxidation, increased HSP70 accumulation in SCs, nor significant differential expression of stress-related genes in the Foxo3−/− cochlea at baseline (Figs. S7, 4, 5). The activation of the oxidative stress pathway was observed most clearly with 4HNE present in the apex of OHCs at 0.5 HPN and around 2 HPN with elevated pcJUN in SC nuclei and expression in OCM-low OHCs in the Foxo3−/− (Fig. 5). Surprisingly, the time course and cellular expression patterns of these factors were similar between genotypes. Immediate cell damage suggests that transcription of protective elements is more crucial at baseline than the transcription of stress responders, which may be a slower process. For example, we did not see a significant downregulation in Bim at 4 HPN (Log FC = 0.10, Log CPM = 4.22, p = 0.547) which is a direct FOXO3 transcriptional target that can activate apoptosis [64]. A limitation of our RNA sequencing analysis is that we used whole cochlea lysates to analyze genetic changes and did not include the 0.5 HPN time point. FOXO3 is normally found in several cell types and its activity may be spatiotemporal. Conditional Foxo3 knock-out lines or single-cell sequencing may provide us with improved control over FOXO3 expression and cell-intrinsic versus extrinsic activity.
FOXO3 may influence OHC viability via extrinsic effects from its presence in SCs. We can draw a comparison to HSP70 which can be secreted immediately after noise or ototoxic insult via exosomes from SCs to hair cells for their protection [65,66,67,68,69,70]. Seeing that Gdpd3, one of the only genes downregulated in the Foxo3−/− cochlea at baseline, is strongly expressed in the SCs (Fig. S9), we speculate that FOXO3 may have a role in its transcription or activation. The fact that LPA was found to delay OCM changes but not completely rescue OCM numbers supports the idea that the LPA-activated ROCK2-RhoA stabilization of actin filaments can mitigate some of the damage in our noise exposure (Fig. 6). However, the differential expression of cytoskeletal genes correlated with Foxo3 deletion (Table 1, S3) may also decrease overall actin stability in the OHCs and/or SCs. Further investigation into cytoskeletal changes including measuring F/G actin ratios will help clarify these observations. To determine if GDPD3 can contribute to NIHL, we would like to evaluate Gdpd3−/− mice for cochlear LPA levels and hearing thresholds after noise exposure. Recapitulation of the Foxo3−/− phenotype would aid in our understanding of the overall mechanism of action for maintaining OHC viability.
In summary, this study provides valuable insight into the mechanism underlying OHC loss in the Foxo3−/− cochlea after mild noise exposure. Future studies will focus on clarifying FOXO3’s cell-intrinsic versus extrinsic functions and its possible influence on cytoskeletal integrity.
Materials and methods
Mice
Mice were housed on a 12-h light/dark cycle and received chow and water ad libitum. Once pups were found, their appearance was compared with the Jackson Labs pups’ age appearance chart and their birth date was designated postnatal day (P) 0. Mice were weaned between P21–28 and no more than five adults were housed in the same cage. They were provided ample nesting materials and small houses within their home cages. Using the NIOSH Sound Level Meter app (Centers for Disease Control, Washington, DC, USA), ambient noise in the mouse colony room was estimated at approximately 70 dB with interior cage levels centered around 58 dB. Cages to be used for experiments were maintained in the center of the holding rack to avoid excess noise.
Mice were genotyped and then randomly assigned to their conditions using a deck of cards. For in-house genotyping, DNA was obtained from 2-mm tail samples digested overnight in Proteinase K (IBI Scientific, Dubuque, IA, USA) solution at 65 °C followed by phenol/chloroform extraction. IProof Taq (BioRad, Hercules, CA, USA) was used in conjunction with a published protocol and primer sequences [71]. For outsourced genotyping, DNA obtained from 2-mm tail samples was processed and verified by Transnetyx Automated PCR Genotyping Services (Transnetyx, Inc., Cordova, TN, USA).
Heterozygous/homozygous FoxO3 (FoxO3+/2) mice (FVB;129S6-Foxo3tm1.1Rdp; RRID:MGI:2668335) were obtained from the Mutant Mouse Regional Resource Center, at the University of California at Davis (Davis, CA, USA; stock no. 016132-UCD) [72]. Originally made on 129 Sv and bred three times to FVB/n, we bred the line a fourth time to FVB/nJ (RRID:IMSR JAX:001800) to obtain heterozygote mutant mice. Heterozygotes with different parents were bred together to obtain both knockout and Foxo3+/+ littermates. Foxo3−/−, Foxo3+/−, and Foxo3+/+ littermates, both male and female, between the ages of P60–90 were used in this study. Foxo3+/+ littermates are denoted as WT mice.
Noise exposure
Mice were exposed to an 8–16 kHz octave noise band at 105 dB for 30 min. This exposure has previously been shown to induce temporary threshold shifts in WT mice and loss of hearing in Foxo3−/− mice after 14 days when measured at five frequencies [20]. Awake P60–90 mice were placed in individual triangular wire mesh cages, 12 cm × 5 cm × 5 cm, in an asymmetric plywood box with a JBL2250HJ compression speaker and JBL2382A biradial horn mounted above. This apparatus was contained within a sound booth. The speaker was driven by a TDT RX6 multifunction processor and dedicated attenuator (Tucker Davis Technologies, Alachua, FL, USA). It was controlled with TDT RPvdsEx sound processing software (Tucker Davis Technologies). All noise exposure equipment was calibrated prior to use via the Quest Technologies portable sound level meter, Model 1900 (TSI Incorporated, Shoreview, MN, USA). The sound level was checked with an iPhone using the NIOSH SLM app and the iPhone’s internal microphone prior to each exposure. The iPhone was previously calibrated with the SoundMeter app software and a solid-state 94 dB source (provided with the Quest sound meter) in a sound booth. Within the sound exposure box, cages were placed in three specific locations at the back of the box where sound levels were highly consistent (+/− <0.5 dB). Mice that escaped or moved their cages from the starting position were excluded. Mice were directly monitored for the first minute of exposure via the plexiglass window on the chamber. A minority of FVB/nJ mice were susceptible to audiogenic seizures [73], normally within the first minute of exposure. These mice were immediately removed from the chamber and excluded. Due to known circadian cycle interactions with noise damage, mice were exposed to noise only between the hours of 9 a.m. and 1 p.m. [74]. Control mice were placed in the apparatus for the same length of time with the power off. After noise exposures, mice were monitored for proper health status and closely examined up until sacrifice. Mice exhibiting signs of pain or distress were euthanized early and excluded from further analysis.
In total, N = 26 noise exposures were performed on groups including all three genotypes of mice. Sixteen exposures were used to gather cochleae for the histology in Figs. 1–3 and 5. Six noise exposures generated cochleae for the RNA sequencing analysis in Fig. 4. Five noise exposures were performed for the LPA studies in Fig. 6. Since noise exposures were performed in open field, one cochlea per animal was used to generate data for each analysis.
ABR and distortion product otoacoustic emission
Initial hearing thresholds were tested between ages P50–55 with additional testing following schedules as described in the text. Testing occurred between the hours of 9 a.m. and 6 p.m. to avoid circadian rhythm effects. Mice were anesthetized with a single intraperitoneal (i.p.) injection of ketamine (80 mg/kg animal weight) and acepromazine (3 mg/kg animal weight) diluted in a sterile saline solution, to provide ~45 min of immobility. Additional anesthetic was administered as needed. A 10B+ OAE microphone was housed in an interaural probe and coupled with the speaker outputs. The probe was placed at the opening of each mouse’s left external auditory meatus. A Smart EP Universal Smart Box (Intelligent Hearing Systems, Miami, FL, USA) with an ED1 speaker (Tucker Davis Technologies) were housed within an anechoic chamber and used for closed field auditory testing. The Quest Technologies portable sound level meter, Model 1900, was used to calibrate the apparatus less than one month prior to beginning each set of experiments.
The ABR consisted of 50-μs click stimuli followed by 1-ms tone pips presented at five frequencies (8, 12, 16, 24, and 32 kHz). Stimuli amplitudes decreased in 5 dB steps from 75 dB sound pressure level to 15–25 dB. The averages of 512 sweeps were recorded for each frequency and amplitude. Three sterilized fine subdermal electrodes were used to record electrical responses (Grass): one inserted at the vertex and one inserted beneath each pinna. Responses were rejected if their peak to trough amplitude was greater than 31 µV at any time between 1.3 and 12.5 ms after stimulus presentation. Well-anesthetized mice typically had a 5–30% rejection rate. ABR thresholds at each of the five frequencies were determined by the last visible trace of wave I (dB). If no waveform was observed, “80 dB” was designated as the ceiling threshold as it can be considered reflective of severe hearing loss and higher intensities might produce acoustic distortions [75, 76].
DPOAEs were measured using the amplitude of evoked otoacoustic emissions to simultaneous pure tones of frequencies f1 and f2, where f1/f2 = 1.2 and the f1 level is 10 dB above f2. Beginning with f1 at 20 dB and ending at 65 dB, 32 sweeps were made in 5 dB steps. The DPOAE threshold was calculated for 3 dB emission. As a second calibration, measurements were collected from a dead mouse and L2 amplitudes with signals above threshold were excluded.
Anesthetized mice were isolated in recovery cages until they woke up. Their arousal levels were monitored, and mice were returned to their home cages after they regained consciousness. The researchers scoring ABRs and DPOAEs were blinded to genotype, condition, and time point.
Tissue preparation for immunostaining
Cochleae were dissected from euthanized mice at designated time points. The stapes were removed, and a small incision was made at the apical tips for proper fluid exchange during immersion fixation with 4% paraformaldehyde (PFA)/1× phosphate-buffered saline (PBS). Tissues were transferred to decalcifying 0.1 M ethylenediaminetetraacetic acid (EDTA) at 4 °C on a rocker for 3 days. For cryosectioning, tissues were submerged in 30% sucrose/1× PBS overnight, embedded in optimal cutting temperature compound (OCT), and frozen with liquid nitrogen. These tissues were sectioned at 20 μm with the 12 and 24 kHz regions visible. The 12 and 24 kHz regions were selected to analyze changes within the noise band (8–16 kHz) and basal to it.
For whole-mount preparations, cochleae were microdissected into three turns (apical, middle, and basal) as previously described [77]. These pieces were frequency mapped using the ImageJ 64 (NIH) plug-in from Massachusetts Eye and Ear Infirmary, and immunostained for quantification and spatiotemporal analysis. Apical IHCs have high levels of OCM [78] and can crowd areas of OCM + OHCs in the apical hook region (<4 kHz), making differentiation difficult. For our cochleograms, we included cell counts from 4 to 86 kHz (0–100% distance from the apex) [79].
Primary antibodies
Primary antibodies are listed in Supplementary Table 1. All antibodies were initially tested using antigen and non-antigen retrieval via freezing and boiling to ensure adequate cellular penetrance and stability. Non-primary and non-secondary antibody control tissues were imaged for autofluorescence levels. Due to a manufacturer’s discontinuation, we utilized two OCM antibodies in this study. Both antibodies were compared with and without antigen retrieval steps on control tissues. No difference in fluorescent antigen binding was noted.
Immunohistochemistry
For immunohistochemical staining on cryosections, sections were washed with 0.5% Tween20/Tris-buffered saline (TTBS), pH 7.4, and then blocked for 1 h at room temperature in 5% normal donkey serum (Jackson ImmunoResearch, West Grove, PA, USA)/0.5% TTBS. For anti-HSP70 immunostaining, antigen retrieval was performed prior to these steps by boiling in 10 mM citric acid, pH 6, for 15 min at 20% microwave power. Antibody incubations were performed overnight at 4 °C in block solution. A complete list of primary antibodies is provided in Supplementary Table 1. Sections were washed in 0.5% TTBS at room temperature prior to secondary antibody incubation overnight at 4 °C in the dark. Alexafluor-conjugated secondary antibodies included 488, 594, and 647 (Thermo Fisher Sci, Waltham, MA, USA) and were diluted at 1:500 each. 4′6-diamidino-2-phenylindole (DAPI) diluted at 1:10,000 was added during the secondary antibody incubation. Sections were washed in 0.5% TTBS and mounted in ProLong Gold Antifade (Thermo Fisher Scientific) on frosted plus microscope slides (Thermo Fisher Scientific).
For whole-mount preparations, tissues were collected in 300 μL dPBS in Eppendorf tubes, flash-frozen with liquid nitrogen, thawed on ice, and transferred to a 40-well plate. Tissues were washed in 0.5% TTBS, blocked for 1 h at room temperature in 5% normal donkey serum/0.5% TTBS, and incubated in 200 μL of primary antibody overnight at 4 °C. Tissues were washed in 0.5% TTBS at room temperature and incubated in 200 μL of secondary antibody solution overnight in the dark at 4 °C. Tissues were washed with 0.5% TTBS prior to mounting in ProLong Gold Antifade between two 50 mm coverslips (Thermo Fisher Scientific).
Confocal and multiphoton microscopy and image processing for figures
Sections were imaged using an Olympus FV1000 (Olympus, Tokyo, Japan) and ZEISS LSM 510 Meta (Carl Zeiss AG, Oberkochen, Germany) laser scanning confocal microscopes. Whole mounts were imaged using the confocal microscopes and Olympus Fluoview FVMPE-RS multiphoton imaging system. All images presented were collected as z-stack projections using one assigned microscopy device for an appropriate comparison. Stitching was performed with Fluoview for multiphoton images or XuvStitch (XuvTools) for confocal images. Composite images of whole mounts were assembled in GIMP 2.10.12 using the layering function to match the frequency regions previously mapped in ImageJ 64 (NIH) after microdissection. For cochleograms, 100 μm bins were measured along the length of the mapped cochlea on the rows of pillar cells. OCM, MYO7a, and Cytochrome-C were used to denote the presence of OHCs and IHCs. Any areas of the cochlea with post-mortem damage to the sensory epithelium were omitted from the analysis. These regions of the scatter plots are empty. In regions with intact sensory epithelium and high OHC loss, an average OHC count determined from the cochlea’s low-frequency regions was used as the denominator [79]. For analysis of the oxidative stress response, normalized fluorescence for sections was performed in ImageJ and relative expression levels were categorized as undetected, low, high, and cells lost. Standardization of counts involved analyzing the same number of cells per section (1 IHC, 1 cluster of IHC synapses and neurites, 3 OHCs, 3 clusters of OHC synapses and neurites, 2 pillar cells, and 3 DCs) before calculating total percentages of those surveyed. For analysis of Hsp70 levels in OHCs and DCs, images were opened in ImageJ, LUT was set to 0–75, and the corrected total cell fluorescence (CTCF) was calculated using the formula CTCF = Integrated Intensity − (area of selected cell × mean fluorescence of background readings).
3-D Reconstructions of OHCs and nuclei
To assess the structural integrity of OHCs and their nuclei following noise damage, 12 and 24 kHz regions from the previously mapped cochleograms were imaged at ×150 on the Olympus FV1000 (n = 3 per genotype/condition/frequency). These frequencies were selected to match the results in cryosection. All regions were standardized in the x, y, and z planes to include the expected three rows of OHCs. Images were converted (.oif to.ims) and imported into Imaris 9.3 Image Visualization & Analysis Software (Oxford Instruments, Abingdon, Oxfordshire, UK) for 3-D reconstruction. All sensory epithelial sections were oriented in the same direction and any remaining tectorial membrane was cropped out of the visualization frame. DAPI (nuclei), OCM (OHCs), and cytochrome-C (OHCs) isosurfaces were appropriately thresholded and constructed. Despite individual isolation attempts, thresholding was incapable of separating all SC nuclei proximate to those of the OHCs. The resulting images have SC nuclei designated as “SC” unless otherwise stated in the figure legend. Total OHC numbers were graphed for cells expressing each fluorescent marker.
RNA sequencing
At P60, WT and Foxo3−/− mice were euthanized under baseline conditions, at 4 h post-noise (HPN), or 24 HPN (n = 6 per genotype/condition). Their cochleae were extracted and processed for RNA sequencing. The 4 HPN time point was selected over 0.5 HPN as it reflected the two-step induction of FOXO3 by AMPK to be used for transcription in glutamate-exposed neurons [19]. We previously observed FOXO3 be responsive to noise [18] via nuclear translocation in SGNs and predicted that this time point would reflect a decrease in FOXO3 target gene transcription. RNA was isolated and purified using the RNeasy Plus Mini Kit (QIAGEN, Hilden, Germany). A 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA) was used to assess quality. The minimum RNA integrity number (RIN) for the group was 8.3 and the average was 9.1. Samples were processed at the University of Geneva, Switzerland. RNA was sequenced on an Illumina HiSeq4000 (Illumina, San Diego, CA, USA). The reads were mapped with TopHat v.2 software (CCB, Johns Hopkins University, Baltimore, MD, USA) to the UCSC mm10 mouse reference (Genomics Institute, University of California Santa Cruz, Santa Cruz, CA, USA). Biological quality control and summarization were performed with RSeQC-2.3.3 and PicardTools1.92. The table of counts was prepared with HTSeq v0.6p1. Differential expression analysis used the statistical analysis R/Bioconductor package EdgeR v. 3.10.5. Counts were normalized according to the library size and filtered. Gene expression clustering was performed using the R/Bioconductor Mfuzz v.2.28 package. These genes were composed in Supplementary Table 3 and their listed functions were adapted from the GeneCards human gene database (Weizman Institute of Science, LifeMap Sciences). Gene ontology analyses on additional gene sets were performed in Ingenuity Pathway Analysis (QIAGEN) and GSEA. Genes expressed in OHCs were identified through comparison with the gEAR portal (He dataset).
Real-time quantitative PCR (RT-qPCR)
At P60, WT and Foxo3−/− mice were euthanized under baseline conditions or at 4 HPN (n = 4–5 per genotype/condition). Cochleae were isolated and total RNA was extracted using the RNeasy Plus Mini Kit (QIAGEN). Total RNA concentration and purity were determined with the NanoDrop 1000 spectrophotometer (NanoDrop, Wilmington, DE, USA). A qScript cDNA Synthesis kit (QuantaBio, Beverly, MA, USA) and T100 Thermal Cycler (Bio-Rad Laboratories, Inc., Hercules, CA, USA) were used to generate the cDNA. RT-qPCR was performed with SYBR Green PCR mix on the Bio-Rad CFX Connect Real-Time PCR System (Bio-Rad). The reaction protocol used was 95 °C for 30 s, followed by 50 cycles of 95 °C for 15 s, 60 °C for 60 s, and 4 °C to hold. Comparative ΔΔCt was performed to determine relative mRNA expression, where the relative abundance of each gene was first internally normalized to the geometric mean Ct for the reference genes Hprt and GusB [80]. The primers are listed in Supplementary Table 2.
Administration of LPA to mice
The selected lysophosphatidic acid (LPA) concentration was considered sufficient for OHC preservation following noise exposure based on previous findings in adult CBA/J mice [52]. LPA (30 mg/kg, Oleoyl-l-α-lysophosphatidic acid sodium salt, Sigma L7260-25MG) was prepared as 0.05 mg/µL stock with CMF-PBS and diluted in 0.9% sterile saline at a final 5 mg/mL concentration. One set of WT (n = 3) and Foxo3−/− mice (n = 5) received a total of three i.p. injections of LPA and were sacrificed at 4 HPN. LPA was injected 24 h and immediately prior to noise exposure, and immediately after noise exposure. Sham mice (WT n = 3, Foxo3−/− n = 4) received three 0.9% sterile saline injections at the same time points. The second set of mice (n = 3 per genotype) was injected four times with LPA and sacrificed at 14 DPN following hearing tests. LPA was administered 24 h and immediately prior to noise, and immediately and 24 h after noise exposure. Sham mice (n = 3 per genotype) received four saline injections at the same time points. No animals displayed ill side effects following injections and all were included in tissue harvesting.
Statistical analyses
Sample sizes were calculated for sufficient power analysis and at least three cochleae of each genotype from individual mice were analyzed for all experimental conditions (80% power). The number of replicates is indicated within each figure legend. The researcher was blinded as to the genotype and condition for the quantification of OHC counts, immunohistochemistry fluorescence labeling, and hearing test analyses. OHC counts in cochleograms were non-normally distributed. Thus, these numbers were analyzed with appropriate rank-sum testing. Unpaired and paired Student’s t-tests or Mann–Whitney U rank-sum tests were used to compare differences across two groups. Two-way ANOVA or Kruskal–Wallis rank-sum tests were used to compare differences across more than two groups. Bonferroni, Tukey’s, Šidák’s, and Welch’s post-tests were used for adjustments. A p value < 0.05 was considered statistically significant. Standard error means (s.e.m.) and variance were calculated as appropriate per data set. Statistics were performed in GraphPad Prism 9.0.0.
Supplementary material
Supplementary information is available at Cell Death & Disease’s website.
Data availability
The datasets used and/or analyzed during the current study may be found through the University of Rochester Research Repository. The archive link is http://hdl.handle.net/1802/36374.
References
Carroll YI, Eichwald J, Scinicariello F, Hoffman HJ, Deitchman S, Radke MS, et al. Vital signs: noise-induced hearing loss among adults—United States 2011–2012. Morb Mortal Wkly Rep 2017;66:139–44.
White PM. Genetic susceptibility to hearing loss from noise exposure. Hear J 2019;72:8–9.
Guo H, Ding E, Bai Y, Zhang H, Shen H, Wang J, et al. Association of genetic variations in FOXO3 gene with susceptibility to noise induced hearing loss in a Chinese population. PLoS ONE 2017;12:e0189186.
Deng A, Ma L, Zhou X, Wang X, Wang S, Chen X. FoxO3 transcription factor promotes autophagy after oxidative stress injury in HT22 cells. Can J Physiol Pharmacol. 2020;99:1–8.
Tsuji T, Maeda Y, Kita K, Murakami K, Saya H, Takemura H, et al. FOXO3 is a latent tumor suppressor for FOXO3-positive and cytoplasmic-type gastric cancer cells. Oncogene 2021;40:3072–86.
Liu S, Zhang X, Sun M, Xu T, Wang A. FoxO3a plays a key role in the protective effects of pomegranate peel extract against amikacin-induced ototoxicity. Int J Mol Med 2017;40:175–81.
Zhang DY, Zhang CF, Fu BC, Sun L, Wang XQ, Chen W, et al. Sirtuin3 protects aged human mesenchymal stem cells against oxidative stress and enhances efficacy of cell therapy for ischaemic heart diseases. J Cell Mol Med 2018;22:5504–17.
Zhang J, Sun H, Salvi R, Ding D. Paraquat initially damages cochlear support cells leading to anoikis-like hair cell death. Hear Res 2018;364:129–41.
Lee N, Tilija Pun N, Jang WJ, Bae JW, Jeong CH. Pitavastatin induces apoptosis in oral squamous cell carcinoma through activation of FOXO3a. J Cell Mol Med 2020;24:7055–66.
Lim HM, Lee J, Nam MJ, Park SH. Acetylshikonin induces apoptosis in human colorectal cancer HCT-15 and LoVo cells via nuclear translocation of FOXO3 and ROS level elevation. Oxid Med Cell Longev 2021;2021:6647107.
Wang Y, Xie Z, Lu H. Significance of halofuginone in esophageal squamous carcinoma cell apoptosis through HIF-1alpha-FOXO3a pathway. Life Sci.2020;257:118104.
Chen R, Morris BJ, Donlon TA, Masaki KH, Willcox DC, Davy PMC, et al. FOXO3 longevity genotype mitigates the increased mortality risk in men with a cardiometabolic disease. Aging (Albany, NY) 2020;12:23509–24.
Davy PMC, Willcox DC, Shimabukuro M, Donlon TA, Torigoe T, Suzuki M, et al. Minimal shortening of leukocyte telomere length across age groups in a cross-sectional study for carriers of a longevity-associated FOXO3 allele. J Gerontol Ser A 2018;73:1448–52.
Flachsbart F, Caliebe A, Kleindorp R, Blanche H, von Eller-Eberstein H, KNikolaus S, et al. Association of FOXO3A variation with human longevity confirmed in German centenarians. PNAS 2009;106:2700–5.
Viatte S, Lee JC, Fu B, Espeli M, Lunt M, De Wolf JN, et al. Association between genetic variation in FOXO3 and reductions in inflammation and disease activity in inflammatory polyarthritis. Arthritis Rheumatol 2016;68:2629–36.
Xu S, Ma Y, Chen Y, Pan F. Role of forkhead box O3a transcription factor in autoimmune diseases. Int Immunopharmacol 2021;92:107338.
Zurawek M, Dzikiewicz-Krawczyk A, Izykowska K, Ziolkowska-Suchanek I, Skowronska B, Czainska M, et al. miR-487a-3p upregulated in type 1 diabetes targets CTLA4 and FOXO3. Diabetes Res Clin Pract 2018;142:146–53.
Gilels F, Paquette ST, Zhang J, Rahman I, White PM. Mutation of Foxo3 causes adult onset auditory neuropathy and alters cochlear synapse architecture in mice. J Neurosci. 2013;33:18409–24.
Davila D, Connolly NM, Bonner H, Weisova P, Dussmann H, Concannon CG, et al. Two-step activation of FOXO3 by AMPK generates a coherent feed-forward loop determining excitotoxic cell fate. Cell Death Differ. 2012;19:1677–88.
Gilels F, Paquette ST, Beaulac HJ, Bullen A, White PM. Severe hearing loss and outer hair cell death in homozygous Foxo3 knockout mice after moderate noise exposure. Sci Rep 2017;7:1054.
Yalcin S, Zhang X, Luciano JP, Mungamuri SK, Marinkovic D, Vercherat C, et al. Foxo3 is essential for the regulation of ataxia telangiectasia mutated and oxidative stress-mediated homeostasis of hematopoietic stem cells. J Biol Chem 2008;283:25692–705.
Marinkovic D, Zhang X, Yalcin S, Luciano JP, Brugnara C, Huber T, et al. Foxo3 is required for the regulation of oxidative stress in erythropoiesis. J Clin Investig 2007;117:2133–44.
Fetoni AR, Paciello F, Rolesi R, Paludetti G, Troiani D. Targeting dysregulation of redox homeostasis in noise-induced hearing loss: oxidative stress and ROS signaling. Free Radic Biol Med 2019;135:46–59.
Ohinata Y, Miller JM, Altschuler RA, Schacht J. Intense noise induces formation of vasoactive lipid peroxidation products in the cochlea. Brain Res 2000;878:163–73.
Ohlemiller KK, Wright JS, Dugan LL. Early elevation of cochlear reactive oxygen species following noise exposure. Audiol Neuro-Otol 1999;4:229–36.
Wu F, Xiong H, Sha S. Noise-induced loss of sensory hair cells is mediated by ROS/AMPKalpha pathway. Redox Biol 2020;29:101406.
Wu PZ, O’Malley JT, de Gruttola V, Liberman MC. Primary neural degeneration in noise-exposed human cochleas: correlations with outer hair cell loss and word-discrimination scores. J Neurosci 2021;41:4439–47.
Bohne BA, Harding GW, Lee SC. Death pathways in noise-damaged outer hair cells. Hear Res 2007;223:61–70.
Bohne BA, Kimlinger M, Harding GW. Time course of organ of Corti degeneration after noise exposure. Hear Res 2017;344:158–69.
Hu BH, Henderson D, Nicotera TM. Extremely rapid induction of outer hair cell apoptosis in the chinchilla cochlea following exposure to impulse noise. Hear Res 2006;211:16–25.
Nicotera TM, Hu BH, Henderson D. The caspase pathway in noise-induced apoptosis of the chinchilla cochlea. J Assoc Res Otolaryngol 2003;4:466–77.
Yang WP, Henderson D, Hu BH, Nicotera TM. Quantitative analysis of apoptotic and necrotic outer hair cells after exposure to different levels of continuous noise. Hear Res 2004;196:69–76.
Tanaka C, Chen GD, Hu BH, Chi LH, Li M, Zheng G, et al. The effects of acoustic environment after traumatic noise exposure on hearing and outer hair cells. Hear Res 2009;250:10–8.
Engel J, Braig C, Ruttiger L, Kuhn S, Zimmermann U, Blin N, et al. Two classes of outer hair cells along the tonotopic axis of the cochlea. Neuroscience 2006;143:837–49.
Fettiplace R, Nam JH. Tonotopy in calcium homeostasis and vulnerability of cochlear hair cells. Hear Res 2019;376:11–21.
Sha SH, Taylor R, Forge A, Schacht J. Differential vulnerability of basal and apical hair cells is based on intrinsic susceptibility to free radicals. Hear Res2001;155:1–8.
Kros CJ, Marcotti W, van Netten SM, Self TJ, Libby RT, Brown SD, et al. Reduced climbing and increased slipping adaptation in cochlear hair cells of mice with Myo7a mutations. Nat Neurosci 2002;5:41–7.
Climer LK, Cox AM, Reynolds TJ, Simmons DD. Oncomodulin: the enigmatic parvalbumin protein. Front Mol Neurosci 2019;12:235.
Tong B, Hornak AJ, Maison SF, Ohlemiller KK, Liberman MC, Simmons DD. Oncomodulin, an EF-hand Ca2+ buffer, is critical for maintaining cochlear function in mice. J Neurosci 2016;36:1631–5.
Wang J, Ruel J, Ladrech S, Bonny C, van de Water TR, Puel JL. Inhibition of the c-Jun N-terminal kinase-mediated mitochondrial cell death pathway restores auditory function in sound-exposed animals. Mol Pharmacol 2007;71:654–66.
Lee HY, Youn SW, Kim JY, Park KW, Hwang CI, Park WY, et al. FOXO3a turns the tumor necrosis factor receptor signaling towards apoptosis through reciprocal regulation of c-Jun N-terminal kinase and NF-kappaB. Arterioscler Thromb Vasc Biol 2008;28:112–20.
Li Z, Zhao J, Tikhanovich I, Kuravi S, Helzberg J, Dorko K, et al. Serine 574 phosphorylation alters transcriptional programming of FOXO3 by selectively enhancing apoptotic gene expression. Cell Death Differ 2016;23:583–95.
Chen FQ, Hill K, Guan YJ, Schacht J, Sha SH. Activation of apoptotic pathways in the absence of cell death in an inner-ear immortomouse cell line. Hear Res 2012;284:33–41.
Han W, Shi X, Nuttall AL. AIF and endoG translocation in noise exposure induced hair cell death. Hear Res 2006;211:85–95.
Jiang H, Sha SH, Forge A, Schacht J. Caspase-independent pathways of hair cell death induced by kanamycin in vivo. Cell Death Differ 2006;13:20–30.
Jiang HY, Yang Y, Zhang YY, Xie Z, Zhao XY, Sun Y, et al. The dual role of poly(ADP-ribose) polymerase-1 in modulating parthanatos and autophagy under oxidative stress in rat cochlear marginal cells of the stria vascularis. Redox Biol 2018;14:361–70.
David KK, Andrabi SA, Dawson TM, Dawson VL. Parthanatos, a messenger of death. Front Biosci (Landmark Ed.) 2009;14:1116–28.
Son YO, Heo JS, Kim TG, Jeon YM, Kim JG, Lee JC. Over-expression of JunB inhibits mitochondrial stress and cytotoxicity in human lymphoma cells exposed to chronic oxidative stress. BMB Rep 2010;43:57–61.
Sunayama J, Tsuruta F, Masuyama N, Gotoh Y. JNK antagonizes Akt-mediated survival signals by phosphorylating 14-3-3. J Cell Biol 2005;170:295–304.
Anttonen T, Herranen A, Virkkala J, Kirjavainen A, Elomaa P, Laos M, et al. c-Jun N-terminal phosphorylation: biomarker for cellular stress rather than cell death in the injured cochlea. ENEURO. 2016;3:0047–16.
Wang F, Marshall CB, Yamamoto K, Li GY, Plevin MJ, You H, et al. Biochemical and structural characterization of an intramolecular interaction in FOXO3a and its binding with p53. J Mol Biol 2008;384:590–603.
Han Y, Wang X, Chen J, Sha SH. Noise-induced cochlear F-actin depolymerization is mediated via ROCK2/p-ERM signaling. J Neurochem 2015;133:617–28.
Naka K, Ochiai R, Matsubara E, Kondo C, Yang KM, Hoshii T, et al. The lysophospholipase D enzyme Gdpd3 is required to maintain chronic myelogenous leukaemia stem cells. Nat Commun 2020;11:4681.
Cho Y, Gong TW, Kanicki A, Altschuler RA, Lomax MI. Noise overstimulation induces immediate early genes in the rat cochlea. Brain Res Mol Brain Res 2004;130:134–48.
Honkura Y, Matsuo H, Murakami S, Sakiyama M, Mizutari K, Shiotani A, et al. NRF2 is a key target for prevention of noise-induced hearing loss by reducing oxidative damage of cochlea. Sci Rep 2016;6:19329.
Heflick CI. Effects of oncomodulin on cytoskeletal actin dynamics. Baylor University; 2019.
Vanden Berghe T, Kaiser WJ, Bertrand MJ, Vandenabeele P. Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol Cell Oncol 2015;2:e975093.
Morrill S, He DZZ. Apoptosis in inner ear sensory hair cells. J Otol 2017;12:151–64.
Olmos Y, Valle I, Borniquel S, Tierrez A, Soria E, Lamas S, et al. Mutual dependence of Foxo3a and PGC-1alpha in the induction of oxidative stress genes. J Biol Chem 2009;284:14476–84.
Salcher S, Hagenbuchner J, Geiger K, Seiter MA, Rainer J, Kofler R, et al. C10ORF10/DEPP, a transcriptional target of FOXO3, regulates ROS-sensitivity in human neuroblastoma. Mol Cancer 2014;13:224.
Yeo H, Lyssiotis CA, Zhang Y, Ying H, Asara JM, Cantley LC, et al. FoxO3 coordinates metabolic pathways to maintain redox balance in neural stem cells. EMBO J. 2013;32:2589–602.
Hagenbuchner J, Kuznetsov A, Hermann M, Hausott B, Obexer P, Ausserlechner MJ. FOXO3-induced reactive oxygen species are regulated by BCL2L11 (Bim) and SESN3. J Cell Sci 2012;125:1191–203.
Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 2007;128:325–39.
Essafi A, Fernandez de Mattos S, Hassen YA, Soeiro I, Mufti GJ, Thomas NS, et al. Direct transcriptional regulation of Bim by FoxO3a mediates STI571-induced apoptosis in Bcr-Abl-expressing cells. Oncogene 2005;24:2317–29.
Breglio AM, May LA, Barzik M, Welsh NC, Francis SP, Costain TQ, et al. Exosomes mediate sensory hair cell protection in the inner ear. J Clin Investig 2020;130:2657–72.
Fairfield DA, Lomax MI, Dootz GA, Chen S, Galecki AT, Benjamin IJ, et al. Heat shock factor 1-deficient mice exhibit decreased recovery of hearing following noise overstimulation. J Neurosci Res 2005;81:589–96.
May LA, Kramarenko II, Brandon CS, Voelkel-Johnson C, Roy S, Truong K, et al. Inner ear supporting cells protect hair cells by secreting HSP70. J Clin Investig 2013;123:3577–87.
Sugahara K, Inouye S, Izu H, Katoh Y, Katsuki K, Takemoto T, et al. Heat shock transcription factor HSF1 is required for survival of sensory hair cells against acoustic overexposure. Hear Res 2003;182:88–96.
Takumida M, Anniko M. Heat shock protein 70 delays gentamicin-induced vestibular hair cell death. Acta Otolaryngol 2005;125:23–8.
Taleb M, Brandon CS, Lee FS, Lomax MI, Dillmann WH, Cunningham LL. Hsp70 inhibits aminoglycoside-induced hair cell death and is necessary for the protective effect of heat shock. J Assoc Res Otolaryngol 2008;9:277–89.
Castrillon DH, Miao L, Kollipara R, Horner JW, DePinho RA. Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science 2003;301:215–8.
Hwang JW, Rajendrasozhan S, Yao H, Chung S, Sundar IK, Huyck HL, et al. FOXO3 deficiency leads to increased susceptibility to cigarette smoke-induced inflammation, airspace enlargement, and chronic obstructive pulmonary disease. J Immunol 2011;187:987–98.
Goelz MF, Mahler J, Harry J, Myers P, Clark J, Thigpen JE, et al. Neuropathologic findings associated with seizures in FVB mice. Lab Anim Sci 1998;48:34–7.
Meltser I, Cederroth CR, Basinou V, Savelyev S, Lundkvist GS, Canlon B. TrkB-mediated protection against circadian sensitivity to noise trauma in the murine cochlea. Curr Biol 2014;24:658–63.
Bisgaard N, Vlaming MS, Dahlquist M. Standard audiograms for the IEC 60118-15 measurement procedure. Trends Amplif 2010;14:113–20.
Willott JF. Measurement of the auditory brainstem response (ABR) to study auditory sensitivity in mice. In: Crawley JN, et al. Current protocols in neuroscience/editorial board. Chapter 8:Unit8 21B, John Wiley & Sons, Inc. Hoboken, NJ, USA 2006.
Montgomery SC, Cox BC. Whole mount dissection and immunofluorescence of the adult mouse cochlea. J Vis Exp. 2016:53561.
Tang F, Chen X, Jia L, Li H, Li J, Yuan W. Differential gene expression patterns between apical and basal inner hair cells revealed by RNA-Seq. Front Mol Neurosci 2019;12:332.
Viberg A, Canlon B. The guide to plotting a cochleogram. Hear Res 2004;197:1–10.
Park JS, Cederroth CR, Basinou V, Meltser I, Lundkvist G, Canlon B. Identification of a circadian clock in the inferior colliculus and its dysregulation by noise exposure. J Neurosci 2016;36:5509–19.
Acknowledgements
We thank the University of Geneva Genomics Core for our RNA sequencing; Dr. Helene McMurray for her assistance in GO analysis; Dr. Paivi Jordan for microscopy training; technical director Dr. V. Kaye Thomas and Julie Zhang of the URMC Center for Advanced Light Microscopy and Nanoscopy, for their assistance in confocal imaging; technical director Dr. Yurong Gao of the URMC Multiphoton and Analytical Imaging Center for help with multiphoton imaging; Drs. Richard Libby, Lin Gan, Ruchira Singh, and Ian Dickerson for equipment access; Drs. Joseph C. Holt, Robert Freeman, Dirk Bohmann, and Amy Kiernan for advice on experimental planning; and Dr. Dorota Piekna-Przybylska, Dr. Hitomi Sakano, and Daxiang Na for editing.
Funding
This work was funded by the National Institute of Health R01 DC014261.
Author information
Authors and Affiliations
Contributions
HJB and PMW performed study concept, design, and analysis of RNA sequencing results; HJB developed the methodology, provided acquisition, analysis and interpretation of data, statistical analysis, and writing, review, and revision of the paper; FG developed the methodology and processed the tissue for the RNA sequencing experiments; JZ, SJ, and PMW scored the hearing tests; JZ provided valuable feedback and assistance on performing experiments; PMW provided technical and material support. All authors read and approved the final paper.
Corresponding author
Ethics declarations
Ethics approval
All experiments were performed under protocol number 2010-011: PI Patricia White, in compliance with the U.S. Department of Health and Human Services and were reviewed by the University of Rochester’s Committee on Animal Resources.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by B. Zhivotovsky
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Beaulac, H.J., Gilels, F., Zhang, J. et al. Primed to die: an investigation of the genetic mechanisms underlying noise-induced hearing loss and cochlear damage in homozygous Foxo3-knockout mice. Cell Death Dis 12, 682 (2021). https://doi.org/10.1038/s41419-021-03972-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-021-03972-6
This article is cited by
-
An update on Glycerophosphodiester Phosphodiesterases; From Bacteria to Human
The Protein Journal (2024)
-
GDE7 produces cyclic phosphatidic acid in the ER lumen functioning as a lysophospholipid mediator
Communications Biology (2023)
