
Abstract
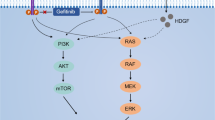
The epidermal growth factor receptor (EGFR) pathway and Hippo signaling play an important role in the carcinogenesis of hepatocellular carcinoma (HCC). However, the crosstalk between these two pathways and its implications in targeted therapy remains unclear. We found that the activated EGFR signaling could bypass RhoA to promote the expression of YAP(Yes-associated protein), the core effector of the Hippo signaling, and its downstream target Cyr61. Further studies indicated that EGFR signaling mainly acted through the PI3K-PDK1 (Phosphoinositide 3-kinase-Phosphoinositide-dependent kinase-1) pathway to activate YAP, but not the AKT and MAPK pathways. While YAP knockdown hardly affected the EGFR signaling. In addition, EGF could promote the proliferation of HCC cells in a YAP-independent manner. Combined targeting of YAP and EGFR signaling by simvastatin and the EGFR signaling inhibitors, including the EGFR tyrosine kinase inhibitor (TKI) gefitinib, the RAF inhibitor sorafenib and the MEK inhibitor trametinib, presented strong synergistic cytotoxicities in HCC cells. Therefore, the EGFR-PI3K-PDK1 pathway could activate the YAP signaling, and the activated EGFR signaling could promote the HCC cell growth in a YAP-independent manner. Combined use of FDA-approved inhibitors to simultaneously target YAP and EGFR signaling presented several promising therapeutic approaches for HCC treatment.
Similar content being viewed by others

Introduction
Hepatocellular carcinoma (HCC), a major malignancy of the liver, is the fifth most common cancer and the third leading cause of cancer-related mortality worldwide. HCC has a poor prognosis, only 15–20% of patients could survive more than 5 years1. Although some targeted therapies, such as sorafenib, can improve the clinical outcome, their effects are limited2,3,4. So there is a great need for more complete understanding of the molecular mechanisms involved in HCC development, which could help us design some new or improve therapeutic strategies for this illness.
Hippo pathway is an evolutionarily conserved mechanism that restricts organ size from drosophila to mammals. The core upstream components of this pathway comprise several tumor suppressors, including Mst1/2, Sav1/WW45, Lats1/2, and Mob1, which act in a kinase cascade that culminate in the phosphorylation and inactivation of YAP/TAZ (transcriptional co-activator with PDZ-binding motif). YAP/TAZ could act as the transcriptional co-activators to promote the expression of their target genes involved in proliferation and survival. Many studies have implicated that Hippo signaling pathway played a vital role in the tumorigenesis of HCC5,6. Conditional over-expression of YAP in transgenic mice or the liver-specific knockout of Mst1/2 or Sav1 could lead to expanded liver size and ultimately induce HCC, and these are the direct evidences for the importance of the Hippo pathway in regulating organ size and liver tumorigenesis7,8,9,10,11. Moreover, many clinical studies have illustrated that over-expression and nuclear accumulation of YAP could also act as an independent prognostic marker for the overall survival and disease-free survival in HCC patients, as well as in several other tumor types12,13,14. RhoA, a small G protein that belongs to the Rho family of Ras GTPase superfamily, plays a vital role in the regulation of many biological activities including actin organization, cell motility, proliferation, apoptosis and development15. Several recent studies indicated that RhoA participated in the activation of YAP through inducing stress fiber formation, and the G-protein-coupled receptor (GPCR) signaling could act through RhoA to regulate the Hippo-YAP pathway16,17.
Epidermal growth factor receptor (EGFR) signaling also plays an important role in hepatocellular carcinogenesis3,18. Several studies have indicated that EGFR was frequently over-expressed and positively correlated with early tumor recurrence in HCC. So anti-EGFR might be a promising therapeutic strategies in HCC19,20,21. Intriguingly, anti-EGFR therapy has achieved a huge success in both of lung cancer and colorectal cancer. However, it failed in HCC and the mechanisms behind remained elusive3,18. Recent reports indicated that the crosstalk between EGFR signaling and Hippo pathway was involved in the carcinogenesis in several other cancers13,22,23,24. However, the crosstalk between these two pathways and its implications in targeted therapy remain unclear in HCC25.
Here, we found that the EGF/EGFR signaling could bypass RhoA, a canonical YAP regulator, to activate YAP signaling in HCC cells26. Further investigation indicated that EGF mainly acted through PI3K/PDK1 pathway, but not the AKT and the MAPK pathway, to regulate YAP signaling. Our study also showed that EGF could promote cell proliferation in a YAP-independent manner. Importantly, our novel therapeutic strategies by simultaneously targeting EGFR signaling and YAP with combined use of the FDA-approved drugs demonstrated synergistic cytotoxicities in HCC cells.
Results
The expression of EGFR and YAP in human HCC cells
To better understand the role of EGFR and Hippo signaling in HCC, western blot (WB) was performed to examine the expression of EGFR and the core members of Hippo signaling, Wnt signaling and Rho in HCC cell lines (Fig. 1a, Suppl. Fig. 1). We verified that EGFR and YAP were simultaneously up-regulated in all the 4 cancer cell lines compared with the normal liver cell HL7702, and only the expression of YAP was positively correlated with EGFR in these cells. In contrast, the expression of other Hippo signaling effectors, including Mst1, Lats1, and TAZ, had no obvious correlation with EGFR (Fig. 1a). We then selected two EGFR high-expressed cells for further investigation. Our studies indicated that activated EGFR signaling could enhance the expression of Lats1 and YAP in HepG2 and SMMC7721 cells in a time-dependent manner (Fig. 1b, c). Our investigation of the effect of EGF on the expression of the core members of EGFR signaling indicated that EGF treatment could stimulate the phosphoralation of EGFR, ERK and Akt1, and enhance the expression of CyclinD1 in HepG2 and 7721 cells (Fig. 1b, c, Suppl. Figs. 2A, B). Further cytoplasmic and nuclear protein extraction assays (Fig. 1d) and immunofluorescence study (Fig. 1e,f) indicated that EGF stimulation could promote the expression and nuclear accumulation of YAP in these two HCC cells.

a WB was used to examine the expression of EGFR and the core effectors of Hippo signaling in HCC cells. b, c Time dependent effect of EGF on the expression YAP and Lats1 in HepG2 and SMMC 7721 cells. d The HepG2 and 7721 cells were plated in the 10 cm dish and then serum-free starved for one night. After 20 ng/ml EGF stimulated for 2 h, the cytoplasmic and nuclear proteins were extracted. The protein samples were subjected to immunobloting with the indicated antibodies. GAPDH and LaminB were as cytoplasm and nucleus loading control, respectively. e, f Immunofluorescence was used to detect the effect of 20 ng/ml EGF stimulation on the expression and localization of YAP in HepG2 and SMMC7721 cells
Activated EGFR signaling could enhance the expression of YAP in a RhoA-independent manner
Several recent studies have indicated that RhoA played a vital role in G protein coupled receptor (GPCR) mediated YAP activation26, while some reports showed that EGF could enhance the activity of RhoA27. Therefore, we tested whether EGFR signaling could act through RhoA to regulate YAP in HCC. The results revealed that EGF could up-regulate the activity and expression of total RhoA in the HepG2 and 7721 cell lines (Fig. 2a, b, Suppl. Fig. 2). Knockdown RhoA could partly inhibit the expression of YAP in HepG2 cells, and significantly reduce the expression of YAP in 7721 cells. However, EGF treatment could still stimulate the expression of YAP in the two HCC cells with RhoA knockdown (Fig. 2c, d). And the treatment had nearly no effect on its phosphorylation level (P-YAP/YAP) (Suppl. Fig. 3).

a, b Rhotekin pulldown assay was performed to examine the effect of 20 ng/ml EGF on the activity of RhoA in HepG2 and SMMC7721 cells. c, d After serum starving for one night, western blot were performed to detect the effect of 20 ng/ml EGF on the expressions of YAP in the cells transfected with SiRhoA in HepG2 and SMMC7721 cells. e, f CCK8 assays were used to detect the effect of RhoA knockdown combined with 50 ng/ml EGF stimulation for 48 h on the proliferation of the two HCC cell lines. g, h WB was used to examine the effect of combined treatment on the core effectors of the EGFR downstream signaling
Previous data suggested a critical role of RhoA in EGF-mediated carcinogenesis27, so we also investigated whether the EGFR signaling could act via RhoA to promote the malignant phenotype of HCC cells. The results indicated that knockdown RhoA could partly inhibit cell proliferation, while EGF treatment could still promote proliferation of the HCC cells with RhoA knockdown (Fig. 2e, f). Further mechanism studies revealed that RhoA knockdown had nearly no effect on the EGFR signaling, but could partly inhibit the expression of C-Myc and CyclinD1. EGF treatment could still activate the EGFR signaling and Cyr61, the main downstream target of YAP, and partly enhance the expression of C-Myc, CyclinD1 and Bcl-xl in HepG2 and 7721 cells (Fig. 2g, h). Together, these results suggested that the activated EGFR signaling could bypass RhoA to activate YAP and promote cell proliferation in HCC cells.
EGF mainly acts through the EGFR-PI3K-PDK1 pathway to regulate YAP in HCC cells
To explore the mechanism that involved in EGF induced YAP up-regulation, we used specific pharmacologic inhibitors to examine which pathway mainly mediated YAP activation in HCC cells. The results indicated that the inhibitors of PI3K(LY294002 and Wortmannin) significantly blocked EGF-mediated YAP over-expression as effectively as the EGFR inhibitor gefitinib. Inhibitors of the PI3K downstream effector PDK1 (GSK2334470 and BX-795) were also able to block YAP enhancement, while inhibitors of another major kinases, including AKT and MEK, had nearly no effect on the expression of YAP (Fig. 3a, d, Suppl. Fig. 4). The present data indicated that EGF mainly acted through the EGFR-PI3K-PDK1 pathway to regulate YAP in HCC cells.

a, b WB was used to detect the effect of EGF treatment for 4 h on the expression of YAP with the inhibitors of EGFR or its downsream members, including the EGFR inhibitor (10uM Gefitinib), PI3K inhibitor (10 uM LY294002, 5 uM Wortmannin), PDK1 inhibitor (10uM GSK2334470, 10 uM BX-795), pan-Akt inhibitor(10 uM MK-2206) and MEK inhibitor(10 uM Trametinib, 10 uM U0126) in the Si RhoA transfected HepG2 and SMMC7721 cells for 48 h in HepG2 and SMMC7721 cells. c, d CCK8 assays were used to detect the effect of YAP knockdown combined with 50 ng/ml EGF stimulation for 48 h on the proliferation of the two HCC cell lines. e, f WB was used to examine the effect of combined treatment on the core effectors of the EGFR downstream signaling. T-test was used to detect the difference. (*P < 0.05, **P < 0.01, ***P < 0.001)
Activated EGFR signaling promotes the proliferation of HCC cells in a YAP independent manner
Since YAP plays an important role in the carcinogenesis of HCC5, we examined whether activated EGFR signaling could act through YAP to regulate the malignant phenotype of HCC cells. YAP siRNA was used to knock-down YAP protein in HepG2 and 7721 cells, while non-targeting siRNA was used as a control. Western blot analysis demonstrated that YAP siRNA successfully reduced YAP protein level in the two HCC cells (Fig. 3c, f). Knockdown YAP could significantly inhibit cell proliferation compared with the non-targeting controls. However, EGF could still promote the proliferation of HCC cells with YAP knockdown (Fig. 3b, e). Further WB results showed that knockdown YAP could inhibit the expression of Cyr61, one of the YAP target genes, but had nearly no effect on the phoshorylation of EGFR and its downstream effector P-ERK and P-AKT. Knockdown YAP could reverse the EGF induced Cyr61 over-expression in HCC cells, indicating that EGF could act though YAP to regulate its downstream target genes of Cyr61. However, EGF treatment could still activate EGFR and its downstream effectors, AKT and ERK, and the expression of C-Myc, CyclinD1and Bcl-xl in HepG2 and SMMC7721 cells with YAP knockdown (Fig. 3c, f). In all, these data indicated that the activated EGFR signaling could bypass YAP to promote the proliferation of HCC cells.
Therefore, taking into consideration that both YAP and EGFR signaling play a vital role in the carcinogenesis of HCC, combined targeting the Hippo and EGFR signaling pathways might provide a novel therapeutic strategy for HCC treatment.
Anti-neoplastic potency of simvastatin in combination with the EGFR-TK inhibitor gefitinib
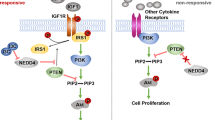
Several pharmacological inhibitors, including verteporfin and statins, have been reported to inhibit the function of YAP in tumorigenesis. Verteporfin could disrupt the YAP-TEAD interaction, however, it has been mainly used to eliminate abnormal blood vessels in the eye28. Statins, primarily used for primary and secondary prevention of cardiovascular diseases worldwide, could inhibit YAP through inhibiting RhoA activity29,30. Several large population-based cohort studies in Asian and Western populations have also shown that statins could reduce the risk of hepatocellular carcinoma31,32,33. So we used simvastatin as a YAP inhibitor in this study (Suppl. Figs. 5, 6).
To check the potency of simultaneous targeting EGFR signaling and Hippo pathway for more effective treatment, we firstly combined EGFR TKI gefitinib with YAP inhibitor simvastatin in HCC cells. The HCC cells were treated with simvastatin alone or in combination with rising concentrations of the gefitinib for 48 h. CCK8 assays showed that simvastatin treatment alone induced a strong growth inhibitory effect on the three HCC cell lines (HepG2, 7402, and 7721) after 48 h of continuous exposure to the drug. Gefitinib’s anti-neoplastic effect was significantly enhanced when the EGFR-TK inhibitor was combined with simvastatin for 48 h in those HepG2, 7402, and 7721cells (Fig. 4a, b, c, Suppl. Table 1). To investigate the coefficient of drug interaction (CDI), HCC cells were treated with a range of simvastatin and gefitinib, alone or in combination with a fixed concentration of ratio (1:1) in HepG2 and 7402 cells, and (2:1) in 7721 cells, after 48 h, cells viability was determined by Cell Counting Kit-8. Then we calculated the combination index (CI) values and the Dm using CompuSyn software (ComboSyn, Inc., Paramus, NJ, USA). According to the method proposed by Chou et al., combination index (CI) values <1, =1, >1, respectively, indicated synergistic, additive and antagonistic effect. The median effect dose (Dm) is the dose required to produce the median effect (analogous to the IC50). Linear regression correlation coefficients (r-values) of the median effect plots reflect that the dose-effect relationships for single treatment and the combination treatment, conform to the principle of mass action (in general, r-values >0.9 confirm the validity of this methodology). The Dm value for simvastatin, gefitinib, and the combination treatment was 8.140, 12.183, and 11.212 μM, respectively, in HepG2 cells, and a summary of the data from the same analysis applied to each of the five simvastatin/gefitinib combinations tested demonstrated that combinations exhibited synergistic therapeutic interactions (CI < 1) across a wide range (~0.55–1) of Fa values (Suppl. Table 2, Suppl. Fig. 10). The Dm value for simvastatin, gefitinib, and the combination treatment was 11.715, 53.759, and 5.962 μM, respectively, in 7402 cells, and the combinations exhibited synergistic therapeutic interactions across a wide range (~0.15–0.85) of Fa values (Suppl. Table 3, Suppl. Fig. 10). The Dm value for simvastatin, gefitinib, and the combination treatment was 65.086, 23.480, and 15.813 μM, respectively, in 7721 cells, and the combinations exhibited synergistic therapeutic interactions across a wide range (~0–0.55) of Fa values (Suppl. Table 4, Suppl. Fig. 10). Colony formation assays revealed that simvastatin combined with gefitinib inhibited the cell survival more significantly than that of simvastatin or gefitinib alone in HCC cells (Fig. 4d, e, f). We also studied the potential mechanism involved in the combined effect. WB results indicated that simvastatin alone could inhibit the expression of YAP, cyclinD1, C-Myc, and Bcl-xl, however, it had nearly no effect on the EGFR signaling, including P-EGFR, P-AKT, and P-ERK. While gefitinib alone could significant inhibit the EGFR signaling pathway, including P-EGFR, P-ERK, and P-AKT, and could also inhibit the expression of CyclinD1, C-Myc, and Bcl-xl, but it had only slightly effect on the expression of YAP. The combination of simvastatin with gefitinib resulted in a significant inhibition of the EGFR signaling, including P-EGFR, P-ERK, and P-AKT, and in an augmented inhibition of CyclinD1, C-Myc, and Bcl-xl (Fig. 4g, h, i, Suppl. Fig. 5).

a, b, c CCK8 assays were used to detect the effect of simvastatin (YAP inhibitor) combined with gefitinib (EGFR inhibitor) on the proliferation in HepG2, Bel 7402 and SMMC7721 HCC cells. d, e, f Colony formation assays were used to detect the effect of simvastatin (YAP inhibitor) combined with gefitinib (EGFR inhibitor) on the survival of HCC cells. g, h, i WB was used to examine the effect of combined treatment on the core effectors of downstream signaling
The effect of simvastatin combined with sorafenib in HCC cells
Sorafenib, the only targeted drug used in HCC, is a multiple kinase inhibitor among its targets, including B-RAF and C-RAF. Sorafenib displays very limited extension of the survival of patients with advanced metastatic HCC, extending their life expectancy by 7.9 to 10.7 months3. So, it is urgent for us to find novel drugs that could enhance the cytotoxicity of sorafenib against HCC. Here, we combined simvastatin with sorafenib to simultaneously target YAP and RAF kinases in HCC cell lines. CCK8 assay indicated that simvastatin could significantly enhance the cytotoxicity of sorafenib in HepG2 and Bel7402 cells (Fig. 5a, b, Suppl. Table 1). The Dm value for simvastatin, sorafenib, and the combination treatment was 9.730, 7.844, and 9.448 μM, respectively, in HepG2 cells, and the combinations exhibited synergistic therapeutic interactions (CI < 1) across a wide range (~0.6–1) of Fa values (Suppl. Table 5, Suppl. Fig. 11). The Dm value for simvastatin, sorafenib, and the combination treatment was 11.715, 13.534, and 11.201 μM, respectively, in 7402 cells, and the combinations exhibited synergistic therapeutic interactions (CI < 1) across a wide range (~0–0.8) of Fa values (Suppl. Table 6, Suppl. Fig. 11). Colony formation assay indicated that the combined group inhibited the colony formation more significantly than that of simvastatin or sorafenib alone in the two HCC cell lines (Fig. 5c, d). Further mechanism study indicated that the combination of simvastatin with sorafenib resulted in a simultaneous inhibition of YAP, P-ERK, P-Akt1, and CyclinD1, C-Myc, and Bcl-xl in the three cell lines (Fig. 5e, f, Suppl. Fig. 6).

a, b CCK8 assays were used to detect the effect of simvastatin combined with sorafenib (Raf inhibitor) on the proliferation in HepG2 and SMMC7721 HCC cells. c, d, Colony formation assays were used to detect the effect of simvastatin combined with sorafenib on the survival of HCC cells. e, f WB was used to examine the effect of combined treatment on the core effectors of downstream signaling
The combination of simvastatin and trametinib in HCC cells
The RAS–RAF-MEK–MAPK pathway plays an important role in the carcinogenesis of diverse cancers34. Though the RAS proteins are undruggable at present, there has been successful drugs targeting RAF(vemurafenib, dabrafenib) and MEK(trametinib), and these have shown substantial clinical activity in melanoma35,36,37. Therefore, we also tested whether the YAP inhibitor simvastatin could potentiate the cytotoxic activity of MEK inhibitor trametinib in HCC. Our results indicated that trametinib alone could inhibit the proliferation of HCC cells, and that simvastatin could enhance the sensitivity of HCC cells to trametinib (Fig. 6a, b, Suppl. Table 1). The Dm value for simvastatin, trametinib, and the combination treatment was 10.629, 686.881, and 16.372 μM, respectively, in HepG2 cells, and the combinations exhibited synergistic therapeutic interactions (CI < 1) across a wide range(~0.1–0.8) of Fa values (Suppl. Table 7, Suppl. Fig. 12). The Dm value for simvastatin, trametinib, and the combination treatment was 64.313, 119.387, and 65.850 μM, respectively, in 7721 cells, and the combinations exhibited synergistic therapeutic interactions (CI < 1) across a wide range (~0.45–1) of Fa values (Suppl. Table 8, Suppl. Fig. 12). Colony formation assay indicated the combined treatment could induce a synergistic cytostatic effects on the HCC cells (Fig. 6c, d). WB results indicated that combined treatment could simultaneously inhibit the expression of YAP and P-ERK, and induce significantly down-regulation of C-Myc and Bcl-xl (Fig. 6e, f).

a, b CCK8 assays were used to detect the effect of simvastatin combined with trametinib (MEK inhibitor) on the proliferation in HepG2 and SMMC7721 HCC cells. c, d, Colony formation assays were used to detect the effect of simvastatin combined with trametinib on the survival of HCC cells. e, f WB was used to examine the effect of combined treatment on the core effectors of downstream signaling
Discussion
Recent studies have demonstrated that YAP, the core effector of the Hippo pathway, was involved in the carcinogenesis of many cancers, including HCC5. YAP activation is an early event in liver tumorigenesis, making which a potential therapeutic target for HCC treatment14. The dysregulated EGFR signaling also plays an important role in the carcinogenesis of HCC19,20. However, the crosstalk between the Hippo pathway and the EGFR signaling and its implication in target therapy is still poorly understood in HCC.
In the current study, we demonstrated that the EGFR signaling could bypass the canonical RhoA signaling and mainly act through PI3K-PDK1 pathway, but not the AKT or the MAPK pathway to activate YAP signaling. Notably, it has been reported that the EGF signaling inhibited the Hippo pathway through activation of PI3-kinase (PI3K) and phosphoinositide-dependent kinase (PDK1), other than that of AKT, thereby leading to inactivation of Lats, dephosphorylation of YAP, YAP nuclear accumulation and transcriptional activation of its target gene of CTGF in immortalized mammary cells24. At this point, our data was partly consistent with that published report. We found that EGF stimulation could enhance total YAP expression and promote its nuclear accumulation in HCC cells, whereas Run Fan et al. reported that the EGF treatment could lead to dephosphorylation of YAP and YAP nuclear accumulation, but had nearly no effect on total YAP expression24. The main reason for the difference might be the use of different cell types in the two studies. Our data also indicated that EGF stimulation could enhance the expression of Lats1, the upstream inhibitor of YAP, and that knockdown YAP could reverse EGF mediated up-regulation of Lats1 (Suppl. Fig. 13). Other studies showed that YAP/TAZ activation resulted in activation of LATS1/2, their upstream negative regulators, to constitute a negative feedback loop of the Hippo pathway in vitro and in vivo38,39. Together, the present data indicated that EGF induced Lats1 up-regulation might be the results of transcriptional induction of up-regulated YAP expression, thus YAP and Lats1 could also form a negative feed-back signaling loop in EGF-treated HCC cell, and the molecular mechanism still need further investigation.
We also found that the activated EGFR signaling could act through YAP to trans-activate Cyr61, one target gene of YAP. This result was similar to Raquel Urtasun et al.’s study which showed that the EGFR signaling could act through YAP to stimulate the expression of another target gene of YAP, CTGF in liver cancer, although, they didn’t illustrate the underlying mechanisms in details25. Other studies reported that the activated YAP and EGFR signaling could form a positive signaling loop to drive cancer progression in several cell types, including cervical cancer, esophageal cancer and breast epithelial cells13,23,40. However, in our study, we didn’t observe that YAP knockdown had any effect on the EGFR signaling. The difference in conclusions of these reports might be that the cell types used in these studies were so different.
The reported YAP inhibitors mainly include veteporfin and statins28,29,30. Several large population-based cohort studies in Asian and Western populations have shown a protective association between the use of statins and the risk of HCC31,32. Recent studies have indicated that statins could down-regulate YAP through inhibiting RhoA activity29,30. Here, we used simvastatin as a YAP inhibitor. Simvastatin could enhance the sensitivity of HCC cells to gefitinib, a EGFR specific TKI. Our results were consistent with a previous work demonstrating that atorvastatin could overcome gefitinib resistance in KRAS mutant human non-small cell lung carcinoma cells41.
The SHARP trial demonstrated that sorafenib could improve survival in patients with advanced metastatic HCC, extending life expectancy from 7.9 to 10.7 months42. Our study indicated that simvastatin could also enhance the cytotoxic activity of sorafenib, the only target drug used in HCC treatment. The targets of sorafenib includes B-Raf and C-Raf both of which act as the critical downstream effectors of EGFR signaling. Notably, one report has demonstrated that the combination of lovastatin and sorafenib produced synergistic cytotoxic effects against renal carcinoma cells43. We also investigated the effect of the combination of simvastatin with trametinib, the first FDA approved MEK inhibitor, on the cell proliferation and survival of HCC. Not surprisingly, the combined therapy produced synergistic lethality in HCC cells. At this point, our results were consistent with a very recent report which revealed that trametinib plus fluvastatin showed synergistic efficacy in both the drosophila Ras-Pten lung cancer model and human lung cancer cell line44.
Several recent studies indicated that YAP could serve as a novel biomarker for cetuximab resistance in colorectal cancer and head and neck cancer12,45, and our unpublished data showed that knockdown YAP could enhance the sensitivity of K-RAS mutant CRC cells to cetuximab. Notably, the YAP inhibitor, veteporfin and simvastatin, could overcome cetuximab resistance in K-RAS mutant CRC cells. Some studies showed that the activated YAP could bypass K-RAS to promote cell proliferation and survival in several tumor types, and that YAP could also promote resistance to RAF-targeted and MEK-targeted cancer therapies46,47,48. These findings indicated that the crosstalk between Hippo pathway and EGFR signaling played a vital role in the carcinogenesis of multiple malignancies, combined targeting YAP and the EGFR signaling might represent a novel therapeutic strategy for the treatment of HCC, as well as several other cancers (Suppl. Fig. 8).
In summary, we demonstrated that EGFR signaling could bypass RhoA and act mainly through the PI3K/PDK1 pathway to activate YAP. The activated EGFR signaling could promote the proliferation of HCC cells in a YAP-independent manner. Our findings also provided several promising poly-therapeutic strategies to enhance the efficacy of HCC treatment through combined targeting of YAP and the EGFR signaling (Fig. 7).

The proposed model of the regulation mechanisms of Hippo pathway by the EGFR signaling
Materials and methods
Cell culture and the inhibitors
The human liver cell line HL7702, and the human HCC cell lines HepG2, Bel7402, Hep3B and SMMC7721 were from ATCC. All five cell lines were maintained in DMEM medium supplemented with 10% fetal bovine serum (HyClone, USA) in 5% CO2 at 37 °C. The inhibitors used in this study include the PI3K inhibitor wortamanin, PDK1 inhibitor GSK2334470 and BX-795, AKT inhibitor MK2206, MEK inhibitor Trametinib(Selleck, USA), EGFR inhibitors Gefitinib, PI3K inhibitor Ly294002, and MEK inhibitor U0126(MCE, USA).
Western-blot
Cells were lysed in RIPA buffer (150 mM NaCl, 1% NP-40, 50 mM Tris-HCl PH 7.4, 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, 1 mM deoxycholic acid and 1 mM EDTA) containing a cocktail of protease inhibitors and phosphatase inhibitors (Calbiochem, Darmstadt, Germany). Equal amounts of protein sample (30–50 μg) were separated by 12% SDS-PAGE and transferred to NC membrane (Millipore, Bedford, MA, USA) using the Bio-Rad wet transfer system. The following antibodies were used for Western blotting: Mst1, YAP, P-YAP(ser127), P-EGFR(1068), P-ERK1/2(202/204), ERK1, P-AKT(ser397), AKT1, C-Myc, CyclinD1 (Epitomics, USA); YAP/TAZ, Lats1, Bcl-xl (CST, USA), EGFR, RhoA (Santa Cruze, USA), Cyr61 (Proteintech, China), GAPDH and β-actin (Biostar, China).
Cytoplasmic and nuclear protein extraction
The serum-starved(one night) HepG2 and 7721 cells were stimulated with EGF(20 ng/ml) for 2 h, then the cytoplasmic and nuclear proteins were extracted using Keygen Nuclear and Cytoplasmic Extraction Kit (Keygen Biotec, Nanjing, China) according to the manufacturer’s protocol.
Pull-down assays
Active RhoA in cell lysates (200 μg) was precipitated with 15 μg GST-RBD (containing amino acids-8–89 of Rhotekin), which was expressed in Escherichia coli and bound to agarose beads. The precipitates were washed three times in washing buffer [50 mmol/L Tris(pH 7.2), 150 mmol/L NaCl, 10 mmol/L MgCl2, 0.1 mmol/Lphenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, and 10 μg/ml leupeptin], and after adding the loading buffer and boiling for 5 min, the bound proteins were resolved in 12% polyacrylamide gels, transferred to NC membranes, and immunoblotted with anti-RhoA antibody as described above.
Fluorescence microscopy
HepG2 and SMMC7721 cells were stimulated by serum-free 20 ng/ml EGF for 4 h before plating into 24-well dishes containing 12 mm glass coverslips. Then cells were fixed by 4% paraformaldehyde for 20 min and incubated in blocking buffer for another 1 h (0.5% triton X-100 and 1% BSA in PBS) prior to primary antibody staining. Both primary and secondary antibodies were diluted in blocking buffer. Coverslips were incubated with primary antibodies for 1 h at room temperature followed by 1 h second antibodies incubation. In the end, diluted DAPI was added on the coverslips, 10 min later, coverslips were mounted on the glass slide. Images were captured under Nikon Eclipse 80i (Nikon, Japan) microscope with NIS-Elements software (version 4.30.01).
Cell transfection
The SiRNAs against RhoA and YAP were transfected into gastric cancer cells, respectively, by Lipofectamine 2000 (Invitrogen, USA), and the non-sense RNA was used as a negative control. The sequence of the two siRNAs were presented in our previous study17.
Cell viability assay
HCC cells were seeded in 96-well plates for one night, then cells were transfected with siRNA or negative control, respectively, for 24 h, next, the cells were treated by different drugs or the combined therapy. Cell growth was measured by Cell Counting Kit-8 (Dojingdo, Kumamoto, Japan). The coefficient of drug interaction (CDI) was used to analyze the synergistically inhibitory effect of drug combinations49. CDI is calculated as follows: CDI = AB/(A*B). According to the absorbance of each group, AB is the ratio of the combination groups to the control group; A or B is the ratio of the single agent groups to the control group. Thus a CDI value less than, equal to or greater than 1 indicates that the drugs are synergistic, additive or antagonistic, respectively. CDI less than 0.7 indicate that the drugs are significantly synergistic.
Colony formation
The 4000–5000 cells were seeded in 3 cm dish for one night, then the cells were incubated with different inhibitors for 4–6 days. Next, cells were washed by PBS and fixed by 4% paraformaldehyde for 20 min, further crystal violet staining was used to observe the effect of treatment on the survival of HCC cells.
Statistical analysis
The qualification of WB was performed using the Image J. All of the statistical calculations were performed using the GraphPad Prism 5. The data were expressed throughout as means ± the standard error of the mean (SEM). Statistical comparisons were performed by one-way ANOVA. Tukey’s post hoc test was used for multiple group comparisons and Student’s t test was used for single comparisons. All of the p-values were two-sided and p < 0.05 was considered to be statistically significant.
Availability of data and materials
Data and materials will be shared.
References
Bruix, J. et al. Conclusions of the Barcelona-2000 EASL conference: European Association for the Study of the Liver. J. Hepat. 35, 421–430 (2001).
Torre, L. A. et al. Global cancer statistics, 2012. Cancer J. Clin. 65, 87–108 (2015).
Llovet, J. M., Villanueva, A., Lachenmayer, A. & Finn, R. S. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat. Rev. Clin. Oncol. 12, 408–424 (2015).
Maluccio, Ma. C. Recent progress in understanding, diagnosing, and treating hepatocellular carcinoma. Cancer J. Clin. 62, 394–399 (2012).
Harvey, K. F., Zhang, X. & Thomas, D. M. The Hippo pathway and human cancer. Nat. Rev. Cancer 13, 246–257 (2013).
Barry, E. R. & Camargo, F. D. The Hippo superhighway: signaling crossroads converging on the Hippo/Yap pathway in stem cells and development. Curr. Opin. Cell. Biol. 25, 247–253 (2013).
Camargo, F. D. et al. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 17, 2054–2060 (2007).
Dong, J. et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130, 1120–1133 (2007).
Zhao, B. et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Gene Dev. 21, 2747–2761 (2007).
Zhou, D. et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell. 16, 425–438 (2009).
Polesello, C. & Tapon, N. Salvador-warts-hippo signaling promotes Drosophila posterior follicle cell maturation downstream of notch. Curr. Biol. 17, 1864–1870 (2007).
Lee, K. W. et al. Significant association of oncogene YAP1 with poor prognosis and cetuximab resistance in colorectal cancer patients. Clin. Cancer Res. 21, 357–364 (2015).
Song, S. et al. The Hippo Coactivator YAP1 mediates EGFR overexpression and confers chemoresistance in esophageal cancer. Clin. Cancer Res. 21, 2580–2590 (2015).
Perra, A. et al. YAP activation is an early event and a potential therapeutic target in liver cancer development. J. Hepat. 61, 1088–1096 (2014).
McBeath, R., Pirone, D. M., Nelson, C. M., Bhadriraju, K. & Chen, C. S. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev. Cell. 6, 483–495 (2004).
Yu, F.-X. et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 150, 780–791 (2012).
Zhang, Y. et al. CD44 acts through RhoA to regulate YAP signaling. Cell. Signal. 26, 2504–2513 (2014).
Tebbutt, N., Pedersen, M. W. & Johns, T. G. Targeting the ERBB family in cancer: couples therapy. Nat. Rev. Cancer 13, 663–673 (2013).
Fuchs, B. C. et al. Epidermal growth factor receptor inhibition attenuates liver fibrosis and development of hepatocellular carcinoma. Hepatology 59, 1577–1590 (2014).
Schiffer, E. et al. Gefitinib, an EGFR inhibitor, prevents hepatocellular carcinoma development in the rat liver with cirrhosis. Hepatology 41, 307–314 (2005).
Minguez, B., Tovar, V., Chiang, D., Villanueva, A. & Llovet, J. M. Pathogenesis of hepatocellular carcinoma and molecular therapies. Curr. Opin. Gastroenterol. 25, 186–194 (2009).
BVVG, Reddy & Irvine, K. D. Regulation of Hippo signaling by EGFR-MAPK signaling through Ajuba family proteins. Dev. Cell. 24, 459–471 (2013).
He, C. et al. The Hippo/YAP pathway interacts with EGFR signaling and HPV oncoproteins to regulate cervical cancer progression. EMBO Mol. Med 7, 1426–1449 (2015).
Fan, R., Kim, N. G. & Gumbiner, B. M. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc. Natl Acad. Sci. USA 110, 2569–2574 (2013).
Urtasun, R. et al. Connective tissue growth factor autocriny in human hepatocellular carcinoma: oncogenic role and regulation by epidermal growth factor receptor/yes-associated protein-mediated activation. Hepatology 54, 2149–2158 (2011).
Yu, F.-X. et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 150, 780–791 (2012).
Deng, W. et al. CD24 associates with EGFR and supports EGF/EGFR signaling via RhoA in gastric cancer cells. J. Transl. Med. 14, 1–13 (2015).
Yi, L. C. et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Gene Dev. 26, 1300–1305 (2012).
Wang, Z. et al. Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc. Natl Acad. Sci. USA 111, 89–98 (2014).
Sorrentino, G. et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Bio. 16, 357–366 (2014).
El–Serag, H. B., Johnson, M. L., Hachem, C. & Morgana, R. O. Statins are associated with a reduced risk of hepatocellular carcinoma in a large cohort of patients with diabetes. Gastroenterology 136, 1601–1608 (2009).
Axel, H., Lai, C. L. & Man-Fung, Y. Statins and the risk of hepatocellular carcinoma in patients with hepatitis B virus infection. J. Clini. Oncol. 31, 623–630 (2013).
Nielsen, S. F., Nordestgaard, B. G. & Bojesen, S. E. Statin use and reduced cancer-related mortality. New Engl. J. Med. 367, 1792–1802 (2012).
Kolch, W. & Pitt, A. Functional proteomics to dissect tyrosine kinase signalling pathways in cancer. Nat. Rev. Cancer 10, 618–629 (2010).
Larkin, J. et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. New Engl. J. Med. 371, 1867–1876 (2014).
Long, G. V. et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet 386, 444–451 (2015).
Long, G. V. et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. New Engl. J. Med. 371, 1877–1888 (2014).
Dai, X. et al. YAP activates the Hippo pathway in a negative feedback loop. Cell. Res. 25, 1175–1178 (2015).
Moroishi, T. et al. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. GeneDev 29, 1271–1284 (2015).
Zhang, J., Ji, J. Y. & Yu, M. et al. YAP-dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nat. Cell. Biol. 11, 1444–1450 (2009).
Chen, J. et al. Atorvastatin overcomes gefitinib resistance in KRAS mutant human non-small cell lung carcinoma cells. Cell Death Dis. 4, e814 (2013).
Bruix, J. et al. Efficacy and safety of sorafenib in patients with advanced hepatocellular carcinoma: Subanalyses of a phase III trial. J. Hepatol. 57, 821–829 (2012).
Bil, J., Zapala, L., Nowis, D., Jakobisiak, M. & Golab, J. Statins potentiate cytostatic/cytotoxic activity of sorafenib but not sunitinib against tumor cell lines in vitro. Cancer Lett. 288, 57–67 (2010).
Levine, B. & Cagan, R. Drosophila lung cancer models identify trametinib plus statin as candidate therapeutic. Cell Rep. 14, 1477–1487 (2016).
Jerhammar, F. et al. YAP1 is a potential biomarker for cetuximab resistance in head and neck cancer. Oral. Oncol. 50, 832–839 (2014).
Kapoor, A. et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 158, 185–197 (2014).
Shao, D. D. et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 158, 171–184 (2014).
Lin, L. et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat. Genet. 47, 250–256 (2015).
Chou, T. C. & Talalay, P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 22, 27–55 (1984).
Acknowledgements
We would thank all of the participants for agreeing to participate in this research. Prof. James Ogg (Purdue University) helped to edit the English of this paper. This work was supported by grants from the National Natural Science Foundation of China (No.81572731, 81702403), the Innovative Research Team Program of National Natural Science Foundation of China (No.81621003), and the National Basic Research Program of China (No. 2011CB935800).
Authors contributions
F. B. and H. X. designed experiments and supervised the project. H. X., X. D., H. Y., S. Z., Z. F., G. W., and Q. T. performed experiments, analyzed data, and wrote the paper. F. B. and Q. G. obtained and provided the fundings.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
This study was approved by the Clinical Research Ethics Committee of West China Hospital, and all experiments were performed in accordance with relevant guidelines and regulations.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xia, H., Dai, X., Yu, H. et al. EGFR-PI3K-PDK1 pathway regulates YAP signaling in hepatocellular carcinoma: the mechanism and its implications in targeted therapy. Cell Death Dis 9, 269 (2018). https://doi.org/10.1038/s41419-018-0302-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-018-0302-x
This article is cited by
-
Role of Hippo pathway dysregulation from gastrointestinal premalignant lesions to cancer
Journal of Translational Medicine (2024)
-
YAP-mediated trophoblast dysfunction: the common pathway underlying pregnancy complications
Cell Communication and Signaling (2023)
-
Insights into recent findings and clinical application of YAP and TAZ in cancer
Nature Reviews Cancer (2023)
-
Non-hippo kinases: indispensable roles in YAP/TAZ signaling and implications in cancer therapy
Molecular Biology Reports (2023)
-
S1PR1 regulates ovarian cancer cell senescence through the PDK1-LATS1/2-YAP pathway
Oncogene (2023)
