
Abstract
Mitochondria form close physical contacts with a specialized domain of the endoplasmic reticulum (ER), known as the mitochondria-associated membrane (MAM). This association constitutes a key signaling hub to regulate several fundamental cellular processes. Alterations in ER–mitochondria signaling have pleiotropic effects on a variety of intracellular events resulting in mitochondrial damage, Ca2+ dyshomeostasis, ER stress and defects in lipid metabolism and autophagy. Intriguingly, many of these cellular processes are perturbed in neurodegenerative diseases. Furthermore, increasing evidence highlights that ER–mitochondria signaling contributes to these diseases, including Parkinson’s disease (PD). PD is the second most common neurodegenerative disorder, for which effective mechanism-based treatments remain elusive. Several PD-related proteins localize at mitochondria or MAM and have been shown to participate in ER–mitochondria signaling regulation. Likewise, PD-related mutations have been shown to damage this signaling. Could ER–mitochondria associations be the link between pathogenic mechanisms involved in PD, providing a common mechanism? Would this provide a pharmacological target for treating this devastating disease?
In this review, we aim to summarize the current knowledge of ER–mitochondria signaling and the recent evidence concerning damage to this signaling in PD.
Similar content being viewed by others


Facts
-
Endoplasmic reticulum (ER) and mitochondria form close associations that constitute key signaling hubs to regulate many cellular processes.
-
ER–mitochondria contacts regulate many different pathways, which are damaged in Parkinson’s disease (PD).
-
ER–mitochondria associations are altered in PD.
Open questions
-
Are ER–mitochondria associations disrupted or upregulated upon PD-related insults?
-
Is ER–mitochondria signaling damage the common link among the different pathways involved in PD?
-
What are the molecular mechanisms implicated in PD-related protein damage to ER–mitochondria associations?
-
Do other PD-related proteins alter ER–mitochondria signaling?
-
Is ER–mitochondria signaling also damaged in sporadic PD?
-
Can ER–mitochondria signaling be targeted therapeutically?
Introduction
Parkinson’s disease (PD) is the most common movement disorder and the second most common neurodegenerative disease after Alzheimer’s disease (AD). PD patients typically experience difficulties with slowness of movements (bradykinesia), involuntary shaking (tremor), increased resistance to passive movement (rigidity) and postural instability. The cardinal motor symptoms of PD are attributable to the progressive degeneration of dopaminergic neurons in the pars compacta of the substantia nigra (SNpc DA). PD is also characterized by the presence of intraneuronal proteinaceous inclusions called Lewy bodies (LB) and abnormal dystrophic neuronal processes termed Lewy neurites in the surviving neurons1.
Although most cases are sporadic, mutations in several genes, the PARK loci, have been unequivocally shown to cause familial parkinsonism in 5–10% of cases. Importantly, the phenotypes of both the sporadic and familial forms are essentially indistinguishable, implying that they might share common underlying mechanisms. Mutations in three genes, SNCA (best known as α-synuclein), LRRK2 (Leucine-rich repeat kinase 2), and VPS35 (Vacuolar protein sorting-associated protein 35), are known to cause a dominant form of PD, whereas mutations in PARK2 (parkin RBR E3 ubiquitin protein ligase, best known as Parkin), PINK1 (PTEN-induced putative kinase 1), and PARK7 (Parkinsonism associated deglycase, best known as DJ-1) cause recessive-inherited forms of the disease2. The discovery of such monogenic forms during the last two decades has significantly advanced our understanding of the pathogenic mechanisms involved in PD, as it allows for the generation of animal and cellular models carrying the mutant gene. Thus, although the precise mechanisms underlying neuronal death in PD remain to be determined, damage to a plethora of cellular processes has been widely reported. These include alterations in Ca2+ homeostasis, cellular proteostasis, axonal transport, mitochondrial function, and neuroinflammation3. Consequently, one of the difficulties in deciphering PD-related toxicity consists of linking these apparently diverse pathological changes to a common disease pathway.
Recently, several indications have argued in favor of the possibility that perturbations in the ER–mitochondrial network have an important role in the pathogenesis of PD4,5. Indeed, ER–mitochondria communication has been demonstrated to be altered in several neurodegenerative diseases, including PD4. This review is mainly devoted to discussing the evidence that ER–mitochondria signaling dysfunction may have a role in PD pathogenesis.
Endoplasmic reticulum–mitochondria associations
In the eukaryotic cell, communication and cooperation between the different membrane-bound organelles must take place to integrate cellular physiology. This integration depends upon effective crosstalk and one way in which this is achieved is through direct membrane contact. Thus, proper endoplasmic reticulum (ER)–mitochondria communication requires the formation of specialized membrane microdomains at the contact sites, defining short distances between membranes to connect them6. The ER and mitochondria association is the most studied and the first described inter-organelle contact7. The ER is closely opposed to 5–20% of the mitochondrial surface. The ER domain specialized in this association is known as mitochondria-associated membranes (MAMs) and can be smooth or ribosome-containing rough ER membranes8,9.
ER–mitochondria tethering complexes
The presence of structures that appear to tether the two organelles has been observed by electron microscopy in many different cell types4,6,10,11,12,13 (Fig. 1). Early studies revealed the proteinaceous nature of the tethers between the two membranes6,14. Studies in yeast revealed the presence of a protein complex, known as ERMES (ER–mitochondria encounter structure)15. However, no mammalian orthologues of ERMES proteins have been identified yet; on the contrary, several different protein complexes have been proposed as ER–mitochondria tethers16. One of these complexes is based on the interaction between the ER Ca2+ channel IP3R (inositol 1,4,5-trisphosphate receptor) and the OMM VDAC1 (voltage-dependent anion channel 1), that is the major mitochondrial Ca2+ transport channel, in a ternary binding complex with the mitochondrial chaperone GRP75 (glucose-regulated protein 75)17. The ER sorting molecule PACS-2 (phosphofurin acidic cluster sorting protein-2) has also been shown to be involved in ER–mitochondria associations18. Similarly, the interaction between the ER protein Bap31 (B-cell receptor associated protein 31) and the mitochondrial fission protein Fis1 has been shown to bridge the mitochondria and the ER and promote apoptosis19.

Multiple structures that tether mitochondria with the mitochondria-associated membranes (MAMs) of endoplasmic reticulum (ER) have been described. Inositol 1,4,5-trisphosphate receptor (IP3R) and voltage-dependent anion channel (VDAC1) interact via GRP75. Synaptojanin 2 binding protein (SYNJ2BP) interacts with the ribosome-binding protein 1 (RRBP1). The outer mitochondrial protein tyrosine phosphatase-interacting protein 51 (PTPIP51) interacts with vesicle-associated membrane proteins-associated protein B (VAPB) or oxysterol-binding protein-related proteins (ORP5/8) at the ER. B-cell receptor associated protein 31 (BAP31) binds to mitochondrial fission 1 protein (Fis1). ER-located mitofusin 2 (MFN2) interacts with mitochondrial MFN1/MFN2. Other proteins, such the ER sorting molecule phosphofurin acidic cluster sorting protein-2 (PACS-2), have been involved in ER–mitochondria association integrity. Yeasts specific proteins have also been described: the ER–mitochondria encounter structure (ERMES) complex, composed of four proteins: the outer mitochondrial membrane proteins Mdm10 and Mdm34, the ER protein Mmm1, and the cytosolic protein Mdm12
Another MAM protein, mitofusin 2 (MFN2), has also been proposed as a tethering complex by establishing homo- and heterotypic interactions with mitochondrial MFN1/220. However, the role of MFN2 as an ER–mitochondria tether has been challenged as several recent studies from different laboratories have now shown that loss of MFN2 leads to an increase and not a decrease in ER–mitochondria contacts21,22,23,24. Thus, whether MFN2 is functionally involved in ER–mitochondria tethering remains to be resolved.
The vesicle-associated membrane proteins-associated proteins (VAPs) are integral ER membrane proteins, which interact with a plethora of proteins to mediate associations between the ER and other membranes25. These include mitochondria but also peroxisomes, the Golgi, the plasma membrane, and the endo-lysosome compartment26,27,28,29,30. Mammals have two homologous VAP proteins, VAPA and VAPB, which share 76% similar or identical amino acid residues31. VAPB binds to the OMM protein, PTPIP51 (protein tyrosine phosphatase-interacting protein 51) to tether ER with mitochondria10,32. Thus, manipulating VAPB or PTPIP51 expression has been shown to induce appropriate changes in ER–mitochondria contacts10,33. An amyotrophic lateral sclerosis (ALS)-related VAPB mutation has been shown to increase the PTPIP51-dependent interaction between the ER and mitochondria32. Regarding PD, a recent study showed that the PD-related protein α-synuclein interacts with VAPB, decreasing the VAPB-PTPIP51 interaction (see below)34.
In addition, PTPIP51 can interact with the oxysterol-binding protein-related proteins ORP5 and ORP8 to tether mitochondria to ER13. ORP proteins have been thought to have a role as sterol sensor or transport proteins35,36. Recently, PTPIP51 has been involved in regulating the interaction of mitochondria with the sarcoplasmic reticulum, a specialized type of ER, in cardiac function37.
Although rough ER–mitochondria contacts have long been observed by electron microscopy6,38,39,40, the above mentioned tethers appear to be specific for ribosome-excluded mitochondria-smooth ER contacts. Interestingly, a recent study has identified novel protein candidates that reside at rough ER–mitochondria contact sites, the OMM protein SYNJ2BP (synaptojanin 2 binding protein), which interacts with the ER protein RRBP1 (ribosome- binding protein 1)41.
Cellular functions regulated by ER–mitochondria signaling
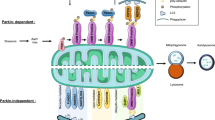
ER–mitochondria contacts are historically linked to lipid metabolism and Ca2+ signaling8,9. Nevertheless, further studies have revealed additional roles for ER–mitochondria signaling in a variety of processes ranging from intracellular trafficking of mitochondria and ER to cell survival, energy metabolism, protein folding and autophagy11,33,42,43,44,45,46. Here, we will give a brief description of the most important ER–mitochondria signaling functions (Fig. 2).

The interaction between mitochondria and the MAMs of ER has been linked with different cellular functions, including inflammasome formation; calcium (Ca2+) signaling, mitochondrial and ER dynamics, autophagy and lipid biosynthesis. AMPK AMP-activated protein kinase, Casp-1 caspase-1, DRP1 dynamin-related protein 1, GRP75 glucose-regulated protein 75, IMM inner mitochondrial membrane, IP3R inositol 145-trisphosphate receptor, MCU mitochondrial calcium uniporter, MFN2 mitofusin 2, NLRP3 NLR family pyrin domain-containing 3, OMM outer mitochondrial membrane, PACS-2 phosphofurin acidic cluster sorting protein-2, PC phosphatidylcholine, PE phosphatidylethanolamine, PEMT2 phosphatidylethanolamine N-methyltransferase 2, PS phosphatidylserine, PSD phosphatidylserine decarboxylases, PTPIP51 protein tyrosine phosphatase-interacting protein 51, ROS radical oxygen species, TCA tricarboxylic acid cycle, UPR unfolded protein response, VAPB vesicle-associated membrane proteins-associated protein B, VDAC1 voltage-dependent anion channel 1
ER–mitochondria contacts serve as a platform for lipid biosynthesis
ER–mitochondria contacts mediate shuttling of lipids between the two organelles, which is necessary for the synthesis of certain lipids such as phosphatidylcholine (PC)47. In fact, this role in the transfer of phospholipids was the first function attributed to ER–mitochondria associations9. For this, phosphatidylserine (PS) is first synthesized in the MAM by the PS synthase 1 and 2; then it is transferred to mitochondria where a decarboxylase (PSD) converts it to phosphatidylethanolamine (PE); PE is crucial for the maintenance of mitochondrial tubular morphology and therefore for mitochondrial functions48,49. PE can be transferred back to the ER, where phosphatidylethanolamine N-methyltransferase-2 (PEMT2) converts it into PC47.
Inflammasome formation
Inflammation is a tightly regulated response of the innate immune system to combat infection or tissue injury and it involves the production of pro-inflammatory cytokines. One of the innate immunity sensors that can orchestrate inflammatory response, by secreting pro-inflammatory cytokines IL-1β and IL-18, are cytosolic multiprotein complexes termed inflammasomes. Upon its activation, the inflammasome complex mediates activation of caspase-1, which represents a crucial step in the secretion of the previously mentioned cytokines and consequently drives the inflammatory response. The NLRP3 inflammasome is the most studied inflammasome and it is formed after the oligomerization of NLRP3 and subsequent recruitment of apoptosis-associated Speck-like protein with a caspase-recruitment domain and pro-caspase-150. One class of these is the NOD-like receptors (NLRs), which sense abnormal cytosolic changes. Upon activation by ROS, some NLRs, including NLRP3, form multiprotein complexes, which redistribute to MAM to activate the inflammasome51.
ER–mitochondria contact sites are crucial for efficient cellular Ca2+ handling and Ca2+-regulated processes
Early studies by Rizzuto and co-workers showed that ER–mitochondria associations mediate Ca2+ transfer from ER to mitochondria8,17,52. These studies demonstrated that the close apposition between the two organelles at contact sites allows the formation of hotspots that meet the low affinity threshold of mitochondrial Ca2+ uptake mechanisms. Consequently, MAM are enriched in proteins associated with Ca2+ handling such as channels and chaperones53. As previously mentioned, the ER Ca2+ channel IP3R contacts VDAC1 through the molecular chaperone GRP75, mediating the Ca2+ transfer from the ER to mitochondria17. In addition, chaperones located at MAM, like calnexin and calreticulin, can interact with the IP3R and the ER Ca2+ transport ATPase SERCA2b to regulate Ca2+ signaling53. Although the ER is the major Ca2+ store, mitochondria are also an important Ca2+ reserve, especially in neurons, so the Ca2+ transfer between them is crucial for the maintenance of cellular Ca2+ homeostasis54.
One of the first roles assigned to the mitochondrial Ca2+ uptake from the ER was the regulation of mitochondrial oxidative metabolism55. Mitochondrial activities are driven in a Ca2+-dependent manner as three dehydrogenases in the tricarboxylic acid cycle (pyruvate-, α-ketoglutarate-, and isocitrate-dehydrogenases) as well as mitochondrial FAD-glycerol phosphate dehydrogenase are activated by Ca2+55. However, prolonged mitochondrial Ca2+ overload compromises mitochondrial function by causing a transient collapse of the mitochondrial membrane potential, leading to necrosis or apoptosis54,56.
Regarding their role in Ca2+ homeostasis and bioenergetics57,58,59, ER–mitochondria associations has been demonstrated to regulate macroautophagy33. Macroautophagy, (hereafter called autophagy), is a lysosomal mechanism of degradation that can be activated during metabolic energy stress, a condition in which the process promotes the recycling of intracellular contents to produce metabolic intermediates60. As mentioned above, mitochondrial Ca2+ uptake through ER–mitochondria contact sites is necessary for ATP production. Consequently, blocking Ca2+ transfer to mitochondria was shown to stimulate autophagy as a physiological response of the cell to the altered bioenergetics57. Recently the ER–mitochondria tethering complex VAPB-PTPIP51 was shown to modulate autophagy, involveing their role in mediating IP3R-mediated delivery of Ca2+ from ER stores to mitochondria33.
Furthermore, ER–mitochondria contacts serve as membrane source for autophagosome formation44. During the autophagy process, specialized double-membrane vesicles, known as autophagosomes, are formed. Autophagosome formation starts with an initial isolation membrane, known as the phagophore, which expands by de novo membrane synthesis and recruitment of lipids and proteins from different membrane sources. Then, the autophagosome engulfs this material for degradation and fuses with the endosomal–lysosomal system where the cargo is degraded and recycled60. Several organelles have been proposed to provide the nucleation site and to contribute to the formation and expansion of the autophagosomal membrane. The involvement of ER and mitochondria to this process has been extensively reported, including the ER–mitochondria contact sites61. Hence, upon autophagy induction, several pro-autophagic proteins relocalize to MAMs to initiate autophagosome formation44,62,63,64.
Regulation of ER and mitochondrial dynamics and homeostasis
ER–mitochondria associations are also important in the movement of both organelles. ER and mitochondria are dynamic organelles transported on cytoskeletal elements. Importantly, ER–mitochondria contacts have been shown to be maintained while both organelles are moving42. This transport involves specialized molecular machinery, as molecular motors such as dynein and kinesin, which are tightly regulated by Ca2+ sensors65,66,67. Hence, a rise in cytosolic Ca2+ concentration has been shown to produce an arrest of the movements of both organelles68,69,70.
Besides participation in mitochondrial motility, MAMs also participate in the regulation of mitochondrial morphology and biogenesis, which is maintained by the balance between fission and fusion events71. Accordingly, MAM is also enriched in proteins related to the control of mitochondrial fission11 and dynamics72. Indeed, mitochondrial fission occurs at positions where ER tubules contact and constrict mitochondria11. These constrictions facilitate the recruitment of DRP1 (dynamin-related protein), a major player in mitochondrial fission73. In addition, ER–mitochondria contacts have a key role during mitophagy, the selective degradation of mitochondria through the autophagy pathway74. During hypoxia, the interaction between the OMM protein FUNDC1 and the ER chaperone calnexin gets disrupted and, as mitophagy proceeds, FUNDC1 preferably recruits DNM1L/DRP1 to drive mitochondrial fission promoting mitophagy64. Importantly, a recent study showed the recruitment into MAM of the PD-related proteins PINK1 and Parkin with downstream effects on ER–mitochondria associations and mitophagy, as explained in detail later in this review62.
The homeostasis of the ER can be altered by several conditions including Ca2+ depletion from its lumen and oxidative stress. These perturbations result in disruption of the folding process in the ER, leading to the accumulation of misfolded/unfolded proteins and ER stress. ER stress then activates the unfolded protein response (UPR), a complex signal-transduction pathway that mediates cellular adaptation to restore ER homeostasis75. A number of ER protein folding chaperones are present in MAM and alterations to ER–mitochondria signaling is linked to UPR53 For example, the structural uncoupling of ER from mitochondria by depletion of PACS-2 or MFN2 was shown to induce ER stress and the UPR18,76,77. Likewise, VAPB also has roles in the UPR78,79.
In consequence, disease-related insults that cause an abnormal tightening or loosening of ER–mitochondria contacts are predicted to be detrimental to cells. Therefore, it is not surprising that alterations in the ER–mitochondria associations have been described in several diseases, including a number of neurodegenerative diseases4,80,81,82.
ER–mitochondria signaling in neurodegeneration
Neurodegenerative diseases including PD, AD, and ALS/FTD (frontotemporal dementia) share several obvious features: they are characterized by progressive nervous system dysfunction, affect millions of people worldwide and there is still no cure for any of them. Furthermore, despite affecting different brain regions PD, AD, and ALS/FTD also share other characteristics suggesting that common cellular processes may converge4.
Thus, whilst the precise mechanisms remain to be determined, a variety of cellular processes are damaged in all of them, including Ca2+ dysregulation, defects in axonal transport, neuroinflammation, loss of cellular proteostasis and mitochondrial dysfunction83,84,85,86,87,88 (Fig. 3). Remarkably, ER–mitochondria associations, regulates all of those processes. The findings that alterations in ER–mitochondria associations occur in neurodegenerative diseases have given rise to the hypothesis that damaged ER–mitochondria signaling is a common potential therapeutic target amongst distinct age-dependent neurodegenerative disorders.

ER–mitochondrial axis appears to be essential for the healthy neurons. Conversely, the disruption of this interaction may involve the develop of some processes as: mitochondrial dysfunction, induction of oxidative stress, calcium (Ca2+) dyshomeostasis, autophagy defects or neuroinflammation, which induce neuronal damage and trigger neurodegenerative diseases as PD
This review focuses on current knowledge of ER–mitochondria signaling in PD. The roles for MAM in other neurodegenerative diseases will be addressed in other chapters of this special issue and have also been recently reviewed in ref.4.
PD and ER–mitochondria signaling
What causes SNpc DA neurons to die in PD?
This is one of the major unresolved questions that has puzzled researchers for many years. Although the mechanisms responsible for the preferential loss of SNpc DA neurons in PD are still a debate, several studies show evidence for a role of Ca2+ signaling in PD pathogenesis89,90. Surmeier et al.91 proposed that the selective vulnerability of SNpc DA neurons relies on their unusual physiological characteristic; adult SNpc DA neurons are autonomously active, this means that they generate action potentials in the absence of conventional synaptic input92. This activity is sustained by their specific voltage-dependent L-type Ca2+ channels, the Cav1.3 channels, which allow Ca2+ influx that contributes to the membrane potential threshold underlying autonomous pacemaking, causing sustained increases in cytosolic Ca2+ concentrations in these cells93,94. As the spatiotemporal pattern of Ca2+ signaling is crucial for the specificity of cellular responses, Ca2+ must be under a tight homeostatic control which requires energy. Consequently, SNpc DA neurons experience a high ATP demand that compromises mitochondrial function and increases the production of reactive oxygen species. These events would have detrimental effects on neuronal viability and could amplify the effects of environmental factors or genetic defects89.
Likewise, both mitochondria and ER have been widely linked to pathogenesis in PD95,96. Toxins that nominally target mitochondria have been shown to induce dopamine cell degeneration96. Furthermore, several studies have evidenced a potential link between proteins known to cause familial PD and defects in mitochondria96.
Apart from a Ca2+ store, the ER is crucial in cellular proteostasis as it is responsible for the production, delivery and degradation of proteins75. Loss of proteostasis is part of the pathogenesis of many neurodegenerative diseases, including PD97. As previously mentioned, one of the hallmarks of PD is the formation of LBs, which reflects a deficiency in proteostasis that is accompanied by signs of ER stress98. As a mechanism for proteostasis, autophagy has a crucial role in the maintenance of protein and organelle homeostasis in the axons, especially in SNpc neurons, which pose an enormous axonal field99. In fact, many studies support a role for autophagy in PD100.
Given its essential role in the above mentioned cellular processes, perturbations in ER–mitochondria associations are expected to be especially detrimental to SNc DA neurons. Several familial PD-related proteins have been shown to cause alterations in ER–mitochondria signaling34,62,101,102,103,104,105,106. However, there is not yet a consensus on the effects of these different PD-associated insults, nor on the mechanisms leading to altered ER–mitochondria associations, which are still unclear. Likewise, whether the disease begins with the dysfunction of ER–mitochondria signaling remains elusive.
Despite the plausible role of ER–mitochondria signaling in PD, ER–mitochondria contacts are poorly characterized in neurons and the exact role of these associations in neuronal (patho)physiology also remains unclear. Several studies have confirmed the presence of ER–mitochondria contacts in neurons107,108,109,110. The presence of these contacts at synapses suggests a role in synaptic activity. In fact, in mouse respiratory neurons, ER–mitochondria axis-mediated Ca2+ handling was shown to determine exocytosis and synaptic activity107. MAMs at synapses may have a critical role in many aspects of mitochondrial biology, which have a direct impact on synaptic activity. As previously mentioned, the accumulation of Ca2+ in the mitochondria leads to the activation of oxidative phosphorylation and to ATP production which is crucial to meet the metabolic demands associated with neuronal activity111. However, the sustained mitochondrial Ca2+ overload driven by the pacemaking activity in SNpc dopaminergic neurons may ultimately compromise ATP production93. Consequently, any types of alteration in ER–mitochondria associations are expected to be potentially damaging to neurons, especially SNpc DA neurons (Fig. 4).

Several PD-associated proteins localize at the ER–mitochondrial (M) axis and have been shown to participate in ER–mitochondria signaling regulation. Proteins such as α-synuclein (α-syn), DJ-1, PINK1 (PTEN-induced putative kinase 1), or Parkin have an important role in the preservation of healthy cells by regulation of calcium (Ca2+) homeostasis and the autophagic responses under different stimulus (a). Dysfunction of these PD-associated proteins leads to a non-efficient interaction between ER and mitochondria that triggers cell damage (b). IP3R inositol 145-trisphosphate receptor, PTPIP51 protein tyrosine phosphatase-interacting protein 51, VAPB VAMP-associated protein B, VDAC1 voltage-dependent anion channel 1
α-Synuclein
α-Synuclein has a central role in the pathogenesis of PD112, however, the normal function of α-synuclein and its precise role in PD remain poorly understood113.
α-Synuclein is a 140 amino acid, lipid-binding protein, which is abundantly expressed in the human, brain and predominantly localized in the presynaptic terminals of neurons. Within neurons, α-synuclein localizes to cytosolic and membrane compartments including synaptic vesicles, mitochondria, and the ER114,115. In this regard, its membrane localization involves targeting to lipid rafts, also known as detergent-resistant membranes, enriched in cholesterol and acidic phospholipids116. Indeed, a subpopulation of α-synuclein is present in MAM34,103,117. Several studies suggest that α-synuclein is involved in modulating synaptic integrity and function118,119. In addition, overexpression of wild-type or familial mutant α-synuclein has been shown to damage a plethora of physiological processes. These include Ca2+ homeostasis101,120, lipid metabolism103, the ER75, autophagy121, and mitochondrial defects96. As mentioned previously, all of these physiological processes are regulated by signaling between ER and mitochondria, so the effects of α-synuclein on ER–mitochondria associations have been investigated.
Until now, three different groups have reported that α-synuclein perturbs ER–mitochondria associations34,101,103. However, the nature of perturbation differs between these studies.
Cali et al.101 reported a role for α-synuclein in modulating ER–mitochondria associations with downstream effects in Ca2+ homeostasis in HeLa cells. Indeed, measurement of Ca2+ exchange between the two organelles is a recognized measurement of MAM activity4. They observed that overexpression of wild-type α-synuclein increases, while downregulation decreases, mitochondrial Ca2+ uptake. The quantification of the ER–mitochondria associations also revealed an increase in the co-localization of ER and mitochondrial markers in cells overexpressing wild-type α-synuclein, suggesting that α-synuclein favors ER–mitochondria contacts. Intriguingly, at high levels of α-synuclein expression, induced by high doses of VPA or TAT α-synuclein fusion protein, there was a drastic reduction in mitochondrial Ca2+ uptake. The authors observed that at those high levels of overexpression α-synuclein re-localizes into cytoplasmic foci. This may reduce the ability of α-synuclein to mediate ER–mitochondria contacts, representing a loss of function.
Later, Guardia-Laguarta et al.103 showed that α-synuclein localizes at MAM and that familial PD mutant α-synuclein associates less than wild-type protein with MAM. This correlates with a decrease in MAM function in cells overexpressing mutant α-synuclein but not wild-type. In this case, the physiological readout of ER–mitochondria associations utilized was the conversion of PS into PE. In these studies, the measurement of ER–mitochondria apposition revealed a lower degree of ER–mitochondria apposition in M17 cells overexpressing familial PD mutant α-synuclein but also in HeLa cells overexpressing the wild-type protein.
Both studies utilized confocal microscopy to analyze ER–mitochondria apposition. However, ER–mitochondria associations are defined by 10–30-nm distances, significantly below of confocal microscopy resolution (∼200 nm)6,16,122,123.
Recently, Paillusson et al.34 also addressed the role of α-synuclein in ER–mitochondria contacts using high resolution techniques such as electron microscopy, structured illumination microscopy, and proximity ligation assays. Such methods afford better resolution for properly quantifying ER–mitochondria associations. They reported that overexpression of either wild-type or familial PD mutant α-synuclein decreases ER–mitochondria contacts. Consequently, these effects disrupt Ca2+ exchange between the two organelles and mitochondrial ATP production. In addition, this study showed that α-synuclein binds to the tethering protein VAPB and decreases the VAPB-PTPIP51 interaction, which is proposed as the mechanism by which it disrupts the contacts. Importantly, this disruption was also seen in neurons derived from induced pluripotent stem cells from familial PD patients harboring pathogenic triplication of the α-synuclein gene34.
PINK1 and Parkin
Loss-of-function mutations in PINK1 or PARK2 genes are associated with juvenile-onset autosomal recessive forms of PD112. Parkin (PARK2 gene expression product) is an ubiquitin E3 ligase that targets specific substrates for degradation. In addition, Parkin has been demonstrated to regulate mitochondrial biogenesis, bioenergetics, dynamics, transport, and degradation124. PINK1 encodes a mitochondrial protein kinase that also protects mitochondrial integrity at different levels. In addition, together with Parkin, PINK1 controls the mechanism of mitophagy125. Therefore, upon conditions of mitochondrial depolarization, PINK1 selectively accumulates on the surface of damaged mitochondria, where it phosphorylates and recruits both ubiquitin and Parkin. Parkin then translocates from the cytosol to the OMM and there ubiquitinates specific substrates (such as MFNs and VDAC1), leading their proteasomal degradation126,127,128. Next, these mitochondria are associated to the forming autophagosome membranes by specific ubiquitin-binding receptor proteins (e.g., p62 and optineurin) and afterwards incorporated within autophagosomes129,130.
As previously mentioned, several pro-autophagic proteins relocalize to MAMs to initiate autophagosome formation44,62,63,64. Similarly, contact regions between the ER and impaired mitochondria have been shown to be prime locations for Parkin-mediated mitophagy and local recruitment of autophagosome precursors131. Recently, Gelmetti et al.62 reported that PINK1 and Parkin are recruited to MAM upon mitochondrial depolarization. PINK1 relocation into MAM seems to be necessary for the recruitment of the autophagy machinery to that area. Furthermore, Parkin translocates into ER–mitochondria contact sites in conditions of excitotoxicity in neurons. However in this case, translocation is not associated with mitophagy and it might be instead related with a distinct unknown pathway that needs to be further investigated104.
Apart from the aforementioned role in mitophagy, several studies have shown that PARKIN accumulates at both mitochondria and ER–mitochondria associations and modulates ER–mitochondrial crosstalk102,105. Once more, the direction of the modulatory effects is controverted.
Cali et al.102 reported that Parkin overexpression enhanced ER–mitochondria coupling and its functions. On the contrary, siRNA loss of Parkin caused a decrease in ER–mitochondria signaling associated with weaker mitochondrial Ca2+ potentials and ATP production.
Conversely, Gautier et al.105 reported that ER–mitochondria associations are instead increased in primary fibroblasts from PARK2 knockout mice and PD patients with PARK2 mutations. This observation correlated with Ca2+ dyshomeostasis and increased levels of MFN2105.
Once more, these controverted finding highlights the difficulties involved in studies of contact sites between the ER and mitochondria. Morphological changes of these organelles, networks, and technical limitations such as the resolution limits of confocal microscopy, may introduce bias into these analyses. For example, the acute siRNA Parkin depletion used for the first study-induced mitochondrial fragmentation102, whereas this is not observed in fibroblasts from PARK2 KO mice or from patients with PARK2 mutations105. Another difficulty with these different models is the possibility of potential compensation mechanisms.
DJ-1
Diverse mutations, including deletions and point mutations, in the DJ-1 gene, have been linked to autosomal recessive early-onset parkinsonism132. DJ-1 protein has a role in the protection against oxidative stress and mitochondria dynamics; however, the mechanism of its protective function is still unknown. Thus, different functions have been suggested for DJ-1, these include characterization as a redox sensor and an antioxidant scavenger, a chaperone with protease activity, or a transcriptional regulator133. DJ-1 is localized in the cytosol and the nucleus. During oxidative stress DJ-1 was shown to translocate to the OMM to maintain a healthy mitochondrial environment133. However, Ottolini et al.106 showed that DJ-1 was localized at the MAMs but not in the pure mitochondrial fraction. This study also showed that DJ-1 overexpression augmented mitochondrial Ca2+ uptake, whereas reduced levels of DJ-1 caused mitochondria fragmentation and decreased mitochondrial Ca2+ uptake. By confocal microscopy studies, they also observed an increased ER–mitochondria association when overexpressing DJ-1. Moreover, its overexpression counteracted p53-mediated effects on mitochondrial deregulation, suggesting that DJ-1 might contribute to maintain ER–mitochondria tethering.
Conclusions and future
Although the exact pathological mechanisms underlying PD remain largely unclear a plethora of cellular pathways are known to be damaged. The discovery that ER–mitochondria signaling, which regulates many of those pathways, are also damaged in PD has highlight the possibility of a common link among them. Therefore, ER–mitochondria signaling may represent a possible drug target upstream of those pathways. However, more research should be done before gaining a clearer understanding of the links between ER–mitochondria signaling and the pathogenesis of PD. Hence, many questions remain unclear. Although the evidence discussed here supports the hypothesis that deregulation of ER–mitochondria signaling has an important role in PD pathogenesis, it is still unclear as to whether ER–mitochondria associations are either upregulated or disrupted upon PD-related insults. Combined, the findings reviewed above highlight the complexity of studying ER–mitochondria associations. Therefore, additional research is needed to gain further insight into the mechanisms of tethering of both organelles, especially in relation to neurons.
Furthermore, investigating whether other PD-related proteins also alter the mitochondria–ER axis or if this is altered in sporadic cases, would be useful to address a possible general pathway for PD. Mutations in LRRK2 are related to both familial and sporadic PD134. Autosomal-dominant mutations in LRRK2 have been shown to cause deficits in intracellular Ca2+ handling, mitochondrial depolarization and increased mitophagy, which can be prevented by L-type Ca2+ channel inhibitors135,136,137. However, whether this is due to altered ER–mitochondria communication remains to be determined.
Another pressing issue is how ER–mitochondria associations can be targeted therapeutically. Likewise, a better understanding in how ER–mitochondria tethers are functionally regulated is crucial to move drug development forward.
In conclusion, more studies are required to enhance our understanding of PD mechanisms and its relation to ER–mitochondria signaling.
References
Kalia, L. V. & Lang, A. E. Parkinson’s disease. Lancet 386, 896–912 (2015).
Lin, M. K. & Farrer, M. J. Genetics and genomics of Parkinson’s disease. Genome Med. 6, 48 (2014).
Poewe, W. et al. Parkinson disease. Nat. Rev. Dis. Primers 3, 17013 (2017).
Paillusson, S. et al. There’s something wrong with my MAM; the ER–mitochondria axis and neurodegenerative diseases. Trends Neurosci. 39, 146–157 (2016).
Rodriguez-Arribas M. et al. Mitochondria-associated membranes (MAMs): overview and its role in Parkinson’s disease. Mol. Neurobiol. 54, 6287–6303 (2016).
Csordas, G. et al. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915–921 (2006).
Copeland, D. E. & Dalton, A. J. An association between mitochondria and the endoplasmic reticulum in cells of the pseudobranch gland of a teleost. J. Biophys. Biochem. Cytol. 5, 393–396 (1959).
Rizzuto, R. et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280, 1763–1766 (1998).
Vance, J. E. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 265, 7248–7256 (1990).
Stoica, R. et al. ER–mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 5, 3996 (2014).
Friedman, J. R. et al. ER tubules mark sites of mitochondrial division. Science 334, 358–362 (2011).
Stoica, R. et al. ALS/FTD-associated FUS activates GSK-3beta to disrupt the VAPB-PTPIP51 interaction and ER–mitochondria associations. EMBO Rep. 17, 1326–1342 (2016).
Galmes, R. et al. ORP5/ORP8 localize to endoplasmic reticulum–mitochondria contacts and are involved in mitochondrial function. EMBO Rep. 17, 800–810 (2016).
Achleitner, G. et al. Association between the endoplasmic reticulum and mitochondria of yeast facilitates interorganelle transport of phospholipids through membrane contact. Eur. J. Biochem. 264, 545–553 (1999).
Kornmann, B. et al. An ER–mitochondria tethering complex revealed by a synthetic biology screen. Science 325, 477–481 (2009).
Rowland, A. A. & Voeltz, G. K. Endoplasmic reticulum–mitochondria contacts: function of the junction. Nat. Rev. Mol. Cell Biol. 13, 607–625 (2012).
Szabadkai, G. et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+channels. J. Cell Biol. 175, 901–911 (2006).
Simmen, T. et al. PACS-2 controls endoplasmic reticulum–mitochondria communication and Bid-mediated apoptosis. EMBO J. 24, 717–729 (2005).
Iwasawa, R., Mahul-Mellier, A. L., Datler, C., Pazarentzos, E. & Grimm, S. Fis1 and Bap31 bridge the mitochondria–ER interface to establish a platform for apoptosis induction. EMBO J. 30, 556–568 (2011).
de Brito, O. M. & Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610 (2008).
Cosson, P., Marchetti, A., Ravazzola, M. & Orci, L. Mitofusin-2 independent juxtaposition of endoplasmic reticulum and mitochondria: an ultrastructural study. PLoS ONE 7, e46293 (2012).
Filadi, R. et al. Mitofusin 2 ablation increases endoplasmic reticulum–mitochondria coupling. Proc. Natl Acad. Sci. USA 112, E2174–E2181 (2015).
Wang, P. T. et al. Distinct mechanisms controlling rough and smooth endoplasmic reticulum contacts with mitochondria. J. Cell Sci. 128, 2759–2765 (2015).
Leal, N. S. et al. Mitofusin-2 knockdown increases ER–mitochondria contact and decreases amyloid beta-peptide production. J. Cell Mol. Med. 20, 1686–1695 (2016).
Prinz, W. A. Bridging the gap: membrane contact sites in signaling, metabolism, and organelle dynamics. J. Cell Biol. 205, 759–769 (2014).
Peretti, D., Dahan, N., Shimoni, E., Hirschberg, K. & Lev, S. Coordinated lipid transfer between the endoplasmic reticulum and the Golgi complex requires the VAP proteins and is essential for Golgi-mediated transport. Mol. Biol. Cell 19, 3871–3884 (2008).
Tavassoli, S. et al. Plasma membrane–endoplasmic reticulum contact sites regulate phosphatidylcholine synthesis. EMBO Rep. 14, 434–440 (2013).
Dong, R. et al. Endosome-ER contacts control actin nucleation and retromer function through VAP-dependent regulation of PI4P. Cell 166, 408–423 (2016).
Costello, J. L. et al. ACBD5 and VAPB mediate membrane associations between peroxisomes and the ER. J. Cell Biol. 216, 331–342 (2017).
Hua, R. et al. VAPs and ACBD5 tether peroxisomes to the ER for peroxisome maintenance and lipid homeostasis. J. Cell Biol. 216, 367–377 (2017).
Levine, T. & Loewen, C. Inter-organelle membrane contact sites: through a glass, darkly. Curr. Opin. Cell Biol. 18, 371–378 (2006).
De Vos, K. J. et al. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 21, 1299–1311 (2012).
Gomez-Suaga, P. et al. The ER–mitochondria tethering complex VAPB-PTPIP51 regulates autophagy. Curr. Biol. 27, 371–385 (2017).
Paillusson S. et al. α-Synuclein binds to the ER–mitochondria tethering protein VAPB to disrupt Ca2+ homeostasis and mitochondrial ATP production. Acta Neuropathol. 134, 129–149 (2017).
Moser von Filseck, J. et al. Intracellular transport. Phosphatidylserine transport by ORP/Osh proteins is driven by phosphatidylinositol 4-phosphate. Science 349, 432–436 (2015).
Chung, J. et al. Intracellular transport. PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma membrane contacts. Science 349, 428–432 (2015).
Qiao, X. et al. PTPIP51 regulates mouse cardiac ischemia/reperfusion through mediating the mitochondria-SR junction. Sci. Rep. 7, 45379 (2017).
Meier, P. J., Spycher, M. A. & Meyer, U. A. Isolation and characterization of rough endoplasmic reticulum associated with mitochondria from normal rat liver. Biochim. Biophys. Acta 646, 283–297 (1981).
Montisano, D. F., Cascarano, J., Pickett, C. B. & James, T. W. Association between mitochondria and rough endoplasmic reticulum in rat liver. Anat. Rec. 203, 441–450 (1982).
Giacomello, M. & Pellegrini, L. The coming of age of the mitochondria–ER contact: a matter of thickness. Cell. Death. Differ. 23, 1417–1427 (2016).
Hung V. et al. Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. Elife 6: e24463 (2017).
Friedman, J. R., Webster, B. M., Mastronarde, D. N., Verhey, K. J. & Voeltz, G. K. ER sliding dynamics and ER–mitochondrial contacts occur on acetylated microtubules. J. Cell Biol. 190, 363–375 (2010).
Kornmann, B., Osman, C. & Walter, P. The conserved GTPase Gem1 regulates endoplasmic reticulum–mitochondria connections. Proc. Natl Acad. Sci. USA 108, 14151–14156 (2011).
Hamasaki, M. et al. Autophagosomes form at ER–mitochondria contact sites. Nature 495, 389–393 (2013).
Raturi, A. & Simmen, T. Where the endoplasmic reticulum and the mitochondrion tie the knot: the mitochondria-associated membrane (MAM). Biochim. Biophys. Acta 1833, 213–224 (2013).
van Vliet, A. R., Verfaillie, T. & Agostinis, P. New functions of mitochondria associated membranes in cellular signaling. Biochim. Biophys. Acta 1843, 2253–2262 (2014).
Flis, V. V. & Daum, G. Lipid transport between the endoplasmic reticulum and mitochondria. Cold Spring Harb. Perspect. Biol. 5, a013235 (2013).
Steenbergen, R. et al. Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J. Biol. Chem. 280, 40032–40040 (2005).
Joshi, A. S., Thompson, M. N., Fei, N., Huttemann, M. & Greenberg, M. L. Cardiolipin and mitochondrial phosphatidylethanolamine have overlapping functions in mitochondrial fusion in Saccharomyces cerevisiae. J. Biol. Chem. 287, 17589–17597 (2012).
Schroder, K., Zhou, R. & Tschopp, J. The NLRP3 inflammasome: a sensor for metabolic danger? Science 327, 296–300 (2010).
Zhou, R., Yazdi, A. S., Menu, P. & Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 (2011).
Szabadkai, G., Simoni, A. M. & Rizzuto, R. Mitochondrial Ca2+ uptake requires sustained Ca2+ release from the endoplasmic reticulum. J. Biol. Chem. 278, 15153–15161 (2003).
Hayashi, T., Rizzuto, R., Hajnoczky, G. & Su, T. P. MAM: more than just a housekeeper. Trends Cell Biol. 19, 81–88 (2009).
Pinton, P., Giorgi, C., Siviero, R., Zecchini, E. & Rizzuto, R. Calcium and apoptosis: ER–mitochondria Ca2 + transfer in the control of apoptosis. Oncogene 27, 6407–6418 (2008).
Griffiths, E. J. & Rutter, G. A. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim. Biophys. Acta 1787, 1324–1333 (2009).
Bononi, A. et al. Identification of PTEN at the ER and MAMs and its regulation of Ca(2+) signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ. 20, 1631–1643 (2013).
Cardenas, C. et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142, 270–283 (2010).
Wong, A., Grubb, D. R., Cooley, N., Luo, J. & Woodcock, E. A. Regulation of autophagy in cardiomyocytes by Ins(1,4,5)P(3) and IP(3)-receptors. J. Mol. Cell Cardiol. 54, 19–24 (2013).
Mallilankaraman, K. et al. MCUR1 is an essential component of mitochondrial Ca2+uptake that regulates cellular metabolism. Nat. Cell Biol. 14, 1336–1343 (2012).
Mizushima, N. Autophagy: process and function. Genes Dev. 21, 2861–2873 (2007).
Tooze, S. A. Current views on the source of the autophagosome membrane. Essays Biochem. 55, 29–38 (2013).
Gelmetti, V. et al. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER–mitochondria tethering and autophagosome formation. Autophagy 13, 654–669 (2017).
Garofalo, T. et al. Evidence for the involvement of lipid rafts localized at the ER–mitochondria associated membranes in autophagosome formation. Autophagy 12, 917–935 (2016).
Wu, W. et al. FUNDC1 regulates mitochondrial dynamics at the ER–mitochondrial contact site under hypoxic conditions. EMBO J. 35, 1368–1384 (2016).
Waterman-Storer, C. M. & Salmon, E. D. Endoplasmic reticulum membrane tubules are distributed by microtubules in living cells using three distinct mechanisms. Curr. Biol. 8, 798–806 (1998).
Anesti, V. & Scorrano, L. The relationship between mitochondrial shape and function and the cytoskeleton. Biochim. Biophys. Acta 1757, 692–699 (2006).
Jung, D. W., Bradshaw, P. C., Litsky, M. & Pfeiffer, D. R. Ca2+transport in mitochondria from yeast expressing recombinant aequorin. Anal. Biochem. 324, 258–268 (2004).
Yi, M., Weaver, D. & Hajnoczky, G. Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J. Cell Biol. 167, 661–672 (2004).
Wang, H. J., Guay, G., Pogan, L., Sauve, R. & Nabi, I. R. Calcium regulates the association between mitochondria and a smooth subdomain of the endoplasmic reticulum. J. Cell Biol. 150, 1489–1498 (2000).
Brough, D., Schell, M. J. & Irvine, R. F. Agonist-induced regulation of mitochondrial and endoplasmic reticulum motility. Biochem. J. 392, 291–297 (2005).
Chan, D. C. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 22, 79–99 (2006).
Schon, E. A. & Area-Gomez, E. Mitochondria-associated ER membranes in Alzheimer disease. Mol. Cell Neurosci. 55, 26–36 (2013).
Ingerman, E. et al. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J. Cell Biol. 170, 1021–1027 (2005).
Bockler, S. & Westermann, B. ER–mitochondria contacts as sites of mitophagosome formation. Autophagy 10, 1346–1347 (2014).
Mercado, G., Valdes, P. & Hetz, C. An ERcentric view of Parkinson’s disease. Trends Mol. Med. 19, 165–175 (2013).
Ngoh, G. A., Papanicolaou, K. N. & Walsh, K. Loss of mitofusin 2 promotes endoplasmic reticulum stress. J. Biol. Chem. 287, 20321–20332 (2012).
Munoz, J. P. et al. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 32, 2348–2361 (2013).
Gkogkas, C. et al. VAPB interacts with and modulates the activity of ATF6. Hum. Mol. Genet. 17, 1517–1526 (2008).
Kanekura, K., Nishimoto, I., Aiso, S. & Matsuoka, M. Characterization of amyotrophic lateral sclerosis-linked P56S mutation of vesicle-associated membrane protein-associated protein B (VAPB/ALS8). J. Biol. Chem. 281, 30223–30233 (2006).
Danese, A. et al. Calcium regulates cell death in cancer: Roles of the mitochondria and mitochondria-associated membranes (MAMs). Biochim. Biophys. Acta 1858, 615–627 (2017).
Marchi S. et al. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium (2017). https://doi.org/10.1016/j.ceca.2017.05.003.
Giorgi, C. et al. Mitochondria-associated membranes: composition, molecular mechanisms, and physiopathological implications. Antioxid. Redox Signal 22, 995–1019 (2015).
Winklhofer, K. F. & Haass, C. Mitochondrial dysfunction in Parkinson’s disease. Biochim. Biophys. Acta 1802, 29–44 (2010).
Moreira, P. I., Carvalho, C., Zhu, X., Smith, M. A. & Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta 1802, 2–10 (2010).
Cozzolino, M. & Carri, M. T. Mitochondrial dysfunction in ALS. Prog. Neurobiol. 97, 54–66 (2012).
Ghavami, S. et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 112, 24–49 (2014).
Wong, E. & Cuervo, A. M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 13, 805–811 (2010).
Millecamps, S. & Julien, J. P. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 14, 161–176 (2013).
Surmeier, D. J. et al. Calcium and Parkinson’s disease. Biochem. Biophys. Res. Commun. 483, 1013–1019 (2017).
Rivero-Rios, P., Gomez-Suaga, P., Fdez, E. & Hilfiker, S. Upstream deregulation of calcium signaling in Parkinson’s disease. Front. Mol. Neurosci. 7, 53 (2014).
Surmeier, D. J., Obeso, J. A. & Halliday, G. M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 18, 101–113 (2017).
Grace, A. A. & Bunney, B. S. Intracellular and extracellular electrophysiology of nigral dopaminergic neurons-3. Evidence for electrotonic coupling. Neuroscience 10, 333–348 (1983).
Guzman, K. M., Jing, L. & Patwardhan, A. Effects of changes in the L-type calcium current on hysteresis in restitution of action potential duration. Pacing Clin. Electrophysiol. 33, 451–459 (2010).
Puopolo, M., Raviola, E. & Bean, B. P. Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J. Neurosci. 27, 645–656 (2007).
Wang, H. Q. & Takahashi, R. Expanding insights on the involvement of endoplasmic reticulum stress in Parkinson’s disease. Antioxid. Redox Signal. 9, 553–561 (2007).
Vila, M., Ramonet, D. & Perier, C. Mitochondrial alterations in Parkinson’s disease: new clues. J. Neurochem. 107, 317–328 (2008).
Nixon, R. A. The role of autophagy in neurodegenerative disease. Nat. Med. 19, 983–997 (2013).
Ryu, E. J. et al. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson’s disease. J. Neurosci. 22, 10690–10698 (2002).
Matsuda, W. et al. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J. Neurosci. 29, 444–453 (2009).
Wang, B., Abraham, N., Gao, G. & Yang, Q. Dysregulation of autophagy and mitochondrial function in Parkinson’s disease. Transl. Neurodegener. 5, 19 (2016).
Cali, T., Ottolini, D., Negro, A. & Brini, M. α-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum–mitochondria interactions. J. Biol. Chem. 287, 17914–17929 (2012).
Cali, T., Ottolini, D., Negro, A. & Brini, M. Enhanced parkin levels favor ER–mitochondria crosstalk and guarantee Ca(2+) transfer to sustain cell bioenergetics. Biochim. Biophys. Acta 1832, 495–508 (2013).
Guardia-Laguarta, C. et al. α-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 34, 249–259 (2014).
Van Laar, V. S. et al. Glutamate excitotoxicity in neurons triggers mitochondrial and endoplasmic reticulum accumulation of Parkin, and, in the presence of N-acetyl cysteine, mitophagy. Neurobiol. Dis. 74, 180–193 (2015).
Gautier, C. A. et al. The endoplasmic reticulum–mitochondria interface is perturbed in PARK2 knockout mice and patients with PARK2 mutations. Hum. Mol. Genet. 25, 2972–2984 (2016).
Ottolini, D., Cali, T., Negro, A. & Brini, M. The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum–mitochondria tethering. Hum. Mol. Genet. 22, 2152–2168 (2013).
Mironov, S. L. & Symonchuk, N. ER vesicles and mitochondria move and communicate at synapses. J. Cell Sci. 119, 4926–4934 (2006).
Hedskog, L. et al. Modulation of the endoplasmic reticulum–mitochondria interface in Alzheimer’s disease and related models. Proc. Natl Acad. Sci. USA 110, 7916–7921 (2013).
Bernard-Marissal, N., Medard, J. J., Azzedine, H. & Chrast, R. Dysfunction in endoplasmic reticulum–mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain 138, 875–890 (2015).
Wu, Y. et al. Contacts between the endoplasmic reticulum and other membranes in neurons. Proc. Natl Acad. Sci. USA 114, E4859–E4867 (2017).
Rangaraju, V., Calloway, N. & Ryan, T. A. Activity-driven local ATP synthesis is required for synaptic function. Cell 156, 825–835 (2014).
Cookson, M. R., Hardy, J. & Lewis, P. A. Genetic neuropathology of Parkinson’s disease. Int. J. Clin. Exp. Pathol. 1, 217–231 (2008).
Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2, a009399 (2012).
Guardia-Laguarta, C., Area-Gomez, E., Schon, E. A. & Przedborski, S. Novel subcellular localization for alpha-synuclein: possible functional consequences. Front. Neuroanat. 9, 17 (2015).
Xu, W., Tan, L. & Yu, J. T. Link between the SNCA gene and parkinsonism. Neurobiol. Aging 36, 1505–1518 (2015).
Fortin, D. L. et al. Lipid rafts mediate the synaptic localization of alpha-synuclein. J. Neurosci. 24, 6715–6723 (2004).
Poston, C. N., Krishnan, S. C. & Bazemore-Walker, C. R. In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). J. Proteomics 79, 219–230 (2013).
Burre, J. et al. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667 (2010).
Burre, J. The synaptic function of α-synuclein. J. Parkinsons Dis. 5, 699–713 (2015).
Hettiarachchi, N. T. et al. α-Synuclein modulation of Ca2+ signaling in human neuroblastoma (SH-SY5Y) cells. J. Neurochem. 111, 1192–1201 (2009).
Winslow, A. R. et al. α-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J. Cell Biol. 190, 1023–1037 (2010).
Helle, S. C. et al. Organization and function of membrane contact sites. Biochim. Biophys. Acta 1833, 2526–2541 (2013).
Filadi, R., Theurey, P. & Pizzo, P. The endoplasmic reticulum–mitochondria coupling in health and disease: molecules, functions and significance. Cell Calcium 62, 1–15 (2017).
McWilliams, T. G. & Muqit, M. M. PINK1 and Parkin: emerging themes in mitochondrial homeostasis. Curr. Opin. Cell Biol. 45, 83–91 (2017).
Scarffe, L. A., Stevens, D. A., Dawson, V. L. & Dawson, T. M. Parkin and PINK1: much more than mitophagy. Trends Neurosci. 37, 315–324 (2014).
Chen, Y. & Dorn, G. W. 2nd PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340, 471–475 (2013).
Gegg, M. E. et al. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 19, 4861–4870 (2010).
Wang, X. et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906 (2011).
Wong, Y. C. & Holzbaur, E. L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl Acad. Sci. USA 111, E4439–E4448 (2014).
Geisler, S. et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131 (2010).
Yang, J. Y. & Yang, W. Y. Bit-by-bit autophagic removal of parkin-labelled mitochondria. Nat. Commun. 4, 2428 (2013).
Bonifati, V. Autosomal recessive parkinsonism. Parkinson. Relat. Disord. 18, S4–S6 (2012).
van der Merwe, C., Jalali Sefid Dashti, Z., Christoffels, A., Loos, B. & Bardien, S. Evidence for a common biological pathway linking three Parkinson’s disease-causing genes: parkin, PINK1 and DJ-1. Eur. J. Neurosci. 41, 1113–1125 (2015).
Chai, C. & Lim, K. L. Genetic insights into sporadic Parkinson’s disease pathogenesis. Curr. Genomics 14, 486–501 (2013).
Papkovskaia, T. D. et al. G2019S leucine-rich repeat kinase 2 causes uncoupling protein-mediated mitochondrial depolarization. Hum. Mol. Genet. 21, 4201–4213 (2012).
Cherra, S. J. et al. Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am. J. Pathol. 182, 474–484 (2013).
Gomez-Suaga, P. & Hilfiker, S. LRRK2 as a modulator of lysosomal calcium homeostasis with downstream effects on autophagy. Autophagy 8, 692–693 (2012).
Acknowledgements
P.G.-S. was funded by Parkinson’s UK (G-1308) and Alzheimer’s Society (287(AS-PG-15b-002)). J.M.B.-S.P. was funded by La Ligue contre le Cancer). J.M.F. received research support from the Instituto de Salud Carlos III, CIBERNED (CB06/05/004) and Instituto de Salud Carlos III, FIS, (PI15/00034). R.A.G.-P. was supported by a “Contrato destinado a la retención y atracción del talento investigador, TA13009” from Junta de Extremadura, as well as research support from the Instituto de Salud Carlos III, FIS, (PI14/00170). M.N.-S. was supported by “Contrato Juan de la Cierva” (JCI-2012-14383) from Ministerio de Economia y Competitividad, Spain. This work was supported also by “Fondo Europeo de Desarrollo Regional” (FEDER), from European Union. We also thank FUNDESALUD for helpful assistance. We thank Naomi Hartopp (King’s College London, UK) for her helpful comments.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interests
The authors declare that they have no competing financial interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by P. Pinton
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gómez-Suaga, P., Bravo-San Pedro, J.M., González-Polo, R.A. et al. ER–mitochondria signaling in Parkinson’s disease. Cell Death Dis 9, 337 (2018). https://doi.org/10.1038/s41419-017-0079-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-017-0079-3
This article is cited by
-
Stay in touch with the endoplasmic reticulum
Science China Life Sciences (2024)
-
Comparative proteomics study of mitochondrial electron transport system modulation in SH-SY5Y cells following MPP+ versus 6-OHDA-induced neurodegeneration
Journal of Analytical Science and Technology (2023)
-
Stable isotope labeling and ultra-high-resolution NanoSIMS imaging reveal alpha-synuclein-induced changes in neuronal metabolism in vivo
Acta Neuropathologica Communications (2023)
-
Keep in touch: a perspective on the mitochondrial social network and its implication in health and disease
Cell Death Discovery (2023)
-
Cell death regulation by MAMs: from molecular mechanisms to therapeutic implications in cardiovascular diseases
Cell Death & Disease (2022)
