
Abstract
Neuroblastoma (NB) is the most common solid childhood tumor outside the brain and causes 15% of childhood cancer-related mortality. The main drivers of NB formation are neural crest cell-derived sympathoadrenal cells that undergo abnormal genetic arrangements. Moreover, NB is a complex disease that has high heterogeneity and is therefore difficult to target for successful therapy. Thus, a better understanding of NB development helps to improve treatment and increase the survival rate. One of the major causes of sporadic NB is known to be MYCN amplification and mutations in ALK (anaplastic lymphoma kinase) are responsible for familial NB. Many other genetic abnormalities can be found; however, they are not considered as driver mutations, rather they support tumor aggressiveness. Tumor cell elimination via cell death is widely accepted as a successful technique. Therefore, in this review, we provide a thorough overview of how different modes of cell death and treatment strategies, such as immunotherapy or spontaneous regression, are or can be applied for NB elimination. In addition, several currently used and innovative approaches and their suitability for clinical testing and usage will be discussed. Moreover, significant attention will be given to combined therapies that show more effective results with fewer side effects than drugs targeting only one specific protein or pathway.
Similar content being viewed by others

Introduction
Neuroblastoma (NB) is the most common solid childhood tumor outside the brain. It originates from primitive cells of the sympathetic nervous system1. NB causes 15% of childhood cancer-related mortality and overall survival rate for metastatic tumors is considerably low, 40% after 5 years2,3. Most incidences are diagnosed during the first year of life, which also gives a better prospect for the outcome, whereas older patients have a poorer diagnosis4,5. In some NB cases, spontaneous regression has also been detected; however, underlying mechanisms remain unclear6,7. Moreover, NB is a complex disease that has high genetic, biological, clinical, and morphological heterogeneity, and is therefore difficult to target for successful therapy8,9,10. Thus, NB is under thorough investigation to better understand its progression and to improve the treatment to increase the survival rate.
Several classification systems have been used in order to improve risk assessment and prognosis of NB. For example, the outcome of the disease can be assessed by the presence or absence of stroma, the degree of differentiation, and the mitosis-karyorrhexis index11. Currently, even more parameters are used for the classification of NBs, such as stage, age, histologic category, grade of tumor differentiation, the status of the MYCN oncogene, chromosome 11q status, and DNA ploidy. These are the most statistically significant and clinically relevant factors in use to describe two stages of localized (L1 and L2) and two stages of metastatic disease (M and MS)12.
The main drivers of NB formation are abnormalities in sympathoadrenal cells that derive from neural crest cells (Figure 1)13. Several germline and sporadic genomic rearrangements have been detected in NB, for example, LIN28B (encoding lin 28 homolog B)14, PHOX2B (paired-like homeobox 2b)15, ALK (anaplastic lymphoma kinase)16, GALNT14 (polypeptide N-acetylgalactosaminyltransferase 14)17, and MYCN18 (Table 1). Around 2% of NB cases appear to be hereditary, with ALK being the first gene identified to be responsible for familial NB16,19. Furthermore, MYCN oncogene amplification is found in 20% of all NB cases, especially in patients who are resistant to therapy and have poor prognosis18,20,21. More than 50% of these high-risk patients relapse even after intensive treatment22. Whole-genome sequencing has been used to identify additional mutations and genes responsible for de novo NB development, but no other specific “NB driver mutations” have been found23,24. Thus, MYCN amplification seems to be the major cause of sporadic NB and other mutations support tumor aggressiveness25. Therefore, investigation of the MYCN gene amplification is considered to be a mandatory step for treatment specification26.

The main drivers of NB formation are neural crest cell-derived sympathoadrenal cells with genetic abnormalities. Several germline and sporadic genomic rearrangements have been detected in NB, for example, in ALK (anaplastic lymphoma kinase) and MYCN genes. These changes are responsible for the suppression of differentiation and apoptosis to support rapid proliferation of the cells
In this review, we provide a thorough overview of how different modes of cell death are exploited or can be employed as treatment for NB. In addition, several novel or already clinically tested drugs against NB and their mechanisms of action are discussed. A special emphasis is also placed on combined therapies that attack many pathways and have been shown to be more effective than drugs targeting only one specific protein or pathway.
Genetic background
Anaplastic lymphoma kinase
Changes in the ALK gene are identified as being responsible for ~ 50% of familial and ~ 1% of all NBs16 (Table 1). ALK is a member of the insulin receptor superfamily of transmembrane RTKs (receptor tyrosine kinase). Mutations and amplifications of the ALK gene can lead to a constitutive activation of ALK that supports cell survival and proliferation in the peripheral neuronal and central nervous system. This can be achieved by the engagement of several pathways, such as Janus kinase–signal transducer and activator of transcription27, PI3K–AKT27 in anaplastic large cell lymphoma, and/or RAS–mitogen-activated protein kinase28 in NB.
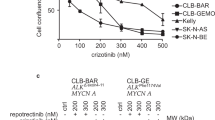
The central role of the ALK in NB development makes it a possible target for NB treatment. For example, NB cell lines with constitutively active or overexpressed ALK are susceptible to RNAi and ALK inhibitors29. For instance, crizotinib30 and entrectinib31 reduce the cells’ proliferation rate and are currently in Phase 1/2 trials (NCT00939770, NCT01606878, and NCT02650401) for relapsed or refractory NB; however, there are problems with their off-target effects and acquired resistance. Therefore, new-generation ALK inhibitors are already been developed and tested for NB therapy, for example, lorlatinib (NCT03107988)32, AZD3463 (ref. 33), and ceritinib (NCT01742286)34. In addition to reducing the proliferation rate, clinical tests have shown that most ALK inhibitors also sensitize NB cells to conventional cytotoxic drugs and their combined use is causing more prominent cell death35,36. On the other hand, this approach is helpful for only ALK-positive tumors and, due to the high heterogeneity of NB, more strategies are needed for successful treatment of NBs carrying other mutations.
MYCN
MYCN is part of the MYC family of transcription factors that regulate several cellular processes including proliferation, cell cycle, glycolysis, glutaminolysis, mitochondrial function, and biogenesis37,38,39. MYCN expression is essential for normal prenatal development and is present until a few weeks after birth40. Amplifications of the MYCN gene are known to be responsible for increased tumor growth, proliferation, and NB development (Table 1)41,42. Deregulation of MYC induces cell proliferation and apoptosis; however, this apoptotic signal is inhibited by reducing p53 activity, overexpressing anti-apoptotic proteins, or downregulating pro-apoptotic proteins43,44. Thus, a combined suppression of MYC-induced apoptosis and MYC-driven proliferative signals supports extensive tumor development.
MYCN usually has a very short half-life, but after amplification it is highly expressed and forms heterodimers with MAX to act as a transcriptional factor and support constant NB tumor growth45. Therefore, downregulation of MYCN is one possible approach to induce apoptosis, decrease NB proliferation, and/or induce neuronal differentiation46. For example, antisense oligonucleotides47 and RNAi48,49,50 have been successfully used for MYCN downregulation in NB that resulted in decreased tumor growth, cellular migration, and invasion. The described approach has proved to be effective in the laboratory; however, off-target effects and clinical delivery of these compounds to the tumor site are still problematic.
Blocking the MYCN/MAX interaction is another option for NB therapy, because unbound MAX homodimerizes and stimulates differentiation51. Several compounds blocking the heterodimerization, such as 10058-F4 (ref. 52,53) and 10074-G5 (ref. 52), have shown cell cycle arrest, apoptosis, and differentiation in vitro, and also increased survival in MYCN transgenic mice. Another approach is to inhibit bromodomain and extra-terminal domain family of transcription-regulating proteins by small molecules such as JQ1 (ref. 54), OTX015 (ref. 55), or I-BET762 (ref. 56), which lead to the suppression of MYCN transcription and proliferation. These compounds can help high-risk patients with MYCN-driven NB; however, thorough clinical testing is still needed. The role of ALK and MYCN in regulation of NB cell fate is shown on Figure 1.
Other genomic abnormalities
Overexpression and amplifications of LIN28B are very common in NB cells and can in turn lead to high MYCN expression (Table 1)14,57. Moreover, whole-genome sequencing revealed that 25% of the patients have rearrangements in TERT (encoding telomerase reverse transcriptase)58,59 promoter and 10% in transcriptional regulator ATRX (encoding the RNA helicase)23, supporting rapid cellular proliferation (Table 1). Chromosomal copy number alterations are also represented in almost all NBs, for example, more than 50% have gain of 17q (ref. 60) and 30% have loss of 1p36 and/or 11q1 (ref. 61) (Table 1). These arrangements have a strong correlation with MYCN amplification and poor prognosis. However, the function of these regions and how they regulate NB formation is still unclear60,61.
Targeting NB via stimulation of various modes of cell death
Apoptosis induction in NB therapy
Apoptosis is essential for the normal growth of an organism, being involved in early embryonic and immune system development. It also has an important role in the maintenance of normal tissue homeostasis and helps to eliminate damaged and harmful cells62. Therefore, misregulation of apoptotic pathways has an important role in cancer development, because mutations or amplifications in the oncogenes (e.g., MYC) can compromise apoptotic pathways. On the other hand, apoptosis induction is the most prominent anticancer strategy.
Targeting p53/MDM2 interaction
The members of the p53 protein family are important regulators of cell cycle and apoptosis in normal and transformed cells63. In addition, p53 as well as p73 act as tumor suppressors. Mutations in the p53 gene that control cell fate occur in more than 80% of tumor cell lines and more than 40% of human cancers64. However, abnormalities of p53 are mostly found in relapsed NB after chemotherapy, but not at the time of the diagnosis65,66. Instead, overexpressed MYCN regulates p53 and MDM2 (murine double minute 2) expression to achieve stringent control over cell death (Figure 2)67,68. Tumors such as NB, which generally have wild-type p53, are likely to induce the degradation of p53 and avoid cell death by overexpression or amplification of MDM2, which is a negative regulator and the primary E3 ubiquitin ligase for p53 (ref. 65,67,69). For instance, MYCN binds to the promoter of MDM2 to induce its expression and vice versa, suggesting that downregulation of MDM2 can also be used to decrease MYCN expression and stabilize p53 to induce apoptosis (Figure 2)67,70,71.

NB cell death can be avoided by overexpression or amplification of MDM2 (murine double minute 2), negative regulator of the p53. The p53–MDM2 pathway may be targeted by small antagonistic molecules, such as nutlin-3, MI-773/219/63, idasanutlin (RG7388), which bind to MDM2, to block its interaction with p53. MYCN also facilitates expression of p53 and FAK (focal adhesion kinase) that in turn interacts with p53 and causes its sequestering in the cytoplasm. Furthermore, MYCN-upregulated MDM2 can in turn bind to MYCN and tumor suppressor TAp73, and decrease its transcription to support resistance to the treatment. MYCN and TAp73 might also directly decrease each other expression and influence the outcome of the NB development and treatment
Understanding these peculiarities of NB and targeting the p53–MDM2 pathway may be helpful in finding better therapeutic treatments for pediatric patients with wild-type p53 (ref. 72,73). For example, small antagonistic molecules, like nutlin-3 (ref. 74,75,76), MI-773/219/63 (ref. 75), and idasanutlin (RG7388)77, which bind to MDM2 to block its interaction with p53, have shown promising results in NB. These inhibitors attenuate the proliferation of MYCN-expressing NB cells and some of them are being tested in clinical trials; however, the development of resistance, toxicity, MDM2 accumulation, and the need for wild-type p53 make the trials challenging78. In addition to the regulation of p53–MDM2, MYCN facilitates an increase in the expression of FAK (focal adhesion kinase), which interacts with p53 and causes its sequestering in the cytoplasm (Figure 2). Interrupting this binding by small molecules or peptides enables p53 to move to the nucleus to induce apoptotic cell death of in vivo breast and colon tumors79.
Furthermore, MYCN-upregulated MDM2 can similarly bind with another member of the p53 family, tumor suppressor TAp73 (p73 locus encodes two isoforms – tumor suppressor (TAp73) and putative oncogene (ΔNp73)) (Figure 2). MDM2 decreases TAp73 transcription and supports resistance to the treatment80,81. It has been discussed that besides regulating p53 and MDM2 levels, MYCN might also directly decrease TAp73 expression and support NB tumor growth82. In addition, there are results showing that overexpression of TAp73 can in turn reduce MYCN expression and induce differentiation of NB cell lines, indicating that the balance between TAp73 and MYCN levels can influence the outcome of the NB development and treatment (Fig. 2)83,84. These new approaches have led to novel combinatorial therapeutic strategies that simultaneously reduce toxicity and enhance the outcome of the treatment and are being tested in preclinical and clinical trials for NB75, melanoma85, prostate cancer86, and renal cell carcinoma87. Although bearing in mind that MYCN has many cellular targets, disrupting its interaction with one of them is probably not enough for successful treatment.
BCL-2 family
Other important apoptosis regulators are B-cell lymphoma/leukemia 2 (BCL-2) family proteins, which are divided into two groups: pro-apoptotic and anti-apoptotic proteins. The main anti-apoptotic proteins are BCL-2, BCL-xL, and myeloid cell leukemia (MCL)-1, which prevent outer mitochondrial membrane (OMM) permeabilization by binding and inhibiting pro-apoptotic proteins. Apoptosis-promoting proteins from this family can in turn be divided into two groups: BH-3 only and effector proteins. The pro-apoptotic BH-3 only proteins (Bid and Bim) respond to apoptotic stimuli and inhibit anti-apoptotic BCL-2 proteins or activate the effector proteins (BAK and BCL-2-associated X protein), which form pores in the OMM to induce cytochrome c release and apoptosis. The balance between pro- and anti-apoptotic proteins determines the fate of the cells through regulation of the mitochondrial apoptotic pathway88,89. As with p53, mutations in BCL-2 are scarce in NB, although dysregulation and increased levels of the BCL-2 gene are frequent90,91,92. Moreover, in B-cell lymphomas a link between MYC and BCL-2 expression has been described, because overexpression of MYC in tumor cells is often found together with rearrangements in the BCL-2 family to support tumor growth and suppress apoptosis93,94. Therefore, therapies that change the balance between pro- and anti-apoptotic proteins are promising strategies for tumor treatment.
One possible approach might be using conventional chemotherapeutics together with inhibitors of anti-apoptotic BCL-2 proteins (e.g., ABT-199)95, although there have been problems with modest outcome, side effects,96 and resistance in relapsed NBs97. This is due to the compensatory upregulation of the anti-apoptotic MCL-1 protein that rescues cells from apoptosis. However, when the MCL-1 inhibitor (e.g., A-1210477) is used in combination with ABT-199, successful induction of NB cell death has been demonstrated98.
Targeting cellular bioenergetics pathways
Considering the key role of mitochondria in various modes of cell death, they might be potential targets for tumor therapy. For instance, many anticancer drugs destabilize mitochondria to induce apoptotic cell death99. Rapidly proliferating tumors easily become hypoxic, which is the reason why the majority of tumors change their source of energy from mitochondrial oxidative phosphorylation (OXPHOS) to glycolysis. These cells usually have lowered amount of mitochondria and/or mutations in one or more OXPHOS complexes100,101,102. In contrast, relapsing cancer cells tend to have increased levels of OXPHOS103,104,105. The role of MYC overexpression in these processes is to increase the expression of mitochondrial complexes and hence mitochondrial respiration38. These metabolic changes help cells to survive in nutrient-deprived environments106. Therefore, to eliminate resistant tumor cells, chemotherapeutic drugs could be used in combination with electron transport chain inhibitors, such as the complex I inhibitors metformin107 or tamoxifen108, to induce leakage of electrons and excessive formation of reactive oxygen species (ROS). In addition, using non-toxic doses of the complex II blockers of the respiratory chain, such as thenoyltrifluoroacetone109 or α-tocopheryl succinate110 together with harmless doses of cytotoxic drugs, synergistically stimulates the formation of ROS and thereby increases the effectiveness of the therapy on breast cancer and NB cell lines.
Fast growth of the tumor cells and poor vascularization leads to hypoxia, which causes the activation of transcription factors, such as hypoxia-inducible factor 1 (HIF-1), that regulate the hypoxic adaptation (Figure 3)111,112,113. Specifically, HIF-1 regulates developmental and physiological pathways that facilitate O2 delivery to the cells or help cells to survive in low O2 conditions. HIF-1 is activated in a hypoxic environment that is very common in solid tumors. HIF-1 expression leads to the activation of glycolysis and angiogenesis, and correlates with aggressive tumors and poor outcome. HIF-1 is a heterodimer consisting of the O2-regulated HIF-1α subunit and a constantly expressed HIF-1β subunit114,115. HIF-1α becomes stabile in a low O2 environment and binds with HIF-1β to form an active HIF-1 complex that has both anti- and pro-apoptotic effects116,117. For instance, severe and continuous hypoxia will result in HIF-1 activation, p53 expression, and apoptosis. On the other hand, simultaneous stabilization of HIF-1 with activation of the PI3K/Akt pathway, survivin, glycolytic enzymes, p21, and/or erythropoietin can inhibit apoptosis and support NB tumor growth118,119.

MYCN overexpression facilitates the fast growth of the tumor cells, leading to poor vascularization and hypoxia. This results in the stabilization of transcription factors HIF-1, inhibition of apoptosis, and stimulation of glycolysis, angiogenesis, and rapid tumor growth. In addition, putative oncogene ΔNp73 is stabilized in O2-deficient environment and supports vascularization. However, TAp73 regulates degradation of HIF-1 and suppression of vascularization in an oxygen-independent manner. Therefore, considering the importance of HIF-1 in tumor progression, several HIF-1 inhibitors, such as topotecan, phenethyl isothiocyanate, and acriflavine, have shown promising results
Furthermore, recent data suggest that the aforementioned TAp73 also regulates the degradation of HIF-1 and the suppression of vascularization in an oxygen-independent manner (Fig. 3)120,121. Therefore, loss of TAp73 activity in MYCN-overexpressed tumors can be associated with increased HIF-1 activity and thereby the stimulation of angiogenesis in tumor cells120,122. Another isoform of p73, NH2 terminally truncated putative oncogene ΔNp73, is also involved in angiogenesis regulation (Fig. 3). In tumor cell lines, ΔNp73 is stabilized in O2-deficient conditions and activates vascularization via vascular endothelial growth factor A expression121, indicating that cellular response to hypoxic conditions and HIF-1 activity is tightly regulated by MYCN and p53 family proteins. Moreover, HIF-1 activity is also associated with low responsiveness to differentiation therapy and the downregulation of HIF-1 can improve the outcome of the NB treatment123. Therefore, taking into account the importance of HIF-1 in NB tumor progression, the search for its inhibitors, such as topotecan124 and acriflavine125, is a promising strategy. Several of these have already been shown to improve the effects of anti-angiogenic drugs in vivo.
Cancer cells modify their metabolism to support their constant proliferation. Adjustments in cancer cells’ metabolism result in excessive glycolytic activity to produce ATP, the Warburg effect, to support rapid cell proliferation. These changes are also seen in aerobic conditions, even though glycolysis generates less ATP than OXPHOS126,127. This decrease in oxygen demand helps tumor cells to survive in hypoxic conditions and continue proliferation due to excessive glycolytic activity128. Such a drastic metabolic change is attained by the activity of various oncogenes and regulatory proteins, such as MYC and HIF-1 (ref. 129,130).
Oncogenic MYC upregulates glucose import (e.g., GLUT1), glycolytic enzymes (e.g., hexokinase 2 (HK2) and PDK1), and mitochondrial biogenesis, thereby ensuring metabolic intermediates that support cell growth131,132. Elevated glucose transport into the cells and glycolysis itself can be targeted for cancer cell-specific therapy133,134. For example, glucose analog 2-DG (2-deoxy-d-glucose) that is phosphorylated by HK2 cannot be metabolized further and accumulates in the cell, leading to the inhibition of glycolysis and tumor growth 135,136,137,138. This approach has been successful in several NB cell lines139 and also in xenograft models,140 regardless of their MYCN status, indicating its potential for clinical significance. Furthermore, the clinical efficacy of 2-DG is enhanced when combined with cytotoxic drugs in breast141, head and neck142, and ovarian143 cancer cell lines.
Another hexokinase inhibitor lonidamine was under clinical trials and revealed promising results in combination therapy for ovarian cancer clinical trial144 and NB cell lines145. Furthermore, HK inhibitor 3-bromopyruvate (3-BrPA) effectively reduces cell growth of leukemia146, breast147, and colon146 cancer cells without any significant toxicity or recurrence146,147. It has been efficient when used alone or in combination with other inhibitors (e.g., rapamycin148,149) or cytotoxic drugs (e.g., platinum-based agents150 and doxorubicin151) for NB, leukemia, breast, lymphatic, colon, and hepatic cancers. There is also a modified version of 3-BrPA named 3-bromo-2-oxopropionate-1-propyl ester, which is a cell-permeable ester that has a strong effect on GLUT1- and MKI67-expressing NB cells, but is less damaging for normal cells152. In addition to HK inhibitors, small-molecule PDK (pyruvate dehydrogenase kinase) inhibitors, such as dichloroacetate (DCA)153,154, or the downregulation of lactate dehydrogenase A (LDHA)155 can also be used to reverse the glycolytic shift by directing pyruvate into mitochondria, to restore the characteristic phenotype of non-malignant cells. For example, DCA has successfully reduced lactate production, proliferation rate, cell viability, and increased respiration in NB cell lines156,157. In addition, LDHA inhibitor FX11 has successfully inhibited aerobic glycolysis and growth of NB cell lines158.
Besides increased glucose metabolism, many tumors, and especially NB, show signs of glutamine dependency159. Glutamine regulates cellular energetics, redox state, amino acid production, cell signaling, and nucleotide synthesis160,161. Therefore, glutamine addiction helps cancer cells to acquire substrates for rapid proliferation and to survive better in complex environments. In tumors, stimulation of glutaminolysis in low glucose and oxygen conditions is mainly induced by MYC, whereas MYC knockdown results in reduced glutamine metabolism in glioblastoma cell line162. Thus, removal of glutamine should lead to the death of addicted cells, whereas oxaloacetate, pyruvate, and α-ketoglutarate can rescue cells from dying, suggesting that MYC-driven glutamine metabolism is a major carbon source for the tricarboxylic acid cycle162,163,164,165. Therefore, targeting glutamine metabolism for MYC-driven tumors is a promising strategy for cancer therapy.
Glutamine depletion results in activating transcription factor 4 (ATF4)-dependent, but p53-independent, apoptosis as a result of the stimulation of expression of the pro-apoptotic BCL-2 family proteins PUMA and NOXA. Therefore, combinations of ATF4 agonists and glutaminolysis inhibitors have shown the induction of apoptosis and a decrease in NB tumor growth164. Inhibitors of glutaminase 1 by small molecules such as 986 (ref. 166) and bis-2-[5-phenylacetamido-1,2,4-thiadiazol-2-yl] ethyl sulfide167,168,169, suppressed cell growth, migration, invasion, and resistance to oxidative stress in MYC-overexpressing tumors. However, MYCN-amplified NB cells that predominantly express GLS2 might be less sensitive to these drugs164,167. Besides GLS blockers, inhibitors of glutamate dehydrogenases, such as epigallocatechin-3-gallate170, or aminotransferases, such as aminooxyacetate171, can be used to block subsequent glutamate processing. However, problems with identifying the predominant pathway in specific cancers make it difficult to predict the NB sensitivity to these drugs.
Autophagy and NB therapy
Autophagy is a catabolic survival mechanism that is activated in somatic cells under metabolic stress, to provide the cell with metabolites and to eliminate damaged organelles, protein aggregates, and infecting organisms. Extensive autophagy can also lead to cell death, but its function is not yet fully understood172,173,174,175. In many solid tumors, including NB, the outcome of the chemotherapeutic agents is also affected by the cellular stimulation/activation of autophagy, which can lead to unexpected consequences and autophagy-mediated cell survival or death176. However, there are ongoing discussions and research to better understand whether extensive activation of autophagy could be used to induce cell death or whether it should be blocked, because it helps cells to survive in extreme environments and therefore support tumor growth.
For example, one of the reasons why previously discussed ALK inhibitors may cause resistance is due to their ability to activate autophagy-mediated cell survival. This can be avoided by using ALK inhibitors together with autophagy inhibitors, such as chloroquine, which have been shown to increase cell death of ALK-positive lung cancer177,178 In addition, research on histone deacetylase 10 has shown its role in autophagy-mediated cell survival and poor outcomes in high-risk NB179. Moreover, BCL-2, a regulator of apoptosis, also controls and inhibits autophagy, which is why it seems to be one of the key factors and a potential target in balancing autophagy and apoptosis180. Therefore, inhibition of autophagy in combination with other apoptosis-inducing drugs is a potential strategy to induce apoptotic cell death of NB cells, especially in resistant tumors181,182.
Targeting PI3K/AKT/mTOR pathway
The PI3K/AKT/mTOR (mechanistic target of rapamycin) signaling pathway is an important regulator of autophagy. In NB, it correlates with a poor outcome and is shown to be upregulated by constitutively activated ALK and MYCN genes183,184,185. The PI3K/AKT/mTOR pathway is regulated by the aforementioned RTKs, which are shown to be involved in malignant NB cell transformation, when mutated and/or amplified. Therefore, several inhibitors of RTK and PI3K/AKT/mTOR pathways have also been tested for NB therapy186,187. However, there are also problems with resistance, as these inhibitors cause secondary mutations and autophagy activation that supports cell survival188,189.
Protein kinase mTOR is considered to be the main inhibitor of autophagy and controller of cellular metabolism190,191,192. Deregulation of mTOR expression is very common in tumor cells and it is targeted in many NB studies, as its inhibition destabilizes MYCN, reduces NB growth, and induces excessive autophagy activation that will result in the stimulation of cell death36,184,193. Although clinical benefits from mTOR inhibitors, when used alone, have been modest, their effectiveness for NB in combination therapies is under investigation194,195,196,197. For example, the mTOR inhibitor temsirolimus (rapamycin analog) has been tested for NB in clinical trials, in combination with standard chemotherapy and monoclonal antibodies (NCT01767194)195. In addition, the combination of mTOR inhibitors, such as dactolisib198, or INK128 (ref. 199), with ALK inhibitors or other conventional chemotherapeutics has shown the ability of the treatment to overcome drug resistance and to prevent NB tumor growth. Moreover, elevated levels of AKT are also very common in NBs185. Studies on combined AKT targeting have shown even more successful results, for example, the combination of AKT inhibitor perifosine and mTOR inhibitor temsirolimus is in clinical testing for pediatric solid tumors (NCT01049841)200. Furthermore, AKT inhibitor MK2206 in combination with etoposide or rapamycin has shown promising results in NB cell lines201. Taken together, targeting the PI3K/AKT/mTOR pathway and thereby inducing excessive autophagy can be used as a strategy for cancer therapy; however, targeting several pathways simultaneously should be used to avoid resistance to treatment.
Necroptosis induction in NB therapy
Cellular stress can activate various caspase- and p53-independent forms of cell death in normal and transformed cells. One of them is necroptosis, which is morphologically similar to inflammation and immune response caused by necrosis202. It is mediated by necrotic death receptors, their ligands, interferons, Toll-like receptors, and the necrosome complex, consisting of receptor-interacting protein kinases 1/3 (RIPK1/3) and mixed lineage kinase domain-like203,204,205,206. Necrosome formation induces mitochondrial ROS production and the release of apoptosis-inducing factor, which are thought to be important executors of necroptosis206,207. Normal cell survival is supported by the inhibition of apoptosis and necroptosis, where apoptosis induction is suppressed by FLICE-inhibitory protein inhibiting caspase-8 (ref. 208) and necroptosis induction is blocked by caspase-8-mediated cleavage of RIPK1/3 (ref. 209). Therefore, the balance between these proteins will determine whether the cell will survive or die and through which pathway. Thus, it is expected that necroptosis has an important role in several human disorders, such as neurodegenerative and inflammatory diseases210. Moreover, necroptotic cell death can be used as a novel approach to modulate antitumor immunity and apoptosis in the treatment of resistant cells211.
As many aggressive NBs do not express caspase-8 and are resistant to apoptosis, inducing necroptotic cell death to eliminate these cells is another strategy to increase the efficiency of treatments212. One way to trigger necroptosis in NB cells is through the increase of cytoplasmic Ca2+ that activates calcium-calmodulin kinase II, which in turn activates RIPK1 (ref. 213). Other agents inducing necroptosis in RIPK3-expressing NB cells are polyphyllin D214 and d-gal215. On the other hand, many NBs have a decreased expression of caspase-8 and low level of proteins involved in necroptosis, especially in the advanced stages, making them also resistant to necroptosis induction216. It is not clear why these genes are downregulated in NB, but epigenetic modifications may be the reason of this outcome. Thus, demethylating drugs and/or histone deacetylase inhibitors217,218 can be used to overcome this issue and support the use of necroptosis as a new approach for NB therapy.
Immunotherapy in NB treatment
Owing to the limitations of current therapies, many immunotherapeutic approaches can be used to induce NB cell death through redirecting the immune system to eliminate the malignant cells and to achieve long-term immunity and protection against relapse. One way is through targeting ALK-positive NBs with antibodies, to inhibit cell growth and induce cytotoxicity219,220. Antibodies can also be used to deliver immunotoxins, radioisotopes, liposomes, or nanoparticles221. This new method of drug delivery has a high potential for very specific on-the-spot effects on tumor cells, at the same time avoiding toxicity on healthy cells.
This approach is also used for other surface epitopes, because NB is derived from embryonic tissue and it expresses surface antigens that are not widespread in non-embryonic tissues, such as L1-cell adhesion molecule (L1-CAM), GD2/3 (disialoganglioside), and B7H3 (ref. 222,223,224). These antigens can be used as biomarkers to target advanced and chemotherapy-resistant NB cells with immunotherapeutic antibodies. The described strategy has shown promising results in preclinical and clinical trials with monoclonal antibodies, such as Hu3F8 (ref. 225,226,227,228) and dinutuximab229,230,231,232, on GD2-positive NB tumors. It has been shown that treatment with these antibodies will lead to cytotoxicity mediated by monocytes, macrophages, granulocytes, the complement system, and natural killer (NK) cells. As anti-GD2 antibodies act via cell-mediated cytotoxicity and NK cell reactivity, NB patients with higher immune activity have better outcomes from this treatment.233,234,235,236,237,238 This method seems to be even more effective when used in combination with cytotoxic chemotherapy, cytokines, adoptive NK cell therapy, and 13-cis-retinoic acid232,239,240,241,242,243,244,245. However, there have been problems with treatment efficiency, pain toxicity, and relapse; attempts to eliminate these issues have not yet been fully successful244. Another problem with this kind of treatment is that, generally, it does not induce immunological memory and other parts of the immune system should be used to achieve long-term effects.
For instance, there is evidence for “natural immunity” against ALK-positive NB cells. This is due to NB’s peculiarity in presenting ALK peptides on human leukocyte antigen I, which is then recognized by T cells246,247. This led to a novel strategy that uses designed and/or activated T cells to induce bio-distributed, long-term, and direct cytotoxicity, which is free of the immunosuppressive influences of the tumor. These designed T cells have a chimeric antigen receptor against GD2, L1-CAM, or ALK, and they have demonstrated safety and no pain toxicity in relapsed NB248,249,250,251,252,253. Another similar approach is to use a peptide vaccine, such as ganglidiximab254, made from the tumor proteins, to activate T cells against the NB255,256,257. These strategies are already in clinical trials and demonstrating high efficiency. However, there are several potential drawbacks with these therapies, starting with the low or altered expression of HLA and its co-stimulatory molecules on the cells, complex and expensive standardization processes, and its requirement to use disease compromised immune system246,258.
Spontaneous regression and TrKA pathway
NB is known for its spontaneous regression by differentiation or reactivated apoptosis, which can be considered as a possible strategy for improved therapy259,260. Experiments with differentiation supporting vorinostat261, a histone deacetylase inhibitor, and didymin262, a citrus-derived compound, have resulted in regression of NB in xenograft models and differentiation in relapsed NB261,262. There are also several other simple compounds, such as all-trans retinoic acid263,264,265,266,267,268,269, nitric oxide270, and phenylacetate267 that trigger the induction of differentiation and inhibition of NB growth by inducing the expression of neural differentiation genes. However, this mechanism is not clear, but there is evidence that NB spontaneous regression caused by retinoids is associated with increased expression of tropomyosin receptor kinase A (TrkA) receptors269,271.
Furthermore, spontaneous regression of NB is correlated with high expression of TrkA and its ligand nerve growth factor (NGF), which protects cells from apoptosis and directs them to differentiation, whereas NGF alone promotes apoptosis272,273,274,275,276,277. Therefore, changing the balance between TrkA and NGF expression can be used for the activation of NB differentiation and apoptosis. For example, re-expression of exogenous TrkA in NB cells guides cells to NGF-induced differentiation.274,277,278,279 Apoptotic cell death can be induced by TrkA inhibitors, like K252a (ref. 280), and GTx-186 (ref. 281) or by downregulating TrkA with miRNA-92a (ref. 282), however, these strategies are not yet clinically tested for NB. NGF can also sensitize TrkA-expressing cells for TRAIL-induced apoptosis and this effect can be further increased by using inhibitors of NF-κB and/or Mcl-1 (ref. 283). However, this approach may work better for the primary NB, but not relapsed NB, which often has mutations in this regulatory pathway.
Another Trk family protein kinase is TrkB, whose expression is correlated with poor NB prognosis and MYCN amplification. For example, TrkB ligands, such as BDNF and NT-4/5, are distributed via autocrine or paracrine signaling to support overall NB viability, drug resistance, and angiogenesis of TrkB-positive tumors284,285,286. Therefore, targeting TrkB may reduce the malignancy of NB with dysregulated TrkB, which can be achieved by the TrkB inhibitors GNF-4256 (ref. 287) or AZD6918 (ref. 288), which have shown promising results alone and in combination in a xenograft mouse model.
Moreover, expression of a homeobox gene HOXC9 is associated with a favorable prognostic outcome and is known as a marker of spontaneous regression in infant NBs, whereas its downregulation is present in advanced-stage NBs. Therefore, re-expression of HOXC9 can be used to induce NB regression or activation of apoptotic cell death in NB cell lines289,290. Based on all of the aforementioned information on spontaneous regression in NB, it is not clear how it is regulated. Regression seems to be as complex mechanism as all the other cellular pathways and it can include a variety of cross-talking cell death mechanisms.
Conclusion
Therapeutics inducing different modes of cell death, mainly apoptosis, have been proved to be successful, but sometimes they demonstrate a modest efficiency and side effects. The main problem with stimulating apoptosis in tumor cells is their ability to compensate for pro-apoptotic signals via upregulating anti-apoptotic agents. Therefore, searching new strategies is crucial to achieve improved outcome of NB therapy. One way to enhance the treatment is to understand better the genetic and metabolic background of NB. This in turn can be used for more specific and even personalized therapy, thereby improving the outcome of the treatment. Moreover, recent developments in NB treatment are directed towards combined therapies that target many pathways, not just different sites of one pathway. Another promising and clinically tested approach is immunotherapy, which can be used to induce NB cell death through redirecting the immune system to eliminate the malignant cells and to achieve long-term immunity and avoid relapse. However, there are several potential drawbacks, starting with the requirement to use healthy and functional immune system, as well as difficult and expensive standardization processes. Thus, there is no easy way to overcome this complex and heterogeneous disease, but step-by-step improvements are bringing us closer to prolonged survival and gain in life quality.
References
Brodeur, G. M. Neuroblastoma: biological insights into a clinical enigma. Nat. Rev. Cancer 3, 203–216 (2003).
Smith, M. A. et al. Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J. Clin. Oncol. 28, 2625–2634 (2010).
Spix, C., Pastore, G., Sankila, R., Stiller, C. A. & Steliarova-Foucher, E. Neuroblastoma incidence and survival in European children (1978-1997): report from the Automated Childhood Cancer Information System project. Eur. J. Cancer 42, 2081–2091 (2006).
Gustafsson G. K. P., Heyman M. (eds). ChildhoodCancer Incidence and Survival in Sweden 1984-2010. Swedish Childhood Cancer Registry. 2013.
Maris, J. M. Recent advances in neuroblastoma. N. Engl. J. Med. 362, 2202–2211 (2010).
Cozzi, D. A. et al. Long-term follow-up of the “wait and see” approach to localized perinatal adrenal neuroblastoma. World J. Surg. 37, 459–465 (2013).
Kushner, B. H. et al. Survival from locally invasive or widespread neuroblastoma without cytotoxic therapy. J. Clin. Oncol. 14, 373–381 (1996).
Bosse, K. R. & Maris, J. M. Advances in the translational genomics of neuroblastoma: From improving risk stratification and revealing novel biology to identifying actionable genomic alterations. Cancer 122, 20–33 (2016).
Luksch, R. et al. Neuroblastoma (Peripheral neuroblastic tumours). Crit. Rev. Oncol. Hematol. 107, 163–181 (2016).
Pinto, N. R. et al. Advances in Risk Classification and Treatment Strategies for Neuroblastoma. J. Clin. Oncol. 33, 3008–3017 (2015).
Shimada, H. et al. Histopathologic prognostic factors in neuroblastic tumors: definition of subtypes of ganglioneuroblastoma and an age-linked classification of neuroblastomas. J Natl Cancer Inst. 73, 405–416 (1984).
Cohn, S. L. et al. TheInternational Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J. Clin. Oncol. 27, 289–297 (2009).
Park, J. R., Eggert, A. & Caron, H. Neuroblastoma: biology, prognosis, and treatment. Hematol. Oncol. Clin. North. Am. 24, 65–86 (2010).
Diskin, S. J. et al. Common variation at 6q16 within HACE1 and LIN28B influences susceptibility to neuroblastoma. Nat. Genet. 44, 1126–1130 (2012).
van Limpt, V. et al. The Phox2B homeobox gene is mutated in sporadic neuroblastomas. Oncogene 23, 9280–9288 (2004).
Mosse, Y. P. et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455, 930–935 (2008).
De Mariano, M. et al. Identification of GALNT14 as a novel neuroblastoma predisposition gene. Oncotarget 6, 26335–26346 (2015).
Schwab, M. et al. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature. 305, 245–248 (1983).
Ogawa, S., Takita, J., Sanada, M. & Hayashi, Y. Oncogenic mutations of ALK in neuroblastoma. Cancer Sci. 102, 302–308 (2011).
Brodeur, G. M. Neuroblastoma: clinical significance of genetic abnormalities. Cancer Surv. 9, 673–688 (1990).
Wang, L. L. et al. Augmented expression of MYC and/or MYCN protein defines highly aggressive MYC-driven neuroblastoma: a Children’s Oncology Group study. Br. J. Cancer 113, 57–63 (2015).
Irwin, M. S. & Park, J. R. Neuroblastoma: paradigm for precision medicine. Pediatr. Clin. North. Am. 62, 225–256 (2015).
Molenaar, J. J. et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483, 589–593 (2012).
Pugh, T. J. et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 45, 279–284 (2013).
Zhu, S. et al. Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer Cell. 21, 362–373 (2012).
Ambros, I. M. et al. Quality assessment of genetic markers used for therapy stratification. J. Clin. Oncol. 21, 2077–2084 (2003).
Palmer, R. H., Vernersson, E., Grabbe, C. & Hallberg, B. Anaplastic lymphoma kinase: signalling in development and disease. Biochem. J. 420, 345–361 (2009).
Eleveld, T. F. et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 47, 864–871 (2015).
Bresler, S. C. et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 26, 682–694 (2014).
Mosse, Y. P. et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 14, 472–480 (2013).
Iyer, R. et al. Entrectinib is a potent inhibitor of Trk-driven neuroblastomas in a xenograft mouse model. Cancer Lett. 372, 179–186 (2016).
Infarinato, N. R. et al. The ALK/ROS1 Inhibitor PF-06463922 Overcomes Primary Resistance to Crizotinib in ALK-Driven Neuroblastoma. Cancer Discov. 6, 96–107 (2016).
Wang, Y. et al. Novel ALK inhibitor AZD3463 inhibits neuroblastoma growth by overcoming crizotinib resistance and inducing apoptosis. Sci. Rep. 6, 19423 (2016).
Nishio, M. et al. Phase I Study of Ceritinib (LDK378) in Japanese patients with advanced, anaplastic lymphoma kinase-rearranged non-small-cell lung cancer or other tumors. J. Thorac. Oncol. 10, 1058–1066 (2015).
Krytska, K. et al. Crizotinib synergizes with chemotherapy in preclinical models of neuroblastoma. Clin. Cancer Res. 22, 948–960 (2016).
Moore, N. F. et al. Molecular rationale for the use of PI3K/AKT/mTOR pathway inhibitors in combination with crizotinib in ALK-mutated neuroblastoma. Oncotarget 5, 8737–8749 (2014).
Grandori, C., Cowley, S. M., James, L. P. & Eisenman, R. N. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 16, 653–699 (2000).
Graves, J. A. et al. Mitochondrial structure, function and dynamics are temporally controlled by c-Myc. PLoS ONE 7, e37699 (2012).
Huang, M. & Weiss, W. A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 3, a014415 (2013).
Zimmerman, K. A. et al. Differential expression of myc family genes during murine development. Nature 319, 780–783 (1986).
Corvi, R., Amler, L. C., Savelyeva, L., Gehring, M. & Schwab, M. MYCN is retained in single copy at chromosome 2 band p23-24 during amplification in human neuroblastoma cells. Proc. Natl Acad. Sci. USA 91, 5523–5527 (1994).
Schwab, M. et al. Chromosome localization in normal human cells and neuroblastomas of a gene related to c-myc. Nature 308, 288–291 (1984).
Eischen, C. M., Weber, J. D., Roussel, M. F., Sherr, C. J. & Cleveland, J. L. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 13, 2658–2669 (1999).
Evan, G. I. et al. Induction of apoptosis in fibroblasts by c-myc protein. Cell 69, 119–128 (1992).
Wenzel, A. & Schwab, M. The mycN/max protein complex in neuroblastoma. Short Rev. Eur. J. Cancer 31A, 516–519 (1995).
Westermark, U. K., Wilhelm, M., Frenzel, A. & Henriksson, M. A. The MYCN oncogene and differentiation in neuroblastoma. Semin. Cancer Biol. 21, 256–266 (2011).
Burkhart, C. A. et al. Effects of MYCN antisense oligonucleotide administration on tumorigenesis in a murine model of neuroblastoma. J. Natl Cancer Inst. 95, 1394–1403 (2003).
Buechner, J. et al. Tumour-suppressor microRNAs let-7 and mir-101 target the proto-oncogene MYCN and inhibit cell proliferation in MYCN-amplified neuroblastoma. Br. J. Cancer 105, 296–303 (2011).
Kang, J. H. et al. MYCN silencing induces differentiation and apoptosis in human neuroblastoma cells. Biochem. Biophys. Res. Commun. 351, 192–197 (2006).
Lynch, J. et al. MiRNA-335 suppresses neuroblastoma cell invasiveness by direct targeting of multiple genes from the non-canonical TGF-beta signalling pathway. Carcinogenesis 33, 976–985 (2012).
Albanus, R. D. et al. Reverse engineering the neuroblastoma regulatory network uncovers MAX as one of the master regulators of tumor progression. PLoS ONE 8, e82457 (2013).
Muller, I. et al. Targeting of the MYCN protein with small molecule c-MYC inhibitors. PLoS ONE 9, e97285 (2014).
Zirath, H. et al. MYC inhibition induces metabolic changes leading to accumulation of lipid droplets in tumor cells. Proc. Natl Acad. Sci. USA 110, 10258–10263 (2013).
Puissant, A. et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 3, 308–323 (2013).
Henssen, A. et al. Targeting MYCN-driven transcription by BET-bromodomain inhibition. Clin. Cancer Res. 22, 2470–2481 (2016).
Wyce, A. et al. BET inhibition silences expression of MYCN and BCL2 and induces cytotoxicity in neuroblastoma tumor models. PLoS ONE 8, e72967 (2013).
Molenaar, J. J. et al. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nat. Genet. 44, 1199–1206 (2012).
Peifer, M. et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 526, 700–704 (2015).
Valentijn, L. J. et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat. Genet. 47, 1411–1414 (2015).
Bown, N. et al. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. N. Engl. J. Med. 340, 1954–1961 (1999).
Attiyeh, E. F. et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N. Engl. J. Med. 353, 2243–2253 (2005).
Kerr, J. F., Wyllie, A. H. & Currie, A. R. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257 (1972).
Murray-Zmijewski, F., Lane, D. P. & Bourdon, J. C. p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ. 13, 962–972 (2006).
Brown, C. J., Lain, S., Verma, C. S., Fersht, A. R. & Lane, D. P. Awakening guardian angels: drugging the p53 pathway. Nat. Rev. Cancer 9, 862–873 (2009).
Carr-Wilkinson, J. et al. High frequency of p53/MDM2/p14ARF pathway abnormalities in relapsed neuroblastoma. Clin. Cancer Res. 16, 1108–1118 (2010).
Vogan, K. et al. Absence of p53 gene mutations in primary neuroblastomas. Cancer Res. 53, 5269–5273 (1993).
Slack, A. et al. The p53 regulatory gene MDM2 is a direct transcriptional target of MYCN in neuroblastoma. Proc. Natl Acad. Sci. USA 102, 731–736 (2005).
Tweddle, D. A. et al. The p53 pathway and its inactivation in neuroblastoma. Cancer Lett. 197, 93–98 (2003).
Corvi, R. et al. Non-syntenic amplification of MDM2 and MYCN in human neuroblastoma. Oncogene 10, 1081–1086 (1995).
Chen, L. et al. p53 is a direct transcriptional target of MYCN in neuroblastoma. Cancer Res. 70, 1377–1388 (2010).
Gu, L. et al. MDM2 regulates MYCN mRNA stabilization and translation in human neuroblastoma cells. Oncogene 31, 1342–1353 (2012).
Barone, G. et al. MDM2-p53 interaction in paediatric solid tumours: preclinical rationale, biomarkers and resistance. Curr. Drug Targets 15, 114–123 (2014).
Wolter, J., Angelini, P. & Irwin, M. p53 family: Therapeutic targets in neuroblastoma. Future Oncol. 6, 429–444 (2010).
Barbieri, E. et al. MDM2 inhibition sensitizes neuroblastoma to chemotherapy-induced apoptotic cell death. Mol. Cancer Ther. 5, 2358–2365 (2006).
Gamble, L. D., Kees, U. R., Tweddle, D. A. & Lunec, J. MYCN sensitizes neuroblastoma to the MDM2-p53 antagonists Nutlin-3 and MI-63. Oncogene 31, 752–763 (2012).
Patterson, D. M. et al. Effect of MDM2 and vascular endothelial growth factor inhibition on tumor angiogenesis and metastasis in neuroblastoma. Angiogenesis 14, 255–266 (2011).
Lakoma A. et al. The MDM2 small-molecule inhibitor RG7388 leads to potent tumor inhibition in p53 wild-type neuroblastoma. Cell Death Discov. 2015;1,15026.
Michaelis, M. et al. Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis. 2, e243 (2011).
Golubovskaya, V. M. et al. A small-molecule inhibitor, 5’-O-tritylthymidine, targets FAK and Mdm-2 interaction, and blocks breast and colon tumorigenesis in vivo. Anticancer Agents Med. Chem. 13, 532–545 (2013).
Balint, E., Bates, S. & Vousden, K. H. Mdm2 binds p73 alpha without targeting degradation. Oncogene 18, 3923–3929 (1999).
Zeng, X. et al. MDM2 suppresses p73 function without promoting p73 degradation. Mol. Cell Biol. 19, 3257–3266 (1999).
Zhu, X. et al. N-myc modulates expression of p73 in neuroblastoma. Neoplasia 4, 432–439 (2002).
De Laurenzi, V. et al. Induction of neuronal differentiation by p73 in a neuroblastoma cell line. J. Biol. Chem. 275, 15226–15231 (2000).
Horvilleur, E. et al. p73alpha isoforms drive opposite transcriptional and post-transcriptional regulation of MYCN expression in neuroblastoma cells. Nucleic Acids Res. 36, 4222–4232 (2008).
de Lange, J. et al. Synergistic growth inhibition based on small-molecule p53 activation as treatment for intraocular melanoma. Oncogene 31, 1105–1116 (2012).
Tovar, C. et al. MDM2 antagonists boost antitumor effect of androgen withdrawal: implications for therapy of prostate cancer. Mol. Cancer 10, 49 (2011).
Vatsyayan, R., Singhal, J., Nagaprashantha, L. D., Awasthi, S. & Singhal, S. S. Nutlin-3 enhances sorafenib efficacy in renal cell carcinoma. Mol. Carcinog. 52, 39–48 (2013).
Cory, S. & Adams, J. M. The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2, 647–656 (2002).
Ola, M. S., Nawaz, M. & Ahsan, H. Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Mol. Cell Biochem. 351, 41–58 (2011).
Castle, V. P. et al. Expression of the apoptosis-suppressing protein bcl-2, in neuroblastoma is associated with unfavorable histology and N-myc amplification. Am. J. Pathol. 143, 1543–1550 (1993).
Goldsmith, K. C. et al. Mitochondrial Bcl-2 family dynamics define therapy response and resistance in neuroblastoma. Cancer Res. 72, 2565–2577 (2012).
Krajewski, S. et al. Immunohistochemical analysis of Bcl-2, Bcl-X, Mcl-1, and Bax in tumors of central and peripheral nervous system origin. Am. J. Pathol. 150, 805–814 (1997).
Martin-Subero, J. I. et al. Amplification of IGH/MYC fusion in clinically aggressive IGH/BCL2-positive germinal center B-cell lymphomas. Genes Chromosomes Cancer 43, 414–423 (2005).
Vita, M. & Henriksson, M. The Myc oncoprotein as a therapeutic target for human cancer. Semin. Cancer Biol. 16, 318–330 (2006).
Delbridge, A. R. & Strasser, A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 22, 1071–1080 (2015).
Souers, A. J. et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 19, 202–208 (2013).
Tanos, R., Karmali, D., Nalluri, S. & Goldsmith, K. C. Select Bcl-2 antagonism restores chemotherapy sensitivity in high-risk neuroblastoma. BMC Cancer 16, 97 (2016).
Bate-Eya, L. T. et al. High efficacy of the BCL-2 inhibitor ABT199 (venetoclax) in BCL-2 high-expressing neuroblastoma cell lines and xenografts and rational for combination with MCL-1 inhibition. Oncotarget 7, 27946–27958 (2016).
Wang, S. S. et al. Destabilization of MYC/MYCN by the mitochondrial inhibitors, metaiodobenzylguanidine, metformin and phenformin. Int. J. Mol. Med. 33, 35–42 (2014).
Calabrese, C. et al. Respiratory complex I is essential to induce a Warburg profile in mitochondria-defective tumor cells. Cancer Metab. 1, 11 (2013).
Feichtinger, R. G. et al. Alterations of oxidative phosphorylation complexes in astrocytomas. Glia 62, 514–525 (2014).
Meierhofer, D. et al. Decrease of mitochondrial DNA content and energy metabolism in renal cell carcinoma. Carcinogenesis 25, 1005–1010 (2004).
LeBleu, V. S. et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 16, 992–1003 (2014). 1-15.
Tan, A. S. et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 21, 81–94 (2015).
Viale, A. et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514, 628–632 (2014).
Caino, M. C. et al. Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. J. Clin. Invest. 123, 2907–2920 (2013).
Hirsch, H. A., Iliopoulos, D., Tsichlis, P. N. & Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 69, 7507–7511 (2009).
Moreira, P. I., Custodio, J., Moreno, A., Oliveira, C. R. & Santos, M. S. Tamoxifen and estradiol interact with the flavin mononucleotide site of complex I leading to mitochondrial failure. J. Biol. Chem. 281, 10143–10152 (2006).
Kruspig, B., Valter, K., Skender, B., Zhivotovsky, B. & Gogvadze, V. Targeting succinate:ubiquinone reductase potentiates the efficacy of anticancer therapy. Biochim. Biophys. Acta 1863, 2065–2071 (2016).
Dong, L. F. et al. Alpha-tocopheryl succinate induces apoptosis by targeting ubiquinone-binding sites in mitochondrial respiratory complex II. Oncogene 27, 4324–4335 (2008).
Gilkes, D. M. & Semenza, G. L. Role of hypoxia-inducible factors in breast cancer metastasis. Future Oncol. 9, 1623–1636 (2013).
Ji, R. C. Hypoxia and lymphangiogenesis in tumor microenvironment and metastasis. Cancer Lett. 346, 6–16 (2014).
Wang, Q. et al. Prognosis value of HIF-1alpha expression in patients with non-small cell lung cancer. Gene 541, 69–74 (2014).
Salceda, S. & Caro, J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J. Biol. Chem. 272, 22642–22647 (1997).
Wang, G. L., Jiang, B. H., Rue, E. A. & Semenza, G. L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl Acad. Sci. USA 92, 5510–5514 (1995).
Cummins, E. P. & Taylor, C. T. Hypoxia-responsive transcription factors. Pflugers Arch. 450, 363–371 (2005).
Licausi, F. et al. Hypoxia responsive gene expression is mediated by various subsets of transcription factors and miRNAs that are determined by the actual oxygen availability. New Phytol. 190, 442–456 (2011).
Chandel, N. S. et al. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl Acad. Sci. USA 95, 11715–11720 (1998).
Zhang, B., Yin, C. P., Zhao, Q. & Yue, S. W. Upregulation of HIF-1alpha by hypoxia protect neuroblastoma cells from apoptosis by promoting survivin expression. Asian Pac. J. Cancer Prev. 15, 8251–8257 (2014).
Stantic, M. et al. TAp73 suppresses tumor angiogenesis through repression of proangiogenic cytokines and HIF-1alpha activity. Proc. Natl Acad. Sci. USA 112, 220–225 (2015).
Dulloo, I., Hooi, P. B. & Sabapathy, K. Hypoxia-induced DNp73 stabilization regulates Vegf-A expression and tumor angiogenesis similar to TAp73. Cell Cycle 14, 3533–3539 (2015).
Amelio, I. et al. TAp73 opposes tumor angiogenesis by promoting hypoxia-inducible factor 1alpha degradation. Proc. Natl Acad. Sci. USA 112, 226–231 (2015).
Edsjo, A., Holmquist, L. & Pahlman, S. Neuroblastoma as an experimental model for neuronal differentiation and hypoxia-induced tumor cell dedifferentiation. Semin. Cancer Biol. 17, 248–256 (2007).
Puppo, M. et al. Topotecan inhibits vascular endothelial growth factor production and angiogenic activity induced by hypoxia in human neuroblastoma by targeting hypoxia-inducible factor-1alpha and -2alpha. Mol. Cancer Ther. 7, 1974–1984 (2008).
Lee, K. et al. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc. Natl Acad. Sci. USA 106, 17910–17915 (2009).
Vander Heiden, M. G., Cantley, L. C. & Thompson, C. B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009).
Warburg, O., Wind, F. & Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 8, 519–530 (1927).
Kruspig, B., Zhivotovsky, B. & Gogvadze, V. Mitochondrial substrates in cancer: drivers or passengers? Mitochondrion 19 Pt A, 8–19 (2014).
Cantor, J. R. & Sabatini, D. M. Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2, 881–898 (2012).
Dang, C. V. Links between metabolism and cancer. Genes Dev. 26, 877–890 (2012).
Osthus, R. C. et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 275, 21797–21800 (2000).
Zhang, H. et al. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 11, 407–420 (2007).
Zhao, F. Q. & Keating, A. F. Functional properties and genomics of glucose transporters. Curr. Genomics 8, 113–128 (2007).
Yeluri, S., Madhok, B., Prasad, K. R., Quirke, P. & Jayne, D. G. Cancer’s craving for sugar: an opportunity for clinical exploitation. J. Cancer Res. Clin. Oncol. 135, 867–877 (2009).
Maher, J. C., Krishan, A. & Lampidis, T. J. Greater cell cycle inhibition and cytotoxicity induced by 2-deoxy-D-glucose in tumor cells treated under hypoxic vs aerobic conditions. Cancer Chemother. Pharmacol. 53, 116–122 (2004).
Pelicano, H., Martin, D. S., Xu, R. H. & Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 25, 4633–4646 (2006).
Woodward, G. E. & Hudson, M. T. The effect of 2-desoxy-D-glucose on glycolysis and respiration of tumor and normal tissues. Cancer Res. 14, 599–605 (1954).
Wick, A. N., Drury, D. R., Nakada, H. I. & Wolfe, J. B. Localization of the primary metabolic block produced by 2-deoxyglucose. J. Biol. Chem. 224, 963–969 (1957).
Chuang, J. H. et al. 2-Deoxyglucose treatment complements the cisplatin- or BH3-only mimetic-induced suppression of neuroblastoma cell growth. Int. J. Biochem. Cell Biol. 45, 944–951 (2013).
Huang, C. C. et al. Glycolytic inhibitor 2-deoxyglucose simultaneously targets cancer and endothelial cells to suppress neuroblastoma growth in mice. Dis. Model Mech. 8, 1247–1254 (2015).
Zhang, F. & Aft, R. L. Chemosensitizing and cytotoxic effects of 2-deoxy-D-glucose on breast cancer cells. J. Cancer Res. Ther. 5(Suppl 1), S41–S43 (2009).
Simons, A. L., Ahmad, I. M., Mattson, D. M., Dornfeld, K. J. & Spitz, D. R. 2-Deoxy-D-glucose combined with cisplatin enhances cytotoxicity via metabolic oxidative stress in human head and neck cancer cells. Cancer Res. 67, 3364–3370 (2007).
Yamada, M. et al. Cellular sensitization to cisplatin and carboplatin with decreased removal of platinum-DNA adduct by glucose-regulated stress. Cancer Chemother. Pharmacol. 44, 59–64 (1999).
De Lena, M. et al. Paclitaxel, cisplatin and lonidamine in advanced ovarian cancer. A phase II study. Eur. J. Cancer 37, 364–368 (2001).
Hagenbuchner, J., Kuznetsov, A. V., Obexer, P. & Ausserlechner, M. J. BIRC5/Survivin enhances aerobic glycolysis and drug resistance by altered regulation of the mitochondrial fusion/fission machinery. Oncogene 32, 4748–4757 (2013).
Xu, R. H. et al. Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 65, 613–621 (2005).
Liu, X. H., Zheng, X. F. & Wang, Y. L. Inhibitive effect of 3-bromopyruvic acid on human breast cancer MCF-7 cells involves cell cycle arrest and apoptotic induction. Chin. Med. J. (Engl). 122, 1681–1685 (2009).
Levy, A. G. et al. The combination of the novel glycolysis inhibitor 3-BrOP and rapamycin is effective against neuroblastoma. Invest. New Drugs 30, 191–199 (2012).
Xu, R. H. et al. Synergistic effect of targeting mTOR by rapamycin and depleting ATP by inhibition of glycolysis in lymphoma and leukemia cells. Leukemia 19, 2153–2158 (2005).
Ihrlund, L. S., Hernlund, E., Khan, O. & Shoshan, M. C. 3-Bromopyruvate as inhibitor of tumour cell energy metabolism and chemopotentiator of platinum drugs. Mol. Oncol. 2, 94–101 (2008).
Bean, J. F. et al. Glycolysis inhibition and its effect in doxorubicin resistance in neuroblastoma. J. Pediatr. Surg. 49, 981–984 (2014). discussion 4.
Matsushita, K. et al. Glycolysis inhibitors as a potential therapeutic option to treat aggressive neuroblastoma expressing GLUT1. J. Pediatr. Surg. 47, 1323–1330 (2012).
Dunbar, E. M. et al. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Invest. New. Drugs 32, 452–464 (2014).
Michelakis, E. D. et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2, 31ra4 (2010).
Fantin, V. R., St-Pierre, J. & Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 9, 425–434 (2006).
Niewisch, M. R. et al. Influence of dichloroacetate (DCA) on lactate production and oxygen consumption in neuroblastoma cells: is DCA a suitable drug for neuroblastoma therapy? Cell Physiol. Biochem. 29, 373–380 (2012).
Vella, S., Conti, M., Tasso, R., Cancedda, R. & Pagano, A. Dichloroacetate inhibits neuroblastoma growth by specifically acting against malignant undifferentiated cells. Int. J. Cancer 130, 1484–1493 (2012).
Rellinger, E. J. et al. FX11 inhibits aerobic glycolysis and growth of neuroblastoma cells. Surgery 161, 747–752 (2017).
Wise, D. R. & Thompson, C. B. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem. Sci. 35, 427–433 (2010).
DeBerardinis, R. J. et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl Acad. Sci. USA 104, 19345–19350 (2007).
Le, A. et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 15, 110–121 (2012).
Wise, D. R. et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl Acad. Sci. USA 105, 18782–18787 (2008).
Dang, C. V. Therapeutic targeting of Myc-reprogrammed cancer cell metabolism. Cold Spring Harb. Symp. Quant. Biol. 76, 369–374 (2011).
Qing, G. et al. ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell 22, 631–644 (2012).
Yuneva, M., Zamboni, N., Oefner, P., Sachidanandam, R. & Lazebnik, Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J. Cell Biol. 178, 93–105 (2007).
Wang, J. B. et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 18, 207–219 (2010).
Robinson, M. M. et al. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem. J. 406, 407–414 (2007).
Seltzer, M. J. et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 70, 8981–8987 (2010).
Ulanet, D. B. et al. Mesenchymal phenotype predisposes lung cancer cells to impaired proliferation and redox stress in response to glutaminase inhibition. PLoS ONE 9, e115144 (2014).
Yang, C. S., Wang, X., Lu, G. & Picinich, S. C. Cancer prevention by tea: animal studies, molecular mechanisms and human relevance. Nat. Rev. Cancer 9, 429–439 (2009).
Thornburg, J. M. et al. Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res. 10, R84 (2008).
Berardi, D. E., Campodonico, P. B., Diaz Bessone, M. I., Urtreger, A. J. & Todaro, L. B. Autophagy: friend or foe in breast cancer development, progression, and treatment. Int. J. Breast Cancer 2011, 595092 (2011).
Kroemer, G. et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 16, 3–11 (2009).
Shen, S., Kepp, O. & Kroemer, G. The end of autophagic cell death? Autophagy 8, 1–3 (2012).
Wang, S. Y., Yu, Q. J., Zhang, R. D. & Liu, B. Core signaling pathways of survival/death in autophagy-related cancer networks. Int. J. Biochem. Cell. Biol. 43, 1263–1266 (2011).
Levine, B. & Kroemer, G. Autophagy in the pathogenesis of disease. Cell 132, 27–42 (2008).
Ji, C. et al. Induction of autophagy contributes to crizotinib resistance in ALK-positive lung cancer. Cancer Biol. Ther. 15, 570–577 (2014).
Aveic, S. & Tonini, G. P. Resistance to receptor tyrosine kinase inhibitors in solid tumors: can we improve the cancer fighting strategy by blocking autophagy? Cancer Cell. Int. 16, 62 (2016).
Oehme, I. et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc. Natl Acad. Sci. USA 110, E2592–E2601 (2013).
Xu, H. D. et al. The pro-survival role of autophagy depends on Bcl-2 under nutrition stress conditions. PLoS ONE 8, e63232 (2013).
Aveic, S. et al. Combating autophagy is a strategy to increase cytotoxic effects of novel ALK inhibitor entrectinib in neuroblastoma cells. Oncotarget 7, 5646–5663 (2016).
Mohan, N., Chakrabarti, M., Banik, N. L. & Ray, S. K. Combination of LC3 shRNA plasmid transfection and genistein treatment inhibited autophagy and increased apoptosis in malignant neuroblastoma in cell culture and animal models. PLoS ONE 8, e78958 (2013).
Izycka-Swieszewska, E. et al. Analysis of PI3K/AKT/mTOR signalling pathway in high risk neuroblastic tumours. Pol. J. Pathol. 61, 192–198 (2010).
Johnsen, J. I. et al. Inhibitors of mammalian target of rapamycin downregulate MYCN protein expression and inhibit neuroblastoma growth in vitro and in vivo. Oncogene 27, 2910–2922 (2008).
Opel, D., Poremba, C., Simon, T., Debatin, K. M. & Fulda, S. Activation of Akt predicts poor outcome in neuroblastoma. Cancer Res. 67, 735–745 (2007).
Mei, H., Wang, Y., Lin, Z. & Tong, Q. The mTOR signaling pathway in pediatric neuroblastoma. Pediatr. Hematol. Oncol. 30, 605–615 (2013).
Segerstrom, L. et al. Effects of small molecule inhibitors of PI3K/Akt/mTOR signaling on neuroblastoma growth in vitro and in vivo. Int. J. Cancer 129, 2958–2965 (2011).
Broekman, F., Giovannetti, E. & Peters, G. J. Tyrosine kinase inhibitors: multi-targeted or single-targeted? World J. Clin. Oncol. 2, 80–93 (2011).
Oxnard, G. R. et al. New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin. Cancer Res. 17, 5530–5537 (2011).
Ganley, I. G. et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 284, 12297–12305 (2009).
Hosokawa, N. et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 20, 1981–1991 (2009).
Jung, C. H. et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 20, 1992–2003 (2009).
Vaughan, L. et al. Inhibition of mTOR-kinase destabilizes MYCN and is a potential therapy for MYCN-dependent tumors. Oncotarget 7, 57525–57544 (2016).
Bagatell, R. et al. Phase 1 trial of temsirolimus in combination with irinotecan and temozolomide in children, adolescents and young adults with relapsed or refractory solid tumors: a Children’s Oncology Group Study. Pediatr. Blood Cancer 61, 833–839 (2014).
Geoerger, B. et al. Phase II trial of temsirolimus in children with high-grade glioma, neuroblastoma and rhabdomyosarcoma. Eur. J. Cancer 48, 253–262 (2012).
Morgenstern, D. A. et al. Phase I study of vinblastine and sirolimus in pediatric patients with recurrent or refractory solid tumors. Pediatr. Blood Cancer 61, 128–133 (2014).
Spunt, S. L. et al. Phase I study of temsirolimus in pediatric patients with recurrent/refractory solid tumors. J. Clin. Oncol. 29, 2933–2940 (2011).
Westhoff, M. A. et al. Sequential dosing in chemosensitization: targeting the PI3K/Akt/mTOR pathway in neuroblastoma. PLoS ONE 8, e83128 (2013).
Zhang, H. et al. mTOR ATP-competitive inhibitor INK128 inhibits neuroblastoma growth via blocking mTORC signaling. Apoptosis 20, 50–62 (2015).
Rodrik-Outmezguine, V. S. et al. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 1, 248–259 (2011).
Li, Z., Yan, S., Attayan, N., Ramalingam, S. & Thiele, C. J. Combination of an allosteric Akt Inhibitor MK-2206 with etoposide or rapamycin enhances the antitumor growth effect in neuroblastoma. Clin. Cancer Res. 18, 3603–3615 (2012).
Pasparakis, M. & Vandenabeele, P. Necroptosis and its role in inflammation. Nature 517, 311–320 (2015).
Cho, Y. S. et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123 (2009).
Holler, N. et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1, 489–495 (2000).
Sun, L. et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 (2012).
Vandenabeele, P., Galluzzi, L., Vanden Berghe, T. & Kroemer, G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell. Biol. 11, 700–714 (2010).
Polito, L. et al. Apoptosis and necroptosis induced by stenodactylin in neuroblastoma cells can be completely prevented through caspase inhibition plus catalase or necrostatin-1. Phytomedicine 23, 32–41 (2016).
Oberst, A. et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367 (2011).
Duprez, L. et al. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 35, 908–918 (2011).
Zhou, W. & Yuan, J. Necroptosis in health and diseases. Semin. Cell. Dev. Biol. 35, 14–23 (2014).
Inoue, H. & Tani, K. Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments. Cell Death Differ. 21, 39–49 (2014).
Zauli, G. et al. TRAIL activates a caspase 9/7-dependent pathway in caspase 8/10-defective SK-N-SH neuroblastoma cells with two functional end points: induction of apoptosis and PGE2 release. Neoplasia 5, 457–466 (2003).
Nomura, M., Ueno, A., Saga, K., Fukuzawa, M. & Kaneda, Y. Accumulation of cytosolic calcium induces necroptotic cell death in human neuroblastoma. Cancer Res. 74, 1056–1066 (2014).
Watanabe, S. et al. a steroidal saponin in Paris polyphylla, induces apoptosis and necroptosis cell death of neuroblastoma cells. Pediatr. Surg. Int. 33, 713–719 (2017).
Li, N. et al. D-galactose induces necroptotic cell death in neuroblastoma cell lines. J. Cell. Biochem. 112, 3834–3844 (2011).
Casciano, I. et al. Expression of the caspase-8 gene in neuroblastoma cells is regulated through an essential interferon-sensitive response element (ISRE). Cell Death Differ. 11, 131–134 (2004).
Bolden, J. E. et al. HDAC inhibitors induce tumor-cell-selective pro-apoptotic transcriptional responses. Cell Death Dis. 4, e519 (2013).
Condorelli, F., Gnemmi, I., Vallario, A., Genazzani, A. A. & Canonico, P. L. Inhibitors of histone deacetylase (HDAC) restore the p53 pathway in neuroblastoma cells. Br. J. Pharmacol. 153, 657–668 (2008).
Carpenter, E. L. et al. Antibody targeting of anaplastic lymphoma kinase induces cytotoxicity of human neuroblastoma. Oncogene 31, 4859–4867 (2012).
Moog-Lutz, C. et al. Activation and inhibition of anaplastic lymphoma kinase receptor tyrosine kinase by monoclonal antibodies and absence of agonist activity of pleiotrophin. J. Biol. Chem. 280, 26039–26048 (2005).
Matthay, K. K., George, R. E. & Yu, A. L. Promising therapeutic targets in neuroblastoma. Clin. Cancer Res. 18, 2740–2753 (2012).
Berois, N. & Osinaga, E. Glycobiology of neuroblastoma: impact on tumor behavior, prognosis, and therapeutic strategies. Front. Oncol. 4, 114 (2014).
Novak-Hofer, I. The L1 cell adhesion molecule as a target for radioimmunotherapy. Cancer Biother. Radiopharm. 22, 175–184 (2007).
Wu, Z. L., Schwartz, E., Seeger, R. & Ladisch, S. Expression of GD2 ganglioside by untreated primary human neuroblastomas. Cancer Res. 46, 440–443 (1986).
Cheung, N. K. et al. Ganglioside GD2 specific monoclonal antibody 3F8: a phase I study in patients with neuroblastoma and malignant melanoma. J. Clin. Oncol. 5, 1430–1440 (1987).
Cheung, N. K. et al. Anti-G(D2) antibody treatment of minimal residual stage 4 neuroblastoma diagnosed at more than 1 year of age. J. Clin. Oncol. 16, 3053–3060 (1998).
Cheung, N. K. et al. Key role for myeloid cells: phase II results of anti-G(D2) antibody 3F8 plus granulocyte-macrophage colony-stimulating factor for chemoresistant osteomedullary neuroblastoma. Int. J. Cancer 135, 2199–2205 (2014).
Saito, M., Yu, R. K. & Cheung, N. K. Ganglioside GD2 specificity of monoclonal antibodies to human neuroblastoma cell. Biochem. Biophys. Res. Commun. 127, 1–7 (1985).
Handgretinger, R. et al. A phase I study of human/mouse chimeric antiganglioside GD2 antibodych14.18 in patients with neuroblastoma. Eur. J. Cancer 31A, 261–267 (1995).
Ladenstein, R. et al. Ch14.18 antibody produced in CHO cells in relapsed or refractory Stage 4 neuroblastoma patients: a SIOPEN Phase 1 study. MAbs 5, 801–809 (2013).
Yu, A. L. et al. Phase I trial of a human-mouse chimeric anti-disialoganglioside monoclonal antibodych14.18 in patients with refractory neuroblastoma and osteosarcoma. J. Clin. Oncol. 16, 2169–2180 (1998).
Yu, A. L. et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N. Engl. J. Med. 363, 1324–1334 (2010).
Barker, E. et al. Effect of a chimeric anti-ganglioside GD2 antibody on cell-mediated lysis of human neuroblastoma cells. Cancer Res. 51, 144–149 (1991).
Castriconi, R. et al. Human NK cell infusions prolong survival of metastatic human neuroblastoma-bearing NOD/scid mice. Cancer Immunol. Immunother. 56, 1733–1742 (2007).
Delgado, D. C. et al. Genotypes of NK cell KIR receptors, their ligands, and Fcgamma receptors in the response of neuroblastoma patients to Hu14.18-IL2 immunotherapy. Cancer Res. 70, 9554–9561 (2010).
Tarek, N. et al. Unlicensed NK cells target neuroblastoma following anti-GD2 antibody treatment. J. Clin. Invest. 122, 3260–3270 (2012).
Venstrom, J. M. et al. KIR and HLA genotypes are associated with disease progression and survival following autologous hematopoietic stem cell transplantation for high-risk neuroblastoma. Clin. Cancer Res. 15, 7330–7334 (2009).
Zeng, Y. et al. Anti-neuroblastoma effect ofch14.18 antibody produced in CHO cells is mediated by NK-cells in mice. Mol. Immunol. 42, 1311–1319 (2005).
Dobrenkov, K. & Cheung, N. K. GD2-targeted immunotherapy and radioimmunotherapy. Semin. Oncol. 41, 589–612 (2014).
Gilman, A. L. et al. Phase I study ofch14.18 with granulocyte-macrophage colony-stimulating factor and interleukin-2 in children with neuroblastoma after autologous bone marrow transplantation or stem-cell rescue: a report from the Children’s Oncology Group. J. Clin. Oncol. 27, 85–91 (2009).
Hank, J. A. et al. Treatment of neuroblastoma patients with antiganglioside GD2 antibody plus interleukin-2 induces antibody-dependent cellular cytotoxicity against neuroblastoma detected in vitro. J. Immunother. Emphasis Tumor Immunol. 15, 29–37 (1994).
Kroesen, M. et al. Anti-GD2 mAb and Vorinostat synergize in the treatment of neuroblastoma. Oncoimmunology 5, e1164919 (2016).
Ozkaynak, M. F. et al. Phase I study of chimeric human/murine anti-ganglioside G(D2) monoclonal antibody (ch14.18) with granulocyte-macrophage colony-stimulating factor in children with neuroblastoma immediately after hematopoietic stem-cell transplantation: a Children’s Cancer Group Study. J. Clin. Oncol. 18, 4077–4085 (2000).
Cheung, N. K. et al. Murine anti-GD2 monoclonal antibody 3F8 combined with granulocyte-macrophage colony-stimulating factor and 13-cis-retinoic acid in high-risk patients with stage 4 neuroblastoma in first remission. J. Clin. Oncol. 30, 3264–3270 (2012).
Neal, Z. C. et al. Enhanced activity ofhu14.18-IL2 immunocytokine against murine NXS2 neuroblastoma when combined with interleukin 2 therapy. Clin. Cancer Res. 10, 4839–4847 (2004).
Airoldi, I. et al. Expression of costimulatory molecules in human neuroblastoma. Evidence that CD40+neuroblastoma cells undergo apoptosis following interaction with CD40L. Br. J. Cancer 88, 1527–1536 (2003).
Ait-Tahar, K., Barnardo, M. C. & Pulford, K. CD4 T-helper responses to the anaplastic lymphoma kinase (ALK) protein in patients with ALK-positive anaplastic large-cell lymphoma. Cancer Res. 67, 1898–1901 (2007).
Gonzalez, S. et al. Genetic engineering of cytolytic T lymphocytes for adoptive T-cell therapy of neuroblastoma. J. Gene Med. 6, 704–711 (2004).
Jena, B., Dotti, G. & Cooper, L. J. Redirecting T-cell specificity by introducing a tumor-specific chimeric antigen receptor. Blood 116, 1035–1044 (2010).
Louis, C. U. et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 118, 6050–6056 (2011).
Park, J. R. et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol. Ther. 15, 825–833 (2007).
Pule, M. A. et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 14, 1264–1270 (2008).
Sun, J. et al. T cells expressing constitutively active Akt resist multiple tumor-associated inhibitory mechanisms. Mol. Ther. 18, 2006–2017 (2010).
Eger, C. et al. Generation and characterization of a human/mouse chimeric GD2-mimicking anti-idiotype antibody ganglidiximab for active immunotherapy against neuroblastoma. PLoS ONE 11, e0150479 (2016).
Fest, S. et al. Survivin minigene DNA vaccination is effective against neuroblastoma. Int. J. Cancer 125, 104–114 (2009).
Krishnadas, D. K., Shapiro, T. & Lucas, K. Complete remission following decitabine/dendritic cell vaccine for relapsed neuroblastoma. Pediatrics 131, e336–e341 (2013).
Shilyansky, J., Jacobs, P., Doffek, K. & Sugg, S. L. Induction of cytolytic T lymphocytes against pediatric solid tumors in vitro using autologous dendritic cells pulsed with necrotic primary tumor. J. Pediatr. Surg. 42, 54–61 (2007). discussion.
Seliger, B., Cabrera, T., Garrido, F. & Ferrone, S. HLA class I antigen abnormalities and immune escape by malignant cells. Semin. Cancer Biol. 12, 3–13 (2002).
Evans, A. E., Gerson, J. & Schnaufer, L. Spontaneous regression of neuroblastoma. Natl Cancer Inst. Monogr. 44, 49–54 (1976).
Pritchard, J. & Hickman, J. A. Why does stage 4s neuroblastoma regress spontaneously? Lancet 344, 869–870 (1994).
Fouladi, M. et al. Pediatric phase I trial and pharmacokinetic study of vorinostat: a Children’s Oncology Group phase I consortium report. J. Clin. Oncol. 28, 3623–3629 (2010).
Singhal, J. et al. Didymin induces apoptosis by inhibiting N-Myc and upregulating RKIP in neuroblastoma. Cancer Prev. Res. (Phila). 5, 473–483 (2012).
Higashi, M. et al. Retinoic acid-induced CHD5 upregulation and neuronal differentiation of neuroblastoma. Mol. Cancer 14, 150 (2015).
Matthay, K. K. et al. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children’s oncology group study. J. Clin. Oncol. 27, 1007–1013 (2009).
Merrill, R. A. et al. All-trans retinoic acid-responsive genes identified in the human SH-SY5Y neuroblastoma cell line and their regulated expression in the nervous system of early embryos. Biol. Chem. 385, 605–614 (2004).
Sidell, N. Retinoic acid-induced growth inhibition and morphologic differentiation of human neuroblastoma cells in vitro. J. Natl Cancer Inst. 68, 589–596 (1982).
Sidell, N. et al. In vitro and in vivo effects of easily administered, low-toxic retinoid and phenylacetate compounds on human neuroblastoma cells. Br. J. Cancer 89, 412–419 (2003).
Thiele, C. J., Reynolds, C. P. & Israel, M. A. Decreased expression of N-myc precedes retinoic acid-induced morphological differentiation of human neuroblastoma. Nature 313, 404–406 (1985).
Yuza, Y., Agawa, M., Matsuzaki, M., Yamada, H. & Urashima, M. Gene and protein expression profiling during differentiation of neuroblastoma cells triggered by 13-cis retinoic acid. J. Pediatr. Hematol. Oncol. 25, 715–720 (2003).
Ciani, E., Severi, S., Contestabile, A., Bartesaghi, R. & Contestabile, A. Nitric oxide negatively regulates proliferation and promotes neuronal differentiation through N-Myc downregulation. J. Cell. Sci. 117, 4727–4737 (2004).
Gao, Q. et al. Elevated TrkA receptor expression is associated with all-trans retinoic acid-induced neuroblastoma differentiation. Genet. Mol. Res. 14, 13195–13202 (2015).
Brodeur, G. M. et al. Trk receptor expression and inhibition in neuroblastomas. Clin. Cancer Res. 15, 3244–3250 (2009).
Kogner, P. et al. Coexpression of messenger RNA for TRK protooncogene and low affinity nerve growth factor receptor in neuroblastoma with favorable prognosis. Cancer Res. 53, 2044–2050 (1993).
Matsushima, H. & Bogenmann, E. Expression of trkA cDNA in neuroblastomas mediates differentiation in vitro and in vivo. Mol. Cell. Biol. 13, 7447–7456 (1993).
Nakagawara, A. et al. Association between high levels of expression of the TRK gene and favorable outcome in human neuroblastoma. N. Engl. J. Med. 328, 847–854 (1993).
Nakagawara, A. Trk receptor tyrosine kinases: a bridge between cancer and neural development. Cancer Lett. 169, 107–114 (2001).
Poluha, W., Poluha, D. K. & Ross, A. H. TrkA neurogenic receptor regulates differentiation of neuroblastoma cells. Oncogene 10, 185–189 (1995).
Eggert, A. et al. Molecular dissection of TrkA signal transduction pathways mediating differentiation in human neuroblastoma cells. Oncogene 19, 2043–2051 (2000).
Lavenius, E., Gestblom, C., Johansson, I., Nanberg, E. & Pahlman, S. Transfection of TRK-A into human neuroblastoma cells restores their ability to differentiate in response to nerve growth factor. Cell. Growth Differ. 6, 727–736 (1995).
Jardi, F., Martinez, V. & Vergara, P. NGF is involved in oral ovalbumin-induced altered colonic contractility in rats: evidence from the blockade of TrkA receptors with K252a. Neurogastroenterol. Motil. 24, e580–e590 (2012).
Narayanan, R. et al. Discovery and preclinical characterization of novel small molecule TRK and ROS1 tyrosine kinase inhibitors for the treatment of cancer and inflammation. PLoS ONE 8, e83380 (2013).
Liao, W. et al. Downregulation of TrkA protein expression by miRNA92a promotes the proliferation and migration of human neuroblastoma cells. Mol. Med. Rep. 10, 778–784 (2014).
Ruggeri, P., Cappabianca, L., Farina, A. R., Gneo, L. & Mackay, A. R. NGF sensitizes TrkA SH-SY5Y neuroblastoma cells to TRAIL-induced apoptosis. Cell Death Discov. 2, 16004 (2016).
Ho, R. et al. Resistance to chemotherapy mediated by TrkB in neuroblastomas. Cancer Res. 62, 6462–6466 (2002).
Matsumoto, K., Wada, R. K., Yamashiro, J. M., Kaplan, D. R. & Thiele, C. J. Expression of brain-derived neurotrophic factor and p145TrkB affects survival, differentiation, and invasiveness of human neuroblastoma cells. Cancer Res. 55, 1798–1806 (1995).
Nakagawara, A., Azar, C. G., Scavarda, N. J. & Brodeur, G. M. Expression and function of TRK-B and BDNF in human neuroblastomas. Mol. Cell. Biol. 14, 759–767 (1994).
Croucher, J. L. et al. TrkB inhibition by GNF-4256 slows growth and enhances chemotherapeutic efficacy in neuroblastoma xenografts. Cancer Chemother. Pharmacol. 75, 131–141 (2015).
Li, Z., Zhang, Y., Tong, Y., Tong, J. & Thiele, C. J. Trk inhibitor attenuates the BDNF/TrkB-induced protection of neuroblastoma cells from etoposide in vitro and in vivo. Cancer Biol. Ther. 16, 477–483 (2015).
Kocak, H. et al. Hox-C9 activates the intrinsic pathway of apoptosis and is associated with spontaneous regression in neuroblastoma. Cell Death Dis. 4, e586 (2013).
Mao, L. et al. HOXC9 links cell-cycle exit and neuronal differentiation and is a prognostic marker in neuroblastoma. Cancer Res. 71, 4314–4324 (2011).
Acknowledgements
We are very grateful to Professor Marie Arsenian-Henriksson for critical reading of the article and valuable discussions. The work in the authors’ laboratories is supported by grants from the Swedish and Stockholm Cancer Societies, the Swedish Childhood Cancer Foundation, the Swedish Research Council. BZ and VG were supported by the Russian Science Foundation (14-25-00056).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Valter, K., Zhivotovsky, B. & Gogvadze, V. Cell death-based treatment of neuroblastoma. Cell Death Dis 9, 113 (2018). https://doi.org/10.1038/s41419-017-0060-1
Published:
DOI: https://doi.org/10.1038/s41419-017-0060-1
This article is cited by
-
Integrating bulk-seq and single-cell-seq reveals disulfidptosis potential index associating with neuroblastoma prognosis and immune infiltration
Journal of Cancer Research and Clinical Oncology (2023)
-
Changes in image-defined risk factors with neoadjuvant chemotherapy in pediatric abdominal neuroblastoma
Abdominal Radiology (2022)
-
Cell death in head and neck cancer pathogenesis and treatment
Cell Death & Disease (2021)
-
Novel therapeutic compound acridine–retrotuftsin action on biological forms of melanoma and neuroblastoma
Journal of Cancer Research and Clinical Oncology (2019)
-
Cell death-based treatment of various diseases: a fifty-year journey
Cell Death & Disease (2018)
