
Abstract
Human death domain superfamily proteins (DDSPs) play important roles in many signaling pathways involved in cell death and inflammation. Disruption or constitutive activation of these DDSP interactions due to inherited gene mutations is closely related to immunodeficiency and/or autoinflammatory diseases; however, responsible gene mutations have not been found in phenotypical diagnosis of these diseases. In this study, we comprehensively investigated the interactions of death-fold domains to explore the signaling network mediated by human DDSPs. We obtained 116 domains of DDSPs and conducted a domain–domain interaction assay of 13,924 reactions in duplicate using amplified luminescent proximity homogeneous assay. The data were mostly consistent with previously reported interactions. We also found new possible interactions, including an interaction between the caspase recruitment domain (CARD) of CARD10 and the tandem CARD–CARD domain of NOD2, which was confirmed by reciprocal co-immunoprecipitation. This study enables prediction of the interaction network of human DDSPs, sheds light on pathogenic mechanisms, and will facilitate identification of drug targets for treatment of immunodeficiency and autoinflammatory diseases.
Similar content being viewed by others
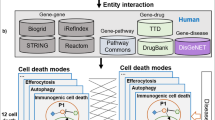


Introduction
The human death domain (DD) superfamily is one of the largest and most studied domain superfamilies. It comprises four subfamilies called the DD subfamily, the death effector domain (DED) subfamily, the caspase recruitment domain (CARD) subfamily, and the pyrin domain (PYD) subfamily [1, 2]. Death domain superfamily proteins (DDSPs) are characterized by containing death-fold domains (DFDs) and function in various signaling pathways involved in apoptosis and inflammation by assembling oligomeric complexes via homotypic binding and inducing caspase and/or kinase activation [3].
Genetic mutations in DFD-containing proteins often cause various immunodeficiency and autoinflammatory diseases [4]. For example, Fas-associated death domain (FADD) interacts with Fas through its DD and recruits pro-caspase‐8 through its DED to form the death-inducing signaling complex (DISC) [5]. Mutations of Fas that lead to the disruption of DISC formation cause autoimmune lymphoproliferative syndrome [6, 7]. NLRP3 interacts with apoptosis-associated speck-like protein containing a CARD (ASC) through its PYD and recruits pro-caspase‐1 through the CARD of ASC to form the inflammasome. Mutations of NLRP3 that lead to constitutive activation of the inflammasome cause an autoinflammatory disease called cryopyrin-associated periodic syndrome [8, 9]. Many autoinflammatory diseases are regarded as rare diseases, with few patients, and their pathogenesis has not been fully elucidated [10, 11].
Although much evidence have been accumulated, no mutations of responsible genes for these diseases have been identified, even in phenotypically diagnosed cases [12]. This prompted us to comprehensively analyze the interactions between all DDSPs, which may provide clues to decipher the pathways and factors associated with immunodeficiency and autoinflammatory diseases.
In this study, we focused on domain–domain interactions rather than on full-length protein–protein interactions. As shown in the diagram in Fig. 1, many DDSPs are inactive in the absence of upstream signals and therefore fail to interact with their partner proteins [13]. However, the domains of DDSPs are theoretically expected to be able to bind to the domains of their partners without interference.

Full-length DDSPs are inactive in the absence of upstream signals. Domains are theoretically expected to bind to their partner domains without interference.
We synthesized FLAG- and biotin-tagged recombinant DDs, DEDs, CARDs, and PYDs using the wheat germ cell-free synthesis system and investigated domain–domain interactions using the amplified luminescence proximity homogeneous assay (ALPHA) to elucidate the interaction network of DDSPs.
Materials and methods
Collection of cDNA clones encoding human DDSP domains
cDNA clones encoding human DDSP domains (Supplementary Table S1) were collected from the cDNA resources of the Kazusa DNA Research Institute [14], the Mammalian Gene Collection [15], and the nonprofit repository of Addgene.
Construction of tagged recombinant cDNA plasmids
Genes encoding DDSP domains were amplified by PCR using cDNA as templates. Overlapping sequences were added at the 5′ and 3′ ends for seamless cloning. DNA fragments encoding 27 DDSP domains were prepared by the GeneArt Gene Synthesis service (Thermo Fisher Scientific, Waltham, MA, USA). Amplified DNA fragments were subcloned into the pEU-E01-GW-FLAG and pEU-E01-GW-bls vectors using Gibson Assembly seamless cloning. After subcloning, pEU expression plasmids were arranged in a 96-well format and stored as glycerol stocks. The glycerol stocks were diluted with TE buffer and used as templates of PCR. Template DNA fragments for transcription were PCR-amplified using the SPu-2 primer (5′-CAGTAAGCCAGATGCTACAC-3′) and AODA2306 primer (5′-AGCGTCAGACCCCGTAGAAA-3′).
Preparation of recombinant DDSP domains using a wheat germ cell-free synthesis system
The tagged recombinant human DDSP domains were synthesized using a wheat germ cell-free synthesis system [16]. Transcription and translation reactions were conducted using a WEPRO7240 Expression Kit (CellFree Sciences, Matsuyama, Japan). The transcription reaction mixture was prepared by mixing 2.5 µL of transcription buffer LM, 1.25 µL of NTP mixture (25 mM each), 0.25 µL of RNase inhibitor, 0.5 µL of SP6 polymerase, and 2.5 µL of PCR product in a 96-well plate. The transcription reaction was incubated at 37 °C for 18 h. Twenty-five microliters of the translation mixture containing 12.5 µL of mRNA, 8 µL of WEPRO 7240 wheat germ extract, 0.1 µL of creatine kinase (20 mg/mL) (Roche Diagnostics, Basel, Switzerland), and 0.5 µL of RNase inhibitor was prepared and overlaid with 125 µL of translation buffer (SUB-AMIX SGC) in a 96-well plate. The biotin ligation site was biotinylated enzymatically by adding BirA biotin ligase and biotin (Sigma-Aldrich, St. Louis, MO, USA) to the translation mixture [17]. The plate containing the translation reaction was incubated at 15 °C for 24 h.
Enzyme-linked immunosorbent assay (ELISA)
Cell-free synthesized DDSP domains were diluted 20-fold, injected into a 96-well MaxiSorp plate (Nunc, Rochester, NY, USA), and incubated overnight at 4 °C. After washing with Tris-buffered saline containing 0.1% Tween 20 (TBST), the plate was blocked with TBST containing 5% skimmed milk for 1 h at room temperature. Next, the plate was incubated with an anti-DYKDDDDK tag monoclonal antibody (012–22384, FUJIFILM Wako Pure Chemical, Osaka, Japan) or anti-biotin antibody (A4541, Sigma-Aldrich) diluted 1:2000 and 1:1000, respectively, in TBST containing 5% skimmed milk for 1 h at room temperature. Thereafter, the plate was washed three times with TBST and incubated with a horseradish peroxidase-conjugated anti-mouse IgG secondary antibody (GE Healthcare, Chicago, IL, USA) diluted in TBST containing 5% skimmed milk for 1 h at room temperature. Finally, 50 µL of tetramethylbenzidine liquid substrate (Sigma-Aldrich) was injected into the plate and incubated for 15–30 min at room temperature. The reaction was terminated by injecting the same volume of 1 M HCl. Absorbance at 450 nm was measured using a SpectraMAX M3 plate reader (Molecular Devices, San Jose, CA, USA).
Amplified luminescence proximity homogeneous assay
All ALPHA reactions were conducted in an AlphaPlate-384 microplate (PerkinElmer, Waltham, MA, USA). All proteins and reagents were diluted in reaction buffer [100 mM Tris-HCl (pH 8.0), 0.01% Tween 20, and 1 mg/mL bovine serum albumin]. Twenty microliters of solution containing 0.4 μL of a biotin-tagged domain in reaction buffer was dispensed into the reaction plate (two domains per plate, 192 replicates) using a Viaflo automated multichannel pipette and the Viaflo Assist system (Integra, Hudson, NH, USA). Next, 0.4 µL of FLAG-tagged domain was transferred to the reaction plate (96 domains per plate, four replicates) using the Janus automated dispensing workstation (PerkinElmer) and Nanohead, a 384-well micro-syringe head (PerkinElmer). This procedure allowed 192 combinations of FLAG- and biotin-tagged domains to be mixed together in duplicate per assay plate. Then, 9.6 μL of detection mixture containing 0.02 μL of an anti-DYKDDDDK tag monoclonal antibody, 0.06 μL of streptavidin-conjugated AlphaScreen donor beads, and 0.06 μL of protein A-conjugated AlphaScreen acceptor beads in reaction buffer was added to each well of the reaction plate using a FlexDrop dropper (PerkinElmer). The detailed dispensing scheme and well layout are shown in Supplementary Fig. S1. After incubation at 25 °C for 24 h, the ALPHA chemiluminescence signal was detected by an EnVision Multilabel Plate Reader (PerkinElmer). The signal data obtained were exported to Microsoft Excel, and the median values of duplicate reactions were calculated. For the heat map, the color scale feature of Microsoft Excel was used to visualize the signal strength. MA plots and bubble charts were drawn using DataGraph (http://www.visualdatatools.com/DataGraph/).
Immunoprecipitation
Genes encoding DDSP domains were inserted into the pcDNA3 mammalian expression vector with a FLAG or a 3×AGIA tag [18] at the C-terminus. HEK293T cells were maintained in Dulbecco’s Modified Eagle’s Medium (Thermo Fisher Scientific) supplemented with 10% heat-inactivated fetal bovine serum, penicillin, and streptomycin. Transfection was performed using the calcium phosphate method. Briefly, plasmids were diluted in 440 μL of distilled water and 60 μL of 2 M CaCl2, mixed with 500 μL of 2× HEPES buffer [50 mM HEPES (pH 7.00), 280 mM NaCl, and 1.5 mM Na2HPO4], and added to each 10 mL well containing HEK293T cells. A total of 1 × 106 HEK293T cells were transfected with 1 μg of each of the following expression plasmid sets: pcDNA3-AIM2-PYD-AGIA and pcDNA3-NLRP9-PYD-FLAG, pcDNA3-AIM2-PYD-AGIA and pcDNA3-ASC-CARD-FLAG, pcDNA3-NLRC4-CARD-AGIA and pcDNA3-ASC-CARD-FLAG, and pcDNA3-NOD2-CARD1-CARD2-AGIA and pcDNA3-CARD10-CARD-FLAG. Transfected cells were lysed in 1000 μL of NP-40 buffer [1% Nonidet P-40, 142.5 mmol/L KCl, 5 mmol/L MgCl2, 10 mmol/L HEPES (pH 7.6), and 1 mmol/L ethylenediaminetetraacetic acid] supplemented with a cOmplete Mini Protease Inhibitor Cocktail tablet (Roche Diagnostics). Cell lysates were centrifuged. Supernatants were mixed with an anti-AGIA tag monoclonal antibody (in-house made) [18] or with an anti-FLAG M2 monoclonal antibody (F3165, Sigma-Aldrich) together with protein A beads (Invitrogen) and incubated for 3 h at 4 °C. The beads were washed with NP-40 buffer and precipitates were subjected to SDS-PAGE and immunoblotting. Blotting membranes were incubated with the anti-FLAG M2 monoclonal antibody or an anti-AGIA tag monoclonal antibody.
Results
Human DDSP domain expression plasmids were constructed
Tagged expression plasmids were constructed for cell-free synthesis of DDSP domains. We first collected the cDNA sequences of human DFD-containing proteins from public databases such as RefSeq (https://www.ncbi.nlm.nih.gov/refseq/) and UniProt (https://www.uniprot.org/). Next, we extracted the DFD fragments using their annotations and domain prediction tools including PROSITE (https://prosite.expasy.org/) and SMART (http://smart.embl-heidelberg.de/). Eventually, we identified 108 DDSP domains, including 36 DDs, 11 DEDs, 39 CARDs, and 22 PYDs. In addition, there were eight DDSP domains containing tandem DFDs, such as CARD–CARD, DED–DED, CARD–DD, PYD–CARD, and DED–DD. Including all such tandem domains, the total number of DDSP domains in this study was 116 (Supplementary Table S1). We constructed cell-free expression plasmids to synthesize all the domains tagged with FLAG or biotin at the C-terminus.
Human DDSP domains were synthesized using the wheat germ cell-free synthesis system
Using the wheat germ cell-free protein synthesis system and expression plasmids, we synthesized 232 recombinant DDSP domains, including 116 FLAG-tagged and 116 biotin-tagged domains. In addition, we used dihydrofolate reductase (DHFR) and Venus fluorescent protein (Venus) tagged with FLAG and biotin respectively as control proteins for cell-free synthesis and ALPHA [19, 20].
To evaluate the expression levels of DDSP domains, ELISAs were performed using an anti-FLAG antibody (Fig. 2A) and an anti-biotin antibody (Fig. 2B), respectively (Supplementary Table S2). All the domains, both FLAG- and biotin-tagged, were expressed at a level equal to or higher than expression of DHFR, which was used as a positive control. Furthermore, the expression levels of the DDSP domains were 50–900% of that of Venus, indicating that all the DDSP domains were sufficiently expressed for the comprehensive interaction assay.

DHFR and Venus were used as positive controls. The longitudinal axis shows the ELISA values representing expression levels and the horizontal axis shows the 116 domains and 2 controls listed in Supplementary Table S2. A Expression levels of 116 FLAG-tagged domains and 2 FLAG-tagged controls. B Expression levels of 116 biotin-tagged domains and 2 biotin-tagged controls.
Human DDSP domain interactions were comprehensively analyzed by ALPHA
A total of 13,924 domain–domain reactions [118 FLAG-tagged domains (116 DDSP domains plus 2 negative controls) × 118 biotin-tagged domains (116 DDSP domains plus 2 negative controls)] were conducted in duplicate in the ALPHA assay.
The results are listed in Supplementary Table S3, which shows the median ALPHA signals detected in two repeats. The highest value was 119,174 relative luminescence units (RLU) (APAF1_CARD-FLAG × CASP9_CARD-biotin) and the lowest value was 166 RLU. The median value was 296 RLU. A total of 799 pairs had signals higher than 1 × 103 RLU, among which 236 pairs had signals higher than 2 × 103 RLU and 95 pairs had signals higher than 5 × 103 RLU (Fig. 3 and Supplementary Table S3).

There were 13,924 reactions between FLAG-tagged (116 domains plus 2 negative controls) and biotin-tagged (116 domains plus 2 negative controls) domains. The longitudinal axis shows FLAG-tagged domains and the horizontal axis shows biotin-tagged domains. The position of each point indicates the specific domain pair. Red points represent positive domain–domain interactions. The shade of red indicates the median value of two repeats in the ALPHA.
To clarify the distribution and strength of the interactions, the values in Supplementary Table S3 were visualized as a heat map (Fig. 3). The longitudinal axis shows FLAG-tagged domains and the horizontal axis shows biotin-tagged domains. The position of each point indicates the combination of each domain–domain pair. The color of each point indicates the strength of ALPHA signals, indicating possible interactions between DDSP domains. Darker shades of red indicate stronger ALPHA signals, which are highly suggestive of interactions between the specific domains. White, which accounts for the majority of the heat map, indicates that an interaction was not detected.
We assessed the reproducibility of the assay between two repeats using an MA plot (Fig. 4). Each point represents the log2 fold change (0.5–2) in two repeats. The red dashed lines represent the fold change threshold (±1). A total of 98.74% of the points were located between the two red dashed lines. This shows that more than 98% of the reactions were reproducible, demonstrating that this assay was highly reproducible and reliable.

An MA plot shows the reproducibility of 13,924 reactions between two repeats in the ALPHA. Each point represents the log2 fold change (0.5–2) in two repeats. The red dashed lines represent the fold change threshold (±1). A total of 98.74% of the points were located between the two red dashed lines, demonstrating that this assay was highly reproducible and reliable.
More positive interactions tend to be from the self-interacting pairs than those nonself-interacting ones
Human DDSP domains function in various signaling pathways involved in cell death and inflammation by oligomerizing with each other, which promotes interactions between proteins [2]. In this assay, therefore, we particularly compared self-interacting and nonself-interacting pairs. The 116 self-interacting pairs are located along the diagonal in Fig. 3, including six DDSPs containing homogeneous tandem DFDs (No. 028, 035, 039, 057, 092, and 102) and three DDSPs containing heterogeneous tandem DFDs (Nos. 010, 041, and 052). Among the 116 self-interacting pairs, the interactions of 38, which accounts for 32.8%, were strong with ALPHA signals higher than 1 × 103 RLU. Meanwhile, among the 13,340 nonself-interacting pairs located on either side of the diagonal, the interactions of 761 (5.7%) were strong with ALPHA signals higher than 1 × 103 RLU. Comparison of the percentages of positive results between self-interacting (32.8%) and nonself-interacting (5.7%) pairs implies that self-interacting pairs, which interact via homotypic binding, tend to interact more readily than nonself-interacting pairs.
Symmetrically distributed nonself-interacting pairs in the heat map confirm previously identified heterotypic interacting pairs and indicate new interacting pairs
We focused on the red points of nonself-interacting pairs symmetrically distributed on both sides of the diagonal in the heat map (Fig. 3, hereafter called double-sided interactions). They correspond to interactions of nonself-interacting pairs that were unaffected by swapping the tag (FLAG and biotin), indicating they are highly reliable interactions. All such double-sided interactions with ALPHA signals higher than 1 × 103 RLU irrespective of the tags used (FLAG × biotin and biotin × FLAG) were extracted and listed in Table 1. Among the 57 pairs, 39 were homotypic and 11 were heterotypic. Thirty-seven (65%) interactions have been previously reported, including 29 homotypic domain interactions and 1 heterotypic domain interaction, demonstrating the high reliability of the results (Table 1). The remaining ten homotypic domain interactions (CARD14_CARD × CARD19_CARD, CARD16_CARD × CARD8_CARD, CARD16_CARD × NLRC4_CARD, CRADD_DD × IRAK1_DD, IRAK1_DD × TNFRSF25_DD, PYRIN_PYD × NLRP4_PYD, PYRIN_PYD × NLRP9_PYD, NLRP14_PYD × NLRP4_PYD, NLRP14_PYD × NLRP9_PYD, and NLRP4_PYD × NLRP9_PYD) and ten heterotypic domain interactions (ANKDD1A_DD × CASP12_CARD, ANKDD1A_DD × NLRC3_CARD, DAPK1_DD × PYDC2_PYD, IRAK1_DD × NLRP4_PYD, IRAK1_DD × NLRP8_PYD, IRAK4_DD × NLRP9_PYD, MYD88_DD × NLRC3_CARD, MYD88_DD × NOD2_CARD2, NLRC3_CARD × NLRP4_PYD, and NLRC5_CARD × NLRP9_PYD) have not been previously reported (Fig. 5). These interactions must be confirmed by in vitro and in vivo studies.

Double-sided interacting pairs are indicated. The bars indicate the ALPHA signals detected using various combinations of FLAG- and biotin-tagged proteins.
Asymmetrically distributed nonself-interacting pairs in the heat map indicate possible interactions that require further confirmation
Some points were asymmetrically distributed across the heat map and there was no corresponding symmetric interaction on the opposite side of the diagonal (Fig. 3, hereafter called one-sided interactions). All such one-sided interactions that had ALPHA signals higher than 2 × 103 RLU with either the FLAG × biotin or biotin × FLAG pair were extracted and listed in Table 2. Among the 129 pairs, 45 were homotypic and 80 were heterotypic. Furthermore, 17 interactions have been previously reported, including 8 homotypic interactions and 4 heterotypic interactions.
The strength of ALPHA signals does not correlate with the expression levels of domains, but with specific pair combinations
To investigate whether the non-normalized expression levels of DDSP domains affect the ALPHA results in an unbiased fashion, we compared the expression levels of the domains and the distribution of positive ALPHA signals. In the bubble chart in Fig. 6, blue bubbles show ALPHA signals. The bigger the bubble area, the stronger the signal. The longitudinal axis shows relative concentrations of FLAG-tagged domains and the horizontal axis shows relative concentrations of biotin-tagged domains. Significantly large bubbles were widely and randomly scattered across the chart, instead of gathering in specific regions, such as the top right where the expressed domains were most abundant. In addition, the sizes of the bubbles showed no linear correlation with the expression levels of domains. These results demonstrate that the strength of ALPHA signals was not correlated with the expression levels of the domains, but with specific pair combinations.

A bubble chart compares the domain expression levels determined by ELISAs and the strength of ALPHA signals. Blue bubbles show ALPHA signals. The bigger the bubble area, the stronger the signal. The longitudinal axis shows relative concentrations of FLAG-tagged domains and the horizontal axis shows relative concentrations of biotin-tagged domains.
Co-immunoprecipitation confirms previously reported interactions and provides hints about new interactions
The ALPHA is an excellent technology to analyze protein–protein interactions because it is homogeneous, highly sensitive, and convenient. However, doubts remain about whether and to what extent the ALPHA results reflect and are consistent with the real situations in natural cells. To validate the accuracy and veracity of the ALPHA results, we performed a co-immunoprecipitation assay.
We selected eight representative pairs of domains from the 116 domains according to the ALPHA (Fig. 7A). Among them, four pairs between AIM2_PYD × NLRP9_PYD and AIM2_PYD × ASC_CARD had low signals, indicating these domains do not interact [13]. The two pairs between (NLRC4_CARD × ASC_CARD) both demonstrated significantly high signals, indicating that these domains interact, which actually was reported as components of NLRC4 inflammasome (Table 1) [21]. A one-sided pair (CARD10_CARD-FLAG × NOD2_CARD1-CARD2-biotin) exhibited an extremely high signal, whereas the other pair with reversed tags did not. An interaction between CARD10_CARD and NOD2_CARD1-CARD2 has not been previously reported and therefore needed to be confirmed in cells.

A ALPHA results of the four domain–domain interaction pairs examined in panels B and C. B, C An AGIA-tagged domain and a FLAG-tagged domain were co-expressed in HEK293T cells. Soluble supernatants generated from whole cell lysates (input) were applied to immunoprecipitation assay using an anti-AGIA or an anti-FLAG antibody, and co-immunoprecipitants were detected by immunoblotting with an anti-FLAG or anti-AGIA antibody, respectively.
The results of the co-immunoprecipitation assay are shown in Fig. 7B, C. As expected, a homotypic domain pair (AIM2_PYD × NLRP9_PYD) that had low ALPHA signals and a known noninteracting heterotopic domain pair (AIM2_PYD × ASC_CARD) did not co-immunoprecipitate [22], whereas a known interacting homotypic domain pair (NLRC4_CARD × ASC_CARD) did [23]. These immunoprecipitation results were consistent with those of the ALPHA assay and previous reports. The homotypic domain pair of CARD10_CARD × NOD2_CARD1-CARD2, which had a high signal in the ALPHA assay, were reciprocally co-immunoprecipitated (Fig. 7B, C).
Discussion
Human DDSPs are involved in assembly of multimeric complexes associated with signaling cascades that lead to cell death and inflammation [3]. Disruption of this assembly or dysregulation of DDSP interactions due to inherited gene mutations causes immunodeficiency and/or autoinflammatory diseases [4]. However, the relationships between the genotypes and phenotypes of inherited immunodeficiency and/or autoinflammatory diseases are not fully understood. In some cases, even though the responsible genes have been reported, mutations of these genes have not been found [12]. Thus, we hypothesize that DDSP interactions are involved in some unknown signaling cascades. In this study, we comprehensively analyzed the interactions of DDSP domains to explore novel interaction pathways.
The greatest difficulties faced when comprehensively investigating the interactions of DDSP domains are the preparation of more than 100 different kinds of proteins and the conduction of a one-against-all domain–domain interaction assay, where conventional methods are commonly used such as yeast two-hybrid assay, pull-down assay, and immunoprecipitation assay. Here, we used the wheat germ cell-free protein synthesis system and ALPHA, which greatly facilitated our study. We successfully synthesized 116 FLAG-tagged and 116 biotin-tagged domains of DDSPs using the wheat germ cell-free protein synthesis system (Supplementary Table S1) and comprehensively analyzed domain–domain interactions using ALPHA (Fig. 3). The ALPHA results of domain–domain interactions were reliable and trustworthy. An MA plot showed high reproducibility (98.74%) between two repeats (Fig. 4). A bubble chart demonstrated that the expression levels of DDSP domains did not affect the strength of ALPHA signals (Fig. 6). Furthermore, many domain interactions detected in this study were previously reported (Tables 1 and 2).
Our data showed that 32.8% of self-interacting pairs and 5.7% of nonself-interacting pairs yielded positive results in the ALPHA, indicating that self-interacting pairs interact more readily than nonself-interacting pairs. Although two domains contained heterogeneous DFDs, all self-interacting pairs exhibited homotypic binding [24].
DDSP domains are widely believed to exert their effects via monovalent, homotypic, subfamily-restricted interactions (DD × DD, CARD × CARD, DED × DED, and PYD × PYD), generating large multi-subunit structures comprising only one type of protein [25]. However, heterotypic binding has been reported for some exceptional DDSP domains [26]. We found ten domain pairs as novel candidates for heterotypic interactions (Fig. 5 and Table 1). Such interactions may help to elucidate the molecular basis of signaling complexes and pathways that regulate cell death and inflammation.
This study shows many novel possible interactions (Tables 1 and 2). Twenty novel double-sided interactions (Fig. 5) were considered reliable because many of the other double-sided interactions identified in this study were previously reported. There were several combinations for which no direct interaction has been previously demonstrated, including CARD16_CARD × NLRC4_CARD, CRADD_DD × IRAK1_DD, IRAK1_DD × TNFRSF25_DD, and MYD88_DD × NOD2_CARD2 (Table 1), even though previous reports suggested their biological and pathological relevance. For example, acute coronary syndrome (ACS) is a disorder in which blood supply to the heart is suddenly blocked, leading to heart attacks and unstable angina. A genome-wide association study of 18,624 patients with ACS identified the associated gene loci in IL-18, NLRC4, and CARD16; however, direct interactions between these proteins have not been demonstrated [27]. Our study identified a double-sided interaction between CARD16_CARD and NLRC4_CARD (Fig. 5 and Table 1). The direct interaction between NLRC4 and CARD16 highlights the role of NLRC4 inflammasome regulation in ACS. Moreover, many studies suggest that Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain 1 (NOD1) and NOD2 synergize with each other to induce production of cytokines and antimicrobial peptides. However, the molecular mechanisms underlying this synergy have not been elucidated [28]. The synergic effect of TLRs and NODs leads to poor outcomes in individuals with septic shock syndrome caused by Gram-positive or -negative bacterial infections [29, 30]. The double-sided interaction between MYD88_DD and NOD2_CARD2 (the second CARD domain from the N-terminus) identified in this study may support the hypothesis that there are direct crosstalk between signaling pathways downstream of NODs and TLRs (Table 1). In addition, our study indicated the direct interaction between IRAK1 and CRADD (Fig. 5 and Table 1), supporting the previous report that IRAK1 functions to inhibit radiation therapy-induced apoptosis mediated by the PIDDosome (PIDD-RAIDD-caspase-2) [31].
The one-sided interactions listed in Table 2, few of which were previously reported, are supposedly less reliable than the double-sided interactions listed in Table 1. The interactions need to be confirmed by co-immunoprecipitation assays in human cell lines. Many immunoprecipitation experiments assessing DDSP interactions have been performed using HEK293T cells. Therefore, we used HEK293T cells and compared the novel interaction between NOD2_CARD1-CARD2 and CARD10_CARD and the known interaction between NLRC4_CARD and ASC_CARD. NOD2_CARD1-CARD2-AGIA was co-immunoprecipitated by CARD10_CARD-FLAG and vice versa when the proteins were co-expressed, which was consistent with the ALPHA results (Fig. 7B, C and Supplementary Table S3), suggesting that CARD10 interacts with NOD2. Consequently, it is worth testing the other one-sided interactions listed in Table 2.
CARD10 (also known as CARD and membrane-associated guanylate kinase (MAGUK) domain-containing protein 3 (CARMA3)) functions as a scaffold and is involved in NF-κB activation in response to various types of upstream innate immune signaling or modulates the interactions of deubiquitinating enzymes such as A20 and CYLD [32]. Several amino acid mutations of CARD10 are reportedly responsible for some inflammatory bowel diseases [33]. NOD2 is a Nod-like receptor that recognizes the bacterial peptidoglycan component muramyl dipeptide, leading to NF-κB activation [34, 35]. Gain-of-function mutations of NOD2 lead to autoinflammatory diseases such as early-onset sarcoidosis and Blau syndrome [36]. Loss-of-function mutations of NOD2 lead to susceptibility to an inflammatory bowel disease called Crohn’s disease [37, 38].
Although the results of this study were highly reproducible, there are several limitations. First, all the recombinant DDSP domains synthesized and used in this study were fused with tags. The type and position of the tag may affect the ALPHA results. Depending on the structure of the domain, the fused tag may be concealed inside the mature domain and inaccessible to the detection antibody or streptavidin. Furthermore, when a domain interacts with partners via amino acid residues close to the terminus, the tag may interfere with the interaction, leading to false ALPHA results. This may explain one of the cause of known pairs of interactors that do not have a high enough ALPHA signal (Supplementary Table S4). Therefore, it is recommended that domains with tags fused at different positions are examined before conducting the ALPHA. The domain interactions were not noticeably affected according to whether the tag was located at the N- or C-terminus. Therefore, we tagged the C-termini of all domains with FLAG or biotin to ensure the domains and tags were fully translated. Second, the recombinant DDSP domains were synthesized using the wheat cell-free synthesis system and therefore their structures may differ from those of proteins expressed in cells. In particular, proteins synthesized using cell-free systems possibly have errors in post-translational modifications. For DDSP domains that require a certain post-translational modification for signal transduction, it is recommended that enzymes that catalyze such modifications, such as protein kinases, are added to the cell-free system. Finally, although we analyzed interactions for all possible DDSP domains combinations, some interactions may not occur in cells. Two DDSPs with different subcellular localizations and expression profiles are very unlikely to encounter each other in cells. Regarding the new interacting pairs identified by our comprehensive analysis, their localizations and interactions should be confirmed and overlap of their temporal and spatial expression patterns should be verified. Therefore, the ALPHA results could not be quantitatively analyzed to compare the strengths of the domain–domain interactions.
In conclusion, we believe that our comprehensive investigation of DDSPs will be helpful for the field of DDSP-related diseases, especially immunodeficiency and autoinflammatory diseases. This work may facilitate future research aiming to identify pharmaceutical targets for drug discovery and to elucidate the pathogenesis of these diseases.
References
Liu T, Rojas A, Ye Y, Godzik A. Homology modeling provides insights into the binding mode of the PAAD/DAPIN/pyrin domain, a fourth member of the CARD/DD/DED domain family. Protein Sci. 2003;12:1872–81.
Weber CH, Vincenz C. The death domain superfamily: a tale of two interfaces? Trends Biochem Sci. 2001;26:475–81.
Lo Y-C, Lin S-C, Wang L, Yang JK, Wu H. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu Rev Immunol. 2007;25:561–86.
Scudiero I, Vito P, Stilo R. The three CARMA sisters: so different, so similar: a portrait of the three CARMA proteins and their involvement in human disorders. J Cell Physiol. 2014;229:990–7.
Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, et al. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995;14:5579–88.
Fisher GH, Rosenberg FJ, Straus SE, Dale JK, Middleton LA, Lin AY, et al. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81:935–46.
Rieux-Laucat F, Le Deist F, Hivroz C, Roberts IA, Debatin KM, Fischer A, et al. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268:1347–9.
Feldmann J, Prieur A-M, Quartier P, Berquin P, Certain S, Cortis E, et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet. 2002;71:198–203.
Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001;29:301–5.
Krainer J, Siebenhandl S, Weinhäusel A. Systemic autoinflammatory diseases. J Autoimmun. 2020;109:102421.
Schnappauf O, Aksentijevich I. Current and future advances in genetic testing in systemic autoinflammatory diseases. Rheumatol (Oxf). 2019;58:vi44–vi55.
Wang W, Yu Z, Gou L, Zhong L, Li J, Ma M, et al. Single-center overview of pediatric monogenic autoinflammatory diseases in the past decade: a summary and beyond. Front Immunol. 2020;11:565099.
Kaneko N, Iwasaki T, Ito Y, Takeda H, Sawasaki T, Morikawa S, et al. Applications of reconstituted inflammasomes in a cell-free system to drug discovery and elucidation of the pathogenesis of autoinflammatory diseases. Inflamm Regen. 2017;37:9.
Nagase T, Yamakawa H, Tadokoro S, Nakajima D, Inoue S, Yamaguchi K, et al. Exploration of human ORFeome: high-throughput preparation of ORF clones and efficient characterization of their protein products. DNA Res. 2008;15:137–49.
Strausberg RL, Feingold EA, Grouse LH, Derge JG, Klausner RD, Collins FS, et al. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc Natl Acad Sci USA. 2002;99:16899–903.
Takai K, Sawasaki T, Endo Y. Practical cell-free protein synthesis system using purified wheat embryos. Nat Protoc. 2010;5:227–38.
Sawasaki T, Kamura N, Matsunaga S, Saeki M, Tsuchimochi M, Morishita R, et al. Arabidopsis HY5 protein functions as a DNA-binding tag for purification and functional immobilization of proteins on agarose/DNA microplate. FEBS Lett. 2008;582:221–8.
Yano T, Takeda H, Uematsu A, Yamanaka S, Nomura S, Nemoto K, et al. AGIA tag system based on a high affinity rabbit monoclonal antibody against human dopamine receptor D1 for protein analysis. PLoS ONE. 2016;11:e0156716.
Yamamoto T, Taira Nihira N, Yogosawa S, Aoki K, Takeda H, Sawasaki T, et al. Interaction between RNF8 and DYRK2 is required for the recruitment of DNA repair molecules to DNA double-strand breaks. FEBS Lett. 2017;591:842–53.
Takeda H, Kawamura Y, Miura A, Mori M, Wakamatsu A, Yamamoto J-I, et al. Comparative analysis of human Src-family kinase substrate specificity in vitro. J Proteome Res. 2010;9:5982–93.
Poyet JL, Srinivasula SM, Tnani M, Razmara M, Fernandes-Alnemri T, Alnemri ES. Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J Biol Chem. 2001;276:28309–13.
Kaneko N, Ito Y, Iwasaki T, Takeda H, Sawasaki T, Migita K, et al. Reconstituted AIM2 inflammasome in cell-free system. J Immunol Methods. 2015;426:76–81.
Masumoto J, Dowds TA, Schaner P, Chen FF, Ogura Y, Li M, et al. ASC is an activating adaptor for NF-κB and caspase-8-dependent apoptosis. Biochem Biophys Res Commun. 2003;303:69–73.
Park Y-H, Jeong MS, Jang SB. Structural insights of homotypic interaction domains in the ligand-receptor signal transduction of tumor necrosis factor (TNF). BMB Rep. 2016;49:159–66.
Park HH. Domain swapping of death domain superfamily: alternative strategy for dimerization. Int J Biol Macromol. 2019;138:565–72.
Lee BL, Mirrashidi KM, Stowe IB, Kummerfeld SK, Watanabe C, Haley B, et al. ASC- and caspase-8-dependent apoptotic pathway diverges from the NLRC4 inflammasome in macrophages. Sci Rep. 2018;8:3788–812.
Johansson Å, Eriksson N, Becker RC, Storey RF, Himmelmann A, Hagström E, et al. NLRC4 inflammasome is an important regulator of interleukin-18 levels in patients with acute coronary syndromes: genome-wide association study in the PLATelet inhibition and patient Outcomes Trial (PLATO). Circ Cardiovasc Genet. 2015;8:498–506.
Pashenkov MV, Murugina NE, Budikhina AS, Pinegin BV. Synergistic interactions between NOD receptors and TLRs: mechanisms and clinical implications. J Leukoc Biol. 2019;105:669–80.
Moreira LO, Smith AM, DeFreitas AA, Qualls JE, Kasmi El KC, Murray PJ. Modulation of adaptive immunity by different adjuvant-antigen combinations in mice lacking Nod2. Vaccine. 2008;26:5808–13.
Kim H, Zhao Q, Zheng H, Li X, Zhang T, Ma X. A novel crosstalk between TLR4- and NOD2-mediated signaling in the regulation of intestinal inflammation. Sci Rep. 2015;5:12018.
Liu PH, Shah RB, Li Y, Arora A, Ung PM-U, Raman R, et al. An IRAK1-PIN1 signalling axis drives intrinsic tumour resistance to radiation therapy. Nat Cell Biol. 2019;21:203–13.
Klei LR, Hu D, Panek R, Alfano DN, Bridwell RE, Bailey KM, et al. MALT1 protease activation triggers acute disruption of endothelial barrier integrity via CYLD cleavage. Cell Rep. 2016;17:221–32.
Friedrichs F, Henckaerts L, Vermeire S, Kucharzik T, Seehafer T, Möller-Krull M, et al. The Crohn’s disease susceptibility gene DLG5 as a member of the CARD interaction network. J Mol Med (Berl). 2008;86:423–32.
Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem. 2003;278:5509–12.
Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278:8869–72.
Miceli-Richard C, Lesage S, Rybojad M, Prieur AM, Manouvrier-Hanu S, Häfner R, et al. CARD15 mutations in Blau syndrome. Nat Genet. 2001;29:19–20.
Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603.
Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–6.
Acknowledgements
Some of the cDNA resources were provided by a collaboration with the Kazusa DNA Research Institute.
Funding
This research was supported by JSPS KAKENHI Grant nos. 20H03719 (JM), and 20K05709 (HT). The establishment and screening of the protein array used in this study were partially supported by the Ministry of Education, Culture, Sports, Science and Technology, Japan (MEXT) and by the Takeda Science Foundation. This research was also supported by the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant no. JP20am0101077.
Author information
Authors and Affiliations
Contributions
JM and HT supervised the study. WZ constructed the human DDSP array. HT, WZ, and TN performed the ALPHA. NK and JM performed the immunoprecipitation assay. WZ analyzed the data. WZ, JM, and HT wrote the paper.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by A. Degterev
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, W., Kaneko, N., Nakagita, T. et al. A comprehensive interaction study provides a potential domain interaction network of human death domain superfamily proteins. Cell Death Differ 28, 2991–3008 (2021). https://doi.org/10.1038/s41418-021-00796-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-021-00796-x
