
Abstract
The transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) is a key regulator of the cellular stress response, but the biological functions of the related Nrf3 protein are largely unknown. Here we demonstrate a novel pro-apoptotic function of Nrf3 in mouse and human keratinocytes. In response to UV irradiation, Nrf3-deficient keratinocytes were protected from apoptosis in vitro and in vivo. The protective function was also seen under oxidative or hyperosmotic stress conditions, but not when apoptosis was induced by disruption of cell–matrix interactions. Mechanistically, we show that Nrf3-deficient keratinocytes exhibit stronger cell–cell and cell-matrix adhesion, which correlates with higher cell surface integrin levels and enhanced activation of focal adhesion kinase. Nrf3-deficient cells also formed more and larger focal adhesions and exhibited a higher motility. These results suggest that the strong expression of Nrf3 in basal keratinocytes promotes their elimination in response to DNA damage-inducing agents, thereby preventing accumulation of mutated stem and transit amplifying cells in the epidermis.
Similar content being viewed by others


Introduction
The skin, as the outermost surface of the body, is frequently challenged by mechanical, chemical, or physical insults. Therefore, it has developed various protective mechanisms, which include formation of a physical protein/lipid barrier, but also expression of proteins involved in the detoxification of compounds or reactive oxygen species (ROS). Of particular importance is nuclear factor-erythroid 2-related factor 2 (Nrf2; NFE2L2), a member of the cap 'n’ collar family of transcription factors, which also includes p45 NF-E2, Bach1, Bach2, Nrf1, and Nrf3 (NFE2L3). Nrf2 controls the expression of ROS- and compound-detoxifying enzymes and other cytoprotective proteins through binding to antioxidant response elements (AREs) in the promoter or enhancer regions of these genes [1, 2,]. Due to its cytoprotective functions, Nrf2 is required for skin protection under stress conditions [3,4,5]. This offers interesting pharmacological perspectives, since topical application of Nrf2-activating compounds protected from inflammation, edema, and carcinogenesis [6, 7,]. In addition, Nrf2 activation promoted wound repair in healing-impaired diabetic mice [8].
In contrast to the extensively studied Nrf2, there is as yet little information on the related Nrf3 protein. Nrf3 knockout (Nrf3-ko) mice do not have an obvious phenotype under unchallenged conditions [9], but they are more susceptible to the development of carcinogen-induced T-cell lymphoblastic lymphoma [10]. The molecular mechanisms underlying the increased lymphoma susceptibility are as yet unknown. A transactivating activity of Nrf3 was described upon binding to the promoter of the NAD(P)H dehydrogenase quinone 1 (Nqo1) gene [11]. Other studies reported, however, that Nrf3 negatively regulates the expression of the Nrf2 target genes Nqo1 and peroxiredoxin 6 (refs. [12, 13,]). Therefore, it is still unclear if Nrf2 and Nrf3 have similar or antagonistic functions or if Nrf3 exerts its biological activities in an Nrf2-independent manner. The latter hypothesis is supported by the identification of Nrf3 targets genes in smooth muscle and colon cancer cells, which have not been described as targets of Nrf2 (refs. [14, 15,]).
Surprisingly, the role of Nrf3 in keratinocytes has not been determined, although it is strongly expressed in these cells in vitro and in normal and wounded skin in vivo [16]. Here we show that Nrf3 is dispensable for wound healing in mice, but its loss protects keratinocytes from apoptosis induced by UV-irradiation or other insults. This occurs in an Nrf2-independent manner and involves Nrf3-mediated alterations in cell–matrix interactions. These results identify an unexpected pro-apoptotic function of Nrf3, which controls the skin’s response to stress conditions.
Results
Nrf3 is dispensable for skin development and homeostasis
To unravel the function of Nrf3 in the skin, we first analyzed its expression and found much higher Nrf3 mRNA levels in the epidermis compared to the dermis of adult mouse skin (Fig. 1a). While Nrf2 establishes a gradient of UVB cytoprotection in the murine epidermis with higher expression in differentiated suprabasal and lower expression in undifferentiated basal cells [3], Nrf3 was expressed at much higher levels in basal compared to suprabasal cells (Fig. 1b). Immunostaining of mouse skin could not be performed due to the lack of a suitable antibody, but staining of human skin confirmed the predominant expression in the basal layer (Fig. 1c). NRF3 is also expressed in cultured human keratinocytes, where it localizes to the endoplasmic reticulum (ER) (Fig. 1d). The specificity of the antibody, which was raised against a peptide located in the middle of the NRF3 protein and should thus detect full-length NRF3, as well as a previously described nuclear cleavage product [14], was verified by staining of cells after siRNA-mediated NRF3 knockdown (Supplementary Fig. S1a-c).

Nrf3 is expressed in basal keratinocytes, but dispensable for skin development and homeostasis. a qRT-PCR of epidermal and dermal RNA for vimentin (Vim), K14 and Nrf3, normalized to Rps29. b qRT-PCR of RNA from basal and suprabasal layers of mouse tail skin for Nrf3. RNAs were pooled from five mice per genotype, two pools were analysed in independent experiments. The efficiency of basal/suprabasal separation in both experiments (Exp1 and Exp2) was verified by analysis of the suprabasal/basal ratio of Sprr2a expression, a marker for differentiated keratinocytes, as indicated. Expression in basal keratinocytes was set to 1. c NRF3 immunofluorescence staining of human skin sections (green), counterstained with DAPI (blue). Bar: 20 μm. d NRF3 immunofluorescence staining of HaCaT keratinocytes (red), counterstained with ER tracker (green) and Hoechst (blue). Note the ER localization of NRF3. e Hematoxylin/eosin (H/E; upper panel) and Ki67 immunohistochemistry staining (lower panel) of sections from back skin of wt and Nrf3-ko mice. Bars: 10 μm (H/E) and 100 μm (Ki67). The indent shows a higher magnification of the area indicated with a rectangle. Quantification of the number of Ki67 positive cells/mm of basement membrane is shown below. f Immunofluorescence staining of back skin sections for involucrin (Inv), K10, K14, or K6 (red), counterstained with DAPI (blue). Bar: 20 μm. Scatter plots in a, e show mean and standard deviation (S.D.). Each data point represents results from an individual mouse
Histological analysis of Nrf3-ko mice [9] did not reveal obvious skin abnormalities, and epidermal thickness, skin morphology, and keratinocyte proliferation were not affected (Fig. 1e). The differentiation-specific proteins keratin 14 (K14), K10, K6, and involucrin were normally expressed (Fig. 1f), and immunostaining, toluidine blue staining and flow cytometry demonstrated similar numbers and frequencies of different types of immune cells in the skin of wt and Nrf3-ko mice (Supplementary Fig. S2a-c).
Nrf3 is dispensable for wound healing in mice
Upon full-thickness excisional wounding, no healing abnormalities were detected in Nrf3-ko mice as shown by morphometric analysis of wound closure, length of the hyperproliferative wound epidermis (HE), area of HE, proliferation analysis of wound keratinocytes, and histopathological evaluation of the granulation tissue at day 3, 5, or 7 after wounding (Fig. 2a–f). The only difference was a slight delay in re-expression of the early differentiation marker K10 in the wound epidermis, while re-expression of the late differentiation marker loricrin was not altered (Supplementary Fig. S3a-c). Delayed K10 expression was, however, not seen in embryonic (E13.5) skin (Supplementary Fig. S3d).

Nrf3 is dispensable for wound healing. a H/E staining of sections from 3-day, 5-day, and 7-day wounds of 8–9-week-old mice. E: Epidermis; Es: Eschar; G: Granulation tissue; HE: Hyperproliferative wound epidermis; HF: Hair follicle. b–d Morphometric analysis of (b) percentage of wound closure, (c) length HE, and (d) area HE of 3-day, 5-day, and 7-day wounds. e, f Immunofluorescence staining for BrdU with arrowheads pointing to representative BrdU-positive cells (e) and quantitative analysis (f) of BrdU-positive cells in the HE of 3-day wounds. Bars: 500 μm (a) and 200 μm (f). Scatter plots show mean and SD in b–d and f. Each data point represents the result from an individual wound
Since the rate of wound healing declines upon aging [17], we also wounded 1-year-old mice. Mice of both genotypes indeed showed delayed wound closure compared to young mice (compare Supplementary Fig. S3e and Fig. 2b), but this was not further affected by Nrf3 deficiency (Supplementary Fig. S3e-g). Overall, these results demonstrate that Nrf3 is dispensable for wound healing in mice.
Nrf3 promotes UV-induced apoptosis of keratinocytes in vivo
Due to the important role of Nrf2 in UV protection [18], we determined if Nrf3 deficiency affects the UV response of keratinocytes. When wt mice were irradiated with a physiologically relevant dose of UVB (100 mJ/cm2), Nrf3 expression rapidly declined in the epidermis, followed by a strong upregulation at 24 h after irradiation (Fig. 3a, left panel). The UVB-induced downregulation of Nrf3 expression was confirmed with primary keratinocytes from wt or heterozygous Nrf3-ko mice (Fig. 3a, right panel).

Loss-of-Nrf3 protects keratinocytes from UVB-induced apoptosis in vivo. a RNA samples from epidermis of mice prior to and post irradiation with 100 mJ/cm2 UVB (a) or from primary keratinocytes of wt and heterozygous or homozygous Nrf3-ko mice prior to and at different time points after irradiation with 10 mJ/cm2 UVB (b) were analyzed by qRT-PCR for expression of Nrf3 relative to Rps29. b–c: Cleaved caspase 3 (b) or TUNEL staining (c; color changed from green to red) using sections from untreated (untr) skin or 1 d or 3 d after irradiation with 100 mJ/cm2 UVB. Nuclei were counterstained with Hoechst (blue). Bar (b, c): 100 µm. Positive cells per length epidermis are shown in the scatter plots. d Electron microscopy of UVB-irradiated epidermis. Upper left: Basal keratinocyte (arrowhead) that has lost cell–cell and cell-matrix contacts and reveals a hyperdense cytoplasm and a condensed nucleus as a sign of early apoptosis. Upper right: Debris of a keratinocyte (arrowhead) at a later stage of apoptosis with complete detachment from the basement membrane. Lower left: Basal keratinocytes (arrowheads) from Nrf3-ko mice show only slight condensation and shrinkage of the nuclei without signs of cytoplasmic alteration. Lower right: Some swollen and hypo-dense keratinocytes with signs of karyolysis (arrowheads) in the basal layer of Nrf3-ko mice, but without detachment from the basement membrane. Bar: 3 µm. e–g Sections from skin 24 h after irradiation with 100 mJ/cm2 UVB were stained with antibodies against γH2AX, p53 or Ly6G, and the numbers of γH2AX- or p53-positive keratinocytes or Ly6G-positive neutrophils per mm epidermis or per area dermis, respectively, were determined. 15–25 microscopic fields of skin sections per mouse were analyzed. Bars: 25 µm (e, f) and 100 µm (g). h RNA from the skin of wt and Nrf3-ko mice prior to and 24 h after UVB irradiation was analyzed by qRT-PCR for Il1b and Il6. RNAs were pooled from 5–6 mice per genotype and treatment group. The result was reproduced with RNAs from an independent experiment with different mice. Arrowheads point to apoptotic keratinocytes (b, c, e) or Ly6G-positive neutrophils (g). E: Epidermis, D: Dermis, HF: Hair follicle. Scatter plots in a–c and e–g show mean and SD. Each data point represents the result from an individual mouse
Extremely few apoptotic cells were detected in non-irradiated skin of wt and Nrf3-ko mice (Fig. 3b). Their numbers increased in mice of both genotypes upon irradiation with UVB, but to a significantly lesser extent in Nrf3-ko mice compared to wt controls as determined by cleaved caspase-3 or TUNEL staining (Fig. 3b, c). This was also reflected by an increased epidermal thickness of Nrf3-ko mice at this time point, while the dermal thickness was not affected (Supplementary Figure S4a-c).
Ultrastructural analysis of UVB-irradiated epidermis confirmed the surprising reduction in apoptosis in Nrf3-ko mice. Keratinocytes with a hyperdense cytoplasm and condensed nuclei that had lost their contact to the basement membrane and to neighboring keratinocytes were seen in wt mice (Fig. 3d, upper left panel), as well as a large number of cells that had completed the apoptotic program and had fully detached from the basement membrane (Fig. 3d, upper right panel). By contrast, most cells in Nrf3-ko mice lacked apoptotic features or only showed a slight condensation of the nucleus without cytoplasmic alterations (Fig. 3d, lower left panel). Signs of karyolysis were observed in some cells without detachment from the basement membrane (Fig. 3d, lower right panel).
The anti-apoptotic effect of Nrf3 deficiency was also observed in response to a high dose of solar light (5 J/cm2 UVA plus 1.5 J/cm2 UVB; Supplementary Fig. S4d). However, the reduced apoptosis of Nrf3-deficient epidermal cells was not associated with a significant reduction in the number of cells with DNA double strand breaks as revealed by immunostaining for phosphorylated histone 2AX (γH2AX) (Fig. 3e). Furthermore, there was no difference in the number of p53-positive keratinocytes (Fig. 3f).
UVB-induced neutrophil infiltration was attenuated in Nrf3-ko mice, and the UVB-induced increase in the mRNA levels of the pro-inflammatory cytokines interleukin (IL)-1β and IL-6 was less pronounced (Fig. 3g, h).
The anti-apoptotic effect of Nrf3 deficiency is cell autonomous and does not depend on altered DNA damage or repair
To determine whether the reduced UV-induced apoptosis of keratinocytes in Nrf3-ko mice is a cell autonomous effect, primary and immortalized keratinocytes were irradiated with UVB. Indeed, we observed a ~40% reduction of cleaved caspase 3-positive cells within 24 h after irradiation of primary keratinocytes from Nrf3-ko mice compared to cells from wt controls (Fig. 4a), and the reduction was even more pronounced for immortalized keratinocytes (70–80% at the 24 h time point) (Fig. 4b). The reduced apoptosis of Nrf3-deficient keratinocytes was observed as early as 1 h following irradiation and lasted for at least 30 h (Fig. 4b). It was confirmed by flow cytometry for annexin V and determination of cell viability using MTT assay (Fig. 4c, d). Similar to the situation in vivo, loss-of-Nrf3 did not affect the number of γH2AX or p53 positive cells within 6–24 h after UVB irradiation (Fig. 4e, f). This finding suggests that DNA damage and/or repair are not affected by Nrf3 deficiency and this was confirmed when DNA double strands were induced by the DNA damage-inducing compounds camptothecin (CPT) or hydroxyurea (HU) (Fig. 4g).

The effect of Nrf3 on UV-induced apoptosis is cell autonomous and independent of DNA damage. Primary (a) or spontaneously immortalized keratinocytes from Nrf3-ko mice or their wt littermates (b–d) were irradiated with 10 mJ/cm2 (a–c) or 20 mJ/cm2 (d) UVB. The percentage of cleaved caspase-3 or annexin V positive cells among all cells was determined by immunofluorescence staining and counting of 3–6 independent microscopic fields in three different dishes (a) or by flow cytometry (b, c). d Cell viability was determined using MTT assay 24 h post irradiation. Values were normalized to the signal obtained prior to UV irradiation and are shown as arbitrary units. e–g Immortalized keratinocytes from Nrf3-ko or wt mice were irradiated with 10 mJ/cm2 UVB (e, f, g) or treated with 0.1 µM camptothecin (CPT) or 3 mM hydroxyurea (HU) (g) and analyzed at different time points after irradiation or addition of the compound by flow cytometry for γH2AX (e, g) or by immunofluorescence staining for p53 (f). Scatter plots show mean and SD. Data points represent results from individual immortalized cell lines derived from different mice. h, i Primary human keratinocytes were transfected with siRNAs against NRF3 or caspase-5 (control) and irradiated with 10 mJ/cm2 UVB. The knockdown was verified by qRT-PCR for NRF3 relative to RPL27 (h). Apoptotic cells were identified by flow cytometry for cleaved caspase 3 (i). j HaCaT keratinocytes were irradiated with 20 mJ/cm2 UVB and cultured in the presence of absence of MG132 as indicated. Prior to and at different time points following irradiation they were co-stained with an NRF3 antibody, Hoechst and Alexa Fluor 488-coupled phalloidin. All results shown are representatives of at least two independent experiments
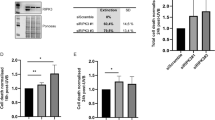
To determine the human relevance of our findings, we performed siRNA-mediated knockdown experiments using primary human foreskin keratinocytes. Although the knockdown efficiency was only 70–80% at the RNA level (Fig. 4h), Nrf3 knockdown cells showed reduced UVB-induced apoptosis compared to cells transfected with siRNA against the unrelated protein caspase-5, which does not affect keratinocyte apoptosis [19] (Fig. 4i). UVB irradiation did not obviously affect the ER localization of NRF3 (Fig. 4j), and we did not even detect nuclear staining in the presence of the proteasome inhibitor MG132, which stabilizes NRF3 and promoted its nuclear translocation in colon cancer cells [14].
Nrf3 deficiency does not affect intracellular ROS levels in keratinocytes and expression of cytoprotective Nrf2 target genes
Since Nrf2 protects keratinocytes from UV-induced apoptosis through its effect on ROS detoxification [18], we determined whether loss-of-Nrf3 affects the levels of intracellular ROS prior to and after UVB irradiation. Upon incubation of cells with 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) and analysis of 2,7-dichlorofluorescein (DCF) fluorescence, we found similar ROS levels in cultured keratinocytes from mice of both genotypes under basal conditions, as well as upon UVB irradiation (Fig. 5a). Together with the finding that UV-induced DNA damage was not affected by Nrf3 deficiency (Figs. 3e, 4e), these data suggest that Nrf3 does not enhance UVB-induced apoptosis via Nrf2 antagonism and thus through suppression of cytoprotective Nrf2 target genes. Indeed, expression levels of Nrf2 and of its target genes Nqo1, glutathione S-transferase A3 (Gsta3), and sulfiredoxin 1 (Srxn1) were similar in UVB-irradiated skin of Nrf3-ko and wt mice. As expected, expression of these genes was reduced in Nrf2-ko mice (Fig. 5b). Most importantly, the expression levels of Nrf2 target genes were similar in Nrf2 single and in Nrf2/Nrf3 double knockout mice (Fig. 5b), further indicating that Nrf3 does not antagonize the activity of Nrf2 under these conditions.

The effect of Nrf3 on UV-induced apoptosis is independent of ROS and Nrf2. a Flow cytometry analysis of DCF in non-irradiated (-UV) immortalized murine keratinocytes from Nrf3-ko and wt mice and 6 h after irradiation with 10 mJ/cm2 UVB. Relative drift of peaks was measured in two independent experiments with two immortalized keratinocyte lines of each genotype. Results from a representative experiment are shown. b Nrf3-ko, Nrf2-ko, Nrf2-ko/Nrf3-ko, and wt mice were irradiated with 100 mJ/cm2 UVB. Epidermal RNA from skin 24 h after UVB irradiation was analyzed by qRT-PCR for expression of Nqo1, Srxn1, GstA3, and Nrf2 relative to Rps29. Scatter plots show mean and SD. Each data point represents the result from an individual mouse
The anti-apoptotic effect of Nrf3 deficiency is dependent on cell-matrix adhesion
Nrf3-deficient keratinocytes were also protected from apoptosis induced by tert-butylhydroquinone, which induces apoptosis through an increase in mitochondrial ROS, or by sorbitol, which induces hyperosmotic stress (Fig. 6a). However, when apoptosis (anoikis) was induced by treatment with EDTA or EGTA, which chelate divalent cations or selectively Ca2+, respectively, and thus affect cell adhesion, the percentage of apoptotic cells was similar for both genotypes. Similar results were obtained with accutase, which detaches cells through its proteolytic/collagenolytic activity (Fig. 6b). This finding suggests that Nrf3 deficiency promotes cell adhesion and thereby protects from apoptosis/anoikis. This was also suggested by the reduced detachment of the Nrf3-deficient keratinocytes in vivo upon UV irradiation that we observed by electron microscopy (Fig. 3d). Adhesion assays with immortalized keratinocytes indeed showed strongly enhanced adhesion of cells from Nrf3-ko mice to collagen I, collagen IV and fibronectin (Fig. 6c). Vice versa, detachment of the cells upon incubation with trypsin was significantly delayed in the absence of Nrf3 (Fig. 6d). Enhanced adhesion was also seen in human primary keratinocytes upon NRF3 knockdown (Fig. 6e), indicating that this is not a secondary effect that results from long-term Nrf3 deficiency. To determine whether the adhesion phenotype results from a difference in the matrix that is deposited by wt versus Nrf3-ko keratinocytes, we performed adhesion assays using wt murine keratinocytes plated on matrix deposited by either wt or Nrf3-ko keratinocytes. However, the enhanced adhesion was not or only mildly affected by the matrix (Fig. 6f), suggesting that Nrf3 controls cell adhesion molecules rather than components of the matrix.

Loss-of-Nrf3 in keratinocytes protects from apoptosis by enhancing cell-matrix and cell–cell adhesion. a, b Immortalized keratinocytes from wt and Nrf3-ko mice were treated with tBHQ, sorbitol, accutase, EDTA, or EGTA or irradiated with 10 mJ/cm2 UVB. At different time points post treatment they were analyzed by flow cytometry for cleaved caspase-3. c Immortalized mouse keratinocytes were analyzed for adhesion on uncoated plastic or glass dishes or on dishes coated with collagen type I (Col I), type IV (Col IV), or fibronectin (FN) in triplicate wells. Adhesion of wt cells on uncoated dishes was set to 1. d Immortalized keratinocytes were analyzed for detachment from uncoated plastic dishes. Values obtained for non-treated wt cells were set to 100. e Human primary keratinocytes were transfected with NRF3 or CASP5 siRNAs. Cell adhesion was analyzed 24 h after transfection and 24 h after seeding. Adhesion of cells transfected with CASP5 siRNA was set to 1. f Immortalized keratinocytes were analyzed for adhesion on dishes coated with matrix deposited by either wt or Nrf3-ko cells. Adhesion of wt cells on dishes coated with matrix of wt cells was set to 1. g Representative pictures of sheets from wt and Nrf3-ko cells after dispase treatment and exposure to mechanical stress (left) and quantification of fragments (right). Scatter plots with mean and SD are shown. Data points represent results from individual immortalized cell lines derived from different mice or from different siRNA transfection experiments. All results shown are representatives of at least three independent experiments
Since the ultrastructural data also suggested a role of Nrf3 in cell–cell adhesion, we tested this possibility by detaching the cells from the dish using dispase, followed by exposure of the cell sheets to mechanical stress. Fragmentation of the sheets was much less pronounced in the absence of Nrf3 (Fig. 6g), demonstrating that Nrf3 deficiency indeed promotes cell–cell adhesion.
Nrf3 deficiency promotes focal adhesion formation and cell migration
Consistent with the enhanced cell-matrix adhesion, the number of focal adhesions (FA) was strongly increased in Nrf3-deficient keratinocytes as determined by staining for F-actin using phalloidin or by vinculin or zyxin immunofluorescence staining. FAs were clearly visible already 1 h after seeding in Nrf3-ko, but not in wt cells (Supplementary Fig. S5a), indicating that their formation is accelerated. At 24 h after seeding, FA numbers were still increased in Nrf3-ko keratinocytes, and these cells exhibited particularly large FAs (Fig. 7a, b). The effect of NRF3 on FA formation is unlikely to result from direct binding of NRF3 to FA components, since NRF3 did not localize to FAs in human keratinocytes (Fig. 7c).

Loss-of-Nrf3 in keratinocytes promotes focal adhesion formation and migration. Immortalized murine keratinocytes were allowed to attach for 24 h on uncoated plates and stained with Alexa Fluor 488-coupled phalloidin and vinculin (a) or zyxin (b) antibodies. Representative cells of the different genotypes are shown. Large FAs in Nrf3-ko cells are indicated by arrowheads (a). Quantification of FAs is shown in the bar graph. All results shown are representatives of at least two independent experiments. c HaCaT keratinocytes were analyzed by NRF3 immunofluorescence. The actin cytoskeleton was stained with Alexa Fluor 488-coupled phalloidin and nuclei were counterstained with Hoechst. Magnification bars: 20 µm (a–c). All results shown are representatives of at least two independent experiments. d–e Immortalized murine keratinocytes were subjected to scratch wounding and analyzed by live cell imaging for 20 h. Five cells from the front row from a minimum of five movies were analyzed for each genotype. All results were reproduced with three independent wt and two Nrf3-ko cell lines in four independent experiments. d Schematic illustration of parameters analyzed for the quantification of cell migration. Migrating immortalized murine keratinocytes were analyzed for velocity (a/time), (b) displacement, (c) perpendicular movement and (d) and persistence coefficient (“b”/”a” in e; [35]. Scatter plots with mean and S.D. are shown. Statistical analysis was performed using 1-way ANOVA with Tukey’s multiple comparison test
Since FA size predicts the migratory capability of cells [20], we performed scratch wounding assays with different lines of spontaneously immortalized keratinocytes from wild-type and Nrf3-ko mice, followed by live cell imaging. Interestingly, velocity, displacement, and perpendicular movement were strongly increased in Nrf3-deficient cells, resulting in a higher persistence coefficient (Fig. 7d, e).
Loss-of-Nrf3 affects adhesion signaling in UV-irradiated keratinocytes
To determine whether the enhanced adhesion of Nrf3-deficient keratinocytes results from upregulation of integrins, we used flow cytometry to determine the total and cell surface levels of several integrin subunits, which are highly expressed by keratinocytes [21]. While surface levels of β1, α6, and αV integrin were not affected by the loss-of-Nrf3 in untreated cells, β1 integrin surface levels were mildly increased after UVB irradiation in Nrf3-ko versus wt cells and this correlated with enhanced activation of β1 integrin (Fig. 8a; Supplementary Fig. S5b, c). However, RT-PCR analysis for β1 integrin and flow cytometry analysis of total cellular β1 integrin did not reveal obvious differences between cells of both genotypes (Supplementary Fig. S5d, Fig. 8a), suggesting that loss-of-Nrf3 does not affect β1 integrin expression, but its internalization, recycling, and/or activation in UV-irradiated keratinocytes.

Loss-of-Nrf3 enhances adhesion signaling prior to and following UVB irradiation. a Immortalized keratinocytes from wt and Nrf3-ko mice were permeabilized (total) or not (extracellular) and stained with antibodies against the integrin subunits β1 or active integrin β1, either prior to or 24 h after irradiation with 10 mJ/cm2 UVB and analyzed by flow cytometry. b–d Immortalized keratinocytes from wt and Nrf3-ko mice were irradiated with 10 mJ/cm2 UVB and analyzed by western blotting prior to and 24 h post irradiation for phosphorylated and total FAK and for α-tubulin (loading control) (b). The ratios of phosphorylated FAK, respectively total FAK to α-tubulin, as well as phosphorylated to total FAK are depicted in the graphs (c). The P-FAK positive control (P-FAK pos. ctrl) was obtained by incubating keratinocytes in fresh EGF-containing culture medium for 20 min immediately before sampling. Alternatively, cells were analyzed by qRT-PCR for expression of Fak relative to Rps29 (d). e Immortalized keratinocytes were treated with 20 µM zVAD before irradiation with 10 mJ/cm2 UVB and analyzed by western blotting 24 h post irradiation for phosphorylated and total FAK and for α-tubulin. The ratios of pFAK, respectively total FAK to α-tubulin, as well as pFAK to total FAK are depicted in the graphs (f). All results shown are representatives of at least two independent experiments
To further test this possibility, we determined whether adhesion signaling is enhanced in the absence of Nrf3 by analysis of focal adhesion kinase (FAK), a major player in integrin signaling, which also promotes adhesion-dependent cell survival [22]. Expression of FAK was mildly reduced in non-treated keratinocytes of Nrf3-ko compared to wt mice at the mRNA and protein level (Fig. 8b–d), while the levels of phosphorylated FAK (Tyr397) were not affected. Consistent with the caspase-mediated cleavage of FAK in apoptotic cells [23, 24,], levels of total and phosphorylated FAK strongly declined upon UVB irradiation in wt keratinocytes, while the reduction was much less pronounced in Nrf3-deficient cells (Fig. 8b, c). This finding most likely reflects the reduced UV-dependent apoptosis in the absence of Nrf3. By contrast, FAK mRNA levels were still lower in Nrf3-ko compared to wt cells upon UVB irradiation (Fig. 8d), suggesting that FAK is mainly controlled at the level of protein stability under these conditions. Importantly, the ratio of phosphorylated (activated) to total FAK was enhanced in Nrf3-deficient keratinocytes prior to and in particular following UVB irradiation.
To determine whether this is only a consequence of reduced caspase-mediated cleavage of FAK or also of enhanced adhesion signaling, we repeated the experiment in the presence of the pan-caspase inhibitor zVAD. The efficacy of the inhibitor was verified by its capability to reduce UV-induced apoptosis of wt keratinocytes (Supplementary Fig. S5e). While total FAK levels were similar in cells of both genotypes in the presence of zVAD due to inhibition of its caspase-mediated cleavage, phosphorylation of FAK was still much higher in keratinocytes from Nrf3-ko mice (Fig. 8e, f). Given the anti-apoptotic function of active FAK [25], this finding may at least partially explain the reduced apoptosis in Nrf3-deficient cells in response to various apoptotic stimuli.
Discussion
We identified Nrf3 as an important player in the UV response of keratinocytes and thus an entirely novel in vivo function of this poorly described protein.
The biological function of Nrf3 had remained fairly elusive, since no obvious abnormalities were observed in unchallenged Nrf3-ko mice [9], and we confirmed this for the skin in this study. In spite of the upregulation of Nrf3 mRNA levels in the wound epidermis [16] and the enhanced migration of Nrf3-deficient cultured keratinocytes (this study), wound healing was not affected by the loss-of-Nrf3. However, preliminary immunofluorescence data of human wounds suggest that NRF3 protein is downregulated in migrating wound keratinocytes, possibly to allow faster migration. Therefore, a further reduction may not have an additional effect.
The most striking finding of our study was the strong reduction in UVB-induced cell death in the absence of Nrf3, indicating that endogenous Nrf3 enhances UVB-induced keratinocyte apoptosis. Therefore, the downregulation of Nrf3 expression that we observed in response to UVB irradiation may prevent excessive UVB-induced apoptosis.
Our results suggest that Nrf3 does not exert its pro-apoptotic function through Nrf2 antagonism, since neither the levels of ROS nor DNA damage were affected by Nrf3 deficiency. In particular, the lack of Nrf3 did not affect the expression of several cytoprotective Nrf2 target genes. This discrepancy to previously published work, which had described positive or negative regulation of Nqo1 by Nrf3 (refs. [11,12,13]), may result from cell type-specific differences or from differences in the experimental conditions. It remains to be determined whether the expression of other Nrf2 target genes is affected by the loss-of Nrf3.
Our data strongly suggest that the effect of Nrf3 on apoptosis is mediated via Nrf3-dependent alterations in cellular adhesion. This hypothesis is supported by (i) the reduced detachment and enhanced adhesion of cultured mouse and human keratinocytes with Nrf3 knockout or knockdown, respectively, (ii) the strong cell–cell and cell-matrix adhesion of Nrf3-deficient keratinocytes in vivo as shown by ultrastructural analysis, and (iii) the protective effect of Nrf3 deficiency from apoptosis by various insults, provided that they do not affect cell adhesion. At the molecular level, loss of Nrf3 resulted in enhanced levels of pFAK in untreated and UV-irradiated keratinocytes and also reduced the UV-induced cleavage of FAK. FAK is involved in the early steps of adhesion-mediated integrin signaling that is crucial for the attachment to the ECM and for cell survival [25]. Furthermore, UV irradiation resulted in caspase-dependent cleavage of FAK in Madin-Darby kidney cells, and overexpression of FAK increased cell survival in response to UV irradiation [26]. Thus, the enhanced levels of activated β1 integrin and the increased levels of total and phosphorylated FAK that we observed in Nrf3-deficient cells after UV irradiation in comparison to wt cells may at least partially explain the attenuated cell death. However, additional mechanisms, such as upregulation/activation of adhesion molecules other than integrins may well contribute to this effect. This seems likely, since the total and surface levels of different keratinocyte integrins, as well as activation of β1 integrin were not altered in non-irradiated Nrf3-deficient cells, although cell–cell and cell-matrix adhesion were enhanced.
In the future, it will be important to identify the targets of Nrf3, which control adhesion signaling and concomitant cell survival. The latter are likely to be transcriptional targets of Nrf3, but non-transcriptional functions of Nrf3 in the control of cell–cell and cell-matrix adhesion cannot be excluded. Consistent with such a possibility, we found that NRF3 mainly or exclusively localizes to the ER in human keratinocytes. Furthermore, both mouse and human NRF3 localize to the ER in other cell types, and nuclear translocation requires proteolytic cleavage [14, 27, 28,]. While our stainings argue for a role of ER-bound NRF3 in its anti-apoptotic effect, we cannot rule out that small amounts of NRF3, which are undetectable by immunostaining, are cleaved and transported to the nucleus, resulting in transcriptional regulation of target genes. In addition, the reactivity of the antibody with the nuclear form may be less pronounced than with the ER form in immunofluorescence experiments.
Independent of the responsible targets, the pro-apoptotic function of Nrf3 described in this study is likely to be of major biological relevance. Thus, we previously showed that expression of Nrf2 and its target genes is low in basal, but much higher in suprabasal cells [3]. In response to UVB irradiation, this distribution allows survival of suprabasal keratinocytes, thereby preserving skin integrity. Concomitantly, basal cells are highly susceptible to UVB-induced apoptosis. This is physiologically important, since it prevents the proliferation and accumulation of mutated stem and transit amplifying cells, which would strongly increase the risk of malignant transformation. This mechanism may be enforced by the high abundance of Nrf3 in basal keratinocytes. Therefore, low levels of Nrf2 combined with higher levels of Nrf3 in stem and transit amplifying cells of the basal layer are likely to confer high susceptibility of these cells to UVB-induced death. This may be particularly important upon irradiation with low doses of UV where the low levels of Nrf2 may be sufficient to allow survival. Future studies will reveal if Nrf3 deficiency affects UV-induced skin carcinogenesis and if Nrf3 activation can be used for skin cancer prevention.
Materials and methods
Genetically modified mice
Nrf2-ko and Nrf3-ko mice (both in C57BL/6 background) were previously described [9, 29,]. Mice were housed and fed according to federal guidelines, and all animal experiments had been approved by the local veterinary authorities of Zurich, Switzerland.
Wounding and preparation of wound tissue
Female mice at the age of 8–9 weeks or ~1 year were anaesthetized, and two full-thickness excisional wounds, 5 mm in diameter, were made on each side of the dorsal midline by excising skin and panniculus carnosus as previously described [30]. Mice were killed at different time points after wounding. For histological analysis, the complete wounds were excised and either fixed overnight in 95% ethanol/1% acetic acid or in 4% paraformaldehyde/PBS followed by paraffin embedding, or directly frozen in tissue freezing medium (Leica Microsystems, Heerbrugg, Switzerland). Sections (7 μm) from the middle of the wounds were stained with hematoxylin/eosin (H/E) or used for immunofluorescence/immunohistochemical analysis. Morphometric analysis of different parameters of the wound healing process was performed using H/E- or immunofluorescence-stained sections.
UVB irradiation of mice and cultured cells
Eight- to nine-week-old mice were irradiated with 100 mJ/cm2 UVB as previously described [3] or with a combination of 5 J/cm2 UVA and 1.5 J/cm2 UVB and killed at different time points after UV irradiation. Cultured cells were irradiated with 10–20 mJ/cm2 UVB. Irradiation was monitored with a UV dosimeter.
Separation of basal and suprabasal keratinocytes
Separation of basal and suprabasal keratinocytes from mouse tail skin was performed using a previously described protocol based on chymotrypsin treatment of epidermal sheets [3].
Immunofluorescence and immunohistochemistry
For immunofluorescence staining of cultured keratinocytes, cells were fixed for 20 min with 4% PFA, washed twice with PBS, and non-specific binding sites were blocked with PBS/12% BSA/0.025% NP-40 for 1 h at room temperature. Subsequently, cells were incubated overnight at 4 °C with the primary antibody (Supplementary Table S1A) diluted in the same blocking solution. After three washes with PBST (1 × PBS/0.1% Tween 20), they were incubated for 1 h with secondary antibodies, Alexa Fluor 488-coupled phalloidin (1:000, Life Technologies, Carlsbad, CA) and Hoechst (1 μg/ml), washed with PBST again and mounted with Vectashield mounting medium (from Vector Laboratories, Burlingame, CA).
Tissue sections (7 μm) were deparaffinized and rehydrated, or fixed with cold methanol (in case of frozen sections). Non-specific binding sites were blocked with PBS/12% BSA/0.025% NP-40 for 1 h at room temperature, and sections were then incubated overnight at 4 °C with the primary antibodies (Supplementary Table S1A) diluted in the same buffer. If needed, antigen retrieval was performed prior to the blocking procedure by cooking in citrate buffer (1 h at 95 °C). For immunohistochemistry, endogenous peroxidase activity was quenched with 3% H2O2 for 10 min prior to blocking. After three washes with PBST (1× PBS/0.1% Tween 20), slides were incubated for 1 h with secondary antibodies, washed with PBST again and mounted with Mowiol (Hoechst, Frankfurt, Germany). All secondary antibodies were from Jackson ImmunoResearch, West Grove, PA. Sections were photographed using a Zeiss Imager.A1 microscope equipped with an Axiocam MRm camera and EC Plan-Neofluar objectives (×10/0.3, ×20/0.5) (Zeiss, Oberkochen, Germany) or a Leica SP8-AOBS microscope (Leica, Wetzlar, Germany) equipped with a sCMOS camera (Hamamatsu Orca Flash 4.0; Hamamatsu Photonics, Hamamatsu, Japan). For data acquisition, we used the Axiovision 4.6 software from Zeiss or the Leica LAS X SP8 Version 1.0.
Analysis of cell proliferation by incorporation of 5-bromo-2′-deoxyuridine (BrdU)
Mice were injected intraperitoneally (i.p.) with BrdU (250 mg/kg in 0.9% NaCl; Sigma) at different time points after wounding and killed 2 h after injection. Skin samples were fixed in 95% ethanol/1% acetic acid. Sections were incubated with a peroxidase-conjugated monoclonal antibody directed against BrdU (Roche, Rotkreuz, Switzerland), and BrdU-positive cells were visualized by staining with diaminobenzidine.
Preparation of keratinocyte lysates and western blot analysis
Murine keratinocytes were lysed in 400 μl (per 10 cm dish) of cell lysis buffer (20 mM Tris-HCl, pH 8.0, 2 mM EDTA, 1% Triton X-100, 137 mM NaCl, 10% glycerol, protease inhibitor (Complete, EDTA-free) and phosphatase inhibitor (PhosSTOP) (both from Roche). Cells were scraped off the dish and sonicated. After centrifugation, the protein concentration was determined using the bicinchoninic acid (BCA) kit (Pierce, Rockford, IL). Proteins were then separated by SDS–PAGE and transferred to a nitrocellulose membrane. Antibody incubations were performed in 5% non-fat dry milk in TBS-T (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.05% Tween 20). Membranes were probed with the primary antibodies (Supplementary Table S1B). Secondary antibodies were from Jackson ImmunoResearch. Equal loading and transfer efficiency were confirmed by Ponceau S staining of the membrane before antibody treatment.
Electron microscopy
UVB-irradiated mice were lethally anesthetized with pentobarbital (700 mg/kg) and perfused with 4% PFA in PBS. Skin samples were kept overnight in fixation solution, rinsed, and stored in PBS. After washing in 0.1 M cacodylate buffer pH 7.2 at 4 °C, the specimens were treated with 2% OsO4 for 2 h. After washing, they were stained in 1% uranyl acetate, dehydrated, and embedded in araldite resin. Ultra-thin sections (30–60 nm) were processed with a diamond knife and placed on copper grids. Transmission electron microscopy was performed using an EM109 electron microscope (Zeiss, Oberkochen, Germany).
Separation of dermis from epidermis of mouse back skin
Separation of epidermis from dermis was achieved either by heat shock treatment (30 s at 55–60 °C followed by 1 min at 4 °C, both in PBS), or by incubation for 50–60 min at 37 °C in 0.143% dispase/DMEM. For dispase treatment the subcutaneous fat was gently scraped off with a scalpel prior to incubation. Isolated epidermis was either directly snap frozen and stored at −80 °C, or homogenized and further processed.
Staining of mast cells with toluidine blue
After deparaffinization and rehydration, skin sections were stained with 0.5% toluidine blue, 0.5 N HCl, pH 2.3 for 30 min. After washing with distilled water, sections were dehydrated and mounted with Eukitt mounting medium (Sigma). Stained mast cells appear violet or purple.
TUNEL staining
TUNEL assays were performed on paraffin sections of 4% PFA-fixed UVB-irradiated skin or cultured keratinocytes using the in situ cell death detection kit (Roche).
Flow cytometry
Dermis and epidermis were separated as described above, further processed into single-cell suspensions and stained as described previously [31]. Immortalized keratinocytes were trypsinized, stained with primary antibodies for 10 min on ice, washed, and stained with secondary antibodies. Dyes and antibodies used for flow cytometry are listed in Supplementary Table S1c. Fluorescence was directly measured by flow cytometry using the BD LSRFortessa or BD ACCURI C6 PLUS (BD Biosciences, San Jose, CA).
Culture of murine primary keratinocytes
Isolation and cultivation of murine primary keratinocytes was performed as previously described using 2- to 4-day-old mice [16]. Cells were seeded at a density of 5 × 104 cells per cm2 on collagen IV (2.5 g/cm2) coated dishes and incubated for 30 min at 37 °C/5% CO2. Thereafter the medium was replaced, and cells were grown to 90% confluency in defined keratinocyte serum-free medium (Invitrogen, Paisley, UK) supplemented with 10 ng/ml epidermal growth factor (EGF) and 10−10 M cholera toxin. Spontaneously immortalized mouse keratinocytes were obtained by serial passaging of the primary cells. This resulted in senescence of most of the cells, but a few foci of immortalized cells appeared, and these cells were further passaged.
Determination of intracellular ROS levels
Intracellular levels of ROS were determined in primary and immortalized keratinocytes using H2DCF-DA (Invitrogen) as previously described [32].
Cell adhesion assay
Immortalized mouse keratinocytes were seeded into 96-well plates coated with bovine serum albumin (BSA) or different concentrations of collagen type I (Roche), collagen type IV (Sigma), or fibronectin (Corning, Corning, NY). The assay was performed as previously described [33].
Preparation of cell-free extracellular matrix from keratinocytes
Immortalized mouse keratinocytes were seeded into 24-well plates and grown to confluency. Forty-eight hours after 100% confluency was reached, cells were incubated with trypsin gold (Promega) for 20 min. Cell-free extracellular matrix-coated plates were washed with PBS and stored at 4 °C.
Cell detachment assay
Immortalized mouse keratinocytes were seeded into 24-well plates and incubated with trypsin gold (Promega) for different time periods. After two washing steps with PBS, the remaining attached cells were stained as described in the cell adhesion assay.
Sheet formation assay
Immortalized keratinocytes were cultured in culture medium enriched with Ca2+ (1.8 mM) in 6-well plates until they reached confluency. After washing cells twice with 1× PBS (enriched with 1.8 mM Ca2+), they were incubated in dispase solution (9 mg/ml dispase in 50 mM HEPES, 150 mM CaCl2, pH 7.4) mixed 1:3 with culture medium. Detached sheets were transferred into 15 ml tubes containing 4 ml of 1× PBS. Cell sheets were exposed to continuous shaking using a shaking rotator (30 U/min for 30 s). Resulting sheet fragments were counted and normalized to initial numbers before rotating.
RNA isolation and qRT-PCR
RNA from tissue was isolated with Trizol followed by purification with the RNeasy Mini Kit, including on-column DNase treatment (Qiagen, Hilden, Germany). RNA from cultured keratinocytes was extracted directly with the RNeasy Mini Kit. cDNA was synthesized using the iScript kit (Bio-Rad Laboratories, Hercules, CA). Relative gene expression was determined using the Roche LightCycler 480 SYBR Green system (Roche). Primers used for qRT-PCR are listed in Supplementary Table S2A.
Isolation, culture, and siRNA transfection of human keratinocytes
Human primary keratinocytes were isolated from foreskin and cultured up to passage 3 in Keratinocyte-SFM (Invitrogen) supplemented with epidermal growth factor and bovine pituitary extract. They were grown to 30% confluence in 3.5 cm dishes and transfected with 50 nM 21-mer siRNA duplexes using INTERFERin (Polyplus, Illkirch, France). Transfection was repeated after 48 h, followed by a 24 h incubation period. The siRNA duplexes used (all from Sigma-Aldrich) are listed in Supplementary Table S2B.
HaCaT keratinocytes [34] were cultured in DMEM supplemented with 10% fetal calf serum and penicillin/streptomycin.
Methylthiazolyldiphenyltetrazolium bromide (MTT) assay
Immortalized keratinocytes were grown to 70–80% confluency and treated with mitomycin C (2 μg/ml medium) for 2 h. Subsequently, cells were irradiated with 20 mJ/cm2 UVB and 24 h later treated with MTT (2 μM) for 30 min at 37 °C. They were lysed with 150 μl of 0.04 M hydrochloric acid in isopropanol. After 10 min at RT, 150 μl dH2O was added and absorption at 590 nm was measured using a SpectraMAX 190 (Molecular Devices, Sunnyvale, CA).
In vitro cell migration assay
Murine keratinocytes were grown to 100% confluency in culture medium, and treated with 10 μg/ml mitomycin C for 2 h. One or several scratches were made into the cell layer using a sterile 200 μl pipette tip. Dead cells and debris were washed off with pre-warmed PBS, and fresh pre-warmed culture medium was added. For live cell imaging, cells were immediately transferred to a microscope equipped with a Zeiss Axiovert 200 M incubator box. Pictures were acquired for up to 20 h using Metamorph 7.53.
Finally, five cells from the front row were analyzed with regard to velocity, displacement (linear distance from starting to end point), persistence coefficient (as described in [35]) and perpendicular movement into the scratch.
Statistical analysis
Statistical analyses were performed using Prism version 6.0 software (GraphPad Software, La Jolla, CA). All data were analyzed using the non-parametric Mann–Whitney U-test for non-Gaussian distributions. Scatter plots with mean values and standard deviation are shown in most figures. *P ≤ 0.05, **P ≤ 0.005, ***P ≤ 0.001.
Change history
22 October 2019
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Motohashi H, Yamamoto M. Carcinogenesis and transcriptional regulation through Maf recognition elements. Cancer Sci. 2007;98:135–9.
Sykiotis GP, Bohmann D. Stress-activated cap’n’collar transcription factors in aging and human disease. Sci Signal. 2010;3:re3.
Schafer M, Dutsch S, auf dem Keller U, Navid F, Schwarz A, Johnson DA, et al. Nrf2 establishes a glutathione-mediated gradient of UVB cytoprotection in the epidermis. Genes Dev. 2010;24:1045–58.
Kawachi Y, Xu X, Taguchi S, Sakurai H, Nakamura Y, Ishii Y, et al. Attenuation of UVB-induced sunburn reaction and oxidative DNA damage with no alterations in UVB-induced skin carcinogenesis in Nrf2 gene-deficient mice. J Invest Dermatol. 2008;128:1773–9.
Saw CL, Huang MT, Liu Y, Khor TO, Conney AH, Kong AN. Impact of Nrf2 on UVB-induced skin inflammation/photoprotection and photoprotective effect of sulforaphane. Mol Carcinog. 2011;50:479–86.
Dinkova-Kostova AT, Jenkins SN, Fahey JW, Ye L, Wehage SL, Liby KT, et al. Protection against UV-light-induced skin carcinogenesis in SKH-1 high-risk mice by sulforaphane-containing broccoli sprout extracts. Cancer Lett. 2006;240:243–52.
Talalay P, Fahey JW, Healy ZR, Wehage SL, Benedict AL, Min C, et al. Sulforaphane mobilizes cellular defenses that protect skin against damage by UV radiation. Proc Natl Acad Sci USA. 2007;104:17500–5.
Long M, Rojo de la Vega M, Wen Q, Bharara M, Jiang T, Zhang R, et al. An essential role of NRF2 in diabetic wound healing. Diabetes. 2016;65:780–93.
Derjuga A, Gourley TS, Holm TM, Heng HH, Shivdasani RA, Ahmed R, et al. Complexity of CNC transcription factors as revealed by gene targeting of the Nrf3 locus. Mol Cell Biol. 2004;24:3286–94.
Chevillard G, Paquet M, Blank V. Nfe2l3 (Nrf3) deficiency predisposes mice to T-cell lymphoblastic lymphoma. Blood. 2011;117:2005–8.
Kobayashi A, Ito E, Toki T, Kogame K, Takahashi S, Igarashi K, et al. Molecular cloning and functional characterization of a new Cap’n’ collar family transcription factor Nrf3. J Biol Chem. 1999;274:6443–52.
Chowdhury I, Mo Y, Gao L, Kazi A, Fisher AB, Feinstein SI. Oxidant stress stimulates expression of the human peroxiredoxin 6 gene by a transcriptional mechanism involving an antioxidant response element. Free Radic Biol Med. 2009;46:146–53.
Sankaranarayanan K, Jaiswal AK. Nrf3 negatively regulates antioxidant-response element-mediated expression and antioxidant induction of NAD(P)H:quinone oxidoreductase1 gene. J Biol Chem. 2004;279:50810–7.
Chowdhury A, Katoh H, Hatanaka A, Iwanari H, Nakamura N, Hamakubo T, et al. Multiple regulatory mechanisms of the biological function of NRF3 (NFE2L3) control cancer cell proliferation. Sci Rep. 2017;7:12494.
Pepe AE, Xiao Q, Zampetaki A, Zhang Z, Kobayashi A, Hu Y, et al. Crucial role of nrf3 in smooth muscle cell differentiation from stem cells. Circ Res. 2010;106:870–9.
Braun S, Hanselmann C, Gassmann MG, auf dem Keller U, Born-Berclaz C, Chan K, et al. Nrf2 transcription factor, a novel target of keratinocyte growth factor action which regulates gene expression and inflammation in the healing skin wound. Mol Cell Biol. 2002;22:5492–505.
Swift ME, Kleinman HK, DiPietro LA. Impaired wound repair and delayed angiogenesis in aged mice. Lab Invest. 1999;79:1479–87.
Schäfer M, Werner S. Nrf2–A regulator of keratinocyte redox signaling. Free Radic Biol Med. 2015;88(Pt B):243–52.
Sollberger G, Strittmatter GE, Grossi S, Garstkiewicz M, Auf dem Keller U, French LE, et al. Caspase-1 activity is required for UVB-induced apoptosis of human keratinocytes. J Invest Dermatol. 2015;135:1395–404.
Kim DH, Wirtz D. Focal adhesion size uniquely predicts cell migration. FASEB J. 2013;27:1351–61.
Moik DV, Janbandhu VC, Fassler R. Loss of migfilin expression has no overt consequences on murine development and homeostasis. J Cell Sci. 2011;124(Pt 3):414–21.
Frisch SM, Vuori K, Ruoslahti E, Chan-Hui PY. Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol. 1996;134:793–9.
Gervais FG, Thornberry NA, Ruffolo SC, Nicholson DW, Roy S. Caspases cleave focal adhesion kinase during apoptosis to generate a FRNK-like polypeptide. J Biol Chem. 1998;273:17102–8.
Grossmann J, Artinger M, Grasso AW, Kung HJ, Scholmerich J, Fiocchi C, et al. Hierarchical cleavage of focal adhesion kinase by caspases alters signal transduction during apoptosis of intestinal epithelial cells. Gastroenterology. 2001;120:79–88.
Gilmore AP, Owens TW, Foster FM, Lindsay J. How adhesion signals reach a mitochondrial conclusion–ECM regulation of apoptosis. Curr Opin Cell Biol. 2009;21:654–61.
Chan PC, Lai JF, Cheng CH, Tang MJ, Chiu CC, Chen HC. Suppression of ultraviolet irradiation-induced apoptosis by overexpression of focal adhesion kinase in Madin-Darby canine kidney cells. J Biol Chem. 1999;274:26901–6.
Nouhi Z, Chevillard G, Derjuga A, Blank V. Endoplasmic reticulum association and N-linked glycosylation of the human Nrf3 transcription factor. FEBS Lett. 2007;581:5401–6.
Zhang Y, Kobayashi A, Yamamoto M, Hayes JD. The Nrf3 transcription factor is a membrane-bound glycoprotein targeted to the endoplasmic reticulum through its N-terminal homology box 1 sequence. J Biol Chem. 2009;284:3195–210.
Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci USA. 1996;93:13943–8.
Antsiferova M, Martin C, Huber M, Feyerabend TB, Forster A, Hartmann K, et al. Mast cells are dispensable for normal and activin-promoted wound healing and skin carcinogenesis. J Immunol. 2013;191:6147–155.
Sulcova J, Meyer M, Guiducci E, Feyerabend TB, Rodewald HR, Werner S. Mast cells are dispensable in a genetic mouse model of chronic dermatitis. Am J Pathol. 2015;185:1575–87.
Durchdewald M, Beyer TA, Johnson DA, Johnson JA, Werner S, auf dem Keller U. Electrophilic chemicals but not UV irradiation or reactive oxygen species activate Nrf2 in keratinocytes in vitro and in vivo. J Invest Dermatol. 2007;127:646–53.
Meyer M, Muller AK, Yang J, Moik D, Ponzio G, Ornitz DM, et al. FGF receptors 1 and 2 are key regulators of keratinocyte migration in vitro and in wounded skin. J Cell Sci. 2012;125(Pt 23):5690–701.
Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988;106:761–71.
Hartwig B, Borm B, Schneider H, Arin MJ, Kirfel G, Herzog V. Laminin-5-deficient human keratinocytes: defective adhesion results in a saltatory and inefficient mode of migration. Exp Cell Res. 2007;313:1575–87.
Acknowledgements
We thank Christiane Born-Berclaz, Hayley Hiebert, Nicole Hallschmid, and Dr. Sabine Dütsch, ETH Zürich, for valuable experimental help and Dr. Josef Jiricny, ETH Zurich, for helpful suggestions. Nrf2 knockout mice were kindly provided by Dr. Yuet-Wai Kan (University of California, San Francisco, CA). This work was supported by the Wilhelm Sander Foundation (to S.W.) and the Swiss National Science Foundation (310030_132884 and 31003A_169204 to S.W.). B.S., C.D., and S.M. are/were members of the Life Science Zurich Graduate School.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Edited by B. Dynlacht
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. If you remix, transform, or build upon this article or a part thereof, you must distribute your contributions under the same license as the original. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/.
About this article

Cite this article
Siegenthaler, B., Defila, C., Muzumdar, S. et al. Nrf3 promotes UV-induced keratinocyte apoptosis through suppression of cell adhesion. Cell Death Differ 25, 1749–1765 (2018). https://doi.org/10.1038/s41418-018-0074-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-018-0074-y
