
Abstract
Desmosome components are frequently mutated in cardiac and cutaneous disorders in animals and humans and enhanced inflammation is a common feature of these diseases. Previous studies showed that inhibitor of Apoptosis Stimulating p53 Protein (iASPP) regulates desmosome integrity at cell–cell junctions and transcription in the nucleus, and its deficiency causes cardiocutaneous disorder in mice, cattle, and humans. As iASPP is a ubiquitously expressed shuttling protein with multiple functions, a key question is whether the observed cardiocutaneous phenotypes are caused by loss of a cell autonomous role of iASPP in cardiomyocytes and keratinocytes specifically or by a loss of iASPP in other cell types such as immune cells. To address this, we developed cardiomyocyte-specific and keratinocyte-specific iASPP-deficient mouse models and show that the cell-type specific loss of iASPP in cardiomyocytes or keratinocytes is sufficient to induce cardiac or cutaneous disorders, respectively. Additionally, keratinocyte-specific iASPP-deficient mice have delayed eyelid development and wound healing. In keratinocytes, junctional iASPP is critical for stabilizing desmosomes and iASPP deficiency results in increased and disorganized cell migration, as well as impaired cell adhesion, consistent with delayed wound healing. The identification of a cell autonomous role of iASPP deficiency in causing cardiocutaneous syndrome, impaired eyelid development and wound healing suggests that variants in the iASPP gene also may contribute to polygenic heart and skin diseases.
Similar content being viewed by others

Introduction
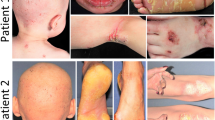
Thousands of human diseases are caused by single gene defects and these diseases affect >25–30 million people in the US alone [1]. Detailed mechanistic insights into how single gene defects can cause rare genetic disorders are critical to advance our understanding of the illness, and may also provide important insights for polygenic diseases, such as cancer and cardiovascular diseases, and physiological processes like development and wound healing. Cardiocutaneous syndromes are examples of disorders that can be caused by single gene defects, and patients with cardiocutaneous syndromes often present with cardiac and epidermal abnormalities that primarily originate from defects in cell junction proteins. For example, Naxos and Carvajal syndromes are cardiocutaneous syndromes caused by mutations in desmosomal intercellular junction genes such as ɣ-catenin and desmoplakin, respectively. Patients with naxos or carvajal syndrome have arrhythmogenic right ventricular cardiomyopathy (ARVC), a type of dilated cardiomyopathy (DCM), along with epidermal abnormalities that include palmoplantar keratoderma and woolly hair [2, 3].
Mutations in components of desmosomes are involved in many cardiocutaneous syndromes [4]. Desmosomes are anchoring junctions predominantly found in cells that endure physical stress, such as cardiomyocytes and epidermal keratinocytes. Desmosomes link intermediate filaments (IFs) to intercellular junctions, forming cellular scaffolding that distributes mechanical forces throughout the tissue [5]. The three classes of desmosome components are: the cadherin family, (i.e., desmoglein (DSG) and desmocollin (DSC)); the armadillo family, (i.e., ɣ-catenin (JUP) and plakophilin (PKP)); and the plakin family (i.e., desmoplakin (DSP)), responsible for anchoring IFs such as desmin and keratin to desmosomes in cardiomyocytes and keratinocytes, respectively [6]. For understanding the mechanisms underlying cardiocutaneous syndromes, an important question is whether the DCM and cutaneous phenotypes are caused by cell intrinsic mechanisms in specific cell types (i.e., compromised desmosome integrity in cardiomyocytes and keratinocytes, respectively) or if the phenotypes are due to extrinsic mechanisms (i.e., paracrine signaling from other cells/tissues). For example, inflammation is an extrinsic mechanism and almost all cardiocutaneous patients exhibit elevated levels of inflammatory cytokines regardless of their gene mutation status [7]. Inflammation is also a key process in several models of DCM [8]. For example, gamma-delta T cells have been reported to induce and/or mediate DCM [9], in part by initiating cardiomyocyte apoptosis [10]. Therefore, it is plausible that the inflammatory response could be involved in cardiac dysfunction in cardiocutaneous patients.
A recently identified single gene mutation that causes cardiocutaneous syndrome (and cardiocutaneous disorder in cattle and mice) is PPP1R13L [11,12,13,14,15], which encodes inhibitor of apoptosis stimulating p53 protein (iASPP), an evolutionarily conserved inhibitor of p53 and p63 [16, 17]. iASPP is a regulator of desmosomes [13], but is also implicated in inflammatory pathways [11, 18]. For example, iASPP was originally identified as a binding partner for NF-κB in a yeast two-hybrid assay [18] and, consistent with this, iASPP-deficiency in patients and mice results in elevated expression of inflammation-related genes, some of which are NF-κB targets [11, 12]. Given that iASPP is ubiquitously expressed and germline mutations in the iASPP gene could affect the function of all iASPP-expressing cells as well as the multiple pathways it regulates, a key challenge is to identify the molecular mechanisms by which the faulty gene causes the observed phenotype.
In addition, the function of iASPP in different cell types seems to depend on its localization. iASPP can localize at cell-cell junctions, the cytoplasm and the nucleus, and can shuttle between different cellular compartments depending on intracellular signals [19,20,21]. For example, in basal keratinocytes of the skin iASPP is in the nucleus where it regulates p63, a master transcription factor of stratified squamous epithelium [17]; in differentiating keratinocytes and in cardiomyocytes iASPP localizes at cell–cell junctions, where it interacts with desmoplakin and IFs to stabilize desmosomes [13]. Therefore, shuttling of iASPP between cell-cell junctions and the nucleus could have profound effects on desmosome stability and nuclear gene expression [20], and loss of both junctional and nuclear iASPP could be important in cardiocutaneous syndrome. For example, loss of nuclear iASPP could result in activation of NF-κB in certain cell types leading to enhanced secretion and activity of inflammatory mediators [11], whereas loss of cell-junctional iASPP could result in desmosome instability [13].
Here, we use specific deletion of iASPP in either cardiomyocytes or keratinocytes to address three main questions: (1) are the cardiac and cutaneous phenotypes due to loss of iASPP in cardiomyocytes and keratinocytes, respectively; (2) what is the potential role of iASPP loss in keratinocytes in development and wound healing; and (3) mechanistically, how can iASPP regulate keratinocyte function? This study provides evidence for an important cell autonomous role of iASPP in vivo.
Results
Cardiomyocyte-specific and keratinocyte-specific iASPP deficiency recapitulates the main features of cardiocutanous syndrome
We previously showed that germline iASPP deficiency in mice results in cardiac [13] and cutaneous disorders [17]. To examine if the observed cardiocutaneous phenotype can be induced by iASPP deficiency in cardiomyocytes or keratinocytes only, we crossed mice with a floxed iASPP gene to mice with Cre recombinase controlled either by a cardiac-specific α-myosin heavy chain (αMyHC) promoter or an epidermis-specific keratin 14 (K14) promoter, respectively (Supplementary Figure 1A; see schematic) [22, 23]. The reported effectiveness of these Cre promoters is quite high, as >90% recombination and gene deletion in cardiac muscle cells has been achieved using the αMyHC promoter [22, 24] and only a small number of cells with incomplete gene deletion in epidermis and hair bulge have been reported using the K14 promoter [25]. We observed comparable deletion of iASPP in our tissue-specific models (Supplementary Figure 1B for keratinocytes; see below for cardiomyocytes).
Similar to germline iASPP-deficient animals [13], αMyHC-Cre iASPP-deficient mice presented with complete penetrance of the myocardial phenotype, including scarring of the myocardium (Fig. 1a). Furthermore, we observed dilation of the left ventricle (LV) with prominent scarring of the septum and LV free wall in adult mice (Supplementary Figure 1C). In contrast, K14-Cre iASPP-deficient mice had no apparent myocardial abnormalities (Fig. 1b), but had corneal opacity, woolly hair and palmoplantar phenotype, similar to the germline iASPP-deficient mice (Fig. 1c, d). Progressive focal palmoplantar skin lesions (Fig. 1d) did not have full penetrance and were observed from 10 weeks of age in less than 50% of both germline and K14-Cre iASPP-deficient mice. Hematoxylin and eosin staining of sections from the paws of adult mice showed focal thickening of the stratum corneum (SC) and stratum spinosum (SS) in both germline iASPP-deficient and K14-Cre iASPP-deficient mice compared to wild-type controls (Supplementary Figure 1D), similar to that seen in patients with palmoplantar keratodermas. In addition, incomplete maturation of keratinocytes in the cornified layer was observed (Supplementary Figure 1D). These results show that intrinsic loss of iASPP in cardiomyocytes or keratinocytes is sufficient to recapitulate heart and skin phenotypes observed with germline iASPP deficiency, supporting the cell autonomous role of iASPP deficiency in causing gross abnormalities of cardiocutaneous disorder. Lack of the myocardial phenotype in the K14-Cre iASPP-deficient mice, and absence of the woolly hair and eye phenotype in the αMyHC-Cre iASPP-deficient mice (Fig. 1a–c), confirm the fidelity of our Cre promoters in generating epidermal- and myocardial-specific iASPP-deficient mice.

iASPP deficiency in cardiomyocytes and keratinocytes leads to cardiocutanous phenotypes. a Representative images of hearts isolated from wild-type (+/+), germline (iASPPΔ8/Δ8) and myocardial-specific iASPP-deficient (αMyHC-iASPPΔ8/Δ8) mice (αMyHC indicated as MHC). All images were taken with comparable magnification, showing larger size of hearts from iASPP-deficient mice. White patchy areas are scarring of the myocardium. b Representative images at the same magnification, showing hearts isolated from wild-type and epidermis-specific iASPP-deficient (K14-iASPP∆8/∆8) mice, with no myocardial abnormalities. c Representative images of eyes and fur of wild-type (+/+), germline iASPP∆8/∆8, αMyHC-iASPPΔ8/Δ8 and K14-iASPP∆8/∆8 adult mice. Sparse wavy coat and eye defects are present in the K14-iASPP∆8/∆8 and iASPP∆8/∆8 mice. d Representative images of the paws of adult wild-type (+/+), germline iASPPΔ8/Δ8 and K14-iASPP∆8/∆8 mice. The paws of iASPPΔ8/Δ8 and K14-iASPPΔ8/Δ8 mice have shedding and peeling of the cornified envelope (black arrows), resembling palmoplantar keratoderma
Cardiomyocyte-specific iASPP deficiency causes defects in intercalated disks and cardiac dysfunction
Defects in the integrity of desmosomes and intercalated disks are known to cause DCM. Our previous studies showed that iASPP locates at intercalated disks in cardiomyocytes and controls desmosome integrity, and germline iASPP-deficient mice suffer from DCM [13]. If iASPP has a cell autonomous role in controlling desmosome integrity in cardiomyocytes, a cardiomyocyte-specific deficiency of iASPP should cause similar phenotypes to those in germline iASPP-deficient mice [13]. To test this hypothesis we studied desmosome integrity in cardiomyocytes of αMyHC-Cre iASPP-deficient mice. iASPP has previously been shown to bind desmoplakin and anchor the cardiomyocyte-specific intermediate filament desmin at the intercalated disks, but not with other intercalated disk structural proteins such as α, β, and γ-catenins, connexin 43, p120, and N-cadherin under the same conditions [13]. Consistent with a cell autonomous role of iASPP, we identified a failure in desmin anchoring at the myocyte-myocyte junctions in the myocardium of αMyHC-Cre iASPP-deficient mice compared to wild-type mice (Fig. 2a, yellow arrowheads denote myocyte-myocyte junctions), similar to results previously observed in germline iASPP-deficient mice [13]. The lack of iASPP expression in cardiomyocytes at the intercalated disks from αMyHC-Cre iASPP-deficient mice (Fig. 2a) also confirms the efficacy of this Cre. As expected, we did not observe loss of desmin at the myocyte-myocyte junctions of myocardium in K14-Cre iASPP-deficient mice (Fig. 2a).

Myocardial-specific iASPP-deficient mice have loss of desmosomes at cell-cell junctions and compromised cardiac function. a Immunofluorescence images of myocardial tissue sections from iASPP (+/+), αMyHC-iASPPΔ8/Δ8 and K14-iASPP∆8/∆8 adult mice stained with antibodies against iASPP LX128.5 (green) and desmin (red). Nuclei shown with DAPI on merge panels. Examples of desmosomes are indicated by yellow arrowheads. b MRI imaging (top panels) in the orthogonal plane of the hearts of iASPP (+/+) and αMyHC-iASPPΔ8/Δ8 mice (see key) at 12 weeks. Right ventricles indicated by white arrowheads; left ventricles (LV) by yellow arrowheads. MRI analysis in the same mice (lower panels) for LV mass, LV end-diastolic volume (LVEDV), LV end-systolic volume (LVESV), LV stroke volume (LVSV) and LV ejection fraction (LVEF). * represents p < 0.05 using unpaired t-test. Each animal is represented by a colored symbol (◊ for males, ○ for females). C is Wild-type controls; KO is iASPP-deficient. Black bar indicates the mean
Hematoxylin and eosin or Masson’s trichrome staining of heart sections from αMyHC-Cre iASPP-deficient and wild-type animals showed that widespread fibrosis is detected in αMyHC-Cre iASPP-deficient hearts, but not in hearts from K14-Cre iASPP-deficient mice (Supplementary Figure 2A). We measured various parameters of ventricular function using magnetic resonance imaging (MRI), which showed that αMyHC-Cre iASPP-deficient mice at 12 weeks have a dilated RV and abnormal LV (Fig. 2b, top; white arrowhead denotes RV free wall and yellow arrowhead denotes LV free wall). MRI analysis of LV function showed that αMyHC-Cre iASPP-deficient mice have a significant increase in LV mass, along with a decrease in LV function (as assessed by an increase in LV end-diastolic volume, LV end-systolic volume and decreased LV ejection fraction) compared to wild-type controls (Fig. 2b, bottom; also see Supplementary Videos 1 and 2). As expected, the αMyHC-Cre iASPP-deficient mice had decreased survival compared to wild-type control mice, with a median survival of 17.3 weeks (121 days) (Supplementary Figure 2B). Therefore, αMyHC-Cre iASPP-deficient mice progress to ventricular failure in a similar manner to germline iASPP-deficient mice. These data support a cell intrinsic role of iASPP in controlling the integrity of desmosomes and intercalated disks in cardiomyocytes and in preventing DCM.
iASPP in keratin 14 keratinocytes is required for mouse eyelid closure independently of p53
We next examined the basis of the eye phenotype in iASPP-deficient mice more closely. In mammals, eyelid closure protects developing ocular structures, particularly the cornea, from environmental insults until the eye becomes functionally mature [26, 27]. Impaired closure causes inflammation and severe corneal opacity; defects in the anterior eye segment and blindness are observed shortly after birth [28,29,30,31]. In humans, eyelid closure occurs in utero [29]. In mice it involves coordinated migration of keratinocytes across the cornea from E14.5 with eyelids fusing by E16.5, and remaining closed until postnatal day 12 [26, 27]. Defects in eyelid closure during development can lead to corneal opacity [32].
Eyelid closure defects at birth and corneal opacity in adult mice are two of the main features of germline iASPP deficiency that occur with full penetrance [12, 33]. Importantly, cloudy cornea has also been reported in 1 of 3 examined patients with an iASPP-null mutation, and eye abnormalities have been found in all 4 examined aborted fetuses with iASPP mutations [11].
As iASPP is an inhibitor of p53, we investigated whether the eyelid closure defect was due to elevated p53 activity. We created germline double knockout mice lacking iASPP and p53 (see Materials and Methods). Consistent with previous findings [17, 33], iASPP deficient E16.6 embryos exhibited open eyelids whereas p53-deficient embryos did not. Intriguingly, p53 and iASPP double knockout embryos retained the open eyelid phenotype (Fig. 3a). This suggests that the open eyelid phenotype in iASPP-deficient embryos occurs independently of p53 activity.

iASPP-deficient mice have impaired embryonic eyelid closure. a Representative histological sections through the eye from E16.5 p53+/+; iASPP+/+, p53+/+; iASPP∆8/∆8, p53-/-; iASPP+/+ and p53-/-; iASPP∆8/∆8 embryos, stained with H&E. Loss of p53 has no effect on eyelid closure in iASPP∆8/∆8 embryos. The eyelid is at the top of the image, extending over the eye in the iASPP wild-type embryos. b Representative sections through the eye from E16.5 iASPP (+/+), iASPP∆8/∆8 and K14-iASPP∆8/∆8 embryos, stained with H&E as in (a)
We then further investigated the intrinsic role of iASPP in keratin 14 expressing keratinocytes in controlling eyelid closure and preventing corneal opacity. We observed impaired extension of eyelid epithelial sheets across the cornea of both K14-Cre and germline iASPP-deficient E16.5 embryos compared to wild-type embryos (Fig. 3b), albeit with lower penetrance of 71% in K14-Cre (10 out of 14 embryos examined) compared to 100% penetrance in germline iASPP-deficient embryos (n = 16). Consistent with this, around 68% of K14-Cre iASPP-deficient mice (53 out of 78 mice examined) exhibited detectable corneal opacity. This suggests that iASPP deficiency in keratin 14 expressing keratinocytes is an important contributing factor to the eyelid closure defects and corneal opacity in germline iASPP-deficient embryos and adult mice.
To investigate the potential cause of eyelid closure defects in germline iASPP-deficient embryos, we assessed cell proliferation and apoptosis in the eyelids. Proliferation was assessed by BrdU incorporation, which labels proliferating S-phase cells, in E14.5, E15.5, and E16.5 embryos and Ki67 in E15.5 embryos (Supplementary Figure 3A-B). The numbers of proliferating cells in the examined eyelid regions of iASPP-deficient and wild-type embryos were similar (Supplementary Figure 3A-B).
Apoptosis in the eyelids of iASPP-deficient and wild-type embryos was examined by TUNEL and cleaved caspase 3 staining (Supplementary Figure 3C-D). Although we consistently observed a slight increase in the number of TUNEL and cleaved caspase 3 positive cells at the leading edge of the eyelids in germline iASPP-deficient embryos compared to wild-type embryos, the difference failed to reach statistical significance. Future studies are needed to confirm whether the observed subtle difference in cell death is sufficient to explain the phenotype.
iASPP is required for desmosome and keratin intermediate filament integrity and epithelial cell adhesion
Patients who present with striate palmoplantar keratoderma (SPKK) associated with desmoplakin mutations have perinuclear aggregation of keratin filaments in their cells [34]. In addition, epidermis-specific deletion of desmoplakin results in morphologically normal but malfunctioning desmosomes as a result of disrupted keratin filaments [35]. Given the observed histological features of palmoplantar keratoderma in iASPP-deficient mice (Fig. 1d, Supplementary Figure 1D) and our recent study describing iASPP as a regulator of desmosomal integrity [13], we examined if iASPP deficiency in mice could lead to disruption of keratin filaments and desmosome adhesion in the epidermis and in differentiating keratinocytes. If iASPP’s role in regulation of desmosomes is cell autonomous, we expect to observe keratin aggregation and desmosome weakness in keratinocytes and other epithelial cell lines lacking iASPP.
High-resolution electron microscopy (EM) of epidermis revealed an increase in keratin aggregates in vivo (Fig. 4a, Supplementary Figure 4A, red arrowheads) and a defect in desmosome and intermediate filament interactions in germline iASPP-deficient mice compared to wild-type mice (Fig. 4a). These findings were confirmed in vitro with immunostaining of Keratin 5 (K5) intermediate filaments in differentiating primary keratinocytes grown in high calcium media for 24 h. K5 intermediate filaments were clearly visible in wild-type primary mouse keratinocytes, while K5 aggregates accumulated in the cytoplasm of iASPP-deficient keratinocytes (Fig. 4b, white arrowheads indicating red foci in cytoplasm). A propensity for keratin aggregation in iASPP-deficient epidermis and keratinocytes agrees with a mechanical role of iASPP in controlling desmosome integrity. We further examined in vitro if the integrity and function of desmosomal adhesion is compromised in the absence of iASPP. In a dispase mechanical dissociation assay, iASPP-deficient keratinocytes or epithelial UT-SCC74a cells had increased fragmentation compared to wild-type or control siRNA-treated cell monolayers (Fig. 4c and Supplementary Figure 4B), which suggests impaired adhesion and decreased resistance to mechanical stress. In a calcium-chelation assay, keratinocytes lacking iASPP lost cell-cell adhesion when calcium was withdrawn, as demonstrated by scattering of cells and lack of desmoplakin at cell-cell contacts (Fig. 4d). This suggests that iASPP has an important role in controlling keratinocyte adhesion, as a loss of iASPP renders cells unable to form hyperadhesive, calcium-independent desmosomes (Fig. 4d).

iASPP is required for keratin intermediate filament integrity and epithelial cell adhesion. a Transmission electron microscopy of epidermal desmosomes from 12 week old iASPP+/+ and germline iASPPΔ8/Δ8 littermates. Red arrowheads indicate keratin aggregates. Yellow arrowheads show examples of desmosomes. Black arrows indicate the interaction between the desmosome plaque and intermediate filaments (IF), with IF being more densely packed at the desmosome interface in the epidermis from iASPP+/+ mice compared to that from iASPPΔ8/Δ8 mice. b (Left panel) Immunofluorescence staining showing increased aggregation of K5 filaments (red) in iASPP-deficient primary mouse keratinocytes compared to control cells. The nuclear stain DAPI is shown in blue. Arrowheads point to dense cytoplasmic K5 aggregates, red foci, that are more abundantly present in iASPP-deficient keratinocytes compared to wild-type keratinocytes as quantified in the right panel. (Right panel) Quantification of percentage of cells with keratin aggregation in four independent experiments with at least 60 cells scored per experimental replicate. Statistical analysis between control and iASPP-deficient keratinocyte groups was performed using unpaired t-test, bars denote ± 2 SEM. c Representative images of iASPP+/+ and iASPPΔ8/Δ8 keratinocyte monolayers before and after subjection to a dispase mechanical dissociation (mechanical stress), as indicated. The bar chart shows the number of fragments released as a result of the mechanical stress with increasing numbers of fragments indicating weaker cell–cell adhesion. The red horizontal bar denotes the group mean. Values are means ± 1SEM (black vertical bars) from three different experiments, where each dot represents individual data point. Statistical analysis between groups was done using unpaired t-test; asterisk denotes p < 0.05. Immunoblot for iASPP and tubulin (as a loading control) expression in lysates from keratinocytes isolated from wild-type and iASPPΔ8/Δ8 epidermis. d Immunostaining for desmoplakin (DSPI/II, white) in wild-type (upper panels) and iASPP-deficient (lower panels) primary keratinocytes cultured at confluence in high calcium medium (HCM) for either 2 or 6 days, with subsequent subjection to calcium depletion for 90 min by switching to low calcium media (LCM) containing 3 mM EGTA. e Representative images of in vitro wound scratch closure by MCF10A cells treated with control or iASPP siRNA; images at indicated time points of timelapse miscroscopy. The efficiency of the iASPP knockdown is shown by immunoblots (right). f Representative Image J tracks of selected iASPP+/+ and iASPPΔ8/Δ8 keratinocytes whose migratory behavior within a cell cluster was examined to determine their tendency to remain and migrate together. Each colored line represents one cell
Defects in desmosomes will affect cell migration in vitro [25, 36, 37], so we assessed migration in the presence or absence of iASPP in the non-tumorigenic desmosome-rich epithelial cell line MCF10A and primary mouse skin keratinocytes, (as eyelid epithelium is composed of keratinocytes equivalent to that of skin that are constituted of K14-positive cells). A scratch wound assay using MCF10A cells treated with control or iASPP siRNA showed increased cell migration in iASPP-deficient compared to control cells (Fig. 4e, Supplementary Figure 4C, right panel, and Supplementary Videos 3 and 4). Similar results were observed in primary mouse keratinocytes from iASPP-deficient mice compared to wild-type mice (Supplementary Figure 4D). Furthermore, we observed a higher degree of disorganized migration in iASPP-deficient MCF10A cells compared to control cells (Supplementary Figure 4C, left panel). Migratory behavior of iASPP-deficient and wild-type keratinocytes within cell clusters of semi-confluent cultures was assessed by pre-treatment with mitomycin C, an agent that blocks cell proliferation. The migration distance was increased and the pattern of migration was more scattered in iASPP-deficient keratinocytes than wild-type cells (Fig. 4f, Supplementary Figure 4E and Supplementary Videos 5 and 6). This suggests that keratinocyte adhesion requires iASPP to withstand mechanical stress and to migrate in a coordinated manner. These data also show that the importance of iASPP in mediating cell-cell adhesion is not limited to keratinocytes, as another iASPP-deficient epithelial cell type also migrated in a more disorganized manner.
Mice with iASPP-deficient K14-expressing keratinocytes have delayed cutaneous wound healing in vivo
Defects in desmosome dynamics and coordinated cell migration are known to disrupt wound healing in vivo [25, 38]. Given our results supporting iASPP’s role in regulating keratinocyte migration and desmosomal adhesion dynamics, we subjected wild-type, K14-Cre, and germline iASPP-deficient mice to full skin thickness punch biopsies and measured the size of the wounds every other day for 14 days. Both germline and K14-Cre iASPP-deficient mice exhibited delayed wound healing up to day 8 compared to control mice (Fig. 5a-b), but by days 10–14 both cohorts of mice had fully healed wounds (Supplementary Figure 5A-B). Wound healing in K14-Cre iASPP-deficient mice was significantly delayed between days 4 and 8, while in germline iASPP-deficient mice the healing delay was between days 2 and 6 (Fig. 5a-b).

Wound healing and cell adhesion is impaired in iASPP-deficient mice. a Left: Representative images of wound healing after skin biopsy in germline iASPP∆8/∆8 adult mice, over the first 6 days. Right: Plot showing the average percentage (mean ± 2 SEM) of wound area closed in iASPP+/+ and iASPP∆8/∆8 mice. **p < 0.001, *p < 0.05. The number of iASPP+/+ and iASPP∆8/∆8 mice are, respectively: 19 and 17 at day 2; 14 and 13 at day 4; 12 and 11 at day 6; 6 and 4 at day 8. Statistical analysis between groups was performed using the Mann-Whitney U test. b Left: Images of wound healing as in panel A, for K14-iASPP+/+and K14-iASPPΔ8/Δ8 mice. Right: Plot showing average percentage (mean ± 2SEM) of wound area closed in K14-iASPP+/+ and K14-iASPPΔ8/Δ8 mice. **p < 0.001, *p < 0.05. The number of K14-iASPP+/+and K14-iASPPΔ8/Δ8 mice are, respectively: 10 and 9 at day 2; 11 and 9 at day 4; 6 and 7 at day 6; 4 and 3 at day 8. Statistical analysis between groups was performed using the Mann-Whitney U test. c H&E staining of sections through the advancing epithelial tongues of wounds from iASPP+/+ and iASPP∆8/∆8 mice at days 2 and 6, as indicated. Black arrowheads point to widened intercellular spaces in iASPP∆8/∆8 wounded epidermis. Black dotted lines indicate the dermal-epidermal boundary
To assess the basis of the delay in wound healing, we performed a BrdU proliferation assay in vivo and detected no differences between keratinocytes at the wound site of iASPP-deficient and wild-type mice (Supplementary Figure 5C). Furthermore, we did not detect any significant differences in apoptosis of keratinocytes at the wound site between iASPP-deficient and wild-type mice as assessed by cleaved caspase 3 staining (Supplementary Figure 5D) and TUNEL assay (Supplementary Figure 5E). We examined the histology of the wound edge epidermis and observed an increase in intercellular spaces between keratinocytes of iASPP-deficient compared to wild-type mice (Fig. 5c). The area of an average intercellular space at the wound edge, as quantified using Image J, was significantly higher in the absence of iASPP (Supplementary Figure 5F). Cell-cell dissociation was also observed at the wound edge of iASPP-deficient mice using immunofluorescence staining for desmosome component Desmoglein I and II (Supplementary Figure 5G). This suggests that in the absence of iASPP, proliferation and apoptosis are unaffected during wound healing but there is disrupted cell adhesion in vivo. This could contribute to a lack of coordinated cell migration, impairing efficient wound re-epithelization in vivo, and this might also explain the faster and disorganized migration observed in vitro (Fig. 4e, f).
Discussion
Here, we show that loss of iASPP specifically in cardiomyocytes or keratinocytes is sufficient to induce phenotypes of cardiocutaneous disorder, including cardiac dysfunction or palmoplantar keratoderma, delayed eyelid development and wound healing, respectively. These findings are consistent with previous studies showing that cardiac-specific or epidermis-specific deletion of desmoplakin, a binding partner of iASPP and an important desmosome component, leads to cardiac dysfunction or keratin abnormalities, respectively [35, 39].
Importantly, we show that tissue-specific deletion of iASPP in cardiomyocytes is sufficient to induce cardiomyopathy with full penetrance, similar to germline deletion of iASPP [13]. We previously showed that RV dilation in germline iASPP-deficient mice can occur as early as day 16.5 in utero with no obvious structural abnormalities in the LV, but that both ventricles were dilated in 12 week-old mice [13]. We predict that RV failure would precede LV failure, as the RV is the predominant ventricle in utero. In this study we analyzed the ventricles of αMyHC-Cre iASPP-deficient postnatal pups and adult mice, which may explain why we observed dilation of both ventricles. The phenotype caused by iASPP deficiency is consistent with that caused by loss of desmin, a component of desmosomes, which also results in biventricular dilation [40] and ARVC [41, 42]. These data suggest that iASPP needs to be added to the list of candidate genes involved in general DCM.
Compared to germline iASPP-deficient embryos, in K14-Cre iASPP-deficient embryos we observed lower penetrance of the open-eyelid and corneal opacity phenotypes (70 vs. 100%) and slightly improved wound closure in K14-Cre iASPP deficient mice. The slightly reduced phenotype penetrance and severity could be caused by incomplete deletion of iASPP by K14-Cre. However, we cannot rule out that a non-cell autonomous role of iASPP may contribute to the penetrance and severity of these phenotypes. Additionally, we observed an intriguing difference in the effects of iASPP deficiency on the degree of structural abnormalities of intercalated disks (IDs) and desmosomes in cardiomyocytes of the heart vs. keratinocytes of the skin. In iASPP-deficient cardiac tissue, there was a significant loss of desmin localization at the IDs (Fig. 2a), whereas desmosome morphology appeared largely unaffected in our electron microscopy analysis of the germline iASPP deficient mouse skin (Fig. 4a). Nevertheless, iASPP-deficiency caused defective keratin IF network (Fig. 4a, b, Supplementary Figure 4A) in the skin and keratinocytes. Similar results have been observed in mice with epidermis-specific deletion of desmoplakin, with the desmosomes appearing morphologically normal but dysfunctional, as shown by the collapse of keratin filaments [35]. It is important to note that the dorsal skin rather than palmoplantar epidermis was used for electron microscopy; the latter may have a stronger phenotype due to increased exposure to mechanical stress. A systemic analysis of both palmoplantar and dorsal skin epidermis is needed to examine the full extent of abnormalities in desmosome morphology and keratin aggregation.
An interesting finding was that the eye phenotypes in both germline and K14-Cre iASPP-deficient mice are similar to those observed in patients and in fetuses aborted between 21 and 31 weeks of gestation with iASPP mutations [11]. The morphogenetic process of eyelid closure in humans parallels that in mice, though in humans this process occurs in utero between weeks 7–25 weeks of gestation. Our results indicate that iASPP expression in K14-expressing cells is important for efficient embryonic eyelid closure and prevention of eye defects.
iASPP is a shuttling multifunctional protein and in the nucleus it is critical for inhibiting p63 function, for example during differentiation of keratinocytes [17] and cardiomyocyte development [13]. p63 regulates target genes involved in adhesion (such as Perp, a desmosome component [43]), and the defects in cell migration in vitro in the absence of a significant change in proliferation or apoptosis and delayed wound healing in vivo that we see with iASPP deficiency are reminiscent of the phenotype observed in Perp deficient cells and animals [25]. Although we have preliminary data that iASPP deficiency does not affect Perp expression (data not shown), it could be argued that iASPP might control keratinocyte function through its ability to regulate p63 transcription. iASPP is also likely to affect keratinocyte function through its cytoplasmic and junctional roles. We have shown previously that iASPP becomes more cytoplasmic/junctional in differentiated keratinocytes or post-mitotic cardiomyocytes [13]. In this study, our data suggest that iASPP can directly influence keratinocyte cell adhesion via stabilizing desmosomes. Therefore, the skin phenotype could be due to junctional iASPP controlling the integrity of desmosome and keratin intermediate filaments, as demonstrated in this study. Notably, iASPP can shuttle between the cytoplasm and nucleus and so may also have a novel role connecting desmosomes and transcriptional programs through its ability to bind transcription factors such as p53, p63, and NF-κB (Fig. 6). Further elucidating how junctional proteins might influence transcriptional programs and cell fate determination is an interesting area for future study. In summary, our results support a cell autonomous role of iASPP in cardiomyocytes and keratinocytes in the regulation of desmosome integrity. This newly identified cell intrinsic role of iASPP adds to our understanding of why iASPP deficiency causes cardiocutaneous disorders in wa3 mice [12], CWH Poll Hereford calves [14], and patients [11].

Working model of iASPP functions in cardiomyocytes and keratinocytes. A schematic model of how iASPP might act at different stages of development in cardiomyocytes and keratinocytes. iASPP (small blue rectangles) shuttles between the nucleus and cytoplasm in dividing cardiomyocytes and keratinocytes during development (e.g., E16.5), and cell junctions (desmosomes) in mature cardiomyocytes (e.g., 12 weeks) and differentiating keratinocytes. At the desmosomes, iASPP is important to maintain the integrity of desmosomal adhesion by interacting with desmoplakin (DSP) and intermediate filaments (Keratin 5 in keratinocytes or Desmin in cardiomyocytes). In the nucleus (e.g., at E16.5) iASPP can interact with transcription factors such as p53 or p63 and trigger cell signaling events. For example, this may lead to inhibition of p53-dependent apoptosis in cardiomyocytes or inhibition of p63-dependent differentiation in keratinocytes (color figure online)
Materials and methods
All animals were handled according to IACUC guidelines and all animal work was approved by the Oxford University ethical review and licenced by the UK home office. Procedures were carried out following the Home Office Animal Scientific Procedures Act 1986 guidelines.
Mouse colonies
Generation of iASPP transgenic mouse in which the loxP-flanked exon8 of iASPP gene, PPP1R13L, is deleted globally by CMV-Cre or tamoxifen inducible R26Cre+ERT, resulting in a frameshift mutation, has been previously described [17]. αMyHC-iASPPΔ8/Δ8 and K14-iASPPΔ8/Δ8 mice were generated in a pure C57BL/6 background. Briefly iASPPLoxP/LoxP mice were backcrossed into a C57BL/6 background strain prior to being crossed with αMyHC-Cre and R26R-EYFP mice (Jackson Laboratory) to generate αMyHC-iASPPΔ8/Δ8 mice, or with K14-Cre mice to generate K14-iASPPΔ8/Δ8 mice. Mice heterozygous for the iASPP LoxP allele were used to generate littermate control and knockout animals for all experiments. Genotyping was done using PCR primers for iASPP wildtype allele I8 (5′CCGAATTGGAGAAGTGAAGC-3′) and E8 (5′-AGAGCAGCCTCAGAGCATGG-3′), loxP allele FLP2 (5′-CCGAATTGGAGAAGTGAAGC-3′) and FRNT9 (5′-GGGTAGGAAAAAGGGCTGAG-3′), and deletion allele I8 and FLP2. Cre transgene was genotyped using Cre-F (5′-CATTTGGGCCAGCTAAACAT-3′) and Cre-R (5′-ATTCTCCCACCGTCAGTACG-3′), and K14 Cre with K14-Cre-F (5′-GCTCTCTGTCACCCTGGCTA-3′) and Cre-R. Balb/C p53+/- mice, as previously described [44], were kindly provided by Guillermina Lozano (MD Anderson Cancer Center) and were crossed with iASPP+/∆8 mice to generate double heterozygotes on a mixed background, which were further crossed to generate compound genotypes presented in this study. Primers: 5′- CACAGCGTGGTGGTACCTTA-3′ (exon 6), 5′-TAAGGATAGGTCGGCGGTTC-3′ (exon 7), and 5′-CATCGCCTTCTATCGCCTTC-3′ (neomycin resistance gene) were used to genotype p53 transgenic mice.
Magnetic resonance imaging (MRI) acquisition and analysis
MRI for LV function assessment was carried out on a 9.4T (400MHz) MRI system (Agilent, Santa Clara, CA) as described previously [45, 46] using a four-channel cardiac array (Rapid Biomedical, Germany). In brief, a double-gated [47], TGRAPPA-accelerated [48], multi-frame gradient echo sequence was applied (TE/TR = 1.79/4.6 ms, field-of-view 30 × 30 mm, slice thickness 1 mm, matrix size 256 × 258, acceleration factor R = 3) in short-axis orientation. Eight to ten slices were acquired covering the heart from apex to base. Image analysis was performed off-line using Amira 5.3.3 (Hillsboro, Oregon, US), and left-ventricular volumes and mass were determined. End-diastolic and end-systolic frames were selected on a slice by slice basis according to maximal and minimal volume of the left ventricle. Epicardial border was manually outlined first. The left-ventricular cavity was then segmented semi-automatically by thresholding, using the Magic Wand tool, built into Amira. Left-ventricular mass, end-diastolic and end-systolic volume were obtained, from which stroke volume volume (LVSV = LVEDV—LVESV) and ejection fraction (LVEF = LVSV/LVEDV * 100%) were calculated (as described in [49]).
Histology and Masson’s trichrome staining
Histology and Masson’s trichrome staining was performed as previously described [13]. Briefly, isolated hearts were processed and cryo-sectioned or paraffin-sectioned in the mid-orthogonal plane to allow for identification of RV, septum and LV prior to staining. RV, septum and LV were then analyzed in detail for structural defects and fibrosis.
Cell culture
Isolation and culture of primary keratinocytes from iASPP+/+, iASPPΔ8/Δ8, and iASPPLoxP/LoxP; Cre+ERT mice was performed by dissecting skin from postnatal day 2 pups and floating it over 0.25% trypsin-EDTA (Gibco) overnight at 4 °C. This allowed the epidermis to be peeled off and cut up in calcium-free EMEM with 8% calcium stripped FBS, 0.05 mM CaCl2 and 50 μg/ml Gentamicin (low calcium media; LCM). After being passed through a 100 µm cell strainer, the cells were plated on a rat collagen type I (BD Bioscience) coated plastic dishes or cover slips. iASPP deletion was induced by adding 1 μM 4-OHT to the cells for 4 days. Induction of cell–cell adhesion of cells was stimulated by supplementing the media with calcium to 1.5 mM final concentration. MCF10a cells were maintained in Dulbecco’s modified Eagle’s medium/F12 containing 5% horse serum, 20 ng/ml EGF, 0.5 mg/ml hydrocortisone, 100 ng/ml cholera toxin, 10 μg/ml insulin, and 100 U/ml penicillin/streptomycin solution.
siRNA-induced gene silencing
For siRNA-induced gene silencing, cell lines were transfected with either a SMART-pool of four siRNAs targeting iASPP (LU-003815-00-002 GE Dharmacon) or control siGENOME RISC-free siRNA (D-001220-01-05 GE Dharmacon). Transfection was carried out using DharmaFECT I transfection reagent (T-2001-02 GE Dharmacon) following manufacturer’s instructions. Cell assays were performed 72 h post-transfection.
In vivo wound healing experiment
Six to eight weeks old wild-type, iASPPΔ8/Δ8 and K14-Cre iASPPΔ8/Δ8 female mice in anagen hair phase were anaesthetized with isoflurane inhalation and subcutaneously injected with 1 mg/kg Metacam. Their dorsal side was shaved and sterilized with ChloraPrep. A full thickness excisional wound was made on the middle paravertebral region of the mouse using a biopsy punch of 6 mm diameter (Stiefel). Wound closure process was monitored by measuring the wound area with calipers every other day and was represented as the percentage of the initial wound area. Data is presented as mean value ± 2 standard errors of the mean (SEM). Statistical analysis between two groups was performed using the Mann-Whitney’s U-test and p < 0.05 was considered statistically significant.
In vitro wound healing assay
MCF10a treated with iASPP or control siRNA and keratinocytes from iASPPLoxP/LoxP Cre+ERT mice treated with vehicle or 4-OHT to induce iASPP deletion were grown to confluence. The confluent monolayers were wounded by scratching with a disposable pipette tip. The cells were washed and incubated in appropriate media. The wound closure was followed, and images were taken every 20 min for up to 72 h using the timelapse video microscopy. The wound healing rate was calculated as the percentage of the remaining wound area versus the original wound area using ImageJ.
In vitro motility behavior assay
Keratinocytes from iASPPLoxP/LoxP Cre+ERT mice were grown in keratinocyte defined medium (Gibco) in presence of 1 μM 4-OHT or 4-OHT solvent ethanol. Under these conditions cells formed cell-cell contacts and cell clusters. The motility behavior of cells was followed for 48 h using timelapse video microscopy. The motility of individual cells was followed using ImageJ cell tracker. To avoid the effect of cell proliferation on cell motility, keratinocytes were pre-treated with 1 ng/ml mitomycin C for 1 h.
In vivo 5-bromo-2’-deoxyuridine (BrdU) incorporation
The same mice used in the wound healing experiment (see above) were intraperitoneally injected with 50 µg/g body weight BrdU 2 h prior to euthanasia to assess proliferation rate. Immunofluorescence technique described below using anti-BrdU antibody was used to localize BrdU positive cells. The number of cells at the epidermal wound edge that had incorporated BrdU were counted using Image J. Wound edge was identified by immunofluorescence staining for the migration marker Keratin 6.
Dispase mechanical dissociation assay
Confluent iASPP+/+ and iASPPΔ8/Δ8 keratinocytes were cultured in 1.5 mM calcium media for 24 h, rinsed in PBS and incubated with 2.4 U/ml dispase (Roche) for 30 min at 37 °C. Released keratinocyte monolayers were gently rinsed in PBS, transferred to 15-ml conical tube containing 2 ml PBS and subjected to orbital rotation (20 rpm) for 5 min. Fragments were imaged in a 6-well dish using HP Scanjet 5590P, and were counted using Image J cell counter function. The fragments smaller than 4 pixels in diameter (~area 1700 µm2) were excluded from the quantification. Statistical analysis was done using unpaired t test and significance was defined as p < 0.05. The same protocol was followed for dispase assay with UT-SCC74A cell line, which was transfected with indicated siRNA for 72 h.
Calcium chelation assay
Confluent vehicle-treated wildtype or 4-OHT-induced iASPPΔ8/Δ8 primary keratinocytes were maintained in 1.5 mM calcium media for either 2 or 6 days, following which the media was replaced with low calcium media containing 3 mM EGTA for 90 min. The cells were subsequently subjected to immunocytochemistry protocol with desmoplakin antibody. Calcium chelation protocol has been described before elsewhere [50].
Antibodies
Antibodies against iASPP—mouse monoclonal LX049.3and LX128.5—have been described previously [17]. Additional antibodies used in this study are: BrdU (ab6326 Abcam), Cleaved Caspase 3 (9661 Cell Signaling), Desmin (ab15200 Abcam), Desmoglein 1&2 (61002 Progen), Desmoplakin 1 & 2 (61003 & 65146 Progen), Keratin 5 (PRB-160P Covance), Keratin 6 (PRB-169P Covance) and Ki67 (ab15589 Abcam). Secondary antibodies carrying a fluorochrome (Alexa Fluor 488, 594 and 647) were purchased from Molecular Probes. Secondary anti-mouse, goat or rabbit horseradish peroxidase (HRP)-labeled antibodies for immunoblots were purchased from DakoCytomation.
Immunofluorescence
Confluent cells seeded on coverslips were fixed in 4% paraformaldehyde (PFA) for 15 min. After three washes in PBS, cells were permeabilized with 0.5% Triton X-100 for 10 min at 4 °C, blocked with 5% normal goat serum in PBS, and subsequently incubated with appropriate primary antibody for 1 h at room temperature. After rinsing in PBS, secondary antibodies (1:400) and DAPI were applied to cells for 45 min. Images were taken using LSM 710 confocal microscopy. Immunostaining of tissue sections was carried out following protocol described in detail elsewhere [17].
TUNEL assay
Apoptosis in eyelid and wound sections was examined by the TUNEL assay using ApopTag®Red In Situ Apoptosis Detection kit (S7165 Millipore) following manufacturer’s instructions.
Immunoblotting
Cell rinsed in ice-cold PBS were lysed in NETN buffer (50 mM Tris pH8.0, 150 mM NaCl, 1 mM EDTA, 1% NP40 + 0.1 mM Na3VO4, protease and phosphatase inhibitors). SDS sample buffer was added to the lysate and samples were boiled for 10 min before being subjected to SDS-PAGE on a 6% gel.
Statistical analyzes
Sample size and statistical tests used to determine statistical significance between samples are indicated in the appropriate figure legends. Two-sided tests were used for all the analyzes. * represents p value < 0.05, while ** represents p < 0.01
References
Brewer GJ. Drug development for orphan diseases in the context of personalized medicine. Transl Res 2009;154:314–22.
McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000;355:2119–24.
Norgett EE, Hatsell SJ, Carvajal-Huerta L, Cabezas JC, Common J, Purkis PE, et al. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet 2000;9:2761–6.
Bardawil T, Khalil S, Bergqvist C, Abbas O, Kibbi AG, Bitar F, et al. Genetics of inherited cardiocutaneous syndromes: a review. Open Heart 2016;3:e000442.
Garrod D, Chidgey M. Desmosome structure, composition and function. Biochim Biophys Acta 2008;1778:572–87.
Franke WW, Borrmann CM, Grund C, Pieperhoff S. The area composita of adhering junctions connecting heart muscle cells of vertebrates. I. Molecular definition in intercalated disks of cardiomyocytes by immunoelectron microscopy of desmosomal proteins. Eur J Cell Biol 2006;85:69–82.
Nitoiu D, Etheridge SL, Kelsell DP. Insights into desmosome biology from inherited human skin disease and cardiocutaneous syndromes. Cell Commun Adhes 2014;21:129–40.
Meng X, Yang J, Dong M, Zhang K, Tu E, Gao Q, et al. Regulatory T cells in cardiovascular diseases. Nat Rev Cardiol 2016;13:167–79.
Takeda N, Seko Y, Oriuchi N, Nagai R. Gamma-delta T-cell-mediated dilated cardiomyopathy. Int J Cardiol 2008;125:130–2.
Huber SA. T cells expressing the gamma delta T cell receptor induce apoptosis in cardiac myocytes. Cardiovasc Res 2000;45:579–87.
Falik-Zaccai TC, Barsheshet Y, Mandel H, Segev M, Lorber A, Gelberg S, et al. Sequence variation in PPP1R13L results in a novel form of cardio-cutaneous syndrome. EMBO Mol Med 2017;9:319–36.
Herron BJ, Rao C, Liu S, Laprade L, Richardson JA, Olivieri E, et al. A mutation in NFkB interacting protein 1 results in cardiomyopathy and abnormal skin development in wa3 mice. Hum Mol Genet 2005;14:667–77.
Notari M, Hu Y, Sutendra G, Dedeic Z, Lu M, Dupays L, et al. iASPP, a previously unidentified regulator of desmosomes, prevents arrhythmogenic right ventricular cardiomyopathy (ARVC)-induced sudden death. Proc Natl Acad Sci USA 2015;112:E973–981.
Simpson MA, Cook RW, Solanki P, Patton MA, Dennis JA, Crosby AH. A mutation in NFkappaB interacting protein 1 causes cardiomyopathy and woolly haircoat syndrome of Poll Hereford cattle. Anim Genet 2009;40:42–46.
Whittington RJ, Cook RW. Cardiomyopathy and woolly haircoat syndrome of Poll Hereford cattle: electrocardiographic findings in affected and unaffected calves. Aust Vet J 1988;65:341–4.
Bergamaschi D, Samuels Y, O’Neil NJ, Trigiante G, Crook T, Hsieh JK, et al. iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat Genet 2003;33:162–7.
Notari M, Hu Y, Koch S, Lu M, Ratnayaka I, Zhong S, et al. Inhibitor of apoptosis-stimulating protein ofp53 (iASPP) prevents senescence and is required for epithelial stratification. Proc Natl Acad Sci USA 2011;108:16645–50.
Yang JP, Hori M, Sanda T, Okamoto T. Identification of a novel inhibitor of nuclear factor-kappaB, RelA-associated inhibitor. J Biol Chem 1999;274:15662–70.
Lu M, Breyssens H, Salter V, Zhong S, Hu Y, Baer C, et al. Restoring p53 function in human melanoma cells by inhibiting MDM2 and cyclin B1/CDK1-phosphorylated nuclear iASPP. Cancer Cell 2013;23:618–33.
Lu M, Muers MR, Lu X. Introducing STRaNDs: shuttling transcriptional regulators that are non-DNA binding. Nat Rev Mol Cell Biol 2016;17:523–32.
Slee EA, Gillotin S, Bergamaschi D, Royer C, Llanos S, Ali S, et al. The N-terminus of a novel isoform of human iASPP is required for its cytoplasmic localization. Oncogene 2004;23:9007–16.
Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA, Schneider MD. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Invest 1997;100:169–79.
Dassule HR, Lewis P, Bei M, Maas R, McMahon AP. Sonic hedgehog regulates growth and morphogenesis of the tooth. Development 2000;127:4775–85.
Chen JF, Murchison EP, Tang R, Callis TE, Tatsuguchi M, Deng Z, et al. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc Natl Acad Sci USA 2008;105:2111–6.
Beaudry VG, Ihrie RA, Jacobs SB, Nguyen B, Pathak N, Park E, et al. Loss of the desmosomal component perp impairs wound healing in vivo. Dermatol Res Pract 2010;2010:759731.
Findlater GS, McDougall RD, Kaufman MH. Eyelid development, fusion and subsequent reopening in the mouse. J Anat 1993;183(Pt 1):121–9.
Harris MJ, McLeod MJ. Eyelid growth and fusion in fetal mice. A scanning electron microscope study. Anat Embryol 1982;164:207–20.
Li G, Gustafson-Brown C, Hanks SK, Nason K, Arbeit JM, Pogliano K, et al. c-Jun is essential for organization of the epidermal leading edge. Dev Cell 2003;4:865–77.
Meng Q, Mongan M, Carreira V, Kurita H, Liu CY, Kao WW, et al. Eyelid closure in embryogenesis is required for ocular adnexa development. Invest Ophthalmol Vis Sci 2014;55:7652–61.
Weng J, Luo J, Cheng X, Jin C, Zhou X, Qu J, et al. Deletion of G protein-coupled receptor 48 leads to ocular anterior segment dysgenesis (ASD) through down-regulation of Pitx2. Proc Natl Acad Sci USA 2008;105:6081–6.
Meng W, Green J, Guest JR. FNR-dependent repression of ndh gene expression requires two upstream FNR-binding sites. Microbiology 1997;143(Pt 5):1521–32.
Luetteke NC, Qiu TH, Peiffer RL, Oliver P, Smithies O, Lee DC. TGF alpha deficiency results in hair follicle and eye abnormalities in targeted and waved-1 mice. Cell 1993;73:263–78.
Toonen J, Liang L, Sidjanin DJ. Waved with open eyelids 2 (woe2) is a novel spontaneous mouse mutation in the protein phosphatase 1, regulatory (inhibitor) subunit 13 like (Ppp1r13l) gene. BMC Genet 2012;13:76.
Wan H, Dopping-Hepenstal PJ, Gratian MJ, Stone MG, Zhu G, Purkis PE, et al. Striate palmoplantar keratoderma arising from desmoplakin and desmoglein 1 mutations is associated with contrasting perturbations of desmosomes and the keratin filament network. Br J Dermatol 2004;150:878–91.
Vasioukhin V, Bowers E, Bauer C, Degenstein L, Fuchs E. Desmoplakin is essential in epidermal sheet formation. Nat Cell Biol 2001;3:1076–85.
South AP, Wan H, Stone MG, Dopping-Hepenstal PJ, Purkis PE, Marshall JF, et al. Lack of plakophilin 1 increases keratinocyte migration and reduces desmosome stability. J Cell Sci 2003;116:3303–14.
Yin T, Getsios S, Caldelari R, Kowalczyk AP, Muller EJ, Jones JC, et al. Plakoglobin suppresses keratinocyte motility through both cell-cell adhesion-dependent and -independent mechanisms. Proc Natl Acad Sci USA 2005;102:5420–5.
Thomason HA, Cooper NH, Ansell DM, Chiu M, Merrit AJ, Hardman MJ, et al. Direct evidence that PKCalpha positively regulates wound re-epithelialization: correlation with changes in desmosomal adhesiveness. J Pathol 2012;227:346–56.
Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, et al. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest 2006;116:2012–21.
Li D, Tapscoft T, Gonzalez O, Burch PE, Quinones MA, Zoghbi WA, et al. Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation 1999;100:461–4.
Klauke B, Kossmann S, Gaertner A, Brand K, Stork I, Brodehl A, et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum Mol Genet 2010;19:4595–607.
Lorenzon A, Beffagna G, Bauce B, De Bortoli M, Li Mura IE, Calore M, et al. Desmin mutations and arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol 2013;111:400–5.
Ihrie RA, Marques MR, Nguyen BT, Horner JS, Papazoglu C, Bronson RT, et al. Perp is a p63-regulated gene essential for epithelial integrity. Cell 2005;120:843–56.
Tordella L, Koch S, Salter V, Pagotto A, Doondeea JB, Feller SM, et al. ASPP2 suppresses squamous cell carcinoma via RelA/p65-mediated repression of p63. Proc Natl Acad Sci USA 2013;110:17969–74.
Schneider JE, Cassidy PJ, Lygate C, Tyler DJ, Wiesmann F, Grieve SM, et al. Fast, high-resolution in vivo cine magnetic resonance imaging in normal and failing mouse hearts on a vertical 11.7 T system. J Magn Reson Imaging 2003;18:691–701.
Schneider JE, Lanz T, Barnes H, Stork LA, Bohl S, Lygate CA, et al. Accelerated cardiac magnetic resonance imaging in the mouse using an eight-channel array at 9.4 Tesla. Magn Reson Med 2011;65:60–70.
Cassidy PJ, Schneider JE, Grieve SM, Lygate C, Neubauer S, Clarke K. Assessment of motion gating strategies for mouse magnetic resonance at high magnetic fields. J Magn Reson Imaging 2004;19:229–37.
Breuer FA, Kellman P, Griswold MA, Jakob PM. Dynamic autocalibrated parallel imaging using temporal GRAPPA (TGRAPPA). Magn Reson Med 2005;53:981–5.
Schneider JE, Wiesmann F, Lygate CA, Neubauer S. How to perform an accurate assessment of cardiac function in mice using high-resolution magnetic resonance imaging. J Cardiovasc Magn Reson 2006;8:693–701.
Merritt AJ, Scothern A, Bhattacharyya T. Assays for the calcium sensitivity of desmosomes. Methods Mol Biol 2006;341:167–83.
Acknowledgements
We would like to thank Indrika Ratnayaka for tissue sectioning, Mark Shipman for technical assistance with microscopy, Leonie Diffley (University of Manchester) for assisting with the MRI experiments, Guillermina Lozano (MD Anderson Cancer Center) for the p53 knockout mice, and Mary Muers for critical reading of the manuscript. This work was predominantly funded by the Ludwig Institute for Cancer Research (LICR) Ltd. J.E.S. is a BHF senior Basic Science Research Fellow (FS/11/50/29038), and acknowledges a Welcome Trust Core Award (090532/Z/09/Z). G.S. acknowledges support from Alberta Innovates Health Solutions.
Author Contributions
Z.D., G.S. and X.L. designed the experiments, analyzed data and wrote the paper. Y.H. initiated junctional iASPP study in keratinocytes. The myocardial-specific iASPP-deficient mice were generated and maintained by E.A.S. and G.S., and the epidermis-specific iASPP-deficient mice by K. C. and Z.D. Figure contributions: Z.D.—1A, B, D; 3; 4B, C, D, F; 5; S1A, S1D; S3; S4B–E; S5, G.S.—1A; 2B; S1C; S2B, Y.H.—4B, 4E, K. C.—1C; 5A-B; S1B; S5A-B, E.A.S.—1C; 2A; S2A, M.J.W.—S1C, F.Y.Z.—S4C; S5F, R.D.G.—S1C, D.J.P.F.—4A; S4A, D.M.—2B, J.E.S.—2B, D.M. and J.E.S. performed and analyzed the experiments in 2B. X.L. supervised the work and provided the funding for the study. All authors edited and approved the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
41418_2017_39_MOESM3_ESM.mov
MRI imaging in the orthogonal plane shows representative cardiac function of a myocardial-specific iASPP-deficient mouse with compromised cardiac function of both the RV and LV
Rights and permissions
About this article

Cite this article
Dedeić, Z., Sutendra, G., Hu, Y. et al. Cell autonomous role of iASPP deficiency in causing cardiocutaneous disorders. Cell Death Differ 25, 1289–1303 (2018). https://doi.org/10.1038/s41418-017-0039-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-017-0039-6
This article is cited by
-
Regulation of immunological tolerance by the p53-inhibitor iASPP
Cell Death & Disease (2023)
-
Novel homozygous stop-gain pathogenic variant of PPP1R13L gene leads to arrhythmogenic cardiomyopathy
BMC Cardiovascular Disorders (2022)
-
ASPP2 maintains the integrity of mechanically stressed pseudostratified epithelia during morphogenesis
Nature Communications (2022)
