
Abstract
Neurodegenerative diseases are a spectrum of chronic, debilitating disorders characterised by the progressive degeneration and death of neurons. Mitochondrial dysfunction has been implicated in most neurodegenerative diseases, but in many instances it is unclear whether such dysfunction is a cause or an effect of the underlying pathology, and whether it represents a viable therapeutic target. It is therefore imperative to utilise and optimise cellular models and experimental techniques appropriate to determine the contribution of mitochondrial dysfunction to neurodegenerative disease phenotypes. In this consensus article, we collate details on and discuss pitfalls of existing experimental approaches to assess mitochondrial function in in vitro cellular models of neurodegenerative diseases, including specific protocols for the measurement of oxygen consumption rate in primary neuron cultures, and single-neuron, time-lapse fluorescence imaging of the mitochondrial membrane potential and mitochondrial NAD(P)H. As part of the Cellular Bioenergetics of Neurodegenerative Diseases (CeBioND) consortium (www.cebiond.org), we are performing cross-disease analyses to identify common and distinct molecular mechanisms involved in mitochondrial bioenergetic dysfunction in cellular models of Alzheimer’s, Parkinson’s, and Huntington’s diseases. Here we provide detailed guidelines and protocols as standardised across the five collaborating laboratories of the CeBioND consortium, with additional contributions from other experts in the field.
Similar content being viewed by others
Facts
-
Mitochondrial dysfunction has been implicated in most neurodegenerative diseases (NDs).
-
It is important to utilise and optimise appropriate cellular models and experimental techniques to determine the contribution of mitochondrial dysfunction to ND.
-
It is currently difficult to compare and contrast the vast volumes of experimental research in cellular models of NDs, due to methodological differences and limited detail in method reporting.
-
Here, we provide guidelines on techniques to investigate mitochondrial function in cellular models of NDs, including detailed protocols for their assessment.
Open questions
-
Can mitochondrial dysfunction be used as an early indicator of neurodegenerative pathology?
-
Are mitochondria-associated dysfunctions viable targets in the treatment of neurodegeneration?
-
By standardising the methods used to assess mitochondrial dysfunction in neurodegenerative diseases, can we improve the translation of in vitro findings to in vivo studies?
Introduction
Neurodegenerative diseases (NDs) comprise a spectrum of heterogeneous pathologies involving the progressive dysfunction, degeneration, and death of neurons, leading to incurable and debilitating conditions. Although some genetic factors have been identified, the underlying pathophysiology behind familial and sporadic NDs remains poorly understood. Of note, nearly all NDs exhibit impaired mitochondrial energy metabolism [1,2,3]. For example, studies have repeatedly demonstrated reduced expression and impaired activity of respiratory chain Complex I in Parkinson’s disease (PD), Complex IV in Alzheimer’s disease (AD), and Complexes II and III in Huntington’s disease (HD), and reduced activity of α-ketoglutarate dehydrogenase in both AD and PD [4]. Drug-induced models of NDs further link respiratory complex deficiency with disease—rotenone and MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), both inhibitors of Complex I, cause Parkinson-related pathology in animal models [5], and 3-nitropropionic acid (3-NP), a Complex II inhibitor, produces a HD phenotype in mice [6]. Several gene mutations associated with NDs affect proteins that contribute to mitochondrial homoeostatic regulation and function (Table 1). Whether these mitochondria-associated dysfunctions are sufficient to cause neurodegenerative pathogenesis, however, remains uncertain, as they may also evolve secondary to a different underlying pathology [7]. Indeed, a decline in mitochondrial function may be a natural process of ageing, but may usually be compensated for by adaptive changes [4, 8, 9]. A loss of these compensatory mechanisms, or a ‘second trigger’ (such as genetic predisposition, endogenous/exogenous toxins, or chronic excitotoxicity), may convert any natural subclinical decline into a clinically relevant neurodegenerative phenotype (‘dual/two/double-hit’ hypotheses [10, 11]). Clearly, extensive work is still required to thoroughly investigate the cause and impact of mitochondrial dysfunction in NDs.
It is currently difficult to compare and contrast the vast volumes of experimental research in cellular models of NDs due to methodological differences—cell and neuron type, age, method of preparation and culture, equipment/assays, experimental set-up, outputs measured—as well as due to limited detail in method reporting. These discrepancies may compound the difficulties in translating experimental findings from in vitro cellular models to in vivo or human studies. Here, we provide guidelines on several experimental methods to assess mitochondrial dysfunction in in vitro cellular models of NDs, with the goal of reducing methodological differences between research laboratories. The CeBioND consortium (www.cebiond.org) is performing cross-disease analyses to investigate mitochondrial bioenergetic dysfunction in cellular models of AD, PD, and HD. Within this consortium, we standardised single-cell fluorescence protocols (TMRM, NAD(P)H autofluorescence) and oxygen consumption rate measurements across five collaborating laboratories. These protocols are detailed here. For this consensus review, we recruited several experts from the field of mitochondrial research to contribute details on additional techniques for a thorough evaluation of mitochondrial function and dysfunction. Collectively, we have developed guidelines that suit most biological laboratories and that provide for a standardised, holistic assessment of mitochondrial function in cellular models of a variety of NDs.
Mitochondrial bioenergetic processes
The components of mitochondrial bioenergetics are described in most biochemistry textbooks, and we provide only a brief introduction here. Although mitochondria are crucially involved in apoptosis, calcium signalling, lipid biosynthesis, synthesis of haem and iron sulphur centres, carbohydrate and fatty acid metabolism, and redox homoeostasis, the most celebrated function of mitochondria is oxidative phosphorylation (OxPhos), the oxygen-dependent production of ATP driven by an electrochemical gradient across the inner mitochondrial membrane. Neurons are heavily dependent on OxPhos, especially in times of increased energy demand [12, 13], and are therefore highly sensitive to loss of substrate or other impairments in the processes underlying OxPhos [14].
Mitochondrial ATP synthesis is driven by the maintenance of a proton circuit comprising the respiratory/electron transport chain, the F1Fo ATP synthase, and proton leaks. The multi-subunit complexes of the respiratory chain (forming the respirasome) reside within the inner mitochondrial membrane and consist of Complex I (NADH:ubiquinone oxidoreductase/NADH dehydrogenase), Complex II (succinate-ubiquinone oxidoreductase/succinate dehydrogenase), Complex III (ubiquinol-cytochrome c oxidoreductase), and the O2 consuming Complex IV (cytochrome c oxidase). Protons (H+) pumped out of the mitochondrial matrix by these respiratory complexes, coupled with electron transport through the complexes, maintain the electrochemical proton-motive force (Δp) which comprises a H+ concentration gradient (ΔpHm) and an electrical gradient (the mitochondrial membrane potential, Δψm) across the inner membrane [15, 16]. These gradients allow H+ to flow back into the matrix through the F1Fo ATP synthase, with the resultant synthesis of ATP from ADP and phosphate. The H+ circuit can also be completed by inducible or non-inducible H+ leaks across the inner membrane, bypassing the F1Fo ATP synthase [17]. Mitochondrial NADH, generated by the tricarboxylic acid (TCA) cycle within the mitochondrial matrix or via the import of NADH-derived reducing equivalents driven by the malate-aspartate shuttle, serves as an electron donor for the respiratory chain. The activity of Complex II, which, through the oxidation of succinate to fumarate reduces ubiquinone to ubiquinol, also enables electron transfer to Complex III. Reactive oxygen species (ROS), produced in mitochondria by the TCA cycle and respiratory chain, are vital second messengers, but their overproduction can lead to oxidative stress.
Methods to assess mitochondrial dysfunction in cellular models of neurodegenerative diseases
Here we provide detailed guidelines on experimental techniques to investigate mitochondrial bioenergetics in in vitro cellular models, including specific protocols to measure the oxygen consumption rate, the mitochondrial membrane potential and mitochondrial NAD(P)H. Analysis of mitochondrial transport, mitophagy, and biogenesis, factors that are also altered in NDs [1, 18,19,20], are beyond the scope of the present work. While cellular models are not optimal experimental systems, as they often lack the multi-cellular environment of intact in vivo models, they provide a relatively simple environment within which complex molecular interactions can be thoroughly studied at a population and single-cell level.
Cellular models (experimental systems) of neurodegenerative diseases
The anatomical, neurochemical, and metabolic uniqueness of primary neuron cultures derived from rodents (mice and rats) offer a currently unparalleled platform for the study of the molecular mechanisms of neurodegeneration. Primary neurons can be cultured from wild-type or transgenic animals to generate relatively homogenous neuronal populations for study in vitro (Fig. 1). Protocols for the preparation and culture of various neuronal populations are available [21,22,23]. Primary neurons from specific brain regions may be the ideal cellular model (e.g. dopaminergic neurons for PD, hippocampal neurons for AD, striatal neurons for HD, motor neurons for ALS), but are less abundant and not always easy to prepare nor culture. Variations in the preparation and culture of primary neuron cultures, such as the age and sex of the animals, seeding density, days in vitro, media/buffer composition and substrate availability, may alter neuronal physiology, mitochondrial function and viability, and contribute to heterogeneity in experimental results [23,24,25,26,27].

a After 10 days in vitro (DIV), cultures were stained with antibodies against the neuron-specific NF200 (red) and the astrocyte-specific GFAP (green), and with the DNA dye Hoechst (blue). Even when moved to a serum-free media quickly after dissociation, neuronal cultures contain a small proportion of astrocytes and other cell types (such as fibroblasts and endothelial cells). Neurons can be morphologically identified when performing single-cell experiments [153], but regular and careful characterisation of cultures is important for cell population assays. b Cortical neurons (after 6 DIV) transfected with a mitochondrial red fluorescent protein highlight the intricate mitochondrial network throughout the neuron
Brain-derived cell lines, such as the human SH-SY5Y or the rodent PC12, N2a, and HT22, can be differentiated to a post-mitotic neuronal state [28,29,30], but they do not exhibit a true neuronal phenotype (e.g. they lack functional NMDA receptors even after differentiation), and their bioenergetics and neurotoxic properties differ from primary neurons [31]. Although their ease of culturing and transfection make them a feasible compromise between convenience and scientific relevance, findings in these cellular models should be complemented with experiments in primary cultures or in vivo models. Neurons can also be differentiated from proliferative cell types, such as neural progenitor cells (NPCs) from the sub-ventricular zone of adult mice. More recently, cellular models of disease have been derived from patient cells. Dopaminergic, motor, striatal, and forebrain neurons derived from induced pluripotent stem cells (iPSCs) reprogrammed from human fibroblasts have been used to investigate PD, ALS, HD, and AD, respectively [32], although iPSCs still require expensive and labour-intensive techniques. It is important to use isogenically-corrected iPSCs as a control when studying the effects of genetic mutations on mitochondrial properties; CRISPR/Cas9, TALEN, or other techniques can be used to correct the mutation [33]. 3-D culture models developed using neural cells can recapitulate multiple aspects of ND pathologies [34]. Primary astrocyte, microglia, and associated cell cultures are also relevant cellular models, specifically to study non-cell autonomous and inflammatory processes, which may contribute significantly to the progression of NDs [35].
The experimental methods described herein focus on primary neuronal cultures, but can be performed in most cellular models. Decisions on specific cellular models and experimental design depend on the research question and on the available equipment, reagents, and expertise. Regardless, conditions should remain as similar as possible for all experiments within a study, and matched experiments (control and treated, or wild-type and transgenic littermates) should always be performed, ideally on the same day. Maintaining consistency both within a study and across platforms will facilitate inter-experiment comparison.
For live cell imaging, neurons should be equilibrated in ‘experimental buffer’ for at least 30 min—we find that (in mM) 120 NaCl, 3.5 KCl, 0.4 KH2PO4, 5 NaHCO3, 20 HEPES, 1.2 Na2SO4, at pH 7.4 (slightly higher than normal brain extracellular pH of ~7.2−7.3) works well for experiments on primary cortical and other central neurons, supplemented with 1–2 mM MgCl2 to reduce neuronal activity, 1.2 mM CaCl2, and the desired energy substrate (e.g. glucose, pyruvate, lactate, glutamine). The type and concentration of energy substrate will critically determine metabolic behaviour, and similar measurements performed with different substrates are not directly comparable. Neurons should be equilibrated at 37 °C, in the dark if utilising fluorescent reporters, and with no CO2 if the buffer contains HEPES or equivalent.
Measurement of the oxygen consumption rate (OCR)
Oxygen consumption is an excellent read-out for mitochondrial respiratory activity. Measurement of the oxygen consumption rate (OCR) is the current experiment of choice to determine underlying mitochondrial dysfunction [36, 37]. Although Complex IV is the only oxygen consumer within the proton circuit, appropriate pharmacological manipulation can isolate different respiratory states and facilitate modularised assessment of the complete circuit, including basal respiration, maximal respiration, proton (H+) leak, and ATP turnover (Fig. 2a).

a Mitochondrial function can be thoroughly investigated in intact cells by measuring the OCR during sequential addition of mitochondrial respiratory inhibitors (marked with grey triangles). The different stages of the experiment (i)–(iv) and the measured parameters (a)–(f) are described in Protocol 1. The addition of pharmacological compounds or fuel substrates prior to oligomycin (not shown here) can capture further detail regarding the OCR. b Illustration of the effects of relevant pharmacological compounds on the mitochondrial respiratory chain, proton (H+) leak across the mitochondrial inner membrane, and the F1Fo ATP synthase, during the experimental stages marked as (i)–(iv) in A. O2 in the mitochondria is consumed by the respiratory chain through the activity of Complex IV. (i) In the basal state, mitochondrial O2 consumption is predominantly driven by H+ flux through the F1Fo ATP synthase. (ii) Inhibition of the F1Fo ATP synthase with oligomycin reduces mitochondrial O2 consumption, with the OCR in this phase predominantly driven by the H+ leak (but also by substrate oxidation). (iii) Addition of an uncoupler such as FCCP or CCCP increases the H+ leak across the inner membrane, creating a H+ short circuit and facilitating the measurement of maximal OCR. The optimal FCCP/CCCP concentration to induce maximal respiration should be determined for each experimental setting (details in Protocol 1), and it is advisable to also assess maximal respiratory capacity in the absence of oligomycin. (iv) Inhibition of respiratory chain activity with Rotenone and/or Antimycin A ablates mitochondrial O2 consumption. Any O2 consumption measured in this phase is due to non-mitochondrial O2 consumption. Rot rotenone, AA antimycin A, Oligo oligomycin, H+ proton, IMS intermembrane space. *Respiration in stage (b) is predominantly driven by H+ leak, but also by substrate oxidation
The OCR has been extensively studied in various cellular models of NDs [7, 38, 39]. It can be measured in isolated mitochondria or permeabilised cells following a slightly altered protocol to the one described below (initial addition of ADP, phosphate, and substrate to initiate pure ‘State 3’ respiration [36]), or in intact cells or brain slices. Mitochondria isolation is a delicate procedure that provides a precise and controllable model at the expense of physiological relevance, while brain slices, which maintain intact neuronal networks, constitute a more complete biological system. In whole cells, optimised permeabilisation of the plasma membrane allows controlled supply of substrate to mitochondria, providing a more controllable model without complete loss of the cytosolic milieu [40,41,42]. The measurement of OCR in permeabilised cells or isolated mitochondria provided with different substrates (e.g. the provision of glutamate/malate to drive flux through Complex I, or succinate to drive flux through Complex II) can isolate specific complex activity and help to identify the molecular origin of a mitochondrial defect [41].
Experimental set-up
Conventional Clark-type oxygen electrode chambers (e.g. Hansatech Oxygraph), which can measure oxygen (O2) in large numbers of cells, isolated mitochondria or tissue homogenates in suspension [43], have been replaced more recently by cell respirometers, which perfuse buffer over live, attached cells within a sealed chamber, a set-up more suitable for use with primary neurons [40, 44]. Chambers mounted on fluorescence microscopes allow simultaneous measurement of other fluorescent indicators [44]. The multi-well plate reader from Agilent Technologies (Seahorse XF Flux Analyser) can simultaneously measure both the OCR and the extracellular acidification rate (ECAR; a read-out that allows calculation of lactate release under certain conditions and therefore the rate of anaerobic glycolysis—more details are provided at the end of Section 5.2.2), although experiments are expensive and limited to non-perfused cell population measurements [45]. The Oroboros Oxygraph-2k system (O2k; Oroboros Instruments) can measure O2 consumption in cell suspensions simultaneously to other parameters, such as the mitochondrial membrane potential or the ADP–ATP exchange rate mediated by the adenine nucleotide translocator (ANT) [46], but it is labour intensive and low-throughput. An overview of these two commercial systems is provided in [47]. Luxcel Bioscience’s MitoXpress® Xtra plate-reader assay allows population-level O2 measurements to be multiplexed with other reporters, such as indicators of cell viability [48].
Regardless of equipment, the OCR is generally inferred by measuring the levels of dissolved O2 in the chamber/well over time, using polarographic O2-sensing electrodes or fluorescent/phosphorescent reporters. The Seahorse system, for instance, utilises solid-state fluorescence-based sensors to measure extracellular O2 levels within a sealed chamber for 2−6 min. The chamber is then unsealed (allowing the O2 to re-equilibrate to atmospheric levels) and re-sealed to repeat the measurement. Intracellular O2−sensing probes include nanoparticles based on the phosphorescent dye Pt(II)-tetrakis(pentafluorophenyl)porphine (PtTFPP; MitoXpress®-Intra, Luxcel Biosciences), which require phosphorescence lifetime measurements and can be detected at single-cell level or on plate readers with time-resolved fluorescence/phosphorescence detection. This probe can provide quantitative intracellular O2 measurements in neurons and brain slices [49, 50].
Experimental protocol
We here describe the most commonly deployed experimental protocol to thoroughly investigate mitochondrial bioenergetic function by measuring the OCR in intact primary neurons (Fig. 2 and Protocol 1). This protocol can be followed independently of the measurement technique.
Protocol 1: Investigating mitochondrial function in primary mouse cortical neurons by measuring the oxygen consumption rate in the presence of various inhibitors of the mitochondrial respiratory chain.
-
Primary cortical neurons preparation and culture
-
Prepare cortical neurons from post-natal (day 0–1) or embryonic (day 16–18) mice of either sex [21, 22].
-
Seed neurons at appropriate density on pre-washed, poly-D-lysine (and/or laminin)-coated dishes suitable for OCR measurements (e.g. 100,000–300,000 cortical neurons/well in 24-well Seahorse cell culture microplates if using Seahorse XF Flux Analyser).
-
Culture neurons in appropriate media. Neurobasal medium 21103-049 is commonly used, supplemented with 0.5 mM l-glutamine and 2% B27. It should be noted that this media contains supraphysiological glucose levels (25 mM).
-
Perform experiments after at least 8 days in vitro.
-
-
Performing the experiment
-
Exchange culture media for appropriate ‘experimental buffer’ (wash neurons once to ensure complete exchange). Example buffer composition (in mM): 120 NaCl, 3.5 KCl, 0.4 KH2PO4, 5 NaHCO3, 20 HEPES, 1.2 Na2SO4, pH 7.4 (NaOH), supplemented with 1.2 CaCl2, 1–2 MgCl2 and desired substrate (e.g. 2.5–5 mM glucose). Equilibrate cells for 1 h at 37 °C with no CO2.
-
Different components of the proton circuit exert varied control over mitochondrial O2 consumption. Sequential addition of specific mitochondrial inhibitors isolates these components. Such an experiment involves several stages ((i)–(iv); refer to Fig. 2):
-
(i) The initial OCR is a measure of mitochondrial (a) and non-mitochondrial (f) O2 consumption, and is predominantly driven by ATP turnover (H+ flow through the F1Fo ATP synthase), and to a lesser extent by H+ leak and substrate oxidation (activity of the respiratory complexes). Differences in basal O2 consumption (in the same cellular microenvironments) can suggest: (1) altered ATP consumption, (2) altered ATP synthesis (F1Fo ATP synthase activity), (3) disrupted transport of adenine nucleotides (ANT) or phosphate between matrix and cytoplasm, (4) altered synthesis or consumption of reducing equivalents within the matrix by substrate oxidation or the respiratory chain, respectively, (5) disrupted substrate supply to the matrix, or (6) disrupted non-mitochondrial O2 consumption. Measurement of the additional parameters below can further elucidate the contributing factors.
-
(ii) Inhibiting the F1Fo ATP synthase with oligomycin allows the measurement of oligomycin-sensitive respiration driven by ATP turnover. Following oligomycin addition, the remaining mitochondrial O2 consumption is predominantly controlled by the H+ leak across the inner membrane, and to a lesser extent by substrate oxidation. As H+ leak itself is voltage-dependent and oligomycin generally hyperpolarises mitochondria, these measurements will tend to over/underestimate the contribution of the H+ leak/ATP turnover, respectively, to O2 consumption. Such errors may significantly impact findings if comparing systems with only subtle differences between them [36, 37]. Differences in the oligomycin-resistant respiration rate can indicate: (1) disrupted H+ leak (accompanied by mitochondrial membrane depolarisation), (2) altered substrate oxidation (accompanied by mitochondrial membrane hyperpolarisation), or (3) disrupted non-mitochondrial O2 consumption.
-
(iii) Addition of an uncoupler (such as FCCP, CCCP or DNP) creates a H+ short-circuit across the mitochondrial inner membrane, decreasing the proton-motive force and allowing respiration to increase. In this state, substrate oxidation is the dominant controller of O2 consumption. This is considered to be maximal respiratory capacity, although this measurement critically depends on the concentration of the uncoupler. Excess uncoupler can inhibit respiration and collapse the proton-motive force, disrupting transport processes facilitated by Δψm (e.g. the malate/aspartate shuttle, Ca2+ transport) or ΔpH (uptake of several metabolites). Cell density and buffer composition can also affect the concentration required to induce maximal respiration (albumin, for instance, can sequester FCCP). Vitally, therefore, the uncoupler concentration should be optimised for each experimental set-up – adding just enough to stimulate uncontrolled respiration while limiting the decrease in Δψm (Table 2; [36, 51]). Discrepancies in maximal respiratory capacity can indicate dysfunction in the respiratory complexes, or in cellular or mitochondrial substrate uptake/supply (processes upstream of the respiratory chain) if measured in intact cells. Be aware that cellular function relies heavily on adequate mitochondrial ATP production, and that the switch to glycolysis on addition of oligomycin can induce energy failure to such an extent that, regardless of uncoupler concentration, subsequent respiration levels are not an accurate measure of maximum respiratory capacity [52]. It is therefore advisable to also assess maximal respiratory capacity in the absence of oligomycin.
Table 2 Drugs targeting the mitochondrial bioenergetic machinery -
(iv) Finally, inhibition of the respiratory complexes (commonly rotenone+antimycin A to inhibit complexes I and III, respectively, although antimycin A is likely sufficient [37]) measures O2 consumption driven by non-mitochondrial processes, such as cytoplasmic NAD(P)H oxidases.
-
The addition of pharmacological compounds or fuel substrates prior to oligomycin can capture further detail [37]. Compounds to induce varying degrees of neuronal stimulation, such as gramicidin (permeabilises plasma membrane to monovalent cations), carbachol (acetylcholine receptor antagonist), or veratridine (inhibitor of Na+ channel inactivation) [53], increase ATP demand and introduce a ‘second hit’ that may be required to unveil underlying deficiencies not apparent in the basal, resting state. Addition of alternative fuel substrates such as ketone bodies or amino acids can help to investigate fuel dependence and metabolic flexibility [54].
-
-
Experiment analysis
-
Measurement of the OCR in this way allows calculation of several experimental parameters ((a)–(f) in Fig. 2a), detailed below.
-
The average OCR during stage (iv), non-mitochondrial O2 consumption (f), is subtracted from the average OCR during all other stages to determine ‘basal respiration’ (a), ‘proton (H+) leak’ (b), and ‘maximal respiration’ (d). Subtraction of non-mitochondrial O2 consumption also removes any background signal [40].
-
ATP turnover, assessed as oligomycin-sensitive respiration (c), is calculated as (a)–(b).
-
Spare respiratory capacity (e), also known as respiratory reserve, is calculated as (d)–(a), and is a measure of the cell’s ability to respond to an increase in energy demand.
-
The calculation of ratios from these parameters can be informative, and provides a form of internal normalisation. The coupling efficiency between ATP turnover and basal respiration is calculated as (c)/(a). The cell respiratory control ratio (RCR; similar but not identical to the RCR measured in isolated mitochondria) is calculated as (d)/(b) [36]. A higher RCR generally indicates more coupled mitochondria and more efficient ATP synthesis [40]. The bioenergetic health index is calculated as (c*e)/(b*f) [55].
-
Further guidelines for interpretation of OCR measurements can be found in [36, 37, 51, 56].
-
-
Data analysis
-
The measured OCR ([O2]/time) can be normalised to cell number, total protein content or to levels of specific proteins of interest, giving final units of [O2]/time/cell number, [O2]/time/μg protein or [O2]/time/band density [37, 41, 51]. The OCR can also be normalised to the activity of citrate synthase, a mitochondrial matrix TCA cycle enzyme commonly assumed to be a measure of mitochondrial abundance [38, 43, 57].
-
Changes in cell viability, mitochondrial density, or protein levels will impact the normalised OCR, and should be reported. In experiments where neurons are exposed to toxic manipulations (e.g. glutamate), it may be particularly important to normalise the OCR to cell viability throughout the experiment [44].
-
As a guideline for multi-well experiments (e.g. for Seahorse measurements), a minimum of three individual wells per condition should be included per plate, with experiments repeated in three independent cultures. Variability between wells and cultures may necessitate increased replicates to identify small effects.
-
One-way analysis of variance with repeated measures can be used to test for differences between OCR measurements at specific time-points.
-
For thorough investigation of the cellular metabolic state and bioenergetic capacity, OCR measurements can be coupled with those of the extracellular acidification rate (ECAR). While lactate release (specifically, protons co-transported with lactate) contributes to ECAR, CO2 formation from mitochondrial oxidative decarboxylation and the oxidative pentose phosphate pathway also contributes. OCR and ECAR measurements can be combined to calculate lactate release under certain conditions (the buffering power of the media must be calculated) [56, 58, 59], but it should be noted that, even after correction, ECAR only determines anaerobic glycolysis (i.e. the portion of pyruvate metabolised to lactate), whereas the true rate of glycolysis (glucose metabolised to pyruvate) would also include pyruvate metabolised to acetyl CoA. Algorithms are also available that utilise OCR and ECAR measurements to accurately calculate mitochondrial and cytosolic ATP production and consumption, providing information on the cellular bioenergetic state, capacity and flexibility [56, 58, 59].
Mitochondrial membrane potential
The mitochondrial membrane potential (Δψm), defined as the difference in electrical potential between the mitochondrial matrix and the cytosol, is commonly considered as a semi-quantitative read-out for the full proton-motive force (Δp), as Δψm dominates over ΔpHm [16] (although changes in ΔpHm do not necessarily parallel with Δψm [60]). While Δψm and ΔpHm both contribute to the regulation of ATP synthesis by Δp, Δψm alone provides the charge gradient to regulate the transport of Ca2+ and other ions across the mitochondrial membrane [60]. It is important to note that Δψm is not necessarily a good indicator of mitochondrial health, as ATP-synthesising mitochondria can have a reduced Δψm to those with ATP synthase switched off. These measurements should therefore be interpreted together with measurements of OCR or NAD(P)H. Small fluctuations in Δψm can indicate disrupted respiration, ATP synthesis, or ionic fluxes across the mitochondrial membrane [61,62,63]. Marked Δψm depolarisation is generally correlated with neuronal death, and may indicate mitochondrial outer membrane permeabilisation during apoptosis or mitochondrial permeability transition during ROS-mediated or Ca2+-mediated injury. In response to oligomycin, Δψm hyperpolarisation indicates that mitochondria were still generating ATP prior to drug exposure (ATP synthase operating in forward mode), while immediate depolarisation indicates that mitochondria had been net ATP consumers (ATP synthase reversal, where glycolytic ATP maintains Δψm) [61, 64]. If depolarisation is preceded by a lag phase, it is likely that oligomycin caused secondary mitochondrial dysfunction, often due to mitochondrial Ca2+ overload followed by opening of the permeability transition pore [65]. Although it is a less sensitive measure than the OCR, the magnitude and direction of Δψm fluctuations can provide complementary information to OCR experiments [36]. The cause of changes in the oligomycin-resistant mitochondrial OCR (Fig. 2a, b), for instance, can be further investigated by measuring Δψm—an increased H+ leak would decrease Δψm, while increased substrate oxidation increases Δψm. High concentrations of a mitochondrial uncoupler that collapse Δψm (Table 2) are commonly added at the end of an experiment as a control for signal specificity.
Experimental set-up
Δψm is commonly measured using membrane-permeant, lipophilic, cationic fluorescent probes which accumulate in the negatively charged mitochondrial matrix in proportion to Δψm. The magnitude of accumulation is described by the Nernst equation [16], but can be affected by mitochondrial binding of the probe [60]. The fluorescent signal can be measured by flow-cytometry or microplate photometers for fixed end-point or population level analyses, respectively. Time-lapse fluorescence imaging in single living cells allows the measurement of intrinsic cell-to-cell heterogeneity and has been applied extensively in the investigation of neuronal metabolism [66]. Δψm can also be monitored in conjunction with other fluorescent reporters to obtain simultaneous measurements of multiple cellular parameters.
All probes are substrates of the multidrug resistance (MDR) transporters. Primary neurons do not generally express these transporters, but they are often over-expressed in cultured cell lines and may limit mitochondrial loading. Cyclosporin A, an inhibitor of the permeability transition pore (PTP [67]), also inhibits MDR transporters, leading to increased mitochondrial probe loading and confounding studies investigating the contribution of the PTP to mitochondrial depolarisation [68]. This issue can be overcome by using Cyclosporin H, which inhibits MDR transporters but not the PTP.
Tetramethylrhodamine methyl ester (TMRM; Molecular Probes) is one of the most commonly used fluorescent reporters of Δψm and is less toxic than TMRE and Rh123 [69]. TMRM can be excited on epi-fluorescence, confocal, or two-photon microscopes. Two-photon imaging on the single mitochondrial level can reduce photo-toxicity and photo-bleaching compared to confocal microscopy [70]. TMRM is predominantly utilised as a single-excitation, single emission dye, and is therefore affected by focus drift, laser intensity fluctuations, photobleaching or changes in cell/mitochondrial volume. Appropriate light control experiments to ensure signal stability are therefore crucial (for a dual-excitation approach in isolated mitochondria, see [69]). Without calibration, TMRM provides a qualitative measure of Δψm. The kinetics of the TMRM signal depend on the relatively slow equilibration of the probe across the plasma membrane (addition of tetraphenylboron can increase the rate of diffusion across membranes [64]), and the contribution of the plasma membrane potential (Δψp) to the TMRM signal (or, indeed, to any cationic probe) cannot be discounted [60, 61, 64]. To overcome this issue, Δψp must be monitored in parallel with Δψm, using fluorescent anionic probes, such as DiBAC4(3) (Thermo Fisher) or a component of the Membrane Potential Assay Kit from Molecular Devices (commonly termed plasma membrane potential indicator, PMPI) [62]. Starting from inferred initial values, relative Δψm and Δψp values can subsequently be calculated using the Nernst equation in Excel [62, 64] or MATLAB [63]. Absolute quantification can be achieved using mathematical models [71], although issues such as dye extrusion from the cell should be considered.
The concentration of TMRM (and other fluorescent cationic probes) is an important consideration in experimental design, and can be classified as either ‘quench’ or ‘non-quench’ mode. High concentrations are classified as ‘quench’ mode, where densely packed TMRM molecules within the mitochondrial matrix are quenched. A transient increase in the whole-cell TMRM signal in this instance therefore indicates Δψm depolarisation—the quenched probe leaves the matrix and is unquenched, leading to a transiently brighter cellular signal until the probe more slowly re-equilibrates with the extracellular space. In contrast, TMRM in ‘non-quench’ mode (<~30 nM) remains fluorescent within the mitochondrial matrix, and Δψm depolarisation leads to a decrease in the fluorescent signal. Non-quench mode is preferable to more reliably estimate slower changes in potential and to compare Δψm between populations, as the number of TMRM molecules and their fluorescence are roughly linearly related [36, 60, 62]. Lower dye concentrations are also more tolerable, and reduce the dye’s impact on mitochondrial function [69]. However, the TMRM signal in non-quench mode does not distinguish between mitochondrial and plasma membrane potential (see earlier) [36]. The precise TMRM concentrations for quench or non-quench mode can be determined using FCCP—in quench mode, addition of ~10 μM FCCP (to collapse Δψm) induces a transient spike in TMRM fluorescence that is absent in non-quench mode [36, 62]. It must be emphasised that this transient response means that quench mode must not be used in flow-cytometry or to compare two cell populations.
Tetramethylrhodamine ethyl ester (TMRE) and rhodamine 123 (Rh123; Molecular Probes) are other cationic fluorescent dyes that can be used to report changes in Δψm, although these dyes may bind to the mitochondrial membrane and affect mitochondrial respiration to a greater extent than TMRM [69]. Rh123 only slowly crosses the plasma membrane and is therefore less affected by Δψp over short timescales. Rh123 is therefore commonly used in short-term experiments (minutes) where ‘quench’ mode is desired, such as when rapid step changes in Δψm are expected [60, 62]. In these instances, Rh123 should be removed from the media and cells washed before imaging [64, 70]. Mitotracker dyes are not suitable to monitor Δψm as mitochondrial binding prevents probe redistribution [68]. Although JC-1 can be ratiometric and has been used in fluorescence-activated cell sorting (FACS), its use (and that of DiOC6) should also be avoided, due to high toxicity, sensitivity to loading concentration, and Δψm-independent fluorescence changes [60]. Practical guides on the use of cationic fluorescent probes to monitor Δψm are provided in [60, 72].
Experimental protocol
In Protocol 2, we provide experimental guidelines to monitor changes in mitochondrial membrane potential in primary cortical neurons exposed to mitochondrial respiratory chain inhibitors. Many of the steps in this protocol can be generalised to other similar experiments.
Protocol 2: Utilising live-cell fluorescence microscopy to monitor changes in mitochondrial membrane potential (by TMRM) in primary mouse cortical neurons exposed to mitochondrial respiratory chain inhibitors.
-
Primary cortical neurons preparation and culture
-
Prepare cortical neurons from post-natal (day 0–1) or embryonic (day 16–18) mice of either sex [21, 22].
-
Seed neurons at appropriate density on pre-washed, poly-D-lysine-coated dishes suitable for microscopy (e.g. 1–2 × 105 cortical neurons/cm2 in 12 or 22 mm aperture glass-bottomed WillCo dishes (WillCo Wells)).
-
Culture neurons in appropriate media e.g. Neurobasal medium 21103-049 supplemented with 0.5 mM l-glutamine and 2% B27. It should be noted that this media contains supraphysiological glucose levels (25 mM).
-
Perform experiments after at least 8 days in vitro.
-
-
Imaging set-up
-
On a confocal microscope, TMRM can be excited with a 543 Helium–Neon (HeNe) excitation laser (3–5% of 1 mW), or similar illumination (peak excitation ~550 nm). Its peak emission is ~576 nm and can be detected using e.g. a 560 nm long-pass filter.
-
On a two-photon microscope, TMRM can be excited at ~830 nm [70].
-
Dichroic beam splitters and filter wheels in the excitation and emission light path, containing filter sets appropriate for TMRM wavelengths, are required.
-
Efforts should be made to reduce phototoxicity—minimise laser excitation power, exposure time and imaging frequency. Photobleaching can decrease the fluorescent signal and phototoxicity leads to excessive neuronal death. Appropriate vehicle controls are necessary to control for effects of phototoxicity on fluorescence.
-
Experiments should be performed at 37 °C, as lower temperatures may alter the TMRM equilibration kinetics and/or neuronal physiology. This requires a temperature controlled microscope stage, chamber and/or objective lens.
-
Imaging set-up (laser and filter settings, imaging frequency, etc.) should remain as similar as possible between experiments, to allow inter-experiment comparison.
-
-
Preparing the cells
-
Exchange culture media for appropriate experimental buffer including 10 nM TMRM (non-quench mode; wash neurons once to ensure complete exchange). Example buffer composition (in mM): 120 NaCl, 3.5 KCl, 0.4 KH2PO4, 5 NaHCO3, 20 HEPES, 1.2 Na2SO4, pH 7.4 (NaOH), supplemented with 1.2 CaCl2, 1–2 MgCl2 and desired substrate (e.g. 2.5–5 mM glucose).
-
Equilibrate neurons in TMRM for 45–60 min prior to imaging (at 37 °C in the dark, no CO2 if using experimental buffer). Baseline recording prior to drug addition can verify dye equilibration.
-
The buffer surface can be covered with mineral oil, or the chamber closed, to minimise evaporation during experiments.
-
TMRM should be kept in the buffer throughout the experiment, as removal will cause re-equilibration of the probe across the plasma and mitochondrial membranes, altering the fluorescent signal (see [60] for potential issues with dye equilibration). In contrast to cancer-derived cell lines, the addition of Cyclosporin H or verapamil (to inhibit the rhodamine-sensitive MDR transporters) is generally not required in primary neurons.
-
-
Performing the experiment
-
Mount chamber/culture dish on microscope stage.
-
Record baseline fluorescence (usually for ~10 min) to ensure signal stability prior to treatment.
-
Image acquisition at 1 min intervals should be acceptable for relatively short-term experiments (<2 h) on an epifluorescence or confocal microscope, dependent on the specific imaging set-up.
-
In the absence of a suitable perfusion system, pharmacological compounds can be added directly to the buffer on stage. Compounds can be pre-prepared in experimental buffer (+TMRM) for ease of mixing.
-
-
Testing respiratory complex activity (Fig. 3)
-
Suggested drug concentrations are listed in Table 2. Drugs can be added alone or in combination.

-
Rotenone and antimycin A inhibit Complex I and Complex III, respectively. In healthy respiring neurons, this inhibits respiration and partially depolarises the mitochondrial membrane (TMRM signal decreases in non-quench mode).
-
Oligomycin inhibits ATP synthase. This may depolarise/hyperpolarise the mitochondrial membrane (decrease/increase TMRM signal in non-quench mode), depending on the prior direction of ATP synthase operation (ATP producing or consuming, respectively) [61, 64].
-
The addition of high concentrations of FCCP (10 μM) at the end of the experiment should rapidly collapse Δψm (and TMRM signal) indicating that the TMRM signal during the experiment emanated from mitochondria. Such high concentrations also more slowly depolarise the plasma membrane [62].
-
Vehicle and light control experiments should also be performed.
-
Sufficient time should be allowed between drug additions for the TMRM signal to stabilise (up to 30 min).
-
Compounds can also be added as in the standard OCR experiment (to complement these measurements) (Protocol 1).
Fig. 3: Representative images and time-series data from TMRM (10 nM) measurements in wild-type mouse cortical neurons exposed to mitochondrial inhibitors 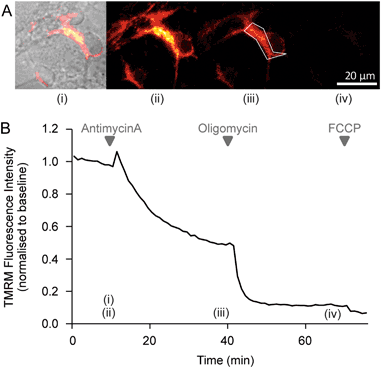
. a Brightfield and TMRM fluorescence images were captured on a Zeiss Axiovert 100 microscope (brightfield and TMRM fluorescence are merged in the left-most image, TMRM only in the other images). b Time-series TMRM fluorescence measurements within the region of interest marked by the white polygon in (A)(ii). The precise time-points of the images in (A) are marked (i)–(iii) on the graph. Baseline fluorescence was recorded for 10 min pre-treatment and used to normalise the signal. Complex III inhibition with antimycin A (1 μM) induced a decrease in TMRM fluorescence, indicating mitochondrial membrane depolarisation. Subsequent oligomycin addition (2 μg/ml) further reduced TMRM fluorescence, indicating that prior to oligomycin addition the mitochondrial membrane potential was being maintained by the F1Fo ATP synthase operating in reverse. The loss of any remaining TMRM fluorescence after FCCP addition (10 μM) indicates mitochondrial membrane potential collapse
-
-
Image processing and analysis
-
Image processing can be performed using tools such as MetaMorph (Molecular Images) or the open-source ImageJ (https://imagej.nih.gov/ij/).
-
Subtract background signal, identify regions of interest (ROI; e.g. individual cells, or mitochondrial-enriched regions), and measure the (thresholded) average signal intensity within the ROI. The total fluorescence within the cell soma can be measured, assuming mitochondria within the cell respond similarly (relatively homogenous response throughout) [60], and provided that mitochondrial loading of the probe has been verified.
-
For each ROI, plot the signal intensity over time (absolute signal intensity, or signal intensity normalised to baseline signal) (Fig. 3).
-
-
Data analysis
-
As a guideline when investigating single-cell behaviour, at least 25 cells from three independent cultures should be analysed, although variability between cells and cultures may necessitate increased replicates to identify small effects. Required cell numbers can be calculated based on the expected effect size and desired power.
-
The response of each neuron can be treated as an independent event in statistical analyses, unless culture-to-culture variability is higher than cell-to-cell variability within cultures.
-
One-way analysis of variance with correction for repeated measures can be used to test for differences between TMRM measurements at specific time-points.
-
As discussed in the main text, the contribution of Δψp to the TMRM signal can be determined by simultaneous use of a Δψp-sensitive probe, allowing relative or absolute quantification of Δψm [62,63,64, 71].
-


Mitochondrial NAD(P)H
Pyridine nucleotides are electron-shuttling agents that act as co-factors in enzymatic reduction–oxidation (redox) reactions. Nicotinamide adenine dinucleotides (NAD+/NADH and the phosphorylated NADP+/NADPH) are pyridine nucleotides that play a central role in mitochondrial energy metabolism and in the maintenance of redox status. In neurons, NADH is the principal electron donor in the respiratory chain, and is oxidised from NADH to NAD+ at complex I to drive mitochondrial OxPhos [73]. Mitochondrial NADH is maintained by the reduction of NAD+ to NADH in the TCA cycle and via the import of NADH-derived reducing equivalents driven by the malate/aspartate shuttle. NAD+ can also be consumed by Poly (ADP-ribose) polymerase (PARP) isozymes and sirtuins [74, 75]. NADP+/NADPH does not contribute directly to mitochondrial OxPhos, but primarily supports the maintenance of reduced glutathione (GSH) and thioredoxin pools [76]. NADPH is maintained by the activity of the NADP+-isocitrate dehydrogenase (ICDH) and the NADP+-malic enzyme, though the mitochondrial NADP+/NADPH pool is often assumed to be largely reduced and invariant compared to the mitochondrial NAD+/NADH pool [61]. Electrons from NADH and NADPH (termed NAD(P)H here) reactions can also be transferred to oxygen molecules (e.g. by NADPH oxidases) to generate reactive oxygen species in the form of superoxide [77]. Measurement of mitochondrial NAD(P)H can be most informative when linked to Δψm measurements, as processes regulating both parameters are tightly linked [36]. A decrease in mitochondrial NAD(P)H can indicate enhanced respiratory chain activity (increased NADH oxidation), reduced TCA cycle activity (decreased NAD+ reduction), or increased NAD+ consumption.
Experimental set-up
NAD(P)H can be monitored by measuring endogenous autofluorescence, through the use of genetically encoded fluorescent proteins, or by high-performance liquid chromatography, labelled spectrometry techniques, or commercially available enzymatic assays (e.g. Promega’s NAD/NADH-GloTM Assay).
Autofluorescence
Both NADH and NADPH autofluoresce in their reduced, but not in their oxidised state, and produce spectrally identical autofluorescence [78]. The cellular NAD(P)H autofluorescence intensity is considered to be dominated by protein-bound mitochondrial NAD(P)H, as the autofluorescence signal co-localises with mitochondrial markers, and NAD(P)H autofluorescence intensity is enhanced when the nucleotides are protein-bound and ‘active’ [73, 79].
NAD(P)H autofluorescence can be detected using epifluorescent illumination or confocal microscopes equipped with UV lasers. It is excited by near ultraviolet wavelength light (340–360 nm), and emission is detected at ~450 nm (excitation/emission spectra similar to Hoechst) [78]. Low excitation wavelengths are required to distinguish NAD(P)H from flavoprotein autofluorescence (which has an excitation peak ~450 nm). While autofluorescence detection is a non-invasive approach, excessive exposure of cells to UV wavelength excitation can be highly phototoxic, and efforts to reduce exposure are required (e.g. optimising detection sensitivity and reducing laser intensity, pixel dwell-time, exposure time and imaging duration/frequency), sometimes resulting in detection difficulties or sub-optimal signal-to-noise levels. Two-photon microscopy (excitation peak ~710 nm; emission ~400–500 nm) has also been used in dissociated cultures [70, 80] and in brain tissue slices [73, 81]. Two-photon NAD(P)H excitation wavelengths are phototoxic and also excite flavoprotein autofluorescence [82]. Semi-quantification and comparison of mitochondrial NAD(P)H levels between experiments can be achieved by determining the autofluorescence dynamic range between maximal and minimal NADH for each sample (neglecting the contribution from NADPH), with maximal oxidation induced by an uncoupler, such as FCCP, and maximal reduction induced by an effective inhibitor of respiration, such as cyanide [77, 83].
Fluorescence lifetime imaging microscopy (FLIM) enables semi-quantitative measurements of free and protein-bound mitochondrial NAD(P)H and may further identify the relative contribution of NADH and NADPH to the autofluorescence signal [77]. FLIM measures the pixel-by-pixel autofluorescence lifetime on a defined sample area, under two-photon pulsed excitation, by recording the delay between the pulse and fluorescence emission (lifetime) [39]. NAD(P)H autofluorescence lifetimes vary based on their binding state—the lifetime of free NAD(P)H molecules (~0.4 ns) is shorter than that of the protein-bound, active molecule (~2–4 ns) [77]. Since the relative amplitude of each lifetime component is proportional to its concentration, FLIM analysis can be used to measure the contribution of active NAD(P)H with respect to its free counterpart. This is particularly important in the study of respiratory chain processes in the mitochondrial compartment. The precise autofluorescence lifetime also depends on the fluorophore’s local environment, and FLIM techniques may therefore provide an alternative measure of characteristics such as pH [79].
Experimental protocol
Protocol 3: Utilising live-cell fluorescence microscopy to monitor NAD(P)H autofluorescence in primary mouse cortical neurons exposed to mitochondrial respiratory chain inhibitors.
-
Prepare and culture primary cortical neurons as described in Protocol 2, performing experiments after at least 8 days in vitro.
-
Imaging set-up
-
The NAD(P)H autofluorescence peak excitation wavelength (~340 nm) and peak emission wavelength (~450 nm) are similar to those of Hoechst. An epifluorescence microscope therefore requires a transmission curve that includes near UV wavelengths. On a confocal microscope, NAD(P)H autofluorescence can be excited with a UV excitation laser (364 nm Argon Gas Ion laser or 355 or 375 nm DPSS laser). The two-photon excitation peak of NAD(P)H autofluorescence ~710 nm.
-
Dichroic beam splitters and filter wheels in the excitation and emission light path, containing filter sets appropriate for NAD(P)H autofluorescence, are required e.g. a 450 nm band-pass filter (30/40 nm bandwidth). It is essential to use a band-pass filter (rather than a long-pass filter) to minimise contamination of the signal by flavoprotein autofluorescence.
-
Phototoxicity is a major concern when exciting neurons at near UV wavelengths—minimise laser excitation power, exposure time, imaging frequency and imaging resolution. The use of UV filters (e.g. neutral density UV filter) can also be considered. Appropriate control experiments should be performed. See also [83].
-
As for TMRM experiments, autofluorescence imaging should ideally be performed at 37 °C.
-
Imaging set-up (laser and filter settings, imaging frequency, etc.) should remain as similar as possible between experiments, to allow inter-experiment comparison.
-
-
Preparing the cells
-
Exchange culture media and equilibrate neurons as for Protocol 2 (simultaneous monitoring of TMRM is also possible).
-
-
Performing the experiment
-
Neurons can be imaged as described in Protocol 2 (with settings specific to detect autofluorescence), although extra care is needed to reduce phototoxicity when exciting and detecting NAD(P)H autofluorescence, so imaging frequency and experiment duration may need to be reduced. NAD(P)H autofluorescence can equilibrate relatively rapidly (within 5 min, Fig. 4) following drug addition.
Fig. 4: Representative images and time-series data from NAD(P)H autofluorescence measurements in wild-type mouse cortical neurons exposed to mitochondrial inhibitors 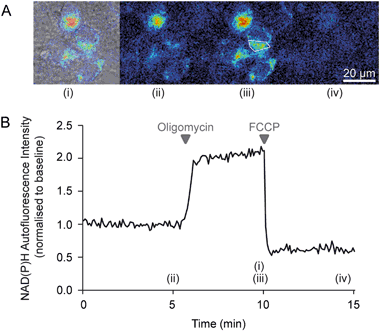
a Brightfield and NAD(P)H autofluorescence images were captured on a Zeiss Axiovert 100 microscope. b Time-series autofluorescence measurements from the region of interest marked within a white polygon in (A)(iii). The precise time-points of the images in (A) are marked (i)-(iv) on the graph. Baseline fluorescence was recorded for 5 min pre-treatment and used to normalise the signal. Inhibition of the F1Fo ATP synthase with oligomycin (2 μg/ml) reduced NADH consumption by the respiratory chain, leading to an increase in the autofluorescence signal. Subsequent mitochondrial uncoupling with FCCP (0.5 μM) increased respiratory NADH oxidation, decreasing autofluorescence
-
-
Testing respiratory complex activity
-
Suggested drug concentrations are listed in Table 2.
-
Oligomycin inhibits ATP synthase and thereby reduces the flux through the respiratory chain. In healthy, unimpaired neurons this decreases NADH consumption and increases autofluorescence (Fig. 4).

-
Mitochondria uncoupling (low concentrations of e.g. FCCP/CCCP, see Table 2) increases NADH oxidation, decreasing autofluorescence ([77], Fig. 4).
-
Inhibition of respiration with rotenone or antimycin A reduces NADH oxidation and increases NAD(P)H autofluorescence [77].
-
Standard calibration of the NAD(P)H autofluorescence dynamic range is performed by inducing maximal NADH oxidation with an uncoupler (e.g. FCCP/CCCP), followed by maximal reduction induced by an inhibitor of respiration such as cyanide or antimycin A.
-
Alternative substrates, modulators or compounds can be added to analyse additional aspects of NAD(P)H processing.

-
Vehicle control experiments should also be performed.
-
-
Image processing and analysis
-
Perform image processing and analysis similar to that described in Protocol 2.
-
Subtract background signal from the image.
-
Identify regions of interest (ROI) for analysis. Areas of brightest autofluorescence can be considered areas of high mitochondrial density (Fig. 4). Selecting a ROI within these areas minimises background signal, which can dilute the dynamic range of the autofluorescence signal.
-
Measure the average signal intensity within the ROI—for autofluorescence measurements, thresholding may reduce the signal dynamic range.
-
For each ROI, plot the signal intensity over time (absolute signal intensity, or signal intensity normalised to baseline signal) (Fig. 4).
-
-
Data analysis
-
Analyse the autofluorescence signal in single neurons as described in Protocol 2.
-



Flavoprotein autofluorescence provides a similar, though not identical, measure of mitochondrial metabolism, and can be especially useful in the absence of available UV excitation or as a complementary measure to NAD(P)H autofluorescence. FAD+ is reduced to FADH2 in the TCA cycle, and FADH2 is oxidised to FAD+ via the activity of Complex II in the respiratory chain. As flavoproteins autofluoresce in their oxidised (FAD+), but not in their reduced (FADH2) form, signal fluctuations are expected to be inverted compared to NAD(P)H autofluorescence. The flavoprotein peak excitation wavelength is ~450 nm, while emission is detected at ~510 nm [39, 73, 82].
Fluorescent reporter proteins
Genetically-encoded fluorescent reporters and their use in biological research are introduced in the section “Fluorescent reporters of mitochondrial ATP, calcium (Ca2+) and pH”. Fluorescent proteins (FPs) to measure mitochondrial NADH or NADH:NAD+ have an improved signal to noise ratio and are less phototoxic than autofluorescence detection methods, but are slower acting, can contribute to NAD(P)H buffering, and require transfection and mitochondrial targeting [84].
Current genetically encoded fluorescent reporters of NADH or of the NADH:NAD+ ratio contain the bacterial Rex protein, a transcription factor that, when bound to NADH, undergoes a conformational change [85, 86]. Frex variants are dual excitation reporters of NADH, comprising a circularly permuted yellow FP (cpYFP) inserted between two Rex proteins, that have been successfully targeted to mitochondria (Frex-Mit/C3L194K) [87]. RexYFP, comprising cpYFP inserted into a Rex homologue, is significantly smaller than Frex, and has also been successfully utilised to report the mitochondrial NAD+:NADH ratio [85]. SoNar, a highly sensitive cpYFP and Rex-based reporter of NADH and NAD+ levels, has yet to be targeted to mitochondria [88]. The use of cpYFP, however, is limited by its pH sensitivity, and any cpYFP reporters need to be used in conjunction with a pH reporter.
Peredox, consisting of two FPs (T-Sapphire and mCherry) and a Rex dimer, reports the free cytosolic NADH:NAD+ ratio with a high affinity for NADH [89]. Peredox is mostly pH insensitive, but thus far has not been successfully utilised to measure mitochondrial NADH:NAD+, likely due to its sub-optimal sensitivity range (NADH:NAD+ ratios are estimated to be >20-fold higher in the mitochondria than in the cytoplasm [89]). Genetically encoded reporters of mitochondrial NAD+ and NADPH have also been developed recently [90, 91]. Because of the predominance of NAD+ over NADH, small changes in NAD+ could be missed using ratio reporters, making NAD+ sensors a promising development, especially considering the recent interest in ageing and NAD+-dependent sirtuin activity [75, 90].
Respiratory complex activity and subunit expression
Reduced expression or activity of respiratory complexes has been reported in most NDs, as described earlier. These changes, however, may not necessarily translate into compromised mitochondrial function, due to variations in rate-limiting factors, the distribution of control along the respiratory chain or the presence of compensatory mechanisms (e.g. between Complex I and II) [36]. For this reason, assays measuring subunit expression or complex activity may be more usefully employed to investigate the specific molecular origin of a known mitochondrial defect, rather than as a general measure of mitochondrial bioenergetic function. Such investigations may be complemented by OCR measurements in permeabilised cells or isolated mitochondria, where the provision of different substrates can isolate specific complex activity.
Experimental protocol
Protein levels of specific subunits of the respiratory complexes (Complexes I–IV and the F1Fo ATP synthase) can be assessed by Western Blotting with individual antibodies or commercially available antibody cocktails (e.g. MitoProfile® from Mitosciences, Abcam). As some antibodies bind to auxiliary rather than catalytic subunits, altered subunit expression in these instances may not correspond to altered complex activity.
The activity of the respiratory complexes can be measured via histochemical staining of fresh frozen brain tissue (e.g. cytochrome c oxidase (COX) and/or succinate dehydrogenase (SDH) staining; [92]), with in-gel enzymatic histochemical reactions [93], or with spectrophotometric assays (commercially available kits, or see below). Complex activity can be measured in tissues, cells, mitochondria-enriched fractions or isolated mitochondria. Activity has also been measured in pre-synaptic nerve terminals (synaptosomes) [94], but these preparations can be hampered by heterogeneity [62]. Although isolating mitochondria from cellular homogenates is a delicate and time-consuming procedure and experimental data can vary based on the quality of the isolation, it is nevertheless required for many assays to reduce the confounding contribution of non-mitochondrial enzymes [57] and to increase assay sensitivity. Measurement of respiratory complex activity also requires appropriate disruption of the mitochondrial membrane. Spectrophotometric assays involve mixing the mitochondrial content with electron donors and acceptors appropriate for the complex being studied, and measuring the change in absorbance at the appropriate wavelength for the specific electron donor/acceptor (Table 3). The activity of the ANT can also be measured in permeabilised cells [46].
Experimental analysis
Enzymatic activity \(\left( {\rm{{nmol.min}}^{ - 1}.mg^{ - 1}} \right)\) is calculated using the Beer–Lambert law
where eλ is the extinction coefficient (mM−1 cm−1), eλ = ~6.2 for NADH at 340 nm, eλ = ~20 for DCPIP at 600 nm, eλ = ~19 for reduced cytochrome c —oxidised cytochrome c at 550 nm (difference extinction coefficient) [43, 57]. V is the volume of sample (ml). Protein conc. = protein concentration of sample (mg ml−1)
Fluorescent reporters of mitochondrial ATP, calcium (Ca2+) and pH
Mitochondrial ATP, Ca2+ and pH, as well as other metabolites and ions, can be monitored using fluorescent dyes or genetically encoded fluorescent reporters. Genetically encoded fluorescent reporters generally comprise one or two fluorescent proteins (FPs) connected to a peptide/protein whose conformation is altered by the target molecule or event [84, 95]. For example, the conformation of the peptide/protein may be altered when bound by a certain ligand (e.g. ATP or glucose), or cleaved or phosphorylated by an enzyme (e.g. caspases or AMPK). This conformational change alters the fluorescent properties of the reporter, providing a detectable measure of the concentration of the ligand, or the activity of the enzyme. Caveats for all genetically encoded fluorescent reporters include; sensitivity to environmental factors such as pH (can be corrected for with simultaneous use of a pH sensor), buffering of the target molecule (especially relevant for low abundance molecules), non-specificity (availability of negative controls is important), variable expression levels across cell types, and the functional impact of the introduction of an exogenous, sometimes large, protein [95].
Förster resonance energy transfer (FRET)-based fluorescence reporters commonly comprise two FPs connected by a linker peptide/protein, with the emission spectra of one FP (donor) overlapping the excitation spectra of the other (acceptor). With both FPs in close physical proximity (<~10 nm), FRET occurs from the donor to the acceptor (energy transferred from the donor excites the acceptor), reducing the donor, and increasing the acceptor, fluorescence intensity (Fig. 5a). Since FRET is proportional to the sixth power of the distance between the FPs, any conformational change in the linker protein that minimally alters the distance between and/or the orientation of the FPs, can induce considerable FRET changes. The occurrence of FRET actually affects several fluorescence parameters, such as the emission intensity of the FPs at constant excitation, and their fluorescence decay time. The former has been used most frequently because of its relative simplicity to measure, limited cost, and high velocity of image acquisition. Fluorescence decay or lifetime imaging microscopy (FLIM) instead requires a dedicated apparatus, but it offers the advantage of being relatively insensitive to some experimental conditions, such as focus drift, bleaching, or excitation light fluctuation. This technique is utilised in enhanced acceptor fluorescence (EAF), where both fluorophores are excited at the same wavelength (~480 nm for GFP–YFP pairs) and the average lifetime of the emitted fluorescence is measured [96]. As YFP has a longer lifetime than GFP, the average lifetime will increase when the two FPs are in close enough proximity for FRET to occur (more YFP contributing to the average lifetime). Ratiometric measurements (such as provided by FRET-based reporters) facilitate intra-experimental normalisation and reduce the effects of volume changes, focus drift and reporter concentration. Fluorescent reporters can be multiplexed to allow simultaneous monitoring of multiple processes (e.g. cyan FP (CFP)/YFP FRET-based reporters in conjunction with red fluorescent reporters, such as TMRM [12]).

[99] a The reporter comprises a linker protein (ε-subunit of a bacterial F1Fo ATP synthase) inserted between a donor CFP and acceptor YFP (enhanced CFP and Venus). ATP binding induces a conformational change in the linker protein, increasing FRET between the two FPs and altering the emitted fluorescence. Ratiometric measurements are obtained by calculating the FRET ratio (CFP/YFP). The acceptor YFP can also be laser-excited at ~488 nm. Fluorescent emissions from both FPs should be monitored, to ensure that any ratio change is due to altered FRET (opposite changes in the fluorescence of the individual FPs), rather than other sources (such as increased auto-fluorescence). Image reproduced with permission from [12]. b Representative images of primary mouse cortical neurons (DIV8) transfected with mitoATeam, stained with TMRM, and imaged on a Zeiss LSM 710 confocal microscope. Mitochondrial localisation of the mitoATeam probe was verified by colocalisation with the TMRM signal (merged TMRM and Mito-ATeam image). Scale bar = 10 μm. DIC differential interference contrast
Genetically encoded fluorescent reporters can be transfected into primary neurons using a variety of techniques, such as lipofection, Ca2+ phosphate co-precipitation or Amaxa nucleofection (Lonza) [97]. Many of these techniques, however, are hampered by high toxicity and low transfection efficiency in primary cells. Lenti-viral or adeno-viral approaches can improve efficiency, increasing the number of cells expressing the reporter [97]. Fluorescent reporters can be modified to target the reporter to the mitochondrial membrane, matrix or inter-membrane space. Targeting fluorescent reporters to mitochondria, however, is not without challenges—the size or complexity of the reporter may limit its ability to translocate into mitochondria, the increased basicity of mitochondria (~pH 8) may impact reporter function, and mitochondrial metabolites may affect reporter readout [95, 98]. Mitochondrial localisation of transfected reporters (and fluorescent dyes) should be verified, by e.g. co-localisation with an established mitochondrial marker. It is also good practice to confirm that the mitochondrial targeting of the fluorescent reporter does not disrupt mitochondrial respiratory function, by e.g. measuring OCR according to Protocol 1.
Accurate calibration of a fluorescent reporter can sometimes provide absolute quantification of metabolite, ion or protein concentrations. Nevertheless, changes in steady-state concentration can be due to any number of processes, including altered import, export, synthesis or consumption—experiments are required to further delineate the contributing factors.
Mitochondrial ATP
The ATeam FRET-based reporters of ATP concentration (Table 4; [99]) comprise the ATP-binding ε subunit of the bacterial F1Fo ATP synthase from Bacillus subtilis inserted between CFP and YFP variants (msECFP and cpVenus). Specific and reversible binding of ATP to the ε subunit induces a conformational change in the ATeam reporter, increasing the FRET-induced fluorescence intensity. The ATeam AT1.03 subtype, with a dissociation constant (Kd) of 3.3 mM for ATP at 37 °C, can detect ATP fluctuations in the mM range (less sensitive to smaller changes), and can be used with the mutated AT1.03R122K/R126K negative control [99]. This affinity is highly temperature dependent, varying five-fold over a range of 10 °C [99]. AT1.03 fluorescence is stable over physiological pH (7.1–8.5; mitochondrial pH ~8.0), but changes outside this range may affect fluorescence. ATeam variants have been localised to mitochondria (mitoATeam) and successfully utilised in neurons (Fig. 5b [99, 100]). The red-shifted mitoGO-ATeam, comprising GFP and orange FP variants (cp173-mEGFP and mKOk), is less phototoxic, less sensitive to pH and enables multiplexing with UV excitable probes, such as the fluorescent Ca2+ indicator Fura-2 (the excitation spectra of CFP, used in the ATeam reporter, is in the UV range) [101, 102].
The ATP/ADP ratio is a more complete measure of cellular energy status than ATP alone [103]. The PercevalHR reporter is a dual-excitation fluorescent reporter of the ATP/ADP ratio that can reliably detect ratios from ~0.4 to 40 [103, 104], with ATP/ADP ~10 in cultured neurons at rest [105]. However, PercevalHR is sensitive to pH changes within the physiological range (6.7–7.8), requiring simultaneous pH monitoring, and to our knowledge has not yet been successfully utilised in mitochondria. pH sensitivity is especially problematic in neurons, where intracellular acidification often occurs with decreases in ATP, such as during excitotoxicity. Other fluorescence-based ATP sensors, such as the dual-excitation, single FP ATeam variant, QUEEN-2m [106], and a potential EAF-based ATP biosensor (developed in [107], although lifetime measurements were not performed), have yet to be targeted to mammalian mitochondria.
Bioluminescence-based probes avoid the excitation light required for fluorescence-based imaging, but emitted bioluminescence is generally weaker than fluorescent signals (necessitating lengthy exposure) and the technique requires the addition of a luciferase substrate (e.g. luciferin or furimazine). The bioluminescence energy transfer (BRET) probe, BTeam [108], comprises the same ATP-binding ε subunit as the ATeam probe, inserted between a non-ATP-consuming NanoLuciferase (NLuc; emission peak ~455 nm), and a YFP variant (mVenus). Changes in the BRET ratio (YFP/Nluc) can report ATP fluctuations between 0 and 10 mM, and the probe can be targeted to mitochondria (mit-BTeam). Similar to the ATeam probe, BTeam measurements are stable within physiological pH (7.1–8.3) but are sensitive to temperature changes [108]. Mitochondria-targeted luciferase chimeras (mtLUC) have also been used to monitor mitochondrial ATP levels in mammalian cells [109].
Mitochondrial ATP can also be measured in isolated mitochondria using a number of single time-point techniques, such as luciferase bioluminescence assays (which also exhibit pH sensitivity), high performance liquid chromatography (HPLC), or 31P NMR [110].
Mitochondrial Ca2+
Mitochondria play a major role in Ca2+ signalling and homoeostasis through its uptake, sequestration, and release [111]. Thanks to the large electrochemical gradient across its inner membrane (proton-motive force), mitochondria can import huge amounts of Ca2+ through the mitochondrial Ca2+ uniporter (MCU) complex, and serve as buffers in periods of high cytoplasmic Ca2+, such as during neuronal excitation or excitotoxicity [61, 112]. Within mitochondria, Ca2+ regulates substrate import, several TCA cycle dehydrogenases, components of the respiratory chain and ATP synthesis, coupling neuronal excitation/activity with bioenergetics [14, 113]. Ca2+ is released from neuronal mitochondria primarily through the Na+/Ca2+ exchanger NCLX [14, 111, 114]. Excessive mitochondrial Ca2+, however, can be toxic, contributing to increased ROS production, activation of the mitochondrial permeability transition pore, delayed Ca2+ deregulation and neuronal death [115]. Such mitochondrial Ca2+ overload and subsequent dysfunction have been implicated in neurodegeneration [116].
In the following discussion, it must be borne in mind that all fluorescent indicators monitor free, rather than total, Ca2+. Under conditions of high matrix loading, for example when modelling excitotoxicity, formation of a Ca2+–phosphate complex can mean that, although total matrix Ca2+ can approach 1 M, free Ca2+ is in the low μM range. Electron probe microanalysis or 45Ca2+ studies are needed to monitor total Ca2+ concentration. It is also important to remember that these reporters act as Ca2+ buffers, and may alter the endogenous ion dynamics [117].
Several fluorescent reporters are available to measure free mitochondrial Ca2+ (Table 5). The positively charged fluorescent Ca2+ indicator Rhod-2 (and its analogues) are imported by mitochondria, but accumulate only incompletely and can alter intracellular Ca2+ dynamics [14, 118, 119]. Although such reporters do not require transfection, they have generally been superseded by genetically encoded Ca2+ indicators that can be targeted to mitochondria.
Cameleons are FRET-based Ca2+ sensors comprising two FPs (the more recent ‘yellow cameleons’ (YC) utilise eCFP and cpVenus [120]) connected by a Ca2+-binding protein, calmodulin (CaM), and a CaM-binding peptide of myosin light-chain kinase, M13. CaM–Ca2+ binding leads to a CaM–M13 interaction that induces a conformational change in the reporter, altering the FRET efficiency and emitted fluorescence [121]. Cameleons have been optimised for brightness and increased dynamic range (YC3.60; [120]), and to reduce off-target interactions (D3cpv; [122]). Replacing CaM with troponin C can also reduce off-target binding in neurons [117]. The mitochondria-targeted 4mtD2cpv–4mtD4cpv series of Cameleons with varying Ca2+ affinities [122] have been successfully used in primary neurons [112].
Smaller, single protein-based Ca2+ indicators may localise more easily to mitochondria [98]. Aequorin, a jellyfish protein that bioluminesces in a Ca2+-dependent manner, can be localised to neuronal mitochondria (mtAEQ) [123, 124], but its low luminescence intensity limits its utility in single-cell measurements [118]. Other single FP-based Ca2+ indicators that have been successfully imaged in neuronal mitochondria (Table 5) include the cpYFP-based Pericam [125] and multiple variants of GCaMP and RCaMP [112, 126,127,128]. GECO Ca2+ reporters [129], variants of GCaMP3, have been used extensively to measure mitochondrial Ca2+ in neurons [53, 102, 119, 130]. CEPIA probes are GCaMP2 variants that can be localised to mitochondria [131]. Luminescence-based methods (BRET) are also promising [132]. Mitochondria-targeted reporters can be co-expressed with fluorescent Ca2+ indicators designed for other subcellular locations (e.g. endoplasmic reticulum or cytoplasm) to simultaneously monitor Ca2+ flux between cellular compartments [119, 130].
These and other Ca2+ reporters are extensively reviewed elsewhere, e.g., [119, 133]. The final choice of reporter depends on the probe’s affinity, dynamic range and available equipment. Reporters should be calibrated to identify their dynamic range in the specific experimental setting—this involves determining Fmax/Fmin or Rmax/Rmin for non-ratiometric and ratiometric dyes, respectively, where Fmax (Rmax) is the fluorescence intensity (ratio) at saturating Ca2+ concentrations, and Fmin (Rmin) is the fluorescence intensity (ratio) at minimal/zero Ca2+ concentrations [122, 134]. Ionomycin, typically used for Fmax/Fmin determination, will take longer to saturate reporters localised within mitochondria. These values can be used, along with the reporter’s dissociation constant (Kd), to calculate relative Ca2+ concentrations [119, 134].
Mitochondrial pH (pHmito)
Knowledge of the mitochondrial matrix pH and ΔpHm (pHmito–pHcyto), in conjunction with Δψm, allows calculation of the proton-motive force (Δp), with ΔpHm contributing ~20–30% of Δp [60]. ΔpHm regulates the flux of several metabolites across the mitochondrial membrane [135], and changes in pHmito often occur with energy metabolism fluctuations. pHmito measurements can also be used to correct for experiments involving genetically encoded FPs or other reporters exhibiting pH-sensitivity. Conversely, many pH-sensitive FPs have themselves been adapted for use as direct pH sensors [136]. For pHmito measurements, reporters with a high acid dissociation constant (pKa > 7.5) are required, appropriate for the higher pH of mitochondria (more alkaline) compared to the cytosol [135, 136].
Although pH-sensitive fluorescent dyes exist (e.g. SNARF, BCECF), they are not specifically targeted to mitochondria, necessitating the concomitant use of a fluorescent mitochondrial marker [135]. pHmito is more commonly measured using genetically encoded FPs (Table 6). YFP variants can be used to measure pHmito, as YFP is more pH-sensitive than GFP. Single FP reporters using EYFP, for example, have been targeted to the mitochondrial intermembrane space (MIMS-EYFP) and matrix (mt-YFP, mitoYFP) in neurons [100, 137, 138]. The pH indicator pHluorin [139] has also been targeted to mitochondria [140]. The pKa of EYFP and pHluorin (~7 and 7.1, respectively), however, are not optimal for measurement of pHmito [136].
SypHer, a dual-excitation ratiometric probe (pKa 8.7; excitation 430, 490 nm; emission ~535 nm [141]), harnesses the inherent pH sensitivity of cpYFP, and was generated from the hydrogen peroxide sensor HyPer. The mitochondria-targeted variant, mitoSypHer, has been used to measure neuronal pHmito [142]. mtAlpHi (mitochondrial alkaline pH indicator, pKa ~8.5), is a non-ratiometric, intensity-based pH indicator that has also been targeted to neuronal mitochondria [143]. mtAlpHi was generated from the Ca2+ indicator Camgaroo2, with CaM replaced by a Ca2+-insensitive aequorin segment. Other pH-sensitive fluorescent indicators suitable for reporting pHmito include pHRed (pKa 7.8), which can also be used for FLIM [144], and pHTomato (pKa 7.8; [145]).
Mitochondrial reactive oxygen species
Reactive oxygen species (ROS) are a group of reactive molecules derived from oxygen, and include superoxide (O2−), hydrogen peroxide (H2O2) and hydroxyl radicals (•OH). Mitochondria, as the primary oxygen consumers within the cell, play a critical role in ROS metabolism, as electrons leaked from components of the OxPhos machinery can react with O2 to form O2− [146, 147]. Mitochondria-derived O2− is rapidly converted (dismutated) to H2O2 by superoxide dismutase (SOD2/MnSOD in the mitochondrial matrix; SOD1 in the inter-membrane space and cytoplasm), but can also be converted to the toxic reactive nitrogen species, peroxynitrite, by reaction with nitric oxide [147, 148]. H2O2 can in turn create more reactive oxygen by-products such as •OH [148]. Glutathione peroxidase catalyses the reduction of H2O2 to H2O via the oxidation of glutathione (GSH) to glutathione disulphide (GSSG). Together, ROS are vital second messengers and play a key role in redox-dependent signalling, but ROS accumulation, such as following hypoxia-ischaemia-reperfusion injury or excitotoxicity, contributes to oxidative stress and has been associated with ageing and neurodegeneration [3, 4]. The precise effects of specific ROS and their associated anti-oxidants vary considerably throughout the cell [149].
The primary sources of mitochondrial O2− are Complexes I and III, and TCA cycle enzymes [14, 94, 147], while the assembly of Complex I into supercomplexes can dictate the rate of ROS formation [150]. Increased respiration and a hyperpolarised mitochondrial membrane can increase ROS production in neurons [61, 151, 152]. Conversely, inhibition of the respiratory complexes can also increase ROS production [94], when the mitochondrial membrane potential may be maintained by reverse activity of the F1Fo ATP synthase and ROS are generated through electron leaks [153, 154]. ROS generation is therefore strongly regulated by respiration, with the underlying factors disrupting respiration likely determining the ROS response [155, 156]. The levels of steady-state mitochondrial ROS depend not only on ROS generation, however, but also on their conversion to other forms, their transport into and out of the mitochondria, and the cellular anti-oxidant activity.
Experimental set-up
ROS levels can be inferred by imaging redox-sensitive fluorophores or genetically-encoded FPs, by measuring the activity of redox-sensitive enzymes such as aconitase, or by detecting probe oxidation products using methods such as electron paramagnetic resonance spectroscopy or HPLC [61, 146, 148, 157]. Recent developments in ROS-sensitive measurements involve the use of inert nanoparticles or carbon nanotubes [157, 158], although these approaches have yet to be tested in neuronal mitochondria.
We strongly stress that current methods for measuring mitochondrial ROS, particularly redox-sensitive fluorophores, are associated with significant difficulties and limitations that must be carefully considered, both when interpreting the existing literature and during experiment design. These difficulties are associated with, among others, signal specificity, identifying the intracellular origin of ROS, and the rapid turnover of ROS within the cell. If performing experiments, stringent control measures must be employed, all findings should be interpreted with caution, and results should be confirmed using alternative techniques, e.g. [159]. Many of these issues have been discussed previously [148, 149, 160, 161], and technical specifications, use guidelines and limitations of several redox-sensitive fluorophores and FPs can be found in [157, 162, 163].
Redox-sensitive fluorophores
Redox sensitive fluorophores can be tracked with live cell microscopy or detected using fixed time-point techniques, such as flow-cytometry or microplate readers. These molecules are frequently targeted to mitochondria by using lipophilic cations, taking advantage of the negative potential across the mitochondrial inner membrane [146]. This means, however, that any readout cannot be reliably attributed to mitochondrial ROS generation in conditions of mitochondrial depolarisation (as is commonly the case in studies of neurodegeneration) without some form of correction [148, 153, 154]. As with all fluorescent dyes, and especially for those expected to localise to mitochondria, cells should be allowed to equilibrate the dye for at least 30 min prior to imaging. Removal of charge-sensitive fluorophores from the media may lead to re-equilibration across membranes and dilution of signal intensity. As the fluorescence of ROS-sensitive fluorophores increases over an experiment (due to accumulation of irreversibly oxidised dye), the initial measured fluorescence is dependent on loading time [164], and laser settings (intensity, gain) should be adjusted for low fluorescence to avoid subsequent saturation. The slope of the increase in fluorescence is calculated as a measure of the steady-state ROS level over the time measured [61, 153]. We here introduce, and discuss the issues associated with, the most commonly utilised redox-sensitive fluorescent probes—an extensive description of other probes is provided in [157].
The blue-fluorescent dihydroethidium (hydroethidine; HEt) is oxidised by O2− to the red fluorescent 2-hydroxyethidium, but can also be oxidised by other ROS to the red fluorescent ethidium and other non-fluorescent by-products [148, 161]. Conjugating HEt to the positively charged, lipophilic triphenylphosphonium cation (TPP+) allows HEt to localise to mitochondria (mito-HEt/MitoSOX Red (Invitrogen/ThermoFisher)). MitoSOX is excited at 543 nm and its emission detected at ~585 nm (although ~408 nm excitation may be somewhat more specific for 2-hydroxyethidium [165]). Unfortunately, a number of issues exist to confound HEt-based mitochondrial ROS measurements. As the fluorescent spectra of 2-hydroxyethidium and ethidium overlap, HEt cannot distinguish between different types of ROS without the aid of additional techniques, such as HPLC [161, 165]. Once oxidised, HEt binds to DNA and RNA, which increases its fluorescence. Positively charged HEt oxidation products can accumulate in the negatively charged mitochondria (dependent on Δψm), regardless of their source [161]. Following mitochondrial depolarisation, these products can also redistribute to the nucleus, significantly increasing fluorescence due to DNA binding and nucleic acid intercalation [148]. Together, this means that the localisation of the fluorescent signal does not inform about where the oxidation took place, even if the nucleus is not imaged. MitoSOX is oxidised by photo-toxicity, so low concentrations (0.1–0.2 μM in several types of cultured neurons) and minimal laser intensities are required [148]. Low concentrations also reduce the binding capacity of HEt to mitochondrial DNA, and the MitoSOX-induced impairment of mitochondrial metabolism [148, 166]. Finally, some drugs interact with HEt-derived compounds, such as high concentrations of the Complex III inhibitor, antimycin A, or the ROS scavenger MnTBAP [148, 161]. Guidelines on the utility, optimisation and imaging of MitoSOX in neurons have been provided recently [148].
Dihydrorhodamine 123 (excitation/emission 500/536 nm), whose emission spectra is also altered by irreversible non-specific oxidation, localises to mitochondria due to its positive charge [167] and is therefore strongly Δψm-dependent. Indeed, its oxidised product, rhodamine 123, is itself used as an indicator of Δψm. Derivatives of dichlorofluorescein (DCF) (excitation/emission 488/510 nm) only penetrate the outer mitochondrial membrane, and are associated with other issues, including a non-linear fluorescence response [160, 164]. MitoPY1 (Mitochondrial peroxy yellow), a hybrid fluorescein/rhodamine reporter, may selectively report mitochondria-derived H2O2 [168]. Similar to HEt-based fluorophores, however, these reporters are associated with significant limitations, such as lack of specificity, auto-oxidation and photo-oxidation, and pH sensitivity [61, 149, 160, 163, 164].
As H2O2 is fully membrane-permeable, release of H2O2 is sometimes used to infer mitochondrial ROS production, especially in isolated mitochondria—Amplex Red (Molecular probes) is a sensitive and stable probe that reacts with H2O2 (in the presence of HRP) to produce the highly fluorescent resorufin, which can be detected by colorimetric or fluorimetric readers (excitation/emission at 570/585 nm) [94]. However, some mitochondrial H2O2 will be consumed prior to its release from mitochondria, and the contribution to the signal from non-mitochondrial sources of ROS is significant and cannot be discounted if performing measurements in intact cells [147].
Redox-sensitive fluorescent proteins
Due to the myriad issues with redox-sensitive fluorophores, the use of mitochondria-targeted redox-sensitive FPs may be more appropriate, although they are prone to similar issues associated with the uncertainty over the origin of ROS, and its rapid turnover. In contrast to fluorophores, redox-sensitive FPs reversibly react with ROS and can therefore be used to monitor ROS production/consumption over longer time periods [157]. Within this set of probes (Table 7), reduction–oxidation-sensitive GFPs (roGFP) are ratiometric, dual-excitation reporters (excitation 395/470 nm, emission ~510 nm), whose fluorescence intensity is sensitive to the thiol redox potential [169]. In addition to their ratiometric measurements, roGFP probes are favoured over the redox-sensitive YFP reporters (rxYFP[170]) as they have higher redox sensitivity and lower pH sensitivity [95, 162]. roGFP has been targeted to neuronal mitochondria in vitro and in vivo (mito-roGFP/roGFPm) [171, 172]. A roGFP variant with Glutaredoxin-1 fused to roGFP (Grx1-roGFP2) reports the glutathione redox potential [173, 174], and roGFP2-Tsa1/2 are peroxiredoxin-based sensors of H2O2, although they remain to be tested in mammals [175]. The mitochondria-targeted HyPer (HyPer-M or mt-HyPer) is specifically sensitive to H2O2 [176], but comprises a strongly pH-sensitive cpYFP, limiting its utility [95, 98].
Redox-sensitive enzymatic assays
Spectrophotometric enzymatic assays have historically been the workhorse assays of traditional biochemists studying redox biology, but in contrast to fluorescence measurements, these assays only provide fixed time-point readouts. The concentration of the anti-oxidant glutathione serves as an indirect measure of ROS, and can be detected using commercial assays [177]. The activity of aconitase, a TCA cycle enzyme that catalyses the isomerisation of citrate to isocitrate, is reversibly inhibited by superoxide, peroxynitrite and H2O2, and decreased aconitase activity is used as an indicator of increased ROS [94, 159]. Aconitase activity spectrophotometric assays are commercially available. Aconitase can also be a source of ROS [94], however, and cytosolic aconitase (mRNA binding regulator of iron homoeostasis) contributes ~15–25% of total cellular aconitase activity [178].
Mitochondrial morphology
Mitochondria, far from being inert, form a dynamic network that constantly undergoes remodelling through fusion and fission events. Changes in mitochondrial morphology or number (e.g. due to fusion/fission, biogenesis or mitophagy) can impact mitochondrial parameters measured at the whole cell level [179], and should ideally be measured in conjunction with any investigation of mitochondrial bioenergetic function. For reviews on mitochondrial morphology and its links with mitochondrial bioenergetics and neurodegeneration, see [18, 20, 180].
Assessment of mitochondrial morphology begins with appropriate labelling. Cationic probes such as TMRM can be used, but these probes are sensitive to changes in Δψm or can dissipate after paraformaldehyde fixation. MitoTrackers insensitive to these issues are now available, and nonyl acridine orange can constitute a good alternative [60, 181]. Targeted fluorescent proteins ensure reliable labelling of mitochondria [182], albeit with the inconveniences associated with transfection, as mentioned earlier. Immunofluorescence techniques can be used to stain mitochondria in fixed cells, though careful selection of the target protein and antibody optimisation is required to ensure a homogenous and specific signal [183]. Finally, mitochondrial morphology can also be assessed via transmission electron microscopy [184].
Following the establishment of appropriate labelling and imaging techniques, the choice of measurement parameters is critical to adequately assess mitochondrial morphology. Many studies limit measurements to the size and number of mitochondria to demonstrate fission/fusion events [179, 184]. Thorough investigation of mitochondrial morphology, however, should include calculation of the form factor and aspect ratio, two geometrical indicators of the branching and elongation state of mitochondria calculated from measurements of mitochondrial axes, perimeters, and diameters [185].
Assessment of neuronal mitochondrial function in vivo
Although in vivo measurements theoretically enable the assessment of mitochondrial function in the multi-cellular environment of complete biological systems, experiments in living organisms, especially in the context of NDs, remain technically challenging. To analyse neuronal mitochondrial function in vivo, genetically encoded imaging tools can be integrated into the living organism—adenoviruses can be used to infect neurons, or mouse models can be generated that constitutively express a fluorescent reporter in specific cell types [174]. Mitochondria-targeted FPs successfully imaged in in vivo neurons include bioluminescent and FRET-based Ca2+ sensors [118, 186], and reporters of mitochondrial pH and redox state [171, 172, 174]. Regarding non-genetically encoded tools, the development of multi-photon imaging potentially enables the utilisation of any chemical dye in in vivo applications [60, 187]—TMRM, for instance, can be loaded directly into the living animal [174]. Live imaging of neurons (or other brain cells) in vivo nevertheless requires surgery to expose the cells/region of interest, followed by two-photon or confocal microscopy of the living (anaesthetised) animal. These issues can be mitigated through the use of simpler in vivo models, such as Drosophila melanogaster [186]. Targeted click chemistry, a novel approach involving the introduction of reporters into living animals and their subsequent detection in isolated tissues via mass spectrometry, has also been used to measure the mitochondrial membrane potential, although not in the brain [188]. Finally, in vivo mitochondrial metabolism in the brain can be assessed by tracking the transport and metabolism of hyperpolarised [2-13C] pyruvate or other substrates, via nuclear magnetic resonance [189].
Additional considerations
We address here, in a non-exhaustive list, some points worth considering when investigating mitochondrial function, with particular relevance to the methods discussed in this article.
-
It is vital to select indicators with affinities/dissociation constants and dynamic ranges appropriate for the desired measurement, and to optimise drug concentrations in each experimental setting. Generally, concentrations between 0.1 and 10 times the reporter’s Kd can be measured most reliably.
-
A useful resource for experiment design utilising fluorescent reporters is provided by ThermoFisher Scientific, which displays the single-photon excitation and emission spectra of a broad array of (commercially available) fluorescent indicators—https://www.thermofisher.com/ie/en/home/life-science/cell-analysis/labelling-chemistry/fluorescence-spectraviewer.html. It is important to ensure minimal spectral overlap if utilising multiple fluorophores simultaneously.
-
Fluorescent dyes should generally be allowed to equilibrate within cells for at least 30 min prior to imaging, and up to 60 min if required—the signal can be measured to ensure it has reached a steady state. Removal of potential-sensitive dye can lead to re-equilibration of the dye across the cell membranes, altering signal intensity. Appropriate baseline and light control experiments should be performed to verify equilibration, as well as to ensure signal stability and avoid artefacts (focal-drift, phototoxicity, etc.).
-
Saturation of fluorescent signal should be avoided and settings should be optimised at the beginning of experiments to allow for the anticipated range of fluorescent signal fluctuations. On this note, optimised microscope settings should remain constant for all similar experiments, to enable inter-experiment comparison.
-
Temperature can significantly alter neuronal physiology and assay performance. The affinity of the ATeam fluorescent reporter of ATP concentration, for instance, can vary five-fold over a temperature range of 10 °C [99]. Where possible, experiments should be carried out at the physiological 37 °C.
-
Bioenergetic parameters are exceptionally sensitive (particularly at steady-state), and experimental measurements may themselves alter intracellular conditions and physiology. For instance, the relatively large GFP molecule may impact mitochondrial transport, and many indicators also act as buffers of the species they are measuring, potentially disrupting physiological dynamics. For this and other reasons, it is good practice to verify results with complementary techniques.
-
Positive and negative controls and appropriate data normalisation are, as with all experiments, critical for correct interpretation of results.
-
Steady-state values (e.g. metabolite or ion concentrations, pH) indicate the balance between the generation/influx and the destruction/metabolism/efflux of the measured component. Direct measurements of steady-state values provide no information on these specific processes, and further experiments are required to elucidate the contributing mechanisms.
Conclusions
In this consensus article we have provided detailed guidelines for the thorough investigation of essential mitochondrial bioenergetic function in cellular models of neurodegenerative diseases, including specific protocols for the measurement of oxygen consumption rate in intact primary neurons, and single-neuron time-lapse fluorescence imaging of the mitochondrial membrane potential and mitochondrial NAD(P)H. These guidelines facilitate analysis of primary and secondary mitochondrial dysfunction in neurodegenerative diseases. Adherence to standardised protocols will enable experimental comparison between laboratories, consolidating this vast area of research and optimising translation of in vitro findings to in vivo studies.
References
Johri A, Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. J Pharmacol Exp Ther 2012;342:619–30.
Pathak D, Berthet A, Nakamura K. Energy failure: does it contribute to neurodegeneration? Ann Neurol 2013;74:506–16.
Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006;443:787–95.
Yin F, Boveris A, Cadenas E. Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid Redox Signal 2014;20:353–71.
Martinez TN, Greenamyre JT. Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxid Redox Signal 2012;16:920–34.
Brouillet E, Conde F, Beal MF, Hantraye P. Replicating Huntington’s disease phenotype in experimental animals. Prog Neurobiol 1999;59:427–68.
Polyzos AA, McMurray CT. The chicken or the egg: mitochondrial dysfunction as a cause or consequence of toxicity in Huntington’s disease. Mech Ageing Dev 2017;161:181–97.
Swerdlow RH, Burns JM, Khan SM. The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim Biophys Acta 2014;1842:1219–31.
Raefsky SM, Mattson MP. Adaptive responses of neuronal mitochondria to bioenergetic challenges: Roles in neuroplasticity and disease resistance. Free Radic Biol Med 2017;102:203–16.
Sulzer D. Multiple hit hypotheses for dopamine neuron loss in Parkinson’s disease. Trends Neurosci 2007;30:244–50.
Zhu X, Lee HG, Perry G, Smith MA. Alzheimer disease, the two-hit hypothesis: an update. Biochim Biophys Acta 2007;1772:494–502.
Connolly NM, Dussmann H, Anilkumar U, Huber HJ, Prehn JH. Single-cell imaging of bioenergetic responses to neuronal excitotoxicity and oxygen and glucose deprivation. J Neurosci 2014;34:10192–205.
Herrero-Mendez A, Almeida A, Fernandez E, Maestre C, Moncada S, Bolanos JP. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol 2009;11:747–52.
Kann O, Kovacs R. Mitochondria and neuronal activity. Am J Physiol Cell Physiol 2007;292:C641–57.
Mitchell P, Moyle J. Estimation of membrane potential and pH difference across the cristae membrane of rat liver mitochondria. Eur J Biochem 1969;7:471–84.
Nicholls DG, Ferguson S. Bioenergetics. Academic Press: London, 2013.
Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Brand MD. Mitochondrial proton and electron leaks. Essays Biochem 2010;47:53–67.
Gao J, Wang L, Liu J, Xie F, Su B, Wang X. Abnormalities of mitochondrial dynamics in neurodegenerative diseases. Antioxidants. 2017;6:25.
Schon EA, Przedborski S. Mitochondria: the next (neurode)generation. Neuron 2011;70:1033–53.
Aouacheria A, Baghdiguian S, Lamb HM, Huska JD, Pineda FJ, Hardwick JM. Connecting mitochondrial dynamics and life-or-death events via Bcl-2 family proteins. Neurochem Int. 2017;109:141–61.
Hilgenberg L-G, Smith MA. Preparation of dissociated mouse cortical neuron cultures. J Vis Exp. 2007;10:562.
Sciarretta C, Minichiello L. The preparation of primary cortical neuron cultures and a practical application using immunofluorescent cytochemistry. Methods Mol Biol 2010;633:221–31.
Fairbanks SL, Vest R, Verma S, Traystman RJ, Herson PS. Sex stratified neuronal cultures to study ischemic cell death pathways. J Vis Exp 2013;82:e50758.
Kleman AM, Yuan JY, Aja S, Ronnett GV, Landree LE. Physiological glucose is critical for optimized neuronal viability and AMPK responsiveness in vitro. J Neurosci Methods 2008;167:292–301.
Zhu J, Aja S, Kim EK, Park MJ, Ramamurthy S, Jia J, et al. Physiological oxygen level is critical for modeling neuronal metabolism in vitro. J Neurosci Res 2012;90:422–34.
Surin AM, Khiroug S, Gorbacheva LR, Khodorov BI, Pinelis VG, Khiroug L. Comparative analysis of cytosolic and mitochondrial ATP synthesis in embryonic and postnatal hippocampal neuronal cultures. Front Mol Neurosci 2012;5:102.
Biffi E, Regalia G, Menegon A, Ferrigno G, Pedrocchi A. The influence of neuronal density and maturation on network activity of hippocampal cell cultures: a methodological study. PLoS One 2013;8:e83899.
Constantinescu R, Constantinescu AT, Reichmann H, Janetzky B. Neuronal differentiation and long-term culture of the human neuroblastoma line SH-SY5Y. J Neural Transm Suppl. 2007;72:17–28.
Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA 1976;73:2424–8.
Tremblay RG, Sikorska M, Sandhu JK, Lanthier P, Ribecco-Lutkiewicz M, Bani-Yaghoub M. Differentiation of mouse Neuro 2A cells into dopamine neurons. J Neurosci Methods 2010;186:60–7.
LePage KT, Dickey RW, Gerwick WH, Jester EL, Murray TF. On the use of neuro-2a neuroblastoma cells versus intact neurons in primary culture for neurotoxicity studies. Crit Rev Neurobiol 2005;17:27–50.
Hung SS, Khan S, Lo CY, Hewitt AW, Wong RC. Drug discovery using induced pluripotent stem cell models of neurodegenerative and ocular diseases. Pharmacol Ther 2017;177:32–43.
Ryan SD, Dolatabadi N, Chan SF, Zhang X, Akhtar MW, Parker J, et al. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1alpha transcription. Cell 2013;155:1351–64.
Choi SH, Kim YH, Quinti L, Tanzi RE, Kim DY. 3D culture models of Alzheimer’s disease: a road map to a “cure-in-a-dish”. Mol Neurodegener 2016;11:75.
Schlachetzki JC, Saliba SW, Oliveira AC. Studying neurodegenerative diseases in culture models. Rev Bras Psiquiatr 2013;35:S92–100.
Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J 2011;435:297–312.
Dranka BP, Benavides GA, Diers AR, Giordano S, Zelickson BR, Reily C, et al. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic Biol Med 2011;51:1621–35.
Rhein V, Song X, Wiesner A, Ittner LM, Baysang G, Meier F, et al. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc Natl Acad Sci USA 2009;106:20057–62.
Chakraborty S, Nian FS, Tsai JW, Karmenyan A, Chiou A, Quantification of the metabolic state in cell-model of Parkinson’s disease by fluorescence lifetime imaging microscopy . Sci Rep. 2016;6:19145
Clerc P, Polster BM. Investigation of mitochondrial dysfunction by sequential microplate-based respiration measurements from intact and permeabilized neurons. PLoS One 2012;7:e34465.
Salabei JK, Gibb AA, Hill BG. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat Protoc 2014;9:421–38.
Divakaruni AS, Rogers GW, Murphy AN. Measuring mitochondrial function in permeabilized cells using the seahorse XF analyzer or a Clark-type oxygen electrode. Curr Protoc Toxicol 2014;60:25 2 1–16.
Barrientos A, Fontanesi F,Diaz F, Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using polarography and spectrophotometric enzyme assays. Current Protocols in Human Genetics 2009;63:19.3:19.3.1–19.3.14, Chapter 19: Unit 19 3.
Jekabsons MB, Nicholls DG. In situ respiration and bioenergetic status of mitochondria in primary cerebellar granule neuronal cultures exposed continuously to glutamate. J Biol Chem 2004;279:32989–3000.
Zhang L, Trushina E. Respirometry in neurons. Techniques to investigate mitochondrial funciton in neurons. Strack, Stefan, Usachev, Yuriy M (eds.), Vol. 123. Springer; 2017. p. 95–113.
Chinopoulos C, Kiss G, Kawamata H, Starkov AA. Measurement of ADP-ATP exchange in relation to mitochondrial transmembrane potential and oxygen consumption. Methods Enzymol 2014;542:333–48.
Horan MP, Pichaud N, Ballard JW. Review: quantifying mitochondrial dysfunction in complex diseases of aging. J Gerontol A 2012;67:1022–35.
Will Y, Hynes J, Ogurtsov VI, Papkovsky DB. Analysis of mitochondrial function using phosphorescent oxygen-sensitive probes. Nat Protoc 2006;1:2563–72.
Dmitriev RI, Borisov SM, Kondrashina AV, Pakan JM, Anilkumar U, Prehn JH, et al. Imaging oxygen in neural cell and tissue models by means of anionic cell-permeable phosphorescent nanoparticles. Cell Mol Life Sci 2015;72:367–81.
Kondrashina AV, Ogurtsov VI, Papkovsky DB. Comparison of the three optical platforms for measurement of cellular respiration. Anal Biochem 2015;468:1–3.
Divakaruni AS, Paradyse A, Ferrick DA, Murphy AN, Jastroch M. Analysis and interpretation of microplate-based oxygen consumption and pH data. Methods Enzymol 2014;547:309–54.
Ruas JS, Siqueira-Santos ES, Amigo I, Rodrigues-Silva E, Kowaltowski AJ, Castilho RF. Underestimation of the maximal capacity of the mitochondrial electron transport system in oligomycin-treated cells. PLoS One 2016;11:e0150967.
Llorente-Folch I, Rueda CB, Amigo I, del Arco A, Saheki T, Pardo B, et al. Calcium-regulation of mitochondrial respiration maintains ATP homeostasis and requires ARALAR/AGC1-malate aspartate shuttle in intact cortical neurons. J Neurosci 2013;33:13957–71. 71a
Divakaruni AS, Wallace M, Buren C, Martyniuk K, Andreyev AY, Li E, et al. Inhibition of the mitochondrial pyruvate carrier protects from excitotoxic neuronal death. J Cell Biol 2017;216:1091–105.
Chacko BK, Kramer PA, Ravi S, Benavides GA, Mitchell T, Dranka BP, et al. The Bioenergetic Health Index: a new concept in mitochondrial translational research. Clin Sci 2014;127:367–73.
Mookerjee SA, Gerencser AA, Nicholls DG, Brand MD. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J Biol Chem 2017;292:7189–207.
Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc 2012;7:1235–46.
Mookerjee SA, Goncalves RL, Gerencser AA, Nicholls DG, Brand MD. The contributions of respiration and glycolysis to extracellular acid production. Biochim Biophys Acta 2015;1847:171–81.
Mookerjee SA, Nicholls DG, Brand MD. Determining maximum glycolytic capacity using extracellular flux measurements. PLoS One 2016;11:e0152016.
Perry SW, Norman JP, Barbieri J, Brown EB, Gelbard HA. Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. Biotechniques 2011;50:98–115.
Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev 2000;80:315–60.
Nicholls DG. Simultaneous monitoring of ionophore- and inhibitor-mediated plasma and mitochondrial membrane potential changes in cultured neurons. J Biol Chem 2006;281:14864–74.
Ward MW, Huber HJ, Weisova P, Dussmann H, Nicholls DG, Prehn JH. Mitochondrial and plasma membrane potential of cultured cerebellar neurons during glutamate-induced necrosis, apoptosis, and tolerance. J Neurosci 2007;27:8238–49.
Ward MW, Rego AC, Frenguelli BG, Nicholls DG. Mitochondrial membrane potential and glutamate excitotoxicity in cultured cerebellar granule cells. J Neurosci 2000;20:7208–19.
Irwin WA, Bergamin N, Sabatelli P, Reggiani C, Megighian A, Merlini L, et al. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat Genet 2003;35:367–71.
Connolly NM, Prehn JH. The metabolic response to excitotoxicity - lessons from single-cell imaging. J Bioenerg Biomembr 2015;47:75–88.
Bonora M, Morganti C, Morciano G, Giorgi C, Wieckowski MR, Pinton P. Comprehensive analysis of mitochondrial permeability transition pore activity in living cells using fluorescence-imaging-based techniques. Nat Protoc 2016;11:1067–80.
Bernardi P, Petronilli V, Di Lisa F, Forte M. A mitochondrial perspective on cell death. Trends Biochem Sci 2001;26:112–7.
Scaduto RC Jr, Grotyohann LW. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys J 1999;76:469–77.
Hayakawa Y, Nemoto T, Iino M, Kasai H. Rapid Ca2+-dependent increase in oxygen consumption by mitochondria in single mammalian central neurons. Cell Calcium 2005;37:359–70.
Gerencser AA, Chinopoulos C, Birket MJ, Jastroch M, Vitelli C, Nicholls DG, et al. Quantitative measurement of mitochondrial membrane potential in cultured cells: calcium-induced de- and hyperpolarization of neuronal mitochondria. J Physiol 2012;590:2845–71.
Joshi DC, Bakowska JC. Determination of mitochondrial membrane potential and reactive oxygen species in live rat cortical neurons. J Vis Exp. 2011;51:2704.
Shuttleworth CW. Use of NAD(P)H and flavoprotein autofluorescence transients to probe neuron and astrocyte responses to synaptic activation. Neurochem Int 2010;56:379–86.
Kahraman S, Siegel A, Polster BM, Fiskum G. Permeability transition pore-dependent and PARP-mediated depletion of neuronal pyridine nucleotides during anoxia and glucose deprivation. J Bioenerg Biomembr 2015;47:53–61.
Fang EF, Scheibye-Knudsen M, Chua KF, Mattson MP, Croteau DL, Bohr VA. Nuclear DNA damage signalling to mitochondria in ageing. Nat Rev Mol Cell Biol 2016;17:308–21.
Lewis CA, Parker SJ, Fiske BP, McCloskey D, Gui DY, Green CR, et al. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol Cell 2014;55:253–63.
Blacker TS, Duchen MR. Investigating mitochondrial redox state using NADH and NADPH autofluorescence. Free Radic Biol Med 2016;100:53–65.
Chance B. Kinetics of enzyme reactions within single cells. Ann N Y Acad Sci 1962;97:431–48.
Ogikubo S, Nakabayashi T, Adachi T, Islam MS, Yoshizawa T, Kinjo M, et al. Intracellular pH sensing using autofluorescence lifetime microscopy. J Phys Chem B 2011;115:10385–90.
Pardo B, Contreras L, Serrano A, Ramos M, Kobayashi K, Iijima M, et al. Essential role of aralar in the transduction of small Ca2+ signals to neuronal mitochondria. J Biol Chem 2006;281:1039–47.
Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, Webb WW. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science 2004;305:99–103.
Huang S, Heikal AA, Webb WW. Two-photon fluorescence spectroscopy and microscopy of NAD(P)H and flavoprotein. Biophys J 2002;82:2811–25.
Blacker TS, Berecz T, Duchen MR, Szabadkai G. Assessment of cellular redox state using NAD(P)H fluorescence intensity and lifetime. Bio Protoc. 2017;7:e2105.
San Martin A, Sotelo-Hitschfeld T, Lerchundi R, Fernandez-Moncada I, Ceballo S, Valdebenito R, et al. Single-cell imaging tools for brain energy metabolism: a review. Neurophotonics 2014;1:011004.
Bilan DS, Matlashov ME, Gorokhovatsky AY, Schultz C, Enikolopov G, Belousov VV. Genetically encoded fluorescent indicator for imaging NAD(+)/NADH ratio changes in different cellular compartments. Biochim Biophys Acta 2014;1840:951–7.
Bilan DS, Belousov VV. Genetically encoded probes for NAD+/NADH monitoring. Free Radic Biol Med 2016;100:32–42.
Zhao Y, Jin J, Hu Q, Zhou HM, Yi J, Yu Z, et al. Genetically encoded fluorescent sensors for intracellular NADH detection. Cell Metab 2011;14:555–66.
Zhao Y, Hu Q, Cheng F, Su N, Wang A, Zou Y, et al. SoNar, a highly responsive NAD+/NADH sensor, allows high-throughput metabolic screening of anti-tumor agents. Cell Metab 2015;21:777–89.
Hung YP, Albeck JG, Tantama M, Yellen G. Imaging cytosolic NADH-NAD(+) redox state with a genetically encoded fluorescent biosensor. Cell Metab 2011;14:545–54.
Cambronne XA, Stewart ML, Kim D, Jones-Brunette AM, Morgan RK, Farrens DL, et al. Biosensor reveals multiple sources for mitochondrial NAD. Science 2016;352:1474–7.
Tao R, Zhao Y, Chu H, Wang A, Zhu J, Chen X, et al. Genetically encoded fluorescent sensors reveal dynamic regulation of NADPH metabolism. Nat Methods 2017;14:720–8.
Ross JM. Visualization of mitochondrial respiratory function using cytochrome c oxidase/succinate dehydrogenase (COX/SDH) double-labeling histochemistry. J Vis Exp. 2011;57:3266.
Jung C, Higgins CM, Xu Z. Measuring the quantity and activity of mitochondrial electron transport chain complexes in tissues of central nervous system using blue native polyacrylamide gel electrophoresis. Anal Biochem 2000;286:214–23.
Sipos I, Tretter L, Adam-Vizi V. Quantitative relationship between inhibition of respiratory complexes and formation of reactive oxygen species in isolated nerve terminals. J Neurochem 2003;84:112–8.
Tantama M, Hung YP, Yellen G. Optogenetic reporters: fluorescent protein-based genetically encoded indicators of signaling and metabolism in the brain. Prog Brain Res 2012;196:235–63.
Harpur AG, Wouters FS, Bastiaens PI. Imaging FRET between spectrally similar GFP molecules in single cells. Nat Biotechnol 2001;19:167–9.
Karra D, Dahm R. Transfection techniques for neuronal cells. J Neurosci 2010;30:6171–7.
De Michele R, Carimi F, Frommer WB. Mitochondrial biosensors. Int J Biochem Cell Biol 2014;48:39–44.
Imamura H, Nhat KP, Togawa H, Saito K, Iino R, Kato-Yamada Y, et al. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc Natl Acad Sci USA 2009;106:15651–6.
Barsukova AG, Bourdette D, Forte M. Mitochondrial calcium and its regulation in neurodegeneration induced by oxidative stress. Eur J Neurosci 2011;34:437–47.
Nakano M, Imamura H, Nagai T, Noji H. Ca(2)(+) regulation of mitochondrial ATP synthesis visualized at the single cell level. ACS Chem Biol 2011;6:709–15.
Rueda CB, Traba J, Amigo I, Llorente-Folch I, Gonzalez-Sanchez P, Pardo B, et al. Mitochondrial ATP-Mg/Pi carrier SCaMC-3/Slc25a23 counteracts PARP-1-dependent fall in mitochondrial ATP caused by excitotoxic insults in neurons. J Neurosci 2015;35:3566–81.
Tantama M, Martinez-Francois JR, Mongeon R, Yellen G. Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat Commun 2013;4:2550.
Berg J, Hung YP, Yellen G. A genetically encoded fluorescent reporter of ATP:ADP ratio. Nat Methods 2009;6:161–6.
Katsura K, Rodriguez de Turco EB, Folbergrova J, Bazan NG, Siesjo BK. Coupling among energy failure, loss of ion homeostasis, and phospholipase A2 and C activation during ischemia. J Neurochem 1993;61:1677–84.
Yaginuma H, Kawai S, Tabata KV, Tomiyama K, Kakizuka A, Komatsuzaki T, et al. Diversity in ATP concentrations in a single bacterial cell population revealed by quantitative single-cell imaging. Sci Rep 2014;4:6522.
Zadran S, Sanchez D, Zadran H, Amighi A, Otiniano E, Wong K. Enhanced-acceptor fluorescence-based single cell ATP biosensor monitors ATP in heterogeneous cancer populations in real time. Biotechnol Lett 2013;35:175–80.
Yoshida T, Kakizuka A, Imamura H. BTeam, a Novel BRET-based biosensor for the accurate quantification of ATP concentration within living cells. Sci Rep 2016;6:39618.
Morciano G, Sarti AC, Marchi S, Missiroli S, Falzoni S, Raffaghello L, et al. Use of luciferase probes to measure ATP in living cells and animals. Nat Protoc 2017;12:1542–62.
Rajendran M, Dane E, Conley J, Tantama M. Imaging adenosine triphosphate (ATP). Biol Bull 2016;231:73–84.
De Stefani D, Rizzuto R, Pozzan T. Enjoy the trip: calcium in mitochondria back and forth. Annu Rev Biochem 2016;85:161–92.
Qiu J, Tan YW, Hagenston AM, Martel MA, Kneisel N, Skehel PA, et al. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat Comms 2013;4:2034.
Llorente-Folch I, Rueda CB, Pardo B, Szabadkai G, Duchen MR, Satrustegui J. The regulation of neuronal mitochondrial metabolism by calcium. J Physiol 2015;593:3447–62.
Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci USA 2010;107:436–41.
Pivovarova NB, Andrews SB. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J 2010;277:3622–36.
Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci 2008;31:454–63.
Grienberger C, Konnerth A. Imaging calcium in neurons. Neuron 2012;73:862–85.
Pozzan T, Rudolf R. Measurements of mitochondrial calcium in vivo. Biochim Biophys Acta 2009;1787:1317–23.
Rysted JE, Lin Z, Usachev YM. Techniques for simultaneous mitochondrial and cytosolic Ca2+ imaging in neurons. Techniques to investigate mitochondrial function in neurons. Stefan S, Yuriy M. Usachev (eds). Springer: New York; Vol. 123. Springer protocols. 2017. p. 151–78.
Nagai T, Yamada S, Tominaga T, Ichikawa M, Miyawaki A. Expanded dynamic range of fluorescent indicators for Ca(2+) by circularly permuted yellow fluorescent proteins. Proc Natl Acad Sci USA 2004;101:10554–9.
Miyawaki A, Griesbeck O, Heim R, Tsien RY. Dynamic and quantitative Ca2+measurements using improved cameleons. Proc Natl Acad Sci USA 1999;96:2135–40.
Palmer AE, Giacomello M, Kortemme T, Hires SA, Lev-Ram V, Baker D, et al. Ca2+indicators based on computationally redesigned calmodulin-peptide pairs. Chem Biol 2006;13:521–30.
Rizzuto R, Simpson AW, Brini M, Pozzan T. Rapid changes of mitochondrial Ca2+revealed by specifically targeted recombinant aequorin. Nature 1992;358:325–7.
Bonora M, Giorgi C, Bononi A, Marchi S, Patergnani S, Rimessi A, et al. Subcellular calcium measurements in mammalian cells using jellyfish photoprotein aequorin-based probes. Nat Protoc 2013;8:2105–18.
Nagai T, Sawano A, Park ES, Miyawaki A. Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proc Natl Acad Sci USA 2001;98:3197–202.
Nakai J, Ohkura M, Imoto K. A high signal-to-noise Ca(2+) probe composed of a single green fluorescent protein. Nat Biotechnol 2001;19:137–41.
Chen M, Wang Y, Hou T, Zhang H, Qu A, Wang X. Differential mitochondrial calcium responses in different cell types detected with a mitochondrial calcium fluorescent indicator, mito-GCaMP2. Acta Biochim Biophys Sin 2011;43:822–30.
Akerboom J, Carreras Calderon N, Tian L, Wabnig S, Prigge M, Tolo J, et al. Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front Mol Neurosci 2013;6:2.
Zhao Y, Araki S, Wu J, Teramoto T, Chang YF, Nakano M, et al. An expanded palette of genetically encoded Ca(2)(+) indicators. Science 2011;333:1888–91.
Wu J, Prole DL, Shen Y, Lin Z, Gnanasekaran A, Liu Y, et al. Red fluorescent genetically encoded Ca2+ indicators for use in mitochondria and endoplasmic reticulum. Biochem J 2014;464:13–22.
Suzuki J, Kanemaru K, Ishii K, Ohkura M, Okubo Y, Iino M. Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat Commun 2014;5:4153.
Suzuki K, Kimura T, Shinoda H, Bai G, Daniels MJ, Arai Y, et al. Five colour variants of bright luminescent protein for real-time multicolour bioimaging. Nat Commun 2016;7:13718.
Pendin D, Greotti E, Filadi R, Pozzan T. Spying on organelle Ca(2)(+) in living cells: the mitochondrial point of view. J Endocrinol Invest 2015;38:39–45.
Rudolf R, Mongillo M, Rizzuto R, Pozzan T. Looking forward to seeing calcium. Nat Rev Mol Cell Biol 2003;4:579–86.
Santo-Domingo J, Demaurex N. Perspectives on: SGP symposium on mitochondrial physiology and medicine: the renaissance of mitochondrial pH. J Gen Physiol 2012;139:415–23.
Bencina M. Illumination of the spatial order of intracellular pH by genetically encoded pH-sensitive sensors. Sensors 2013;13:16736–58.
Porcelli AM, Ghelli A, Zanna C, Pinton P, Rizzuto R, Rugolo M. pH difference across the outer mitochondrial membrane measured with a green fluorescent protein mutant. Biochem Biophys Res Commun 2005;326:799–804.
Bolshakov AP, Mikhailova MM, Szabadkai G, Pinelis VG, Brustovetsky N, Rizzuto R, et al. Measurements of mitochondrial pH in cultured cortical neurons clarify contribution of mitochondrial pore to the mechanism of glutamate-induced delayed Ca2+ deregulation. Cell Calcium 2008;43:602–14.
Miesenbock G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 1998;394:192–5.
Vijayvergiya C, De Angelis D, Walther M, Kuhn H, Duvoisin RM, Smith DH, et al. High-level expression of rabbit 15-lipoxygenase induces collapse of the mitochondrial pH gradient in cell culture. Biochemistry 2004;43:15296–302.
Poburko D, Santo-Domingo J, Demaurex N. Dynamic regulation of the mitochondrial proton gradient during cytosolic calcium elevations. J Biol Chem 2011;286:11672–84.
Breckwoldt MO, Armoundas AA, Aon MA, Bendszus M, O’Rourke B, Schwarzlander M, et al. Mitochondrial redox and pH signaling occurs in axonal and synaptic organelle clusters. Sci Rep 2016;6:23251.
Abad MF, Di Benedetto G, Magalhaes PJ, Filippin L, Pozzan T. Mitochondrial pH monitored by a new engineered green fluorescent protein mutant. J Biol Chem 2004;279:11521–9.
Tantama M, Hung YP, Yellen G. Imaging intracellular pH in live cells with a genetically encoded red fluorescent protein sensor. J Am Chem Soc 2011;133:10034–7.
Li Y, Tsien RW. pHTomato, a red, genetically encoded indicator that enables multiplex interrogation of synaptic activity. Nat Neurosci 2012;15:1047–53.
Dickinson BC, Srikun D, Chang CJ. Mitochondrial-targeted fluorescent probes for reactive oxygen species. Curr Opin Chem Biol 2010;14:50–6.
Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 2009;417:1–13.
Polster BM, Nicholls DG, Ge SX, Roelofs BA. Use of potentiometric fluorophores in the measurement of mitochondrial reactive oxygen species. Methods Enzymol 2014;547:225–50.
Murphy MP, Holmgren A, Larsson NG, Halliwell B, Chang CJ, Kalyanaraman B, et al. Unraveling the biological roles of reactive oxygen species. Cell Metab 2011;13:361–6.
Lopez-Fabuel I, Le Douce J, Logan A, James AM, Bonvento G, Murphy MP, et al. Complex I assembly into supercomplexes determines differential mitochondrial ROS production in neurons and astrocytes. Proc Natl Acad Sci USA 2016;113:13063–8.
Bindokas VP, Jordan J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci 1996;16:1324–36.
Garcia O, Almeida A, Massieu L, Bolanos JP. Increased mitochondrial respiration maintains the mitochondrial membrane potential and promotes survival of cerebellar neurons in an endogenous model of glutamate receptor activation. J Neurochem 2005;92:183–90.
Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci 2007;27:1129–38.
Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, et al. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci 2009;12:857–63.
Adam-Vizi V, Chinopoulos C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol Sci 2006;27:639–45.
Brand MD. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med 2016;100:14–31.
Woolley JF, Stanicka J, Cotter TG. Recent advances in reactive oxygen species measurement in biological systems. Trends Biochem Sci 2013;38:556–65.
Uusitalo LM, Hempel N. Recent advances in intracellular and in vivo ROS sensing: focus on nanoparticle and nanotube applications. Int J Mol Sci 2012;13:10660–79.
Vincent AM, McLean LL, Backus C, Feldman EL. Short-term hyperglycemia produces oxidative damage and apoptosis in neurons. FASEB J 2005;19:638–40.
Chen X, Zhong Z, Xu Z, Chen L, Wang Y. 2’,7’-Dichlorodihydrofluorescein as a fluorescent probe for reactive oxygen species measurement: forty years of application and controversy. Free Radic Res 2010;44:587–604.
Zielonka J, Kalyanaraman B. Hydroethidine- and MitoSOX-derived red fluorescence is not a reliable indicator of intracellular superoxide formation: another inconvenient truth. Free Radic Biol Med 2010;48:983–1001.
Booth DM, Joseph SK, Hajnoczky G. Subcellular ROS imaging methods: relevance for the study of calcium signaling. Cell Calcium 2016;60:65–73.
In: Can K, Kügler S, Müller M, Strack S, Usachev YM, editors. Techniques to investigate mitochondrial function in neurons. New York, NY: Springer; 2017. p. 179–97. .
Hempel SL, Buettner GR, O’Malley YQ, Wessels DA, Flaherty DM. Dihydrofluorescein diacetate is superior for detecting intracellular oxidants: comparison with 2’,7’-dichlorodihydrofluorescein diacetate, 5(and 6)-carboxy-2’,7’-dichlorodihydrofluorescein diacetate, and dihydrorhodamine 123. Free Radic Biol Med 1999;27:146–59.
Robinson KM, Janes MS, Beckman JS. The selective detection of mitochondrial superoxide by live cell imaging. Nat Protoc 2008;3:941–7.
Roelofs BA, Ge SX, Studlack PE, Polster BM. Low micromolar concentrations of the superoxide probe MitoSOX uncouple neural mitochondria and inhibit complex IV. Free Radic Biol Med 2015;86:250–8.
Dugan LL, Sensi SL, Canzoniero LM, Handran SD, Rothman SM, Lin TS, et al. Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-D-aspartate. J Neurosci 1995;15:6377–88.
Dickinson BC, Lin VS, Chang CJ. Preparation and use of MitoPY1 for imaging hydrogen peroxide in mitochondria of live cells. Nat Protoc 2013;8:1249–59.
Hanson GT, Aggeler R, Oglesbee D, Cannon M, Capaldi RA, Tsien RY, et al. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J Biol Chem 2004;279:13044–53.
Ostergaard H, Henriksen A, Hansen FG, Winther JR. Shedding light on disulfide bond formation: engineering a redox switch in green fluorescent protein. EMBO J 2001;20:5853–62.
Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, et al. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010;468:696–700.
Wagener KC, Kolbrink B, Dietrich K, Kizina KM, Terwitte LS, Kempkes B, et al. Redox indicator mice stably expressing genetically encoded neuronal roGFP: versatile tools to decipher subcellular redox dynamics in neuropathophysiology. Antioxid Redox Signal 2016;25:41–58.
Gutscher M, Pauleau AL, Marty L, Brach T, Wabnitz GH, Samstag Y, et al. Real-time imaging of the intracellular glutathione redox potential. Nat Methods 2008;5:553–9.
Breckwoldt MO, Pfister FM, Bradley PM, Marinkovic P, Williams PR, Brill MS, et al. Multiparametric optical analysis of mitochondrial redox signals during neuronal physiology and pathology in vivo. Nat Med 2014;20:555–60.
Morgan B, Van Laer K, Owusu TN, Ezerina D, Pastor-Flores D, Amponsah PS, et al. Real-time monitoring of basal H2O2 levels with peroxiredoxin-based probes. Nat Chem Biol 2016;12:437–43.
Belousov VV, Fradkov AF, Lukyanov KA, Staroverov DB, Shakhbazov KS, Terskikh AV, et al. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat Methods 2006;3:281–6.
Wilkins HM, Kirchhof D, Manning E, Joseph JW, Linseman DA. Mitochondrial glutathione transport is a key determinant of neuronal susceptibility to oxidative and nitrosative stress. J Biol Chem 2013;288:5091–101.
Liang LP, Ho YS, Patel M. Mitochondrial superoxide production in kainate-induced hippocampal damage. Neuroscience 2000;101:563–70.
Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Graber S, et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J 2006;25:3900–11.
Galloway CA, Lee H, Yoon Y. Mitochondrial morphology-emerging role in bioenergetics. Free Radic Biol Med 2012;53:2218–28.
Chazotte B. Labeling mitochondria with MitoTracker dyes. Cold Spring Harb Protoc 2011;2011:990–2.
Rizzuto R, Brini M, De Giorgi F, Rossi R, Heim R, Tsien RY, et al. Double labelling of subcellular structures with organelle-targeted GFP mutants in vivo. Curr Biol 1996;6:183–8.
Yamano K, Fogel AI, Wang C, van der Bliek AM, Youle RJ. Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. Elife 2014;3:e01612.
Wang X, Yan MH, Fujioka H, Liu J, Wilson-Delfosse A, Chen SG, et al. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum Mol Genet 2012;21:1931–44.
Koopman WJ, Verkaart S, Visch HJ, van der Westhuizen FH, Murphy MP, van den Heuvel LW, et al. Inhibition of complex I of the electron transport chain causes O2-. -mediated mitochondrial outgrowth. Am J Physiol Cell Physiol 2005;288:C1440–50.
Lark AR, Kitamoto T, Martin JR. In vivo functional brain imaging approach based on bioluminescent calcium indicator GFP-aequorin. J Vis Exp. 2016;107:e53705.
Tada M, Takeuchi A, Hashizume M, Kitamura K, Kano M. A highly sensitive fluorescent indicator dye for calcium imaging of neural activity in vitro and in vivo. Eur J Neurosci 2014;39:1720–8.
Logan A, Pell VR, Shaffer KJ, Evans C, Stanley NJ, Robb EL, et al. Assessing the mitochondrial membrane potential in cells and in vivo using targeted click chemistry and mass spectrometry. Cell Metab 2016;23:379–85.
Park JM, Josan S, Grafendorfer T, Yen YF, Hurd RE, Spielman DM, et al. Measuring mitochondrial metabolism in rat brain in vivo using MR Spectroscopy of hyperpolarized [2-(1)(3)C]pyruvate. NMR Biomed 2013;26:1197–203.
Gotz J, Ittner LM. Animal models of Alzheimer’s disease and frontotemporal dementia. Nat Rev Neurosci 2008;9:532–44.
Harvey BK, Richie CT, Hoffer BJ, Airavaara M. Transgenic animal models of neurodegeneration based on human genetic studies. J Neural Transm 2011;118:27–45.
Duan W, Guo Z, Mattson MP. Participation of par-4 in the degeneration of striatal neurons induced by metabolic compromise with 3-nitropropionic acid. Exp Neurol 2000;165:1–11.
Bizat N, Galas MC, Jacquard C, Boyer F, Hermel JM, Schiffmann SN, et al. Neuroprotective effect of zVAD against the neurotoxin 3-nitropropionic acid involves inhibition of calpain. Neuropharmacology 2005;49:695–702.
Chinopoulos C, Gerencser AA, Mandi M, Mathe K, Torocsik B, Doczi J, et al. Forward operation of adenine nucleotide translocase during F0F1-ATPase reversal: critical role of matrix substrate-level phosphorylation. FASEB J 2010;24:2405–16.
Duregotti E, Negro S, Scorzeto M, Zornetta I, Dickinson BC, Chang CJ, et al. Mitochondrial alarmins released by degenerating motor axon terminals activate perisynaptic Schwann cells. Proc Natl Acad Sci USA 2015;112:E497–505.
Acknowledgements
We acknowledge the support of the CeBioND EU Joint Programme for Neurodegenerative Disease Research (JPND; www.jpnd.eu). The programme is supported through the following national funding organisations: Canada, CIHR; Germany, BMBF; Ireland: Science Foundation Ireland (14/JPND/ B3077); Italy: MIUR; Sweden: VR.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Edited by G. Melino
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Connolly, N.M.C., Theurey, P., Adam-Vizi, V. et al. Guidelines on experimental methods to assess mitochondrial dysfunction in cellular models of neurodegenerative diseases. Cell Death Differ 25, 542–572 (2018). https://doi.org/10.1038/s41418-017-0020-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-017-0020-4
This article is cited by
-
A partial Drp1 knockout improves autophagy flux independent of mitochondrial function
Molecular Neurodegeneration (2024)
-
N1-methylation of adenosine (m1A) in ND5 mRNA leads to complex I dysfunction in Alzheimer’s disease
Molecular Psychiatry (2024)
-
The landscape of programmed cell death-related lncRNAs in Alzheimer’s disease and Parkinson’s disease
Apoptosis (2024)
-
BAP1 promotes osteoclast function by metabolic reprogramming
Nature Communications (2023)
-
Theaflavin mitigates acute gouty peritonitis and septic organ injury in mice by suppressing NLRP3 inflammasome assembly
Acta Pharmacologica Sinica (2023)
