
Abstract
Background
Screening programmes utilising blood-based multi-cancer early detection (MCED) tests, which can detect a shared cancer signal from any site in the body with a single, low false-positive rate, could reduce cancer burden through early diagnosis.
Methods
A natural history (‘interception’) model of cancer was previously used to characterise potential benefits of MCED screening (based on published performance of an MCED test). We built upon this using a two-population survival model to account for an increased risk of death from cfDNA-detectable cancers relative to cfDNA-non-detectable cancers. We developed another model allowing some cancers to metastasise directly from stage I, bypassing intermediate tumour stages. We used incidence and survival-by-stage data from the National Cancer Registration and Analysis Service in England to estimate longer-term benefits to a cohort screened between ages 50–79 years.
Results
Estimated late-stage and mortality reductions were robust to a range of assumptions. With the least favourable dwell (sojourn) time and cfDNA status hazard ratio assumptions, we estimated, among 100,000 screened individuals, 67 (17%) fewer cancer deaths per year corresponding to 2029 fewer deaths in those screened between ages 50–79 years.
Conclusion
Realising the potential benefits of MCED tests could substantially reduce late-stage cancer diagnoses and mortality.
Similar content being viewed by others

Introduction
Cancer caused 10 million deaths worldwide in 2020 [1]. It is the leading cause of death in most high income countries [2] including England, where it resulted in ~140,000 deaths in 2020 in a population of ~57 million [3, 4]. Early cancer detection is strongly associated with more treatment options for patients, leading to improved survival [5].
A key commitment of the National Health Service (NHS) in England’s Long Term Plan is to ensure that by 2028, 75% of cancers are diagnosed early, defined as at stage I or II [6]. Despite this, early cancer diagnosis has remained low, at ~54% [7]. England currently has nationally organised and quality-assured cancer screening programmes, all of which are free at the point of care, for bowel, breast, and cervical cancer, and is currently in the process of introducing a lung cancer screening programme. However, most cancers remain unscreened; over 80% of cancer deaths in the UK can be attributed to cancer types for which no population screening is currently available [8]. This gap suggests that innovations such as multi-cancer early detection (MCED) tests are needed to meaningfully improve cancer outcomes.
A screening programme using an MCED test has greater scope for benefits beyond single-cancer screening [9]. The low prevalence of individual cancer types makes it hard to develop single-cancer screening strategies in which the benefits outweigh the potential harms and costs. MCED tests are designed to test for multiple types of cancer with a fixed false-positive rate, in contrast to false-positive rate accumulation across multiple different single-cancer screening tests [10]. One blood-based MCED test (Galleri®) utilises circulating cell-free DNA (cfDNA) methylation patterns to detect a shared cancer signal from more than 50 cancer types and predict the cancer signal of origin (CSO) [11]. MCED screening has the potential to be a clinically- and cost-effective addition to the current screening programmes in the UK [12]. The randomised controlled NHS-Galleri trial (NCT05611632) aims to evaluate the clinical utility of this MCED test in an asymptomatic population [13].
Previous modelling demonstrated that a single screen with sensitivity as estimated in a case-control study [11] could result in 177–220 fewer late-stage diagnoses per 100,000 persons (there are currently 409 without MCED screening), corresponding to 74–91 fewer cancer deaths per 100,000 persons within five years (currently 393 without MCED screening), depending on modelling assumptions [14]. Recent research suggests that DNA shedding may be a marker of tumour aggressiveness [15]. Here, we built on this work to develop a model which (1) considers the differential survival of cfDNA-detectable (cfDNA+; i.e. shedding) cancers versus cfDNA-non-detectable (cfDNA–) cancers, and (2) is well-calibrated to current population cancer incidence and survival data in England, permitting an exploration of potential late-stage reduction and mortality benefits of MCED-based screening in England. We considered both the immediate impact of screening in a mixed-age cohort and the long-term impact in a cohort 50 years of age who were ‘aged’ through annual screening until they were 79 years old (national screening programme).
Methods
Data
This study used data provided by patients and collected by the NHS as part of their care and support. The data are collated, maintained and quality-assured by the National Cancer Registration and Analysis Service, which is part of NHS Digital [16, 17]. Crude incidence rates and net survival were calculated for 24 main cancer types and an additional ‘other’ category (see Table S1 for definitions) based on 50–79-year-olds diagnosed with cancer between 2013 and 2018 for each stage and by five-year age band. The age range is aligned with the 50–77-year age criterion for enrolment in the NHS-Galleri trial (NCT05611632) [13], allowing an additional two years of screening while enroled in the trial; this reflects the intended roll-out age group of an annual screening programme. For the purposes of the model, lymphoid leukaemia, myeloid neoplasm and plasma cell neoplasm were considered to be unstageable and there is no modelled mortality advantage from early detection. Incidence rates per 100,000 persons were provided for all cancers, apart from sex-specific cancers (ovary, cervix, uterus, prostate) for which person rates were created by multiplying the rate by the proportion of the relevant sex in the five-year age band. Age-specific net survival was estimated using a period approach with the Pohar Perme estimator [18], capped at 99.9%. The analysis was censored on 5 January 2020, providing a minimum of one year of follow-up for all patients. Aggregate incidence and survival data used in the analysis are provided in Tables S2, S3.
Interception model
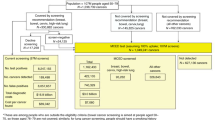
Hubbell and colleagues [14] first presented the interception model (Fig. 1). We extended this model to (1) accommodate emerging evidence regarding the prognostic significance of cfDNA+ versus cfDNA– cancers [19] and (2) estimate the impact of an annual national screening programme (30 years of screening in a cohort aged 50 years until 79 years of age) by incorporating attrition from the programme due to mortality and cancer diagnosis. Additionally, we completed a sensitivity analysis to model the impact of non-sequential stage progression (varying proportions of tumours metastasising early, with direct progression from stage I to IV, for a subset of aggressive cancers) by reducing stage II and III dwell times to zero (Analysis S4).

Before a single prevalent round of screening, cancer a was shedding cell-free DNA (cfDNA) at stage I and progressed to stage III, when it was intercepted; b became cfDNA shedding at stage II and progressed to stage IV, when it was intercepted; c became cfDNA shedding at stage II, when it was intercepted; d was not shedding cfDNA at stage I when it was screened in a prevalent screening round, and progressed to stage III before it was intercepted in the incident screening round; e became cfDNA shedding at stage I and did not progress before being intercepted at the incident screening round; f never became cfDNA-detectable, was not intercepted by MCED screening, and presented in usual care at stage IV; and g became cfDNA-detectable at stage II and progressed to stage III between screening rounds, when it was detected via usual care.
The interception model starts by considering a population of incident cancers under usual care. We calculated the number of these cancers present (but undetected) at earlier stages in prior years using estimates of dwell (sojourn) times with exponential distributions. Dwell time corresponds to the amount of time a tumour spends in each stage before progressing to the next stage. Mean dwell times (for each stage in each cancer type) were estimated based on expert elicitation for the interception model published by Hubbell and colleagues [14]. In typical situations, cancers are treated shortly after diagnosis, therefore there is no reliable data on the time it takes for cancer to progress between stages. Dwell times cannot be inferred from cancer patients who are not treated, as these patients cannot be considered representative; other comorbid conditions and frailty complicate the clinical picture. Mean dwell times are therefore intrinsically uncertain, so we have chosen to model a range of scenarios. Lastly, we calculated the number of cancers intercepted at each stage, based on cancer type- and stage-specific sensitivity of an MCED test (Galleri®) which were estimated in a case-control study [11] and adjusted using isotonic regression so that test sensitivity did not decrease by stage (Table S5). Throughout the manuscript, we use the generic term MCED screening to reflect that this model could be used with different cancer type- and stage-specific sensitivity estimates, though the results estimated in this paper are based on Galleri® test sensitivity estimates [11]. We used dwell times specific to each cancer type and stage, as previously described by Hubbell and colleagues [14], and the individual variation in cancers of the same type was modelled using an exponential distribution (Table S6).
Interception occurs if an individual is screened when a cancer is present and detectable, which in turn depends on the sensitivity of the test (Method S7). The detection of detectable cancers depends on the time interval between screening rounds relative to stage dwell times. The proportion of cancers that are intercepted increases as the interval between screening rounds is shortened, with a maximum proportion equal to stage-specific test sensitivity. Additionally, in modelled scenarios with longer (slower) dwell times, cancers are less likely to progress through stages in the window of time between screening rounds, and more are intercepted at earlier stages. For cancers that are unstageable, a single sensitivity value is assumed, from which the proportion of detected cancers is calculated. Therefore, these cancers do not contribute to the estimation of late-stage cancer reduction or consequent mortality, but only to the proportion of cancers detected via the MCED screening route. We imputed cancers with missing stage information by proportionately increasing the incidence in each stage. Given staging procedures are often not undertaken as it is anticipated the patient will not benefit from them, it is anticipated many missing stage cancers would be late-stage cancers. As such, this imputation is conservative as it reduces the number of late-stage cancers available to be intercepted. Results presented here are for a cohort of individuals who participate in screening. Diagnostic resolution was assumed to follow shortly after screen detection, and we did not model the impact of returning ‘cancer signal origin’ results to the diagnosing physician.
Mortality modelling
After computing the stage shift based on the interception model, we estimated mortality benefits by comparing the difference in stage-specific survival between stage distributions with and without MCED screening in addition to usual care. Specifically, for instance, a breast cancer that would have been clinically diagnosed at stage IV, following interception at stage II, would be assigned the survival of a stage II breast cancer. This within-cancer-type modelling approach allows for prognostic heterogeneity between cancer types for different stages. We used a two-population model for survival, stratified by stage at diagnosis, to model differential survival depending on cfDNA detectability status (cfDNA+ or cfDNA–). Based on previous findings regarding the increased risk of death from cfDNA+ cancers [19], we modelled cfDNA+ cancers as having double (hazard ratio [HR] = 2) or triple (HR = 3) the stage-specific risk of cancer death compared with cfDNA– cancers of the same cancer type. Net survival was adjusted for both cfDNA+ and cfDNA– status to ensure that, given the population fraction of each cancer type at each stage, average survival would match that observed in the population, resulting in reduced mortality for cfDNA– cases compared with population net survival-by-stage estimates. This adjustment was applied to all cfDNA+ cancers, intercepted or not; thus, in the resultant survival data for both cfDNA+ and cfDNA–, the total mortality in the overall population was preserved. For comparison, we also modelled a scenario similar to Hubbell and colleagues [14] in which survival for cfDNA+ and cfDNA– cancers was identical (HR = 1).
For both survival populations, we modelled lead-time until date of diagnosis via usual care, during which individuals cannot die from their cancer (to avoid a situation in which early detection causes cancer death before a symptomatic tumour) [20]. We then applied the HR to net survival-by-stage to estimate cancer-specific mortality (so that expected mortality was calculated for years following diagnosis under usual care, regardless of interception occurrence). Expected cancer mortality within five years of the date of diagnosis under usual care was calculated per 100,000 persons screened. The difference in cancer mortality with MCED screening compared with usual care is the estimated mortality benefit with MCED screening.
Modelled scenarios
Open cohort
This modelled scenario is comparable to the NHS-Galleri trial, run in a population attending their first (prevalent) screen at ages 50–79 years, followed by subsequent (incident) annual screening tests. This model assesses how the removal of previously intercepted cancers from the screened population impacts late-stage cancer incidence and cancer mortality rates.
National screening programme cohort
This modelled scenario enabled comparisons between the presence and absence of an MCED-based screening programme. For both incidence and survival data, participants entered the cohort at 50 years of age, and were ‘aged’ through the programme until they turned 80 years old, died, or emigrated, with different age-specific incidence rates and net survival probabilities applied in the interception model at each of the 30 annual screening rounds. Figure S8 shows the cohort weights applied at each age, corresponding to movement out of the cohort.
The cumulative benefits of this ‘national screening programme’ scenario were calculated using a life table to adjust for attrition from all-cause mortality [21] and a competing cancer diagnosis. The weighted average of male and female mortality was used to generate a mortality rate for all persons. Participants did not re-enter the screening cohort after a cancer diagnosis, even though many will survive their cancer diagnosis. This is a simplifying assumption; for some cancer types, cancer survivors are at greater risk for the development of subsequent cancers [22]. However, survival for some early-stage cancers (e.g. breast and prostate cancer) is very high, and people who survive these cancers may go on to develop a fatal cancer that is not captured in the model, because they have been censored out. This assumption equates the risk of developing a cancer in the population without a competing cancer diagnosis to that observed in the whole England population. It is beyond the scope of the current paper to consider these complexities of screening cancer survivors.
Results
NCRAS data for England demonstrated an average annual cancer incidence of 1213 per 100,000 persons in 50–79 year olds in the period 2013–2018. Of these cancers, 1164 per 100,000 were stageable and 973 per 100,000 were staged, with 450 per 100,000 diagnosed at a late stage (III or IV).
Open cohort model: overall late-stage incidence and mortality reductions
Table 1 shows the number of cancers (per 100,000 persons screened) found via usual care alone and when MCED screening was added to usual care, and the subsequent reduction in late-stage cancer incidence with the addition of MCED screening.
When MCED screening was first added to usual care (prevalent round), cancers which would otherwise have been diagnosed in future years are intercepted, increasing incidence for that year. Per 100,000 individuals screened, 502 of the 1023 originally late-stage (III + IV) cancers (fast), and 1443 of the 2168 originally late-stage cancers (slow) were found instead at an early stage (Table 1). This indicates a strong influence of dwell times on the impact of screening in the prevalent round.
Incident screening rounds were less susceptible to changes in dwell times, and correspondingly we observed a narrower range in the number of cancers found via usual care with and without MCED screening (Table 1). The annual reductions in late-stage incidence with MCED screening added to usual care were predicted to be between 180 and 222 per 100,000 persons screened. These rates were considerably lower than in the prevalent screen, reflecting the fact that cancers diagnosed via screening are removed from the available population each year.
In Table 2 we present five-year mortality rates per 100,000 persons screened with and without MCED screening added to usual care, and the resulting mortality rate differences with MCED screening. The effect of different dwell times on mortality reduction in the prevalent screening round was estimated to be greater than the effect of different HRs for cfDNA+ versus cfDNA– cancers. The estimated mortality rate reduction when MCED screening was added to usual care was between 192 (fast dwell time, HR = 3) and 622 (slow dwell time, HR = 1) deaths per 100,000 persons screened in the prevalent screening round (Table 2), and between 67 (fast dwell time, HR = 3) and 97 (slow dwell time, HR = 1) deaths per 100,000 persons in the incident screening round (Table 2).
Open cohort model: late-stage incidence and mortality rate reductions by cancer type
The potential late-stage incidence reduction benefit varied by cancer type (Table 3). For colon/rectum, head and neck, liver/bile duct, lung, lymphoma, ovary and pancreas cancers (Table 3), the reduction in late-stage incidence with MCED screening was estimated to be substantial (>10 per 100,000 persons screened), reflecting high test sensitivity and current adverse stage profiles for these cancers. Although breast cancer is predominantly diagnosed at an early stage, the reduction of 7 per 100,000 in late-stage incidence reflects the potential to diagnose this high-incidence cancer at stage I or II, despite the relatively poor sensitivity at earlier stages.
In contrast, MCED screening did not appear to contribute to earlier diagnosis for melanoma, thyroid and urothelial tract cancers, and for prostate cancers, the proportion of cancers shifted from late to early stage was small. This reflects both lower MCED test sensitivity at early stages for these cancers and that the majority of them are already diagnosed at an early stage.
For breast, colon/rectum, head and neck, kidney, liver/bile duct, lung, lymphoma, ovary and prostate, sarcoma and uterus cancers, the reduction in late-stage incidence translates into >10% reductions in the five-year cancer mortality rate (deaths from cancer within five years of diagnosis, based on when the diagnosis would have happened in the absence of MCED screening), which reflects that early-stage survival is considerably better than late-stage survival for these cancer types. The substantial modelled mortality reduction for prostate cancer in the context of minimal late-stage incidence reduction highlights that the majority of prostate cancer mortality is observed among those diagnosed at stage IV, and therefore the impact of intercepting these cancers prior to progression to stage IV is proportionally large. For several relatively low-incidence cancer types including ovary, liver/bile duct, pancreas and oesophagus, the absolute number of deaths avoided with MCED is small; however, the proportional mortality benefits for these cancer types are substantial, as is the aggregate total mortality rate reduction of these low-incidence cancer types combined.
For a subset of cancers stratified by stage, Fig. 2 shows modelled five-year survival for each cfDNA detectability status when HR = 3, along with observed survival based on NCRAS data. By cancer type and stage, when test sensitivity is either very low or high, the impact of having cfDNA+ status on survival is minimal. This is because the majority of cancers are cfDNA+ or cfDNA–, such that the modelled survival curves do not separate markedly from observed survival. The impact of low test sensitivity is demonstrated in early-stage breast and prostate cancers, for which all three survival curves closely overlap. For cancer types in which the majority of cancers are cfDNA+ even at early stages, such as liver/bile duct cancers and lung cancer from stage II onwards, the impact of cfDNA+ status is also minimal, as the modelled survival for these cancers closely follows observed survival. Conversely, the modelled survival for cfDNA– cancers at these sites is considerably better, as almost all of the deaths over the period are among those with cfDNA+ cancers. The impact of cfDNA detectability status is more critical when the MCED test has a less extreme sensitivity, therefore it adds more prognostic information at a population level. This pattern is observed in stage II and III gallbladder cancer and sarcoma. Figure S9 shows the impact of cfDNA status by cancer type for all cancers included in this study.

This figure shows observed five-year survival data for cancers diagnosed between 2013 and 2018, as registered by the National Cancer Registration and Analysis Service (NCRAS), and modelled five-year survival data based on cfDNA detectability status (hazard ratio [HR] = 3). For visualisation purposes, we plotted net survival for a 65-year-old in the middle of the screening programme, and data has been smoothed. Where the lines representing cfDNA+/− survival are not visible, they are (almost) identical to that of original survival. For a small number of cancers at specific stages, adjustment of survival data for both cfDNA-detectable and cfDNA-non-detectable cases to reflect the observed survival in the population created implausible results, in which later-stage survival for cfDNA-detectable cancers was favourable compared with that of earlier-stage cancers. This occurred when there was a large difference in test sensitivity between one stage and the next, such that applying the HR to the different cfDNA populations moved the survival curves further out from the observed mean. We did not correct for this anomaly, as it was only ever a difference of a few percentage points, and it would have decreased the average observed survival-by-stage.
National screening programme: late-stage incidence and mortality rate reductions
In the national screening programme modelled scenario (Table 4), the incidence of cancer diagnosed through MCED screening was estimated to be between 9766 and 13,084 per 100,000 persons (for fast and slow dwell time assumptions, respectively), corresponding to a proportion of between 31 and 41% of cancers detected through MCED screening. Thus, the estimated reduction in the incidence rate of late-stage cancer was between 5468 and 7032 per 100,000 persons in the screening programme (40–50%). The reduction in five-year cancer mortality was between 2029 and 2581 per 100,000 persons in the screening programme (between 17 and 21%) in the fast dwell time scenario with HR = 3 and slow dwell time scenario with HR = 1, respectively. Figure S10 demonstrates how the incidence rate of MCED screen-detected cancers changes with age for each of the dwell time scenarios.
Results of the non-sequential stage progression model (Supplementary Analysis S6) demonstrate that the benefits of MCED screening may be reduced in scenarios where greater proportions of cancers show non-sequential progression and have a shorter dwell time in stage I.
Discussion
In this study, we extended the interception model first described by Hubbell et al. [14], to explore the benefits of MCED screening, with sensitivity as estimated in a case-control study [11], under a range of model assumptions. Overall, the reduction in late-stage cancer incidence and mortality rates were substantial under a range of assumptions regarding dwell times and differential survival between cfDNA-detectable and non-detectable cancer. Even with the fastest dwell times, the proportion of cancers screen-detected with MCED was estimated to be 31%. Given that in England, only ~6% of cancers are currently diagnosed via screening [23], widespread adoption of MCED screening among 50–79 year olds would improve the cancer diagnostic landscape. This has the potential to contribute to the NHS Long Term Plan goal of achieving 75% of cancers diagnosed at an early stage [6]; our model estimates that 49–56% of cancers are diagnosed at an early stage via usual care, which could be increased to 73–79% (74–78% in incident rounds of screening) among those who are screened. MCED screening (based on parameters of one commercially available test) has the greatest potential to reduce late-stage diagnoses and consequent mortality among cancers currently diagnosed predominately at late stages, and for which the MCED test has high sensitivity. Thus, MCED screening may contribute to the early detection, and, potentially, mortality reduction of cancers for which there is substantial unmet need. Even achieving the lower bound of mortality reduction via MCED screening would mean that cancer was no longer the leading cause of mortality among screened individuals in England [24].
The areas of uncertainty in the modelling will be resolved, in part, by the results of the NHS-Galleri trial, which will directly estimate the reduction in the incidence of late-stage cancers resulting from annual MCED screening in 50–79 year olds across three annual screening rounds [13]. Beyond late-stage incidence reduction, the trial results will help to inform model structure and parameter values in future iterations of the model presented here, which could in turn be used to extrapolate from NHS-Galleri trial results. Should the interception model be well-calibrated to the trial outcomes, it could be used to examine scenarios not directly tested in the trial, including different screening intervals and age cut-offs for the screening programme.
Results from the NHS-Galleri trial, as well as other emerging evidence, may also challenge structural aspects of the model. For example, non-sequential stage progression may be more common than previously thought [25]. If this is the case, then other credible alternative model structures, such as the non-sequential stage progression model detailed in the Supplementary Information (Analysis S10), may provide a better fit to the trial data. This model demonstrates that greater proportions of cancers showing non-sequential progression and a short (6 month) dwell time in stage I have the potential to reduce the benefits of MCED screening.
Chabon et al. [26] found that in lung cancer, cfDNA+ cancers are enriched for subclinical micrometastases that are invisible to current scanning techniques, suggesting that cfDNA+ cancers may be more lethal than cfDNA– cancers, irrespective of their apparent stage at diagnosis. Here we estimated that MCED screening is beneficial even when cfDNA+ cancers had a three-fold greater relative mortality risk. However, longer-term follow-up is required to fully characterise the consequences of cfDNA detectability, both in case-control and screening settings in which these cancers are shifted to an earlier stage. There are no current data to suggest that cfDNA+ cancers are incurable, and there is some evidence to suggest that ~40% of circulating tumour DNA+ (ctDNA+) early-stage (I–III) non-small-cell lung cancers diagnosed through usual care remain free from recurrence up to five years after the end of treatment [27]. For breast, colorectal and lung cancer, cfDNA concentration has been linked to both tumour volume and surrogate biomarkers of clinically significant prognosis, including depth of invasion (colorectal), mitotic (breast), and metabolic activity [15], and adjuvant chemotherapy resistance (lung) [28]. This suggests inter-cancer heterogeneity in the mechanisms by which cfDNA enters the tumour microenvironment and subsequently the bloodstream (for a review of the issue, see ref. [29]). A greater understanding of these mechanisms is required to understand the extent to which cfDNA+ cancers are likely to be treatment-resistant, and how this corresponds to outcome heterogeneity. Further clinical trials may also be required to examine the efficacy of alternative or escalated systemic anti-cancer therapy regimens for such cancers, and/or de-escalated treatment regimens for cfDNA– cancers. In this case, the MCED test may confer additional benefits as a risk stratification tool. Preliminary evidence also supports the use of ctDNA as a marker for molecular residual disease, to help identify when further treatment may help avoid recurrence [25, 27].
The performance of the MCED test was previously estimated in case-control settings to have a test sensitivity of 51.5% [11]. In a population screening trial such as the NHS-Galleri trial, in which participants are invited from the community, the performance of the test may differ. If cancers tend to shed cfDNA only shortly before becoming symptomatic, or if cfDNA-shedding cancers progress very rapidly, then patients would need to be tested more frequently to diagnose cancers earlier, limiting the test’s utility as a population-level screening tool. However, there is currently no evidence directly linking cfDNA shedding to disease progression or symptom development. Indeed, for lung cancer, detectable levels of cfDNA may be reached in some patients prior to tumour sizes that would likely result in symptomatic presentation [30].
Unlike conventional screening techniques, especially those based on imaging or direct visualisation, the MCED screening signal is based on cancer biology; thus, overdiagnosis (of indolent lesions) is less of a concern, particularly compared with multiple separate conventional screening techniques for individual cancers, in which the risks associated with each screening mode accumulate [31]. Poor sensitivity to cancers with known overdiagnosis issues, including early-stage breast, prostate, and thyroid cancers [32], provides reassurance that MCED screening will not contribute to overdiagnosis. As such, overdiagnosis was not incorporated in the current model. However, this is the first time that a candidate screening technology has existed for most cancer types, and the understanding of cfDNA is nascent. The possibility of overdiagnosis will be investigated in the NHS-Galleri trial [13], which may inform the evidence base on the natural history of cancer. Future iterations of the model will incorporate these considerations.
We used five-year net survival from the date of usual-care diagnosis in this study [33], but some cancer patients are still at increased risk of death ten years after their initial diagnosis [34]. While a longer follow-up period may allow estimation of survival over ten or more years, survival with more historical cancer cases would not capture current best treatment practices, and the availability of historic staging data is limited.
Additionally, we only considered an idealised 100% screening attendance scenario with rapid subsequent diagnostic resolution. The harms associated with screening such as psychological distress, diagnostic work-up of false-positive results, and healthcare resource utilisation should be considered in future studies on real-world attendance, and the feasibility and implications of national MCED screening.
Some other potential benefits of MCED screening are not captured in the study. MCED screening may lead to within-stage changes in cancer detection that are prognostically relevant and confer mortality benefits, but do not lead to stage shift. There are treatments for patients with oligometastatic lesions with the potential for long-term survival that are not an option for patients with widely disseminated metastases [35,36,37,38,39,40]. Within-stage reduction in breast tumour size due to screening has been shown to be associated with clinically relevant reductions in mortality [41]. Diagnosis via a managed (rather than emergency) pathway is also associated with improved survival [42]. A higher level of granularity in the anticipated prognosis could be gained by including additional tumour characteristics or substages in the model. We accounted for this to some extent by allowing for differential stage-specific hazards in cfDNA+ and cfDNA– cancers, but the model could be modified with further empirical data. Lastly, potential morbidity benefits to patients were not considered in this study. Cancers curable at a late stage may be shifted to earlier stages with MCED screening, and patients with these cancers are likely to experience quality of life benefits from less toxic treatments, even if no mortality benefit is observed [43].
In summary, we built upon the interception model to incorporate recent evidence regarding the prognostic significance of cfDNA detectability status and estimate the longer-term benefits of a national screening programme in England, from where the NHS-Galleri trial has enroled participants. Despite uncertainty regarding this intervention, we demonstrated that the potential benefits are robust to a range of model parameters, and that MCED screening may contribute to reduction in the incidence of late-stage cancers and consequent mortality, particularly for cancers with a current adverse staging profile. The magnitude of the benefits estimated here supports the launch of the NHS-Galleri trial.
Data availability
These data can be made publicly available on request.
Code availability
The code used to generate the results in this study are available on request from the corresponding author.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Office for National Statistics. Population estimates. https://www.ons.gov.uk/peoplepopulationandcommunity/populationandmigration/populationestimates. Accessed 22 November 2022.
Office for National Statistics. Total cancer deaths in the UK in 2019 and 2020. https://www.ons.gov.uk/aboutus/transparencyandgovernance/freedomofinformationfoi/totalcancerdeathsintheukin2019and2020. Accessed 23 November 2022.
NHS Digital. Cancer survival in England, cancers diagnosed 2015 to 2019, followed up to 2020. https://digital.nhs.uk/data-and-information/publications/statistical/cancer-survival-in-england/cancers-diagnosed-2015-to-2019-followed-up-to-2020. Accessed 22 November 2022.
National Health Service. The NHS long term plan. https://www.longtermplan.nhs.uk/wp-content/uploads/2019/08/nhs-long-term-plan-version-1.2.pdf. Accessed 22 November 2022.
National Cancer Registration and Analysis Service. Cancer data: Percentage of stageable cancers diagnosed at stage 1 and 2 in England. https://www.cancerdata.nhs.uk/stage_at_diagnosis. Accessed 22 November 2022.
Cancer Research UK. Cancer mortality statistics. https://www.cancerresearchuk.org/health-professional/cancer-statistics/mortality/common-cancers-compared. Accessed 22 November 2022.
Liu MC, Oxnard GR, Klein EA, Swanton C, Seiden MV, Cummings SR, et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann Oncol. 2020;31:745–59.
Clarke CA, Hubbell E, Ofman JJ. Multi-cancer early detection: a new paradigm for reducing cancer-specific and all-cause mortality. Cancer Cell. 2021;39:447–8.
Klein E, Richards D, Cohn A, Tummala M, Lapham R, Cosgrove D, et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann Oncol. 2021;32:1167–77.
Hackshaw A, Cohen SS, Reichert H, Kansal AR, Chung KC, Ofman JJ. Estimating the population health impact of a multi-cancer early detection genomic blood test to complement existing screening in the US and UK. Br J Cancer. 2021;125:1432–42.
Neal RD, Johnson P, Clarke CA, Hamilton SA, Zhang N, Kumar H, et al. Cell-free DNA–based multi-cancer early detection test in an asymptomatic screening population (NHS-Galleri): design of a pragmatic, prospective randomised controlled trial. Cancers. 2022;14:4818.
Hubbell E, Clarke CA, Aravanis AM, Berg CD. Modeled reductions in late-stage cancer with a multi-cancer early detection test. Cancer Epidemiol Biomark Prev. 2021;30:460–8.
Bredno J, Lipson J, Venn O, Aravanis AM, Jamshidi A. Clinical correlates of circulating cell-free DNA tumor fraction. PLoS ONE. 2021;16:e0256436.
National Cancer Registration and Analysis Service. Staging data in England. https://www.cancerdata.nhs.uk/stage_at_diagnosis. Accessed 1 November 2022.
National Cancer Registration and Analysis Service. Incidence and Mortality. https://www.cancerdata.nhs.uk/incidence_and_mortality. Accessed 7 December 2022.
Perme MP, Stare J, Estève J. On estimation in relative survival. Biometrics. 2012;68:113–20.
Chen X, Dong Z, Hubbell E, Kurtzman KN, Oxnard GR, Venn O, et al. Prognostic significance of blood-based multi-cancer detection in plasma cell-free DNA. Clin Cancer Res. 2021;27:4221–9.
Wever EM, Draisma G, Heijnsdijk EAM, de Koning HJ. How does early detection by screening affect disease progression?: Modeling estimated benefits in prostate cancer screening. Med Decis Mak. 2011;31:550–8.
Office for National Statistics. National life tables, United Kingdom, 1980–1982 to 2017–2019. https://www.ons.gov.uk/peoplepopulationandcommunity/birthsdeathsandmarriages/lifeexpectancies/datasets/nationallifetablesenglandreferencetables/current. Accessed 22 November 2022.
Schacht DV, Yamaguchi K, Lai J, Kulkarni K, Sennett CA, Abe H. Importance of a personal history of breast cancer as a risk factor for the development of subsequent breast cancer: results from screening breast MRI. Am J Roentgenol. 2014;202:289–92.
National Cancer Registration and Analysis Service. National Cancer Intelligence Network, routes to diagnosis (2006–2017). https://www.gov.uk/government/statistics/routes-to-diagnosis-2006-to-2017-results/routes-to-diagnosis-2006-to-2017-results. Accessed 29 November 2022.
Office for National Statistics. Mortality statistics. https://www.nomisweb.co.uk/query/construct/summary.asp?mode=construct&version=0&dataset=161#. Accessed 22 November 2022.
Riggio AI, Varley KE, Welm AL. The lingering mysteries of metastatic recurrence in breast cancer. Br J Cancer. 2021;124:13–26.
Chabon JJ, Hamilton EG, Kurtz DM, Esfahani MS, Moding EJ, Stehr H, et al. Integrating genomic features for non-invasive early lung cancer detection. Nature. 2020;580:245–51.
Gale D, Heider K, Perry M, Marsico G, Ruiz-Valdepeñas A, Rundell V, et al. Residual ctDNA after treatment predicts early relapse in patients with early-stage NSCLC. J Clin Oncol. 2021;39:8517–8517.
Abbosh C, Birkbak NJ, Wilson GA, Jamal-Hanjani M, Constantin T, Salari R, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 2017;545:446–51.
Bredno J, Venn O, Chen X, Freese P, Ofman JJ. Circulating Tumor DNA allele fraction: a candidate biological signal for multicancer early detection tests to assess the clinical significance of cancers. Am J Pathol. 2022;192:1368–78.
Avanzini S, Kurtz DM, Chabon JJ, Moding EJ, Hori SS, Gambhir SS, et al. A mathematical model of ctDNA shedding predicts tumor detection size. Sci Adv. 2020;6:eabc4308.
Croswell JM, Kramer BS, Kreimer AR, Prorok PC, Xu JL, Baker SG, et al. Cumulative incidence of false-positive results in repeated, multimodal cancer screening. Ann Fam Med. 2009;7:212–22.
Welch HG, Black WC. Overdiagnosis of cancer. J Natl Cancer Inst. 2010;102:605–13.
Hubbell E, Clarke C. Detecting cancer when it can be cured: The potential for cure across all stageable cancers. Cancer Res. 2022;82:2239.
National Cancer Registration and Analysis Service. 10-year survival by stage for the East of England. http://www.ncin.org.uk/about_ncin/10yearsurvival. Accessed 22 November 2022.
Hanagiri T, Takenaka M, Oka S, Shigematsu Y, Nagata Y, Shimokawa H, et al. Results of a surgical resection for patients with Stage IV non–small-cell lung cancer. Clin Lung Cancer. 2012;13:220–4.
Liu S, Zhou F, Liu Z, Xiong A, Jia Y, Zhao S, et al. Predictive and prognostic significance of M descriptors of the 8th TNM classification for advanced NSCLC patients treated with immune checkpoint inhibitors. Transl Lung Cancer Res. 2020;9:1053–66.
David EA, Clark JM, Cooke DT, Melnikow J, Kelly K, Canter RJ. The role of thoracic surgery in the therapeutic management of metastatic non–small cell lung cancer. J Thorac Oncol. 2017;12:1636–45.
Lo Dico R, Faron M, Yonemura Y, Glehen O, Pocard M, Sardi A, et al. Combined liver resection and cytoreductive surgery with HIPEC for metastatic colorectal cancer: results of a worldwide analysis of 565 patients from the Peritoneal Surface Oncology Group International (PSOGI). Eur J Surg Oncol. 2021;47:89–100.
Zou X, You R, Liu H, He YX, Xie GF, Xie ZH, et al. Establishment and validation of M1 stage subdivisions for de novo metastatic nasopharyngeal carcinoma to better predict prognosis and guide treatment. Eur J Cancer. 2017;77:117–26.
Pagani O, Senkus E, Wood W, Colleoni M, Cufer T, Kyriakides S, et al. International guidelines for management of metastatic breast cancer: Can metastatic breast cancer be cured? J Natl Cancer Inst. 2010;102:456–63.
Elkin EB, Hudis C, Begg CB, Schrag D. The effect of changes in tumor size on breast carcinoma survival in the U.S.: 1975–1999. Cancer. 2005;104:1149–57.
McPhail S, Swann R, Johnson SA, Barclay ME, Abd Elkader H, Alvi R, et al. Risk factors and prognostic implications of diagnosis of cancer within 30 days after an emergency hospital admission (emergency presentation): an International Cancer Benchmarking Partnership (ICBP) population-based study. Lancet Oncol. 2022;23:587–600.
Carreira H, Williams R, Dempsey H, Stanway S, Smeeth L, Bhaskaran K. Quality of life and mental health in breast cancer survivors compared with non-cancer controls: a study of patient-reported outcomes in the United Kingdom. J Cancer Surviv. 2021;15:564–75.
Acknowledgements
Emma B. Saxon (Nottingham, England) provided medical writing support in the development of this manuscript. PosterDocs (Oakland, California) provided graphics assistance. Ewan Gray provided support with model development and interpretation of results in the early stages of the publication.
Funding
This study was funded by GRAIL, LLC, Menlo Park, CA.
Author information
Authors and Affiliations
Contributions
Conceived and/or designed the work that led to the submission of this manuscript: CS, EH, PS, RS. Acquired data: EH, JB. Interpreted the results: CS, EH, PS, RDN, RS. All authors were involved in the drafting and revision of the manuscript. All authors have read and approved this version of the manuscript for publication and agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Competing interests
PS is a paid member of GRAIL’s Scientific Advisory Board and the Director of the The Cancer Research UK and King’s College London Cancer Prevention Trials Unit that is contracted by GRAIL, LLC, to run the NHS-Galleri trial, is on the Medical Advisory Committee for NSV, and has received consultant fees from Roche. RS and EH are employees of GRAIL Bio UK Ltd, and hold stock in Illumina, Inc.; RS also has contingent value rights in GRAIL. JB is employed by NHS Digital and is an Associate Research Fellow (honorary) at the Nuffield Department for Population Health, University of Oxford. RDN provides his services as Co-Chief Investigator to the NHS-Galleri trial through university consultancy funded by GRAIL, LLC, to the University of Exeter; both he and the University of Exeter financially benefit from this partnership. CS provides his services as Co-Chief Investigator to the NHS-Galleri trial through university consultancy funded by GRAIL, LLC, to University College London Business, for another study; is an AstraZeneca advisory board member and Chief Investigator for the AZ MeRmaiD 1 and 2 clinical trials; CS is a paid member of GRAIL’s Scientific Advisory Board (SAB); received grant funding from AstraZeneca, Boehringer-Ingelheim, Bristol Myers Squibb, Pfizer, Roche-Ventana, Invitae, and Ono Pharmaceutical; receives consultant fees from Achilles Therapeutics (also SAB member), Genentech, Medixci, Roche Innovation Centre Shanghai, Bicycle Therapeutics (also SAB member), Metabomed (until July 2022), and the Sarah Cannon Research Institute; has received honoraria from Amgen, AstraZeneca, Pfizer, Novartis, GlaxoSmithKline, MSD, Bristol Myers Squibb, Illumina, and Roche-Ventana; had stock options in Apogen Biotechnologies and GRAIL until June 2021; currently has stock options in Epic Bioscience, Bicycle Therapeutics, and Achilles Therapeutics; and is co-founder of Achilles Therapeutics.
Ethics approval and consent to participate
Not applicable.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sasieni, P., Smittenaar, R., Hubbell, E. et al. Modelled mortality benefits of multi-cancer early detection screening in England. Br J Cancer 129, 72–80 (2023). https://doi.org/10.1038/s41416-023-02243-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-023-02243-9
