
Abstract
Background
Patients with triple-negative breast cancer (TNBC) develop early recurrence. While PARP inhibitors (PARPi) have demonstrated potential in BRCA1/2-mutant (BRCAMUT) TNBC, durable responses will likely be achieved if PARPi are used in combination. It is plausible that sequential administration of a potent PARPi like talazoparib in combination with carboplatin can enhance primary tumour and metastasis inhibition in BRCAMUT and BRCA1/2 wild-type (BRCAWT) TNBCs, and decrease toxicity.
Methods
We evaluated the impact of the concurrent combination of talazoparib and carboplatin on cell survival in 13 TNBC cell lines. We compared the concurrent and sequential combination upon fork replication, migration and invasion. We also used three orthotopic xenograft models to evaluate primary tumour growth, distant metastasis, and toxicity.
Results
Concurrent talazoparib and carboplatin was synergistic in 92.3% of TNBC cell lines, independent of BRCA1/2-mutation status. The sequential combination decreased fork speed in normal cells, but not in TNBC cells. The talazoparib-first sequential combination resulted in a strong reduction in migration (70.4%, P < 0.0001), invasion (56.9%, P < 0.0001), lung micrometastasis (56.4%, P < 0.0001), and less toxicity in a BRCAWT model.
Conclusion
The sequential combination of talazoparib and carboplatin is an effective approach to inhibit micrometastatic disease, providing rationale for the use of this combination in early TNBC patients.

Similar content being viewed by others


Background
Triple-negative breast cancer (TNBC) is the most aggressive subtype of breast cancer with the poorest outcomes. Representing 15–20% of all breast cancers, TNBCs lack oestrogen/progesterone receptor (ER/PR) and human epidermal growth factor receptor 2 (HER2) overexpression. Most patients receive four or five types of chemotherapy yet still suffer from rapid disease progression with significant toxicity. An orally available family of targeted therapeutic agents, Poly (ADP-Ribose) Polymerase inhibitors (PARPi), was approved by the FDA for metastatic and early TNBC patients with germline mutations in BRCA1/2 (gBRCAMUT). However, patients with gBRCAMUT constitute about 11–20% of all TNBCs [1,2,3,4].
The efficacy of PARPi monotherapy in gBRCAMUT patients was demonstrated in the metastatic, neoadjuvant (pre-surgery), and adjuvant (post-surgery) settings [5,6,7,8]. In particular, talazoparib and olaparib demonstrated an improvement in progression-free survival (PFS) and less toxicity in comparison to standard chemotherapy in the metastatic setting [5, 6]. Talazoparib was also associated with a pathologic complete response (pCR) in 53% of gBRCAMUT patients in the neoadjuvant context [7]. Importantly, in the adjuvant setting, in comparison to placebo, olaparib improved 3-year distant disease-free survival (87.5% versus 80.4%) [8], suggesting that olaparib can inhibit the development of micrometastatic disease.
PARPi target PARP1/2 enzymes and have two main mechanisms of action: synthetic lethality and PARP-DNA trapping [9, 10]. Synthetic lethality occurs in the context of BRCA1/2 mutations, whereby PARPi prevent the release of PARP1 from DNA, leading to an accumulation of double-stranded DNA breaks, resulting in complex chromatid rearrangements and cell death [11, 12]. Trapped PARP-DNA complexes form DNA lesions that are not bypassed by replication forks and induce cytotoxicity [10, 13, 14]. While various PARPi demonstrate similar catalytic activity, they differ in their capacity for PARP-DNA trapping, with the most potent PARPi being talazoparib, then niraparib, followed by olaparib, and finally veliparib [15].
Despite PARP1 being widely known for its activities in DNA repair, PARP1 has also been implicated in several other cancer cell functions [16]. PARP1 regulates chemokine signalling, facilitating tumour dissemination at several key steps of metastasis, including angiogenesis, adherence of tumour cells to endothelium, cell migration, and cancer cell extravasation at the metastatic site [17]. PARP1 has also been shown to promote lung metastasis using a mechanism independent of DNA repair [18]. Furthermore, alterations in DNA repair genes in distant metastasis have also been detected in melanoma and colon cancer [19], all pointing towards a plausible role of PARPi in inhibiting the development of distant metastasis through DNA repair-dependent and independent mechanisms.
While PARPi have mainly been used in gBRCAMUT breast cancer patients, recent clinical trials have observed the efficacy of PARPi beyond patients with gBRCAMUT [2, 20, 21]. We and others have demonstrated preclinical efficacy of PARPi in TNBCs that are both BRCAMUT and BRCA1/2 wild type (BRCAWT) [22,23,24]. Efficacy in BRCAWT tumours is likely due to BRCAness, a phenotype similar to BRCAMUT, with a defect in homologous recombination repair, but actually lack the mutation in BRCA1/2 [25]. However, one of the clinical challenges is that patients treated with PARPi monotherapy often develop therapeutic resistance.
Hence, to improve overall survival, it is likely that PARPi will need to be administered in combination. Carboplatin, an alkylating agent, is one such promising chemotherapeutic agent. While one trial did not demonstrate the added benefit of low-dose veliparib to carboplatin and chemotherapy [26], higher-dose veliparib in combination with carboplatin and paclitaxel resulted in a more durable response in BRCAMUT metastatic patients [27]. Little is known about the role of the combination of a potent PARPi plus carboplatin in preventing the development of metastatic breast cancer in BRCAWT patients.
While toxicity was a concern with the concomitant and continuous administration of talazoparib and carboplatin [28], it is plausible that a sequenced and intermittent dosing regimen may decrease toxicity. Pre-treatment with carboplatin was previously shown to be a plausible approach to increase cellular uptake of olaparib and increase olaparib clearance, which may improve efficacy and decrease toxicity [29]. Furthermore, lower endogenous replication stress in normal cells, in comparison to cancer cells, helped to explain the increased toxicity observed with concomitant PARPi combination with a targeted therapeutic agent [30].
In this study, our goals were to determine the impact of different dosing strategies of the combination of a potent PARPi, talazoparib and carboplatin, on tumour efficacy, toxicity, and development of metastasis. We accomplished this by using a panel of TNBC cell lines, characterising cell proliferation, the sustained DNA damage response and apoptosis. We further evaluated the replication fork speed and DNA damage response in both TNBC and normal cells. Moreover, we utilised three orthotopic xenograft models, including BRCAMUT and BRCAWT tumours, to evaluate the influence of different sequencing strategies on primary tumour growth, mice weight, haematologic toxicity, and distant metastasis. We additionally evaluated the impact of the sequencing strategies on cell migration, invasion, differential expression of chemokines, and gene expression in metastatic lung tissue.
Materials and methods
A detailed list of all reagents and resources, including cell lines with sources, are provided in Supplementary Methods Table S1.
Cell lines
All cell lines were validated by DNA fingerprinting using short-term repeat (STR) analysis done by Genome Quebec (microsatellite geneprint 10) (Montreal, Canada), last performed March 2021, and were mycoplasma free, tested using Mycoalert mycoplasma detection kit (LT07, Lonza).
Ten-day chemosensitivity assay and immunofluorescence staining, analysis and visualisation were performed as previously described [22] and explained in Supplementary Methods.
High-content imaging
High-content imaging was done using Operetta (PerkinElmer) with a ×20 objective and filter sets for Alexa 488, Alexa 647, and DAPI. We scanned 47 images per well and performed image analysis with Harmony High-Content Imaging and Analysis Software (version 4.1, PerkinElmer Inc.).
Combination index values
Cells were treated with six concentrations with twofold dilutions of either carboplatin, talazoparib alone or concomitant combination using the 10-day chemosensitivity assay. Cells were fixed, stained with DAPI, imaged using Operetta (PerkinElmer), and enumerated using Harmony High-Content Imaging and Analysis Software (version 4.1, PerkinElmer Inc.). Combination Index (CI) and Dose Reduction Index (DRI) values were calculated at Fa = 0.50, using the Chou-Talalay method with Compusyn software (ComboSyn Incorporated, Paramus, NJ, USA). CI values between 0.9 and 1.10 are considered nearly additive; 0.85–0.7 demonstrate moderate synergism, and 0.3–0.7 indicate synergism [31].
See Supplementary Methods for details regarding drug treatment strategies.
Flow cytometry
Cells were fixed/permeabilized in ice-cold 70% ethanol overnight. Samples were washed, blocked with PBS, Triton 0.2%, BSA 1% solution for 15 min at room temperature. Cells were then incubated with gamma H2AX-AlexaFluor 647 (1:100; #613408, BioLegend), phospho-Histone H3- AlexaFluor 488 (Ser-10) (1:100; #3465, Cell Signaling), cleaved-caspase 3-Pacific Blue™ (1:100; #8788, Cell Signaling), and 20 µg/mL propidium iodide (PI) (#P3566, Invitrogen) with 200 µg/mL RNase A (#10109142001, Roche®), for 1 h at room temperature. Samples were analysed with FACS BD LSRFortessa™ cell analyzer (BD Biosciences). At least 10,000 events were assessed per measurement. Staining was analysed with FlowJo™ (BD Biosciences). Values were obtained from triplicate assays performed in triplicate wells. Gating strategy is presented in Supplementary Methods.
DNA fibre assay
Cells were sequentially labelled with 25 µM of 5-Chloro-2′-deoxyuridine (CldU) (#C6891, Sigma-Aldrich) and then 250 µM of 5-Iodo-2’-deoxyuridine (IdU) (#I7125, Sigma-Aldrich) for 30 min each [32]. Cells were resuspended in ice-cold PBS at 5 × 105 cells/mL. Four drops of 2.5 µL of cell suspension was pipetted in staggered rows onto a microscope slide (Cat #4951PLUS602811, Fisherbrand™). In all, 6 µL of spreading buffer (200 mM Tris-HCl, 50 mM EDTA, 0.5% SDS, pH 7.4) was added to cell suspension drops. DNA was allowed to run down the slide, slowly tilting slides at 15–45°. DNA was air-dried and fixed in methanol/acetic acid (3:1) for 10 min. DNA was denatured in 2.5 M HCl for 45 min. Slides were incubated in blocking solution (PBS, 2% BSA, 0.1% Tween) for 1 h, followed by rat anti-BrdU [clone BU1/75 (ICR1)] (1:100, #ab6326, Abcam) and Mouse anti-BrdU [clone B44] antibodies (1:20, #347580, BD Biosciences), overnight at 4 °C. Slides were incubated with anti-Rat AlexaFluor 568 (1:500, #A11077, Invitrogen) and anti-Mouse AlexaFluor 488 (1:500, #A21202, Invitrogen) antibodies, for 1 h at room temperature. Slides were mounted in ProLong™ Gold antifade Mountant (#P36930, Invitrogen) and analysed using a Zeiss Axio Observer Z1 microscope with ×63 oil objective. Replication fork speed was calculated using the CldU + IdU track length/60 min * 2.59 kb/µm.
Orthotopic xenografts
All animal experiments were approved by the Institutional Animal Protection Committee (CIPA) of the Centre de Recherche de Centre hospitalier de l’Université de Montréal (CRCHUM) under protocol C17017SHs. Either 5 million MDAMB231 or 2 million MX1/HCC1806 cells were resuspended in 50% Matrigel Matrix Phenol Red Free (#CB40234C, Fisher Scientific), 25% PBS and 25% collagen type 1 (#08-115, Millipore Sigma) solution [33]. Using 7-week-old NOD-SCID gamma (NSG) female mice (#005557, Jackson Laboratory), 0.2 mL cell suspension was surgically implanted. Once tumours reached an average volume of 150 mm3, mice were randomised into treatment groups based on tumour volume volumes and weight. Our sample size was based on the experiments previously described [34]. Except for the control group for MDAMB231, which had 14 mice, each treatment group consisted of 8–10 mice. See Supplementary Methods for further details.
Cell migration and invasion assays
In all, 3 × 104 or 5 × 104 cells were suspended in serum-free media in 8-μm transwell inserts (#83.3932.800, Sarstedt), for 24-h migration or 48-h invasion assays, respectively. For the invasion assay, the upper chamber was pre-coated with 1/9 dilution of Matrigel (Cultrex Reduced Growth Factor Basement Membrane Extract, #3433-005-01, R&D Systems). After incubation, inserts were washed with PBS. Migrated or invaded cells on the underside of the membrane were fixed with 4% paraformaldehyde and stained with hematoxylin (Hematoxylin QS Counterstain, #H-3404, Vector Laboratories). Migrated or invaded cells were counted from six representative fields from the inverted EVOS XL core microscope (Thermo Fisher Scientific).
Chemokine array and ELISAs
In total, 200 μg of protein was extracted from cell lysates from MDAMB231 cells to detect the expression of 31 chemokines using the human chemokine array kit (#ARY017, R&D Systems). Images of membranes were acquired with ChemiDoc (BioRad) and dots were quantified using QuickSpots (Western vision) software. At the same time point, supernatants of MDAMB231 and HCC1395 cells were collected to detect MCP-1 levels with the human CCL2/MCP-1 Immunoassay (#SCP00, R&D Systems).
See Supplementary Methods for details regarding RNA-seq analysis.
Statistical analysis
Normality of each dataset was first verified before performing Kruskal–Wallis ANOVA or one-way ordinal ANOVA with multiple-comparison post test. Two-way ANOVAs with post-tests were performed for grouped conditions. Data are represented as mean + /− SEM, except for fork speed, in which median values with interquartile ranges are presented. Data were analysed and plotted using GraphPad Prism software 8. P < 0.05 is considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Results
Talazoparib and carboplatin synergise in most TNBC cell lines
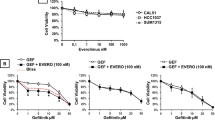
We determined the IC50 values of talazoparib and carboplatin as single agents in a panel of 13 TNBC cell lines (Fig. 1, Supplementary Table 1 and Supplementary Fig. 1). Talazoparib demonstrated a larger dynamic range, with IC50 values ranging from 0.0003 to 0.44 μM, in comparison to carboplatin, with IC50 values ranging from 0.09 to 4.0 μM (Fig. 1a, b). Using the median IC50 values, we set a threshold of 0.0036 μM for talazoparib and 1.25 μM for carboplatin to define sensitivity and resistance. Concurrent administration of talazoparib and carboplatin resulted in synergy, with Combination Index (CI) values <1 in 92.3% (12/13) of cell lines (Fig. 1c). Stronger synergy was observed mainly in BRCAWT cell lines and those that were PARPi-resistant. Dose-reduction indices (DRI) were calculated to determine the dose-fold reduction of each therapeutic agent to achieve synergy. DRI for talazoparib ranged from 1.5 to 3.8, while the DRI for carboplatin ranged from 2.1 to 7.1. This is suggestive that synergy can be accomplished with important dose reductions of both drugs, with greater reductions in carboplatin versus talazoparib commonly observed in 61.5% (8/13) of cell lines.

IC50 values for (a) talazoparib, and (b) carboplatin in TNBC cell lines. Data are mean + /− SEM. The dashed line indicates the median IC50 value for all cell lines for that therapeutic agent. Effect of the combination of talazoparib and carboplatin demonstrated in (c) by combination index values reported at an FA of 0.5. Under each cell line is the dose-reduction index (DRI) for talazoparib and carboplatin. Teal green bars/dots represent BRCAWT cell lines, and pink bars/dots represent BRCAMUT cell lines.
Lower concentrations of talazoparib and carboplatin required for DNA damage and cell death in PARPi-resistant TNBC cell lines
We determined the impact of 9 increasing concentrations (1 refers to the lowest, and 9 refers to the highest concentration) of talazoparib (T1-9), carboplatin (C1-9), or the concomitant combination of talazoparib and carboplatin (TC1-9) upon 53BP1 response and apoptosis using a 10-day chemosensitivity assay in 14 TNBC cell lines (Fig. 2). DNA damage was quantified using a 53BP1 product score (Fig. 2a), calculated from the product of mean number of nuclear 53BP1 foci (Fig. 2c and Supplementary Fig. 2A) and percentage of cells positive for 53BP1. We identified a dose-dependent response of the 53BP1 product score to talazoparib. The combination of talazoparib and carboplatin had the greatest impact in the PARPi-resistant cell lines, where lower concentrations of each of the drug induced a DNA damage response that otherwise required higher drug concentrations as monotherapy. We also evaluated apoptosis by calculating percent cells positive for cleaved-PARP (cl-PARP+ ) (Fig. 2b, d and Supplementary Fig. 2B). While cell death required higher concentrations for talazoparib (mainly T8, 9) or carboplatin alone (mainly C7, 8, 9), combination therapy induced apoptosis at lower concentrations, starting at TC5, and was strongly present in 10/14 cell lines.

Heatmaps of normalised values of either (a) 53BP1 product score (product of mean 53BP1 foci per nuclei and percentage of cells positive for 53BP1) (upper panel) and b percentage of cleaved (cl)-PARP positive (+) cells (second panel). Along x axis are increasing drug concentrations, 1–9, for talazoparib, T; carboplatin, C, and the concurrent combination of talazoparib and carboplatin, TC. Along the y axis are TNBC cell lines, of which * indicates BRCAMUT cell lines. Representative images at 20x objective of 53BP1 foci in MDMAB231 are shown in (c), and cl-PARP+ cells in HS578T are shown in (d). MDAMB231 and HS578T cells are treated with talazoparib (tala) (left column), carboplatin (carbo) (middle column), and the combination of tala + carbo (right column). Blue represents nuclear staining, pink represents 53BP1 foci, and green represents cl-PARP.
Sequential combination comparable to concurrent combination in TNBC cells but decreases replication fork speed in normal cells
We first evaluated the impact of three different combination strategies including concurrent administration of talazoparib and carboplatin (conc.T + C) and two sequential approaches: carboplatin first followed by talazoparib (seq.C→T), and talazoparib run-in approach followed by carboplatin (seq.T→C) upon cell survival using the 10-day chemosensitivity assay (Fig. 3a). We selected three BRCAWT cells, including two PARPi-resistant cell lines, HCC1143 and MDAMB231, a PARPi-sensitive yet carboplatin-resistant cell line, HCC1806; and one PARPi-sensitive cell line that is both BRCA1-deleted and BRCA2-mutated, MX1. In all four cell lines, cell survival was comparable between the concomitant and sequential combination approaches, with either talazoparib or olaparib as the PARPi backbone (Fig. 3b–e and Supplementary Fig. 7A, B).

Impact of the combination of talazoparib and carboplatin using concurrent or sequential approaches on cell proliferation, fork speed, and DNA damage. Using a 9-day (216-hour) treatment assay (a), cell survival was determined for b HCC1806, c MDAMB231, d HCC1143 and e MX1 TNBC cell lines. Using a 72-h treatment assay (a), impact of treatment on fork speed (DNA fibre assay) and DNA damage (flow cytometry) was shown in TNBC cell lines (f–i), and normal cell lines (l–o). f, g, l, m Representative images of DNA fibres with DNA labelling with CldU (magenta) and IdU (green) thymidine analogues. Images were taken at ×63 objective. Scale bars, 10 microns. h, i, n, o Dot plots of fork speed (kb/min), with a mean count of 313 DNA fibres/condition. Line represents median values with interquartile range. Impact of treatments on DNA damage represented by γ-H2AX+ cells in (j, k, p, q) Data are represented as mean +/– SEM. ANOVA with post test for multiple comparisons was performed. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05.
To better understand the impact of the combination approaches on in vitro toxicity, we compared fork replication speed and DNA damage response between TNBC and normal cells. We used two TNBC cell lines, HCC1806 and MDAMB231, and two human normal cell lines including MCF10A (breast epithelial cell line) and BM-MSC (bone marrow-derived mesenchymal stem cells) (Fig. 3f–q). We determined that the mean endogenous replication fork speed was 0.56 kb/min for the TNBC cells in comparison to 0.8 kb/min for the normal cells.
Treatment with talazoparib for 72 h increased fork speed in all four cell lines, which is similar to what was previously reported when talazoparib was administered for 2 or 24 h [35, 36]. In comparison to control, the concomitant combination increased fork speed by 86.0% (P < 0.0001), 48.9% (P < 0.0001), 59.2% (P < 0.0001), and 59.9% (P < 0.0001) in MDAMB231, HCC1806, MCF10A and BM-MSC cells, respectively (Fig. 3h, i, n, o and Supplementary Figs. 3–6). The sequential approaches resulted in comparable or increased fork speeds in comparison to the concomitant approach in HCC1806 and MDAMB231. Similarly, there were no statistically significant differences in γ-H2AX+ cells between concurrent or sequential combination approaches in HCC1806, MDAMB231 (Fig. 3j, k), or MX1 (Supplementary Fig. 7D).
However, in normal cells, sequential treatments decreased fork speed in comparison to concomitant treatments. Seq.T→C demonstrated a 10.3% (P = 0.0004) and 14.8% (P = 0.0005) reduction in median fork speed in comparison to the concomitant combination, in MCF10A and BM-MSC cells, respectively. Conc.T + C resulted in the highest proportion of γ-H2AX+ cells in MCF10A (70.4%, P < 0.0001) and BM-MSC (51.4%, P < 0.0001) in comparison to control (Fig. 3p, q). In BM-MSC, seq.T→C reduced γ-H2AX + cells by 44.9% (P = 0.001), in comparison to conc.T + C. We also evaluated apoptosis in MCF10A and BM-MSC cells (Supplementary Fig. 7F, G). Of all the combination approaches, in comparison to control, conc. T + C resulted in the highest proportion of cl-Caspase 3 cells, with 43.8% (P < 0.0001), and 31.7% (P < 0.0001) for MCF10A and BM-MSC, respectively.
We and others previously demonstrated that PARPi resulted in S/G2 arrest [22, 30], and carboplatin also induced G2/M arrest [37, 38], and so we evaluated the impact of the combination on cell cycle changes. Greater accumulation of cells in G2 phases was observed with the concomitant and carboplatin-first combination approaches in MDAMB231, HCC1806, and MX1 (Supplementary Fig. 7H–L). Comparatively, treatment-induced cell cycle changes were less pronounced in normal cells in comparison to TNBC cells (Supplementary Fig. 7K, L), yet conc.T + C resulted in the greatest accumulation of cells in the S/G2 phases of BM-MSC, which was less distinct in the sequential approaches.
In summary, concurrent and sequential combination approaches resulted in similar cell proliferation, fork speeds and DNA damage responses in TNBC cells. However, in normal cells, sequential combination approaches resulted in decreased fork speed, decreased DNA damage and cell death.
Concurrent and sequential combination have comparable primary tumour growth and tumour volume inhibition in three xenograft models
To evaluate the impact of the combination approaches in vivo, we selected three orthotopic xenograft models comprising MX1, HCC1806 and MDAMB231 (Fig. 4a). In the MX1 and MDAMB231 xenografts, statistically significant differences in in vivo normalised tumour volumes were observed with different treatments from day 8 and day 13 until necropsy, respectively, while only trends were observed with HCC1806 (Fig. 4b–d).

Sequential versus concurrent combination treatment strategies were tested in three orthotopic xenografts with different tumour kinetics (a). Three TNBC models included MX1 (top row), HCC1806 (middle row), and MDAMB231 (bottom row). N = 8–14 mice per treatment group. Primary tumour growth rates are shown in (b, c, d). The x axis indicates days post treatment, where day 0 indicates the day before treatment. The y axis indicates normalised tumour volumes in which ratios of the tumour volumes on day x were divided by the tumour volumes on day 0 for that mouse. Ex vivo primary tumour volumes are quantified in (e, g, i). Data are represented as mean +/– SEM. ANOVA with post test for multiple comparisons was performed. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05. Representative images of ex vivo tumours are shown in (f, h, j). i.p. intraperitoneally, o.g. oral gavage.
At necropsy, combination approaches inhibited MX1 ex vivo primary tumour volumes by 79.9% (P < 0.0001), 75.5% (P = 0.0001), 71.7% (P = 0.002), by the conc.T + C., seq.C→T, and seq.T→C strategies, respectively, in comparison to control (Fig. 4e, f). For the HCC1806 xenograft, tumour volumes decreased by 54.4% (P = 0.002) with conc. T + C, 46.6% (P = 0.04) with seq.C→T, and 62.0% with seq.T→C (P < 0.0001) in comparison to control (Fig. 4g, h). For the MDAMB231 cohort, seq.C→T and seq.T→C demonstrated comparable effects with 68.9% (P < 0.0001), and 66.7% (P < 0.0001) reductions in tumour volume, respectively, which was more remarkable than the 51.4% reduction (P = 0.08) with conc.T + C, in comparison to control (Fig. 4i, j). There were no statistically significant differences in ex vivo primary tumour volumes between any of the combination groups within the three xenograft models.
The sequential combination associated with less weight loss but no difference in haematologic toxicity in comparison to the concurrent combination in vivo
To evaluate toxicity, percent change in weight in comparison to pre-treatment weight was calculated for each orthotopic xenograft model (Fig. 5). The greatest decrease in weight by the combination approaches was demonstrated by MX1 and MDAMB231 cohorts, reaching a maximum of 5.2% decrease within 7 days of treatment. However, all mice gained weight by the time of necropsy, albeit 16 days (for MX1/MDAMB231) or 4 days (for HCC1806) after the termination of treatment. Nonetheless, at necropsy, the combination approach that consistently demonstrated minimal weight gain was conc.T + C, which was 3.7% for MX1, 2.3% for HCC1806, and 0.22% for MDAMB231 (Fig. 5a–c).

The impact of different treatment strategies was evaluated in three orthotopic xenograft models including MX1 (top row), HCC1806 (middle row) and MDAMB231 (bottom row). Kinetics of percent change in weight is shown in (a–c). Percent change in mice weight was calculated by normalising the weight on a given day by pre-treatment weight. x axis represents days post treatment until necropsy, and y axis represents percent change in weight. Impact of different treatments on white blood cell count (WBC) are shown in (d–f); neutrophil count shown in (g–i), and haemoglobin levels shown in (j–l). Sample sizes for each treatment are annotated at the top of each bar. Data are represented as mean +/– SEM. ANOVA with post test for multiple comparisons was performed. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05.
For the MX1 cohort, statistically significant changes in weight were first identified on day 4 post treatment, with smaller changes in weight gain demonstrated by conc.T + C and seq.T→C (Fig. 5a). At necropsy (16 days post treatment), in comparison to control, conc.T + C was the only group that differed in weight (P = 0.046), while seq.C→T demonstrated the greatest gain in weight at 10.4%. In the HCC1806 cohort (Fig. 5b), on the day of necropsy (4 days post treatment), weight gain was less than 3% for all combination approaches.
In the MDAMB231 cohort, one mouse that was treated with the conc.T + C approach had to be sacrificed 5 days prior to the rest of the cohort due to toxicity. The mouse was experiencing abdominal pain, with macroscopic and histologic analysis consistent with the diagnosis of haemorrhagic pancreatitis. On days 13 and 16 post treatment, seq. T→C demonstrated the greatest weight gain (4.6–6.7%) (Fig. 5c).
Haematologic toxicity was also evaluated in each xenograft model with plasma analysis at the time of necropsy. In the MX1 cohort, combination approaches decreased white blood cell counts with mean values of 1.6 (P = 0.002), 1.5 (P = 0.0003), and 1.7 (P = 0.0098), for conc.T + C, seq.C→T, and seq.T→C respectively, in comparison to control (mean 4.9) (Fig. 5d). Similarly, neutropenia was observed for conc.T + C (mean 0.95, P = 0.002), seq.C→T (mean 1.15, P = 0.01) and seq.T→C (mean 0.78, P < 0.0001), in comparison to control (mean 4.3) (Fig. 5e). No statistical significance was observed between the combination approaches. A similar trend, with greater leukopenia and neutropenia with the combination of talazoparib and carboplatin was observed with HCC1806 and MDAMB231 (Fig. 5f–i).
No changes in haemoglobin (Hgb) levels were observed with treatment in the MX1 cohort (Fig. 5j). While the three combination approaches led to a mean reduction in Hgb levels of 36.2% in the HCC1806 cohort (Fig. 5h), the mean reduction was only 9.5% in the MDAMB231 cohort (Fig. 5l). While the moderate anaemia in HCC1806 could partly be considered an adverse event from talazoparib alone and partly due to a shorter recovery period post-completion of treatment, the mild anaemia with the MDAMB231 cohort is probably not “clinically” meaningful.
Sequential combinations inhibit distant metastasis and migration, invasion
To further evaluate the impact of the combination of talazoparib and carboplatin on distant metastasis, we used an orthotopic xenograft model of MDAMB231, which is known for its high metastatic efficiency [33]. In comparison to control, both seq.C→T resulted in a 44.7% (P = 0.003) and seq.T→C led to a 56.4% (P < 0.0001) reduction in lung micrometastases, which was not observed with the concomitant combination (19.6% reduction, P = NS) (Fig. 6a, b). The seq.T→C approach also inhibited lung micrometastases by 40.8% in comparison to carboplatin (P = 0.01), and 36.9% in comparison to the concomitant approach (P < 0.05). Similar trends were also observed with liver micrometastasis (Fig. 6c). In comparison to control, seq. C→T resulted in a 72.3% (P = 0.03) reduction in micrometastasis, which was similar to the 76.3% (P = 0.02) reduction with the seq. T→C combination.

Orthotopic xenograft of MDAMB231 was used to histologically evaluate (a, b) lung micrometastasis and (c) liver micrometastasis. b Representative images of H&E-stained sections of lung tissue, using ×20 objective. Metastatic cancer cells are demarcated from normal cells with a dashed black line and asterisks. The impact of different treatment strategies using a 24-h assay (d) in MDAMB231 cells on migration (e, f) and invasion (g) using transwell inserts + /− matrigel. f Representative images taken at 20x of MDAMB231 migrated cells that have been fixed and stained with hematoxylin. Secreted MCP-1 levels post treatment were quantified using an ELISA, in (h) MDAMB231 and (i) HCC1395 cells. Data are represented as mean +/– SEM. ANOVA with post test for multiple comparisons was performed. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05.
To further dissect the metastatic process, we evaluated the impact of talazoparib as monotherapy and in combination on cell migration and invasion in vitro (Fig. 6d–g). In MDAMB231 cells, as single agents, talazoparib and carboplatin resulted in a 24.0% (P = 0.05) and 30.8% (P = 0.01) reduction in cell migration, respectively. Once again, the seq.T→C demonstrated the most striking inhibition in cell migration (Fig. 6e, f), with 70.4% (P < 0.0001) reduction in comparison to control, 61.1% reduction (P < 0.0001) in comparison to talazoparib, and 39.0% reduction (P = 0.008) in comparison to conc.T + C. The other combination strategies also inhibited migration with 51.5% (P < 0.0001) reduction for the concomitant approach, and 38.9% (P < 0.0001) for the seq.C→T approach. The treatments as single agents or in combination demonstrated a similar trend with HS578T (Supplementary Fig. 8A). In comparison to control, seq.T→C also resulted in 56.8% (P < 0.0001) reduction in cell migration.
For cell invasion, in MDAMB231 cells, the seq.T→C approach also led to the strongest inhibition with 56.9% (P < 0.0001), 47.0% (P < 0.0001), 47.5% (P < 0.0001), and 41.3% (P = 0.0005) reduction in comparison to control, talazoparib, carboplatin, seq.C→T, and conc.T + C, respectively (Fig. 6g). The concomitant combination resulted in a 41.2% (P = 0.001) inhibition in cell invasion in comparison to control. In HS578T, the greatest inhibition was also demonstrated by seq.T→C with a 26.7% (P < 0.0001), 18.9% (P = 0.0002), and 21.3% (P < 0.0001) reduction in cell invasion in comparison to control, talazoparib, and carboplatin, respectively (Supplementary Fig. 8B). Conc.T + C demonstrated a 21.0% (P = 0.001) reduction in cell invasion, in comparison to control. Similar results were obtained with other PARPi. Olaparib or niraparib resulted in the greatest reduction in migration or invasion when administered witih the sequential PARPi-first combination approach (Supplementary Fig. 9).
Since PARPi have been shown to regulate cytokine signalling [39], we hypothesised that sequential talazoparib and carboplatin can enhance the inhibition of tumour secretion of chemokines which can explain its reduced migration capacity. We performed a chemokine array to profile 31 chemokines from MDAMB231 cell lysates (Supplementary Fig. 10A). In comparison to control, noteworthy reductions for seq. T→C were identified for CCL (C-C Motif Ligand) 2, CXCL (C-X-C Motif Ligand) 7, CCL18, CXCL9, CXCL1 and CCL7 (Supplementary Fig. 10B).
CCL2/MCP (Monocyte chemoattractant protein)-1 drives migration and invasion, and is implicated in cancer cell homing at the metastatic site [17, 39]. PARP1 knockdown was previously shown to significantly reduce levels of MCP-1 [39]. Interestingly, carboplatin upregulates MCP-1 [40], and blocking MCP-1 can improve the anti-tumour efficacy of carboplatin [41]. Therefore, it is plausible that pre-treatment with talazoparib can downregulate MCP-1 levels, enhancing the anti-migration potential of carboplatin. Hence, we further evaluated tumour secretion of MCP-1 in MDAMB231, and in a cell line known to express high levels of MCP-1, HCC1395 [42] (Fig. 6h, i). In MDAMB231, seq.T→C resulted in a 14.2% reduction in MCP-1 in comparison to control (P = 0.001), 30.7% reduction in comparison to carboplatin (P < 0.0001), and a 11.8% reduction in comparison to conc.T + C (P = 0.03). In HCC1395, seq.T→C also reduced MCP-1 levels by 31.5% in comparison to control (P < 0.0001), and by 33.9% in comparison to carboplatin (P < 0.0001). Conc.T + C also reduced MCP-1 levels by 19.5% (P = 0.049), and seq.C→T reduced MCP-1 levels by 21.7% (P = 0.01). Since the greatest downregulation of MCP-1 levels was demonstration by seq. T→C in comparison to carboplatin, this is suggestive that the sequential combination of talazoparib and carboplatin may be an important approach to inhibit MCP-1 levels that may otherwise be upregulated with carboplatin.
To better understand the underlying mechanisms for enhanced inhibition of metastasis, we also performed gene expression analysis of the metastatic lung tissue of the MDAMB231 xenograft to determine if there are differentially expressed pathways in the human cancer tissue between the treatment groups (Supplementary Figs. 11 and 12). We found that treatment groups clustered together, including talazoparib alone with carboplatin alone and control, while conc. T + C and seq. C→T also clustered together (Supplementary Fig. 11A). Principal component analysis showed the seq. T→C was distinct from the other combination groups. (Supplementary Fig. 11B). Pre-ranked gene set enrichment analysis (GSEA) demonstrated Seq.T→C had 9 significant gene sets that were downregulated in comparison to control, including pathways involved with the DNA damage response and metabolism, which were not identified with conc. T + C (Supplementary Fig. 11C–E). Furthermore, seq. T→C demonstrated downregulated gene sets in the DNA damage response including homologous recombination and mismatch repair, metabolism, and angiogenesis in comparison to conc. T + C and seq. C→T (Supplementary Fig. 12). Furthermore, in the mouse microenvironment, in comparison to control, seq. T→C demonstrated a downregulation of the CCR5 pathway, in which MCP-1 was a core-enriched gene (data not shown).
Altogether, the sequential administration of talazoparib and carboplatin resulted in a noteworthy reduction in distant metastasis, cell migration and invasion. Our results are suggestive that the PARPi-first combination may have a distinct mechanism of action in comparison to other combination approaches, which can significantly enhance the efficacy of each therapeutic agent.
Discussion
Preclinical studies have commonly focused on the combination of PARPi and platinum-based therapy using BRCAMUT models [43, 44]. To our knowledge, we are the first to evaluate the combination of a potent PARPi, talazoparib, with carboplatin in a large panel of TNBC cell lines. We identified synergy with the concurrent combination in 92.3% of TNBC cell lines comprising both BRCAMUT and BRCAWT subtypes. Talazoparib and carboplatin demonstrated a mean dose-reduction index of 2.8- and 3.7-fold, respectively. Similarly, sustained DNA damage and apoptosis responses were observed at lower drug concentrations of the combination, particularly in the PARPi-resistant cell lines. These results could partly explain the toxicity observed with the concomitant combination of talazoparib and carboplatin in a phase I trial, in which only a 0.25-fold reduction in talazoparib and carboplatin doses were utilised [28].
Sequential talazoparib and carboplatin was associated with a reduction in fork speed, DNA damage, and cell death in normal cells with, in comparison to TNBC cells. This is similar to what was previously reported: higher endogenous fork speeds correlated with lower endogenous replication stress in normal cells, which explained the reduced toxicity observed with the sequential PARPi combination approaches [30]. In our in vivo models, while the concomitant dosing tended to demonstrate the greatest toxicity (least weight gain), the sequential treatment approach had a different contextual impact. Whereas the least toxicity was observed with the carboplatin-first approach in the BRCAMUT model, MX1, the least weight gain and haematologic toxicity was demonstrated by the talazoparib-first approach in the BRCAWT model, MDAMB231. Since the same background strain was used, one plausible explanation for the differential toxicity could be different inflammatory responses induced by the BRCAMUT versus BRCAWT tumours [45, 46], which may in turn be modulated in different ways by the sequential administration of talazoparib and carboplatin. Therefore, our results are suggestive that a carboplatin-first approach in the BRCAMUT context and talazoparib-first approach in the BRCAWT context may offer patients the greatest safety profiles.
For our in vivo experiments, we based our dosing schedule of one dose of carboplatin and nine days of talazoparib on a xenograft model of subcutaneous tumour fragment implantation [34]. Our results showed that using orthotopic xenograft models with different tumour kinetics, that pausing the treatment for 4–16 days, allows for greater opportunity for the mice to regain weight and possibly mitigate anaemia, suggesting the importance of an intermittent dosing schedule. Indeed, clinical trials that have tested PARPi in combination have also shown improved efficacy and less toxicity with an intermittent dosing schedule [2]. For example, two trials dosed olaparib and veliparib on days 1–7 of 21-day cycles [47, 48]. Interestingly, the BROCADE3 trial also used a 2-day run in with veliparib for 7 days, and carboplatin administered on day 3 for a 21-day cycle. The combination of veliparib and carboplatin resulted in an improvement in PFS, with similar toxicity as chemotherapy [27].
While we understand some of the limitations of cells lines and their derived models [49], the strength of our study was the panel of TNBC cell lines, the use of three orthotopic xenograft models, including a BRCAWT model with high metastatic efficiency. We showed comparable efficacy of the concurrent and sequential approaches for primary tumour inhibition. However, to our knowledge, we are the first to report that the sequential combination approaches strongly inhibit migration, invasion, and distant metastasis. The talazoparib-first combination demonstrated the greatest reduction in cell migration and invasion at 70.4% and 56.9%, respectively, with an enhanced effect in comparison to concurrent combination. In vivo, the talazoparib-first approach resulted in a 56.4% and 76.3% inhibition in lung and liver metastasis, respectively. Since we did not identify a differential impact on the tumour volumes with the sequential or concomitant combination approach, it is plausible that the impact upon migration and metastasis is independent of treatment effects on the primary tumour.
Furthermore, the talazoparib-first combination was associated with decreased expression of seven chemokines. The expression of one such chemokine, MCP-1, was downregulated the greatest with the talazoparib-first combination. Interestingly, chemotherapy including cisplatin has been shown to promote secretion of MCP-1, recruit inflammatory monocytes enriched with the receptor of MCP-1, CCR2, forming pre-metastatic niches, thus promoting distant metastasis in breast and lung cancer [50, 51]. Such chemotherapy-induced responses need to be countered immediately, prior to the development of distant metastasis. Since MCP-1 blockade can enhance carboplatin efficacy [41], it is plausible that pre-treatment with talazoparib decreases MCP-1 levels to enhance carboplatin sensitivity. However, since the magnitude of reduction of MCP-1 is less than the magnitude of migration reduction with different treatment combination approaches, it is likely that multiple factors may be contributing to inhibition of migration. Furthermore, stromal MCP-1 from the microenvironment was shown to more effective in reducing tumour burden than blockade of MCP-1 induced by the proper tumour itself [41]. While we did identify a downregulation of the MCP-1 pathway in the microenvironment, the therapeutic impact on the microenvironment needs to be further explored.
Finally, we evaluated the pathways that were differentially expressed by the different treatment groups using metastatic lung tissue. We found that DNA damage response pathways were distinctly downregulated with seq.T→C in comparison to control and other combination groups. We and others have previously shown that the downregulation of these pathways allow for enhanced sensitivity to PARPi [22, 52], suggesting that the sequential approach is enhancing PARPi sensitivity, particularly in this BRCAWT model.
Taken together, we have provided a comprehensive preclinical analysis of the combination of talazoparib and carboplatin in TNBC. We have demonstrated that the combination is synergistic in most TNBC cell lines. While the sequence of administration does not impact tumour proliferation or tumour growth, sequential administration of talazoparib and carboplatin can decrease toxicity and significantly inhibit migration, invasion, and distant metastasis. Therefore, our results lead the way to future clinical trials with the evaluation of the sequential combination of talazoparib and carboplatin in early breast cancer, potentially providing an effective approach to eradicate micrometastatic disease in TNBC patients.
Data availability
All data and materials are available upon request.
References
Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13:4429–34.
Yordanova M, Hubert A, Hassan S. Expanding the use of PARP inhibitors as monotherapy and in combination in triple-negative breast cancer. Pharmaceuticals. 2021;14:1270.
U.S. Food and Drug Administration. FDA approves olaparib for adjuvant treatment of high-risk early breast cancer 2022 [updated 11-Mar-2022. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-adjuvant-treatment-high-risk-early-breast-cancer.
Denkert C, Liedtke C, Tutt A, von Minckwitz G. Molecular alterations in triple-negative breast cancer—the road to new treatment strategies. Lancet. 2017;389:2430–42.
Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. New Engl J Med. 2017;377:523–33.
Litton JK, Rugo HS, Ettl J, Hurvitz SA, Goncalves A, Lee KH, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. New Engl J Med. 2018;379:753–63.
Litton JK, Scoggins ME, Hess KR, Adrada BE, Murthy RK, Damodaran S, et al. Neoadjuvant talazoparib for patients with operable breast cancer with a germline BRCA pathogenic variant. J Clin Oncol. 2020;38:388–94.
Tutt ANJ, Garber JE, Kaufman B, Viale G, Fumagalli D, Rastogi P, et al. Adjuvant olaparib for patients with BRCA1- or BRCA2-mutated breast cancer. New Engl J Med. 2021;384:2394–405.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21.
Murai J, Huang S-YN, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588–99.
Lord CJ, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat Med. 2013;19:1381–8.
Lord CJ, Tutt ANJ, Ashworth A. Synthetic lethality and cancer therapy: lessons learned from the development of PARP inhibitors. Annu Rev Med. 2015;66:455–70.
Murai J, Huang S-YN, Renaud A, Zhang Y, Ji J, Takeda S, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther. 2014;13:433–43.
Rose M, Burgess JT, O’Byrne K, Richard DJ, Bolderson E. PARP inhibitors: clinical relevance, mechanisms of action and tumor resistance. Front Cell Dev Biol. 2020;8:564601.
Pommier Y, O’Connor MJ, de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med. 2016;8:362ps17.
Weaver AN, Yang ES. Beyond DNA repair: additional functions of PARP-1 in cancer. Front Oncol. 2013;3:290.
Demény MA, Virág L. The PARP enzyme family and the hallmarks of cancer part 2: hallmarks related to cancer host interactions. Cancers. 2021;13:2057.
Choi EB, Yang AY, Kim SC, Lee J, Choi JK, Choi C, et al. PARP1 enhances lung adenocarcinoma metastasis by novel mechanisms independent of DNA repair. Oncogene. 2016;35:4569–79.
Corcoran NM, Clarkson MJ, Stuchbery R, Hovens CM. Molecular pathways: targeting DNA repair pathway defects enriched in metastasis. Clin Cancer Res. 2016;22:3132–7.
Eikesdal HP, Yndestad S, Elzawahry A, Llop-Guevara A, Gilje B, Blix ES, et al. Olaparib monotherapy as primary treatment in unselected triple negative breast cancer. Annal Oncol. 2021;32:240–9.
Chopra N, Tovey H, Pearson A, Cutts R, Toms C, Proszek P, et al. Homologous recombination DNA repair deficiency and PARP inhibition activity in primary triple negative breast cancer. Nat Commun. 2020;11:2662.
Hassan S, Esch A, Liby T, Gray JW, Heiser LM. Pathway-enriched gene signature associated with 53BP1 response to PARP inhibition in triple-negative breast cancer. Mol Cancer Ther. 2017;16:2892–901.
Beniey M, Haque T, Hassan S. Translating the role of PARP inhibitors in triple-negative breast cancer. Oncoscience. 2019;6:287–8.
Evans KW, Yuca E, Akcakanat A, Scott SM, Arango NP, Zheng X, et al. A population of heterogeneous breast cancer patient-derived xenografts demonstrate broad activity of PARP inhibitor in BRCA1/2 wild-type tumors. Clin Cancer Res. 2017;23:6468–77.
Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16:110–20.
Loibl S, O’Shaughnessy J, Untch M, Sikov WM, Rugo HS, McKee MD, et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): a randomised, phase 3 trial. Lancet Oncol. 2018;19:497–509.
Dieras V, Han HS, Kaufman B, Wildiers H, Friedlander M, Ayoub JP, et al. Veliparib with carboplatin and paclitaxel in BRCA-mutated advanced breast cancer (BROCADE3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21:1269–82.
Dhawan MS, Bartelink IH, Aggarwal RR, Leng J, Zhang JZ, Pawlowska N, et al. Differential toxicity in patients with and without DNA repair mutations: phase I study of carboplatin and talazoparib in advanced solid tumors. Clin Cancer Res. 2017;23:6400–10.
Lee JM, Peer CJ, Yu M, Amable L, Gordon N, Annunziata CM, et al. Sequence-specific pharmacokinetic and pharmacodynamic phase I/Ib study of olaparib tablets and carboplatin in women’s cancer. Clin Cancer Res. 2017;23:1397–406.
Fang Y, McGrail DJ, Sun C, Labrie M, Chen X, Zhang D, et al. Sequential therapy with PARP and WEE1 inhibitors minimizes toxicity while maintaining efficacy. Cancer Cell. 2019;35:851–67. e7.
Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55.
Halliwell JA, Gravells P, Bryant HE. DNA fiber assay for the analysis of DNA Replication progression in human pluripotent stem cells. Curr Protoc Stem Cell Biol. 2020;54:e115.
Iorns E, Drews-Elger K, Ward TM, Dean S, Clarke J, Berry D, et al. A new mouse model for the study of human breast cancer metastasis. PLoS ONE. 2012;7:e47995.
Shen Y, Rehman FL, Feng Y, Boshuizen J, Bajrami I, Elliott R, et al. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin Cancer Res. 2013;19:5003–15.
Maya-Mendoza A, Moudry P, Merchut-Maya JM, Lee M, Strauss R, Bartek J. High speed of fork progression induces DNA replication stress and genomic instability. Nature. 2018;559:279–84.
Cong K, Peng M, Kousholt AN, Lee WTC, Lee S, Nayak S, et al. Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Mol Cell. 2021;81:3128–44. e7.
Wang S, Zhang H, Cheng L, Evans C, Pan C-X. Analysis of the cytotoxic activity of carboplatin and gemcitabine combination. Anticancer Res. 2010;30:4573–8.
Shah MA, Schwartz GK. Cell cycle-mediated drug resistance: an emerging concept in cancer therapy. Clin Cancer Res. 2001;7:2168–81.
Dutta P, Paico K, Gomez G, Wu Y, Vadgama JV. Transcriptional regulation of CCL2 by PARP1 is a driver for invasiveness in breast cancer. Cancers. 2020;12:1317.
Geller MA, Bui-Nguyen TM, Rogers LM, Ramakrishnan S. Chemotherapy induces macrophage chemoattractant protein-1 production in ovarian cancer. Int J Gynecol Cancer. 2010;20:918–25.
Moisan F, Francisco EB, Brozovic A, Duran GE, Wang YC, Chaturvedi S, et al. Enhancement of paclitaxel and carboplatin therapies by CCL2 blockade in ovarian cancers. Mol Oncol. 2014;8:1231–9.
Dutta P, Sarkissyan M, Paico K, Wu Y, Vadgama JV. MCP-1 is overexpressed in triple-negative breast cancers and drives cancer invasiveness and metastasis. Breast Cancer Res. Treatment. 2018;170:477–86.
Clark CC, Weitzel JN, O’Connor TR. Enhancement of synthetic lethality via combinations of ABT-888, a PARP inhibitor, and carboplatin in vitro and in vivo using BRCA1 and BRCA2 isogenic models. Mol Cancer Ther. 2012;11:1948–58.
Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci USA. 2008;105:17079–84.
Heijink AM, Talens F, Jae LT, van Gijn SE, Fehrmann RSN, Brummelkamp TR, et al. BRCA2 deficiency instigates cGAS-mediated inflammatory signaling and confers sensitivity to tumor necrosis factor-alpha-mediated cytotoxicity. Nat Commun. 2019;10:100.
Zhang S, Iyer S, Ran H, Dolgalev I, Gu S, Wei W, et al. Genetically defined, syngeneic organoid platform for developing combination therapies for ovarian cancer. Cancer Discov. 2021;11:362–83.
Lee J-M, Hays JL, Annunziata CM, Noonan AM, Minasian L, Zujewski JA, et al. Phase I/Ib study of olaparib and carboplatin in BRCA1 or BRCA2 mutation-associated breast or ovarian cancer with biomarker analyses. JNCI: J Natl Cancer Inst. 2014;106:1–11.
Appleman LJ, Beumer JH, Jiang Y, Lin Y, Ding F, Puhalla S, et al. Phase 1 study of veliparib (ABT-888), a poly (ADP-ribose) polymerase inhibitor, with carboplatin and paclitaxel in advanced solid malignancies. Cancer Chemother Pharmacol. 2019;84:1289–301.
Hassan S, Heiser LM, Gray J. Systems biology and genomics. In: Bast RJ, Croce CM, Hait WN, Hong WK, Kufe DW, Piccart-Gebhart M, et al., editors. Holland-Frei cancer medicine. 9th edn. New Jersey, USA: Wiley Blackwell; 2017. p. 247–54.
Keklikoglou I, Cianciaruso C, Güç E, Squadrito ML, Spring LM, Tazzyman S, et al. Chemotherapy elicits pro-metastatic extracellular vesicles in breast cancer models. Nat Cell Biol. 2019;21:190–202.
Bertolini G, Cancila V, Milione M, Lo Russo G, Fortunato O, Zaffaroni N, et al. A novel CXCR4 antagonist counteracts paradoxical generation of cisplatin-induced pro-metastatic niches in lung cancer. Mol Ther. 2021;29:2963–78.
Fleury H, Carmona E, Morin VG, Meunier L, Masson JY, Tonin PN, et al. Cumulative defects in DNA repair pathways drive the PARP inhibitor response in high-grade serous epithelial ovarian cancer cell lines. Oncotarget. 2017;8:40152–68.
Acknowledgements
We thank Pfizer Canada for the 2017 ASPIRE award, the Fonds de recherche du Québec–Santé (FRQ-S) for the Operating Grant for Young Clinician Investigators, and the Scotiabank Chair in the diagnosis and treatment of breast cancer, the Department of Surgery, Université de Montreal for providing research funds for this project. We also thank FRQ-S for the Chercheur-Boursier Clinicien (Clinician-Scientist) salary award to SH, the Fondation les Roses de l’Espoir–La Vie en Rose, and Institute of Cancer of Montreal (Bourse Canderel) for the salary bursaries to MB and AH. We also thank the RAMQ Bursary from the Programme Chercheur-Clinicien for salary awards to MB. We thank Dr. Laura Heiser and Dr. Joe W. Gray for some of the TNBC cell lines as part of a collaboration. Graphical abstract was created with BioRender.com. We thank Raphaelle Lambert and Patrick Gendron from the Genomics and Bioinformatics Facility of the Institut de Recherche en Immunologie et Cancérologie (IRIC) for the RNA-Seq analysis. We also thank the Animal, Molecular Pathology, Cellular Imaging, and Cellular Physiology Platforms of the Centre de Recherche de Centre hospitalier de l’Université de Montréal (CRCHUM).
Funding
This research project was funded by Pfizer Canada for the 2017 ASPIRE, an independently peer-reviewed grant, the Fonds de recherche du Québec–Santé (FRQ-S) Operating Grant for Young Clinician Investigators, File No. 265384, and funds from the Scotiabank Chair in the diagnosis and treatment of breast cancer, Department of Surgery, Université de Montreal. The project design, data collection, analysis, and manuscript writing were conducted by the authors. The final manuscript was approved by Pfizer Canada. The following salary awards were attributed to SH by the Fonds de recherche du Québec for the Chercheur-Boursier Clinicien; MB and AH by the Fondation les Roses de l’Espoir–La Vie en Rose; and M.B. by the RAMQ Bursary from the Programme Chercheur-Clinicien.
Author information
Authors and Affiliations
Contributions
MB and AH share the first-authorship role. The order of the first authors was decided on an alphabetical basis. Conceptualisation: SH; methodology: MB, AC, TH and SH; investigation: MB, AH, TH, AC, NB, XZ and DT-T; visualisation: AH, TH and XZ; formal analysis: AH, TH, AC and SH; writing—original draft: AH and SH; writing—review and editing: SH and TH; funding acquisition: SH; resources: SH; project administration, SH; supervision: SH. All authors have seen and accepted the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
SH received research funding from Pfizer Canada to conduct this project. SH also received research funding from Exact Science, which is not related to this project. SH was on advisory committee meetings for Merck Global and Exact Sciences, which is not related to this project. MB, AH, TH, AC, NB, XZ and DT-T declare no competing interests.
Ethics approval and consent to participate
All animal experiments were approved by the Institutional Animal Protection Committee (CIPA) of the Centre de Recherche de Centre hospitalier de l’Université de Montréal (CRCHUM) under protocol C17017SHs.
Consent for publication
Not applicable.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Beniey, M., Hubert, A., Haque, T. et al. Sequential targeting of PARP with carboplatin inhibits primary tumour growth and distant metastasis in triple-negative breast cancer. Br J Cancer 128, 1964–1975 (2023). https://doi.org/10.1038/s41416-023-02226-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-023-02226-w
This article is cited by
-
Nanoemulsion potentiates the anti-cancer activity of Myricetin by effective inhibition of PI3K/AKT/mTOR pathway in triple-negative breast cancer cells
Medical Oncology (2024)
-
Recent advances in targeted strategies for triple-negative breast cancer
Journal of Hematology & Oncology (2023)
