
Abstract
Background
Genomic alterations of the anaplastic lymphoma kinase gene (ALK) occur recurrently in neuroblastoma, a pediatric malignancy of the sympathetic nervous system. However, information on their development over time has remained sparse.
Methods
ALK alterations were assessed in neuroblastomas at diagnosis and/or relapse from a total of 943 patients, covering all stages of disease. Longitudinal information on diagnostic and relapsed samples from individual patients was available in 101 and 102 cases for mutation and amplification status, respectively.
Results
At diagnosis, ALK point mutations occurred in 10.5% of all cases, with highest frequencies in stage 4 patients <18 months. At relapse, ALK alteration frequency increased by 70%, both in high-risk and non-high-risk cases. The increase was most likely due to de novo mutations, frequently leading to R1275Q substitutions, which are sensitive to pharmacological ALK inhibition. By contrast, the frequency of ALK amplifications did not change over the course of the disease. ALK amplifications, but not mutations, were associated with poor patient outcome.
Conclusions
The considerably increased frequency of ALK mutations at relapse and their high prevalence in young stage 4 patients suggest surveying the genomic ALK status regularly in these patient cohorts, and to evaluate ALK-targeted treatment also in intermediate-risk patients.
Similar content being viewed by others
Background
Neuroblastoma is the most frequent extracranial solid cancer in childhood [1]. The tumor presents as clinically heterogeneous disease with courses that range from spontaneous regression to fatal progression despite multimodal treatment. Major risk factors for high-risk disease are genetic alterations that lead to telomere maintenance in the tumor cells, such as amplification of the proto-oncogene MYCN, genomic rearrangements of the TERT locus [2], and activation of the alternative lengthening of telomeres pathway [3]. In the presence of telomere maintenance mechanisms, mutations of genes of the RAS/MAPK pathway are associated with devastating outcome [3]. The most frequent mutations that lead to activation of the RAS/MAPK pathway in neuroblastoma affect the gene that encodes for anaplastic lymphoma kinase (ALK). The ALK protein is a receptor tyrosine kinase that is frequently activated in pediatric malignancies, such as anaplastic large cell lymphoma, inflammatory myofibroblastic tumor, rhabdomyosarcoma, and neuroblastoma [4]. In neuroblastoma, ALK mutations are mostly activating missense single nucleotide variants (SNV) leading to increased kinase activity by disrupting the auto-inhibited conformation of the kinase [5, 6], thereby promoting tumor growth, proliferation, and migration [7]. ALK point mutations have been reported in 6–17% of all cases of sporadic neuroblastoma [8,9,10,11,12,13,14,15]. The vast majority of ALK mutations cluster at three hotspot positions within the tyrosine kinase domain (R1275, F1174, and F1245), accounting for about 85% of all ALK variants in neuroblastoma [8, 12, 16]. Mutations at position F1174 and F1245 are almost exclusively found in sporadic disease, whereas mutations at R1275 are also present as germline variants in familial cases. Autosomal dominant gain-of-function ALK mutations are reported in about 50% of familial neuroblastoma cases [17]. In addition to point mutations, focal genomic amplification of ALK has been reported in 2–10% of neuroblastoma [5, 11, 12, 15, 17,18,19]. Such alterations occur almost always in combination with amplification of MYCN, and mutually exclusive with ALK point mutations [18], with few exceptions [15]. Patients whose tumors harbor ALK amplification have poor outcomes [12]. The prognostic significance of somatic ALK SNV, however, has been discussed controversially, as poorer survival has been reported in the subgroups of intermediate and high-risk patients in some studies [12, 15], while others did not find prognostic relevance of ALK mutations [11, 19]. In contrast to SNV and amplification, translocation of ALK has been reported only occasionally in neuroblastoma [20, 21].
ALK is a tractable target in tumors bearing activating ALK alterations, and various ALK inhibitors have been developed. The ALK inhibitors crizotinib and ceritinib have been evaluated in early clinical trials in neuroblastoma and other pediatric malignancies [4, 22, 23], showing promising clinical activity in a fraction of cases. Third-generation ALK inhibitors, such as lorlatinib, may overcome primary resistance in ALK-mutated neuroblastoma, and are currently evaluated in clinical trials.
Although the overall frequency of ALK alterations in neuroblastoma at diagnosis has been well documented, little is known about their prevalence and prognostic impact in specific patient subgroups, still, and in patients with relapsed or progressive disease in particular. Since ALK inhibitory therapies are primarily considered for patients with relapsed or progressive disease in current treatment strategies, it appears to be essential to gain detailed knowledge of the frequencies and spectrum of ALK alterations in this cohort. We, therefore, set out to determine ALK alteration frequencies and associations with clinical variables in a large cohort of diagnostic and relapsed neuroblastomas that were obtained from patients treated within successive trials and registries of the Gesellschaft für Pädiatrische Onkologie (GPOH).
Methods
Patients
Patients included in this study were diagnosed with neuroblastoma and covered the entire spectrum of the disease. Patients were selected based on availability of tumor ALK mutation and/or ALK amplification status and corresponding clinical follow-up data. Thereby, reports on ALK testing were collected retrospectively. Furthermore, tumor specimens were investigated for the purpose of this study in the lab of experimental pediatric oncology in Cologne to validate ALK alterations found previously and to understand their dynamics over the course of disease. Of note, the cohort of samples with ALK amplification status was biased towards MYCN-amplified tumors, as ALK amplification status is routinely assessed in MYCN-amplified tumors. Two sub-cohorts had been published previously (n = 263 [13] and n = 436 [3]). Patients included in this study were diagnosed between 1989 and 2020, and treated according to the successive German trial protocols NB85 (n = 1), NB90 (NCT00002802, n = 24), NB95-S (NCT00002803, n = 2), NB97 (NCT00017225, n = 249), NB2004/NB2004-HR (NCT00410631/NCT03042429, n = 551), and NB2016-Registry (DRKS00023442, n = 97) with informed consent by patients or their legal guardians. The trials were conducted in accordance with the Declaration of Helsinki and Good Clinical Practice and were approved by the institutional ethical review boards of the University of Cologne. Further, a cohort of patients not included in any clinical trial but treated accordingly in Cologne (n = 19), was considered for analyses. Additionally, a cohort of patients, registered in the INFORM registry trial was included in this study (Supplementary Table 1). All samples were checked histologically for tumor cell content by an experienced pathologist.
In total, 943 patients were included in the study (Fig. 1, Supplementary Table 1). Primary tumor samples were taken at time of initial diagnosis or during first line treatment and relapsed samples were taken at the time of relapse or during relapse treatment. For a sub-cohort we investigated paired samples, taken at diagnosis and at relapse from the same patient. For ALK mutation status 717 primary tumors samples and 198 samples taken at relapse were analyzed. Of the primary tumor samples, 352 were derived from high-risk tumors, defined by MYCN amplification, or stage 4 disease for patients ≥18 months, and 365 were derived from non-high-risk tumors, defined as localized disease, or stage 4 disease in patients <18 months, each without amplification of MYCN. Tumor stages ranged from INSS stage 1–4 and 4S [24, 25]. 172 of the primary tumors showed amplification of MYCN, whereas 545 were MYCN non-amplified. For ALK amplification, we investigated 298 samples taken at diagnosis. From these, 256 were high-risk, and 42 non-high-risk tumors, 186 showed amplification of MYCN, whereas 112 were MYCN non-amplified.

Flow diagram of patient inclusion in this study. Patients that are listed in the category “ALK alteration status” represent the overlap of the cohorts for which ALK mutation and for which ALK amplification status was available. ddPCR, digital droplet PCR.
At relapse, we investigated 198 samples for ALK mutations (Fig. 1, Supplementary Table 1). 151 of these were high-risk and 47 were non-high-risk tumors, 64 showed amplification of MYCN, and 134 were MYCN non-amplified. ALK amplification status at relapse was determined in 172 samples. 119 of these were high-risk tumors, 53 were non-high-risk, and 53 showed amplification of MYCN whereas 119 were MYCN non-amplified.
Detection of ALK alterations
Neuroblastoma samples were analyzed for ALK mutations by various methods: dideoxy-sequencing (exons 21–25, n = 303), whole-exome or whole-genome sequencing (WES, WGS, n = 306), panel next-generation sequencing (panel NGS, n = 124) or a combination of at least two of the respective methods (n = 185, Supplementary Table 1). For one sample, method for detection of ALK mutations remained unknown. Information on germline variants was not available for all samples, thus we did not distinguish between somatic and germline ALK mutations. ALK amplifications were analyzed by Fluorescence in-situ hybridization (FISH, n = 401), low-coverage whole genome sequencing (WGS, n = 16) or by a combination of both methods (n = 53, Supplementary Table 1). ALK amplification in FISH analyses was defined as >4 copies of ALK in relation to copies of chromosome 2 reference. For cases analyzed by low-coverage WGS we defined amplification as copy number of ALK locus >10. Samples with ambiguous copy numbers [3,4,5,6,7,8,9,10] in low-coverage WGS were only included when material for validation by FISH was available.
Fluorescence in situ hybridization (FISH)
FISH was performed using a dual-color probe (Zytolight Spec ALK/2q11 Dual Color, Zytovision) for chromosome 2q11 as reference and a probe specific for the ALK gene locus to count the number of ALK copies in relation to the number of chromosomes 2 copies as described previously [26].
DNA extraction
DNA extraction was performed according to the manufacturers’ instructions, using the Puregene Core Kit A (Qiagen) for fresh frozen and Maxwell® 16 FFPE Plus LEV DNA Purification (Promega) for paraffin-embedded tumor material.
Dideoxy-sequencing
DNA was amplified with primer pairs for ALK exons 21–25 (Supplementary Table 2). Dideoxy sequencing was performed as described previously [27]. Sequencing analyses were carried out on the eight capillary electrophoresis system 3500 Genetic Analyzer (Life Technologies).
Whole exome, whole genome, and low-coverage whole genome sequencing
Whole genome and whole exome sequencing was performed and analyzed as described previously [2, 3]. Cancer cell fractions and copy number values were estimated with Sclust [28]. The inference of mutational clusters and their dynamics over the development of the tumor was performed as described previously [29]. A sub-cohort of samples evaluated in this study was produced and kindly provided by the INFORM program [30, 31].
Amplicon-based next-generation sequencing
Mutational analysis for low input DNA was performed by next-generation sequencing (NGS) using an Ion AmpliSeq Custom DNA Panel (Thermo Fisher Scientific) and the Ion AmpliSeq Library Kit 2.0 (Thermo Fisher Scientific) according to the Ion AmpliSeq Library Preparation User Guide (Thermo Fisher Scientific). After multiplex PCR, libraries were generated by adapter ligation and target enrichment using the Gene Read DNA Library I Core Kit, the Gene Read DNA I Amp Kit (Qiagen), and the NEXTflex DNA Barcodes (Bio Scientific). 12 pM of the constructed libraries were sequenced on the MiSeq platform (Illumina) with a MiSeq reagent kit V2 (Illumina) with 300 cycles following the manufacturer’s recommendations. Data analysis and mutation calling were performed as previously described [32]. The following genes and gene regions were evaluated for mutations: ALK (Exons 21–25), AKT1 (Exon 4), BRAF (Exons 11 and 15), CTNNB1 (Exon 3), DDR2 (Exons 3–18), EGFR (Exons 18–21), HER2 (Exons 19 and 20), KRAS (Exons 2 and 3), MEK1 (MAP2K1) (Exon 2), MET (Introns 13/14, Exon 14), NRAS (Exons 2 and 3), PIK3CA (Exons 9 and 20), PTEN (Exons 1–9), TP53 (Exons 5–8).
A subset of samples was further analyzed with the QIAseq targeted DNA panel for human lung cancer (NGHS-005X-96) with the GeneRead DNAseq Panel PCR Kit V2 (Qiagen). Libraries were prepared using the Gene Read DNA Library I Core Kit and the Gene Read DNA I Amp Kit (Qiagen) according to manufacturer’s protocol. Barcoded libraries were amplified, final library products were quantified, diluted, and pooled in equal amounts. Finally, 1, 2 pM of the final libraries were sequenced on a NextSeq Sequencer (Illumina, San Diego, CA, USA) with the NextSeq 500 Mid Output Kit v2 following manufacturer’s recommendations. The following regions were analyzed: ALK (Exons 2–15, 17–29), ATM (Exons 2–13, 15–45, 47–63), BRAF (Exons 2–4, 6–18), EGFR (Exons 2–26, 28), ERBB2 (Exons 1, 3, 5–6, 8–12, 14–26), ERBB4 (Exons 1–6, 8–13, 15–28), FGFR1 (Exons 2, 4–10, 12–18), FGFR2 (Exon 2–18), KDR (Exon 2–20, 22–30), KEAP1 (Exon 3–6), KIT (Exon 2–21), KRAS (Exons 2–6), MET (Exons 2–21), NFE2L2 (Exons 2–5), PDGFRA (Exons 2–23), PIK3CA (Exons 2–10, 12–16, 18–21), PIK3CG (Exons 3–10), RET (Exons 2–10, 12–20), ROS1 (Exons 1–43), SMARCA4 (Exons 2–5, 7–17, 19–27, 29, 30, 32–35), STK11 (Exons 1–2, 4–8), TP53 (Exons 2, 4–11).
The generated libraries were equimolarly pooled for amplicon sequencing to a concentration of 3 nM of each sample to counterbalance differences in sample quality. Sequencing was performed on an Illumina MiSeq benchtop sequencer (Illumina, San Diego, USA). Results were visualized in the Integrative Genomics Viewer (IGV) and manually analyzed with an in-house pipeline. Results were only interpreted if the coverage was >200.
Digital droplet PCR
In a digital droplet PCR (ddPCR), the reaction is split into ~12–15,000 individual droplets, each containing zero or one or more DNA molecules. During the readout, each droplet is individually counted and accessed for fluorescence. In each experiment, a DNA sample with the confirmed ALK mutation served as a positive control. The number of wildtype DNA molecules was determined in the same reaction using a second probe complementary to the wildtype sequence of the tested gene. Amplifications were carried out in duplicates and a reaction volume of 20 μL on the QX200 Droplet Digital PCR System (Bio-Rad). Each PCR reaction contained 3 μL PCR grade water, 10 μL Bio-Rad PCR mix for probes, 1 μL of each (target and reference) amplification primer/probe mix, and 10 ng of genomic DNA. PCR cycling was performed on a C1000 thermo cycler (Bio-Rad) according to manufacturer’s instructions. A minimum of 200 wildtype droplets was necessary for analyses. Results were analyzed with Quantasoft v.1.3.2 software (Bio-Rad) and reported as allelic fractions.
Statistics
Statistical analyses were performed using IBM SPSS (version 27) and R (version 4.1.2) [33]. Fisher’s exact test (two-sided) was used to assess occurrence of ALK alterations in different subgroups if the test was performed between two groups, or Chi-squared test for comparisons between more than two groups. Median, first and third quartile of age at diagnosis in different subgroups are presented as boxplots with whiskers displaying the minimum and maximum of the data within 1.5 times the IQR (interquartile range). A Wilcoxon-Mann-Whitney (two-sided) test was used to compare median ages. Kaplan-Meier curves showing survival estimates were compared using the log-rank test in case the proportional-hazards assumption hold and Gehan-Breslow (generalized Wilcoxon) test otherwise. Additionally, probabilities of 5-year event-free survival (EFS) and overall survival (OS) along with their 95% confidence intervals basing on the cumulative hazard were extracted from the Kaplan-Meier estimates. Results of statistical tests with P values below 0.05 are regarded as being significant.
Results
To comprehensively determine the spectrum of ALK alterations in neuroblastomas at diagnosis and at relapse, we retrospectively collected data on ALK mutations and/or amplifications from tumors of 943 neuroblastoma patients. Longitudinal samples from the same patient were available for subcohorts. Patients were treated in successive GPOH trials and covered the entire spectrum of the disease, including high-risk and non-high-risk patients, as well as all stages of the International Neuroblastoma Staging System (INSS; Fig. 1, Supplementary Table 1).
ALK alterations in neuroblastoma at diagnosis
In neuroblastomas obtained at diagnosis, we detected SNV in the ALK kinase domain in 10.5% (75/717) of the cases (Fig. 2a). These mutations were equally distributed across the high-risk and non-high-risk groups (10.8% versus 10.1%, P = 0.808), and mutation frequencies did also not differ in MYCN-amplified and non-amplified tumors (12.2% versus 9.9%, P = 0.393). ALK mutations were significantly enriched in stage 4 tumors compared to non-stage 4 tumors (13.6% versus 7.8%, P = 0.014), with lowest frequencies occurring in stage 1 (4.5%) and stage 4S (7.6%) tumors (Fig. 2a). ALK mutation frequencies did not differ in patients <18 months and ≥18 months of age (9.7% versus 11.1%, P = 0.544, Fig. 2a). We noted, however, that ALK mutations occurred significantly more frequently in stage 4 tumors of patients <18 months than of patients ≥18 months (21.7% versus 11.5%, P = 0.046, Fig. 2a), with highest frequencies in MYCN non-amplified tumors of the former subgroup (9/35 cases, 25.7%; Supplementary Table 1). In general, age was not associated with ALK mutation status in the entire cohort (Fig. 2b). By contrast, patients of the non-high-risk cohort were significantly older when having ALK-mutated tumors in comparison to ALK wildtype tumors (15.8 versus 10.6 months, P = 0.038, Fig. 2b).

a ALK mutation frequencies in neuroblastoma obtained at diagnosis. b Age at diagnosis of patients with ALK-mutated versus non-mutated neuroblastoma obtained at diagnosis. c ALK amplification frequencies in neuroblastoma obtained at diagnosis. d Age at diagnosis of patients with ALK-amplified versus non-amplified neuroblastoma obtained at diagnosis. Boxplots show the median, first and third quartile (boxes), with whiskers indicating the minimum and maximum of the data within 1.5× the interquartile range. P values for a and c were calculated using Fisher’s exact test or Chi-squared test where appropriate, and for b and d using Wilcoxon–Mann–Whitney test. HR, high-risk; NHR, non-high-risk; ampl, amplified; m, months; St4, stage 4; loc, localized disease.
ALK amplification was detected in 4.7% of neuroblastomas obtained at diagnosis (14/298, Fig. 2c). However, this cohort was biased towards high-risk patients and may thus overestimate the overall frequency of such alterations in neuroblastoma. In the high-risk cohort and in MYCN-amplified tumors, ALK amplifications occurred in 5.5% (14/256) and 7.6% (14/185), respectively. ALK amplifications were not detected in non-high-risk tumors, nor in tumors without MYCN amplification. ALK amplifications occurred only in stage 3 (4/28, 14.3%) and stage 4 (10/234, 4.3%) neuroblastomas, and were significantly enriched in stage 3 compared to non-stage 3 tumors (14.3% versus 3.7%, P = 0.032, Fig. 2c). Age at diagnosis of patients having ALK-amplified tumors did not differ from that of patients with non-amplified tumors in the entire cohort (22.2 versus 28.2 months, P = 0.110, Fig. 2d), however, patients with high-risk disease showing ALK amplification were significantly younger than high-risk patients with tumors lacking ALK amplification (22.2 versus 29.9 months, P = 0.046).
ALK alterations in neuroblastoma at relapse
To assess ALK alterations in neuroblastoma at relapse or progression, we investigated ALK SNV and amplification in 198 and 172 tumors, respectively (Fig. 1). We found ALK SNV in 17.7% of the cases (35/198, Fig. 3a). The ALK mutation frequency at relapse did not differ between high-risk and non-high-risk patients (17.2% versus 19.1%, P = 0.827). We also did not observe differences in ALK mutation frequencies at relapse between MYCN-amplified and MYCN non-amplified tumors (12.5% versus 20.1%, P = 0.234), between tumors at stage 4 and other stages (19.7% versus 11.8%, P = 0.286), and between tumors from patients of different ages (Fig. 3a, b).

a ALK mutation frequencies in relapsed neuroblastoma. b Age at diagnosis of patients with ALK-mutated versus non-mutated relapsed neuroblastoma. c ALK amplification frequencies in relapsed neuroblastoma. d Age at diagnosis of patients with ALK-amplified versus non-amplified relapsed neuroblastoma. Boxplots show the median, first, and third quartile (boxes), with whiskers indicating the minimum and maximum of the data within 1.5× the interquartile range. P values for a and c were calculated using Fisher’s exact test or Chi-squared test where appropriate, and for b and d using Wilcoxon–Mann–Whitney test. HR, high-risk; NHR, non-high-risk; ampl, amplified; m, months; St4, stage 4; loc, localized disease.
ALK amplification was detected in 3.5% (6/172) of all relapsed tumors (Fig. 3c). In the high-risk subgroup, 5.0% (6/119) harbored amplification of ALK, all of which occurred in MYCN-amplified tumors (6/53) and were thus significantly enriched in this subgroup (11.3% versus 0%, P = 0.001). All patients with tumors showing ALK amplification at relapse had stage 4 disease (6/122 patients with stage 4 disease, 4.9%). Patients with tumors bearing ALK amplification at relapse were significantly younger than patients with ALK non-amplified tumors in the entire cohort (23.0 versus 42.7 months, P = 0.040, Fig. 3d) and in the subgroup of high-risk patients (23.0 versus 44.6 months, P = 0.009), while it did not differ between ALK-amplified and non-amplified tumors in the subgroup of patients with MYCN-amplified neuroblastoma (23.0 versus 29.0 months, P = 0.156). We noted, however, that ALK amplification frequencies at relapse were significantly elevated in patients <18 months with stage 4 disease as compared to patients ≥18 months with stage 4 neuroblastoma (16.7% versus 2.9%, P = 0.041, Fig. 3c).
Differences of ALK alteration frequencies in neuroblastoma at diagnosis and relapse
Evaluation of changes in ALK mutation and amplification frequencies over the course of disease revealed a significant increase of ALK SNV at relapse in the entire cohort (10.5% versus 17.7%, P = 0.009, Fig. 4a). A similar increase in potential de novo mutations was observed in the subgroup of paired samples (n = 101; 9.9% versus 16.8%, P = 0.214). Both tumors of high-risk (10.8% versus 17.2%, P = 0.057, Fig. 4b) and non-high-risk patients (10.1% versus 19.1%, P = 0.082, Fig. 4c), comprising both low- and intermediate-risk cases, were affected by the increment of mutations over time, at almost identical rates.

a ALK mutation frequencies in neuroblastoma obtained at diagnosis versus relapse in the entire cohort (left) and in the subgroup of patients of whom paired samples were available (right). b ALK mutation frequencies in neuroblastoma obtained at diagnosis versus relapse in high-risk tumors (HR) in the entire cohort (left) and in the subgroup of patients of whom paired samples were available (right). c ALK mutation frequencies in neuroblastoma obtained at diagnosis versus relapse in non-high-risk tumors (NHR) in the entire cohort (left) and in the subgroup of patients of whom paired samples were available (right). d ALK amplification frequencies in neuroblastoma obtained at diagnosis versus relapse in the entire cohort (left) and in the subgroup of patients of whom paired samples were available (right). e ALK alteration frequencies in stage 4 neuroblastoma of patients <18 months versus ≥18 months at diagnosis in the cohort of tumors obtained at diagnosis (left) and at relapse (right). f Schematic representation of the ALK tyrosine kinase domain (TKD) and the amino acid positions and frequencies of mutations detected in neuroblastoma samples at diagnosis and at relapse. g Frequencies of mutation types at position F1174 at diagnosis and at relapse. h Frequencies of mutation types at position F1245 at diagnosis and at relapse. i Frequencies of mutation types at position R1275 at diagnosis and at relapse. Of note, mutation type of one mutation detected in a tumor at relapse had remained unknown. P values were calculated using Fisher’s exact test.
ALK amplification frequencies did neither differ between tumors at diagnosis and relapse in the entire cohort (4.7% versus 3.5%, P = 0.639; Fig. 4d), nor in the subgroup of MYCN-amplified tumors (7.0% versus 11.3%, P = 0.386). In line with that finding, no de novo amplifications were found in the subgroup of paired samples obtained from 102 patients (ALK amplification in 4/102 cases, 3.9%). Information on both ALK mutation and amplification status was available for 310 samples (167 at diagnosis, 143 at relapse), 58 of which showed any type of ALK alteration (18.7%). In this cohort, we observed a particular high frequency of ALK alterations in tumors of patients with stage 4 disease <18 months of age, with 25% (5/20) of tumors being affected at diagnosis and 50% (6/12) at relapse (Fig. 4e). ALK mutations and amplifications occurred mutually exclusive in all samples investigated.
Allelic fractions were available for 62/76 and 24/36 ALK mutations detected at diagnosis and relapse, respectively, as some samples had been analyzed by dideoxy-sequencing only (Supplementary Table 1). At diagnosis, 2/62 mutations (3.2%) were detected at allelic fractions below 5%, and 15 (24.2%) at allelic fractions ≤20% (Supplementary Fig. 1a). At relapse, all mutations were detected at allelic fractions >5%, and 6/24 (25.0%) were found at allelic fractions ≤20% (Supplementary Fig. 1b). Allelic fractions (>20% versus ≤ 20%) were not associated with prognostic variables, such as MYCN amplification status (MYCN-amplified versus non-amplified, P = 0.332), risk group (high-risk versus non-high-risk, P = 1), stage (stage 4 versus stage 1–3/4S, P = 0.362) and age (≥18 versus < 18 months, P = 0.235, Supplementary Fig. 1c). Allelic fractions did also not differ between patients with stage 4 disease <18 months versus ≥ 18 months (P = 0.443, Supplementary Fig. 1d).
The vast majority (88.2%, 67/76) of ALK mutations at diagnosis and all mutations at relapse (100%, 36/36) were detected at the three common hotspot positions within the tyrosine kinase domain, i.e., R1275, F1174, and F1245 (Fig. 4f). In tumors at diagnosis, mutations also rarely affected other positions within the kinase domain, i.e., T1151, I1170, L1196, R1231, L1240, and Y1278 (Fig. 4f). F1174 was the position most frequently affected by mutations in tumors at diagnosis (28/76), showing an exchange of phenylalanine to leucine in most cases (Fig. 4g). The majority of mutations at position F1245 led to an exchange of phenylalanine to valine (6/13, Fig. 4h). Mutations at R1275 resulted in arginine to glutamine substitutions in all but one case and were the most frequent SNV in relapsed tumors (19/36, Fig. 4i). Of note, the frequency of R1275Q mutations increased significantly at relapse (32.9% versus 52.8%, P = 0.001, Fig. 4i).
Impact of ALK alterations on neuroblastoma patient outcome
The presence of ALK SNV in tumors obtained at diagnosis was not prognostic for both OS and EFS in the entire cohort (Fig. 5a, Supplementary Fig. 2a), the non-high-risk subgroup (Supplementary Fig. 2b, c), and the high-risk subgroup (Supplementary Fig. 2d, e). Strikingly high ALK mutation frequencies were found in tumors of patients with stage 4 disease younger 18 months of age, however, survival of these patients did not differ from that of corresponding patients lacking ALK mutations (Supplementary Fig. 3a, b). We also found no difference in outcome of patients with tumors bearing ALK mutations at allelic fractions >20% versus ≤ 20% versus no mutation in the entire cohort (Supplementary Fig. 4a, b). In high-risk patients, however, OS of patients with tumors harboring ALK mutations at allelic fractions >20% was significantly worse than that of patients whose tumors lacked an ALK mutation (Fig. 5b). EFS of high-risk patients with tumors harboring ALK mutations at allelic fraction ≤20% was significantly better than that of patients with tumors harboring ALK mutations at allelic fractions >20% or without ALK mutations (Fig. 5c). OS of patients with ALK-amplified tumors obtained at diagnosis was significantly poorer than that of patients with ALK non-amplified tumors (Fig. 5d), with a similar trend in the high-risk subgroup (Supplementary Fig. 5a), whereas ALK amplification was not prognostic for EFS in the entire cohort and in the high-risk subgroup (Supplementary Fig. 5b, c). As ALK amplification exclusively occurred in MYCN-amplified tumors, we also examined patient survival in this subgroup. We found that patients with ALK-amplified tumors appear to have poorer OS and EFS than those without ALK amplification, however, the differences did not reach significance (Supplementary Fig. 5d, e). We also assessed the prognostic relevance of ALK alterations, i.e., both mutations and amplification, within the subgroup of high-risk patients, however, we did not find significant impact on OS and EFS (Supplementary Fig. 6a, b).

a OS of patients with ALK-mutated tumors versus patients with ALK wildtype tumors at diagnosis (5-year OS, 66% versus 76%). b OS of high-risk patients with tumors harboring ALK mutations at allelic fractions >20% versus ≤ 20% versus ALK wildtype (5-year OS, 28% versus 57% versus 54%). c EFS of high-risk patients with tumors harboring ALK mutations at allelic fractions >20% versus ≤ 20% versus ALK wildtype (5-year EFS, 17% versus 57% versus 33%). d OS of patients with ALK-amplified tumors versus ALK non-amplified tumors at diagnosis (5-year OS, 22% versus 60%). e OS of patients with ALK-amplified versus non-amplified tumors at relapse (5-year OS, 0% versus 57%). f OS of patients with ALK-altered versus ALK-non-altered tumors at relapse (5-year OS, 37% versus 61%). P values were calculated by log-rank and, in case of non-proportional hazards, Gehan-Breslow test. OS, overall survival; y, years; ampl, amplification; AF, allelic fraction.
In patients with relapsed tumors, ALK mutations did not significantly affect OS or EFS in the entire cohort (Supplementary Fig. 7a, b), the non-high-risk subgroup (Supplementary Fig. 7c, d), and the high-risk subgroup (Supplementary Fig. 7e, f). The presence of ALK amplification at relapse was associated with fatal outcome, since all such patients had died. In detail, patients with ALK-amplified tumors at relapse had significantly poorer OS (Fig. 5e) and EFS in the entire cohort (Supplementary Fig. 8a), as well as in the high-risk subgroup (Supplementary Fig. 8b, c). In the subgroup of patients with MYCN-amplified tumors, ALK amplification at relapse was significantly associated with poorer EFS (Supplementary Fig. 8d), but not OS (Supplementary Fig. 8e). We found that genomic ALK alterations (i.e., both mutations and amplification) at relapse were associated with impaired OS and EFS in the entire group (Fig. 5f and Supplementary Fig. 9a, respectively), and with impaired EFS, but not OS, in the subgroup of high-risk patients (Supplementary Fig. 9b, c, respectively). ALK alterations at relapse were also associated with poorer secondary OS, calculated from the timepoint of relapse, in the entire cohort (Supplementary Fig. 10a), but not in the subgroup of high-risk patients (Supplementary Fig. 10b).
Outcome of neuroblastoma patients did not significantly depend on the type of ALK mutations, although OS of patients with F1174L mutations tended to be slightly worse than that of patients with tumors harboring other ALK mutations or ALK wildtype tumors (Supplementary Fig. 11a). We did not observe any associations of specific ALK mutations with MYCN amplification in tumors obtained at diagnosis (Supplementary Fig. 11b, c). In relapsed tumors, however, R1275Q mutations were predominantly found in MYCN non-amplified tumors (Supplementary Fig. 11b), and F1174L mutations tended to be associated with MYCN amplification (Supplementary Fig. 11c).
Dynamics of ALK alterations in neuroblastoma over the course of disease
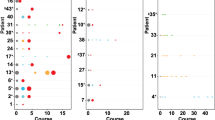
To investigate the dynamics and clonal evolution of ALK aberrations over the course of disease, we determined ALK alterations in longitudinal samples, taken at diagnosis and at relapse or progression. ALK mutations were analyzed in 202 samples (101 diagnostic/relapse pairs) by panel NGS (n = 71), dideoxy-sequencing (n = 27), WES (n = 44), or a combination of at least two of these methods (n = 60). As outlined above, we found ALK SNV in 10/101 tumors (9.9%) at diagnosis, and in additional 7 tumors (total, 17/101 cases, 16.8%) at relapse (Fig. 4a). To further assess clonal development of mutations detected within this cohort, allelic fractions of mutations were determined by mutation-specific ddPCR. We found that allelic fractions detected by ddPCR matched well with allelic fractions determined by massively parallel sequencing (Fig. 6a). In addition, this approach validated all mutations identified by dideoxy-sequencing or panel NGS, except in one sample (patient P7). In this case, an R1275Q mutation detected at diagnosis was not detected by panel NGS but by ddPCR at relapse at an allelic fraction of 0.35% (Fig. 6a), which might be due to low tumor infiltration in the relapse sample (5%). In two cases, we found two distinct ALK mutations within the same tumor. One tumor harbored an F1174L mutation at diagnosis and at relapse, and an additional F1245L mutation at low allelic fraction at diagnosis, which had disappeared at relapse (patient P2, Fig. 6a). In another tumor, two different ALK de novo mutations occurred during the course of the disease (R1275Q and F1174L, patient P13, Fig. 6a, see below). Potential de novo ALK mutations at relapse were mostly R1275Q mutations (n = 4), while F1174L, F1174C, F1245L, and F1245C mutations were detected in one sample each. Allelic fractions of R1275Q mutations were higher than those of other de novo mutations, however, the number of cases was too small to draw final conclusions (Supplementary Fig. 11d).

a Comparison of allelic fractions of ALK mutations detected at diagnosis and relapse by NGS versus ddPCR. In two patients (P2 and P13), two different ALK mutations were detected in the same tumor samples. X indicates that no material was available, check marks indicate that mutations were detected by dideoxy-sequencing only, thus leaving their allelic fraction unknown. b Longitudinal monitoring of allelic fractions (as determined by ddPCR) of two different ALK mutations (R1275Q and F1174L) over the course of disease in an individual patient. The allelic fraction of the HRAS mutation occurring at week 275 was determined by panel NGS. ALK inhibitory treatment and the clinical course of disease are shown at the top, with arrowheads indicating progression or relapse of disease. Timepoints and localizations of biopsies are indicated by arrows; biopsies of metastasis were taken from the same maxillary metastasis, except the last biopsy, which was derived from bone marrow. c Schematic diagram for the clonal evolution of cancer cell populations reconstructed from whole-exome sequencing data in patient P13. T1–T6 represent the consecutive biopsies that were taken from the patient. Gray boxes highlight clones that were private to the respective biopsy. ALK and HRAS mutations are indicated as they appeared in chronological order. d Clonal composition of each biopsy. The ancestral clone C0 is illustrated on the left to indicate its presence in every biopsy. AF, allelic fraction; NGS, next generation sequencing; ddPCR, digital droplet PCR; mut, mutation; D, diagnosis; R, relapse; met, metastasis; PT, primary tumor; BM, bone marrow.
Patient P13 was particularly informative for analyzing the clonal evolution of ALK mutations in neuroblastoma, as tumor material of six different timepoints was available for monitoring allelic fractions over the course of disease by ddPCR (Fig. 6b). While ALK was wild-type in the metastatic tumor at diagnosis, an F1174L mutation occurred at low allelic fractions in the second biopsy which was derived from the primary tumor. This mutation was also found in the fourth biopsy, obtained from a maxillary metastasis, but not at any other timepoint. In addition, an R1275Q mutation occurred in the fourth biopsy with an allelic fraction of 40.5%, which gradually increased to nearly 80% in the sixth biopsy. The patient received treatment with the ALK inhibitor ceritinib [4] after the fourth biopsy, which was changed to lorlatinib after the fifth biopsy. In the sixth biopsy, the tumor had acquired an additional HRAS mutation at codon 61 (Q61K), and the patient died shortly afterwards due to disease progression [34]. Reconstruction of the clonal evolution of this tumor revealed that all ALK mutations and the HRAS alteration had developed from the same ancestral clone C0 (Fig. 6c). Surprisingly, the two clones C2 and C7 that harbored subclonal F1174L mutations did not share common ancestors besides C0, indicating that the two F1174L mutations had developed independently. By contrast, the last three timepoints were dominated by clones that carried the R1275Q mutation, with C6 representing the ancestor of all subsequent clones (Fig. 6c, d). Together, the chronology of the occurrence of mutations in this tumor indicates that ALK F1174L mutations may not necessarily prevail in neuroblastoma, both in the absence and presence of a concurrent ALK R1275Q mutation.
Discussion
We here provide a detailed view on the development of genomic ALK alterations in neuroblastoma over the course of the disease. We found an overall ALK mutation frequency of 10.5% in a large and representative cohort of neuroblastomas obtained at diagnosis, which is largely in the range of ALK mutation frequencies reported previously [8,9,10,11,12,13,14,15]. In our study, the prevalence of ALK mutations did not differ between clinical risk groups, which has been controversially discussed in earlier studies, some of which had suggested that ALK mutations may be more prevalent in high-risk group tumors [5, 12, 18, 19, 35]. In addition, we did not find enrichment of ALK mutations in MYCN-amplified neuroblastomas [15, 19]. We noticed, however, that ALK mutation frequencies were highest in stage 4 tumors, and in stage 4 tumors of patients younger than 18 months of age in particular. The enrichment of ALK mutations in the latter subgroup, which is considered as intermediate risk if MYCN amplification is absent, has not been reported in previous studies [11, 12, 18, 19, 36,37,38]. ALK mutations were not prognostic in our study, except in high-risk patients when allelic fractions were taken into account. In the latter analysis, ALK mutations were associated with poorer outcome when they occurred at allelic fractions >20%, which is in line with previous results [15].
ALK amplification was found in 4.7% of all tumors and 5.5% of high-risk tumors at diagnosis, which is in line with the frequency of ALK amplifications reported previously [5, 11, 12, 15, 17,18,19]. Similar to other studies, ALK amplification occurred exclusively in MYCN-amplified neuroblastomas and were mutually exclusive with ALK point mutations [5, 19, 39]. In addition, we noted that ALK amplification was associated with young patient age and with stage 3 in tumors obtained at diagnosis. Patients whose tumors harbored ALK amplification had worse overall survival, supporting the notion that these tumors constitute a highly aggressive subtype of neuroblastoma [12].
Only few studies with limited patient numbers have reported on ALK mutation and amplification frequencies in relapsed or progressive neuroblastoma [10, 40, 41], despite their potential relevance as therapeutic targets for these patients with poor survival expectations. In a cohort of roughly 200 cases, we found ALK point mutations in 17.7% of tumors at relapse or progression, which was a significant increase compared to cases at diagnosis. Of note, the mutation frequency of the entire cohort closely matched the frequency detected in relapsed tumors of the sub-cohort of patients for whom paired samples from diagnosis and relapse were available (n = 101), suggesting that ALK mutations may occur de novo in 7% of relapsed or progressive neuroblastoma. We also found that mutation frequencies increased at similar rates in high-risk and non-high-risk tumors, indicating that ALK mutations may develop under various conditions of selective pressure, ranging from limited to high treatment intensities. In a previous study of 54 paired neuroblastoma samples, an increase of the ALK mutation frequency from 16.7% at diagnosis to 25.9% at relapse has been reported [10]. A deep genome sequencing approach in this study revealed de novo ALK mutations in only 3.7% at relapse, whereas some ALK mutations were found at sub-clonal levels already at diagnosis. This finding is in contrast to our observations, as we could not detect putative de novo mutations of relapse samples in their corresponding diagnostic counterparts with highly sensitive mutation-specific ddPCR assays. It has to be noted, though, that we cannot formally exclude that mutations had been present already at the time of diagnosis in very minor subclones or in tumor regions other than the biopsy sample. Another study of 23 paired samples revealed ALK mutations in 30.4% at diagnosis and 43.5% at relapse, however, the small patient number limits its informative value on ALK mutation frequencies in relapsed neuroblastoma, and no attempts to assess sub-clonality had been made [40].
We found that the type of mutation occurring most frequently in relapsed neuroblastomas was R1275Q, with a significant increase of such mutations compared to tumors at diagnosis. The abundance of these mutations at relapse was probably caused by de novo occurrence during the course of disease, as we detected R1275Q mutations in four of the seven tumors that harbored ALK mutations only at relapse. In addition, we did not find clear associations of mutation types with patient outcome, arguing against significant enrichment effects of R1275Q mutations through disproportionally high relapse rates in tumors bearing such mutations. We also noted that R1275Q mutations at relapse were more prevalent in MYCN non-amplified tumors, while F1174L tended to be associated with MYCN-amplified disease, as described previously [9, 11, 15]. Detailed analyses of clonal evolution of two ALK mutations that emerged in an individual tumor revealed F1174L mutations at low allelic fractions in the second and fourth biopsy, and an R1275Q mutation at high allelic fractions from the time of fourth biopsy to the time of final progression. These data demonstrate that (i) the presence of an ALK mutation may not always provide selective advantage in neuroblastoma, and (ii) the mutation with the putatively highest oncogenic potential F1174L [11] does not necessarily prevail over the R1275Q mutation, both under conventional treatment and ALK inhibitor therapy.
Treatment strongly affects neuroblastoma patient outcome and may therefore be a confounding factor in prognostic biomarker studies. The fact that patients from successive GPOH trials or registries with evolving treatment strategies had been analyzed in this study may thus have biased the results. It has to be noted, though, that the vast majority of patients had been treated within the trial NB2004 and the subsequent registry NB2016, which kept the therapeutic concept of NB2004, resulting in homogeneous treatment of most patients included in this study. Heterogeneity of diagnostic procedures used for detection of ALK mutations may be another limiting factor of this study. A fraction of cases had been analyzed by dideoxy-sequencing only, which has a lower sensitivity than NGS-based approaches [8, 9]. Sequencing results from 125 samples examined by both dideoxy-sequencing and approaches with higher sensitivity (i.e., NGS or ddPCR), however, were concordant in 99.2% (124/125), with dideoxy-sequencing detecting ALK mutations at surprisingly low allelic fractions going below 5% (Patient P652, Supplementary Table 1). In fact, we found only one mutation by ddPCR at an allelic fraction of 2.3% that had been missed by dideoxy-sequencing, suggesting that mutation frequencies reported in this study are accurate. Nevertheless, we would prefer NGS-based methods for ALK diagnostics in future studies, as they provide comprehensive genetic information with high sensitivity and precise allelic fractions of mutations in a single approach. Other factors that may have affected the detection sensitivity of ALK alterations are low tumor cell contents in some samples and spatial heterogeneity of genetic alterations within the tumor. Finally, small sample sizes in specific subgroups, such as the cohorts of ALK amplified tumors or young patients with stage 4 disease, may limit interpretation of the study results.
Considering the high incidence of putative de novo ALK point mutations at relapse and their potential therapeutic consequences, we suggest to determine the genomic ALK status in all neuroblastoma cases at relapse or progression. Extensive testing might be even more important as we found that R1275Q mutations were most frequently detected at relapse, a mutation that has been reported to be sensitive to ALK inhibitory treatment [4, 42]. By contrast, assessment of ALK amplification in relapsed disease may be dispensable if already determined at diagnosis, as novel ALK amplification was not detected at relapse. In addition, we suggest to also assess genomic ALK alterations systematically in intermediate-risk patients at diagnosis, which have been underappreciated in previous studies. The finding that intermediate-risk neuroblastomas of young stage 4 patients at diagnosis harbor ALK mutations at high frequencies provides a starting point for targeted treatment of these children. While these patients have overall favorable outcome with current therapies [43, 44], it has to be noted that they receive quite intensive cytotoxic treatment, which is associated with substantial toxicities and long-term sequelae. We therefore suggest to evaluate the therapeutic benefit of ALK inhibitors in intermediate-risk patients with ALK-mutated neuroblastoma in prospective clinical trials.
Data availability
Patient whole-genome and whole-exome sequencing data have been published previously and are deposited at the European Genome-phenome Archive (https://ega-archive.org) under study accession numbers EGAS00001001308 and EGAS00001003244, respectively. Sequencing data of INFORM patients is available via accession number EGAS00001005112. Due to the sensitive nature of these patient datasets, the WGS data is subject to approval by the data provider. Please see the corresponding EGA data access committee (DAC) for more details on the procedure (https://ega-archive.org/dacs/EGAC00001000361).
References
London WB, Castleberry RP, Matthay KK, Look AT, Seeger RC, Shimada H, et al. Evidence for an age cutoff greater than 365 days for neuroblastoma risk group stratification in the Children’s Oncology Group. J Clin Oncol. 2005;23:6459–65.
Peifer M, Hertwig F, Roels F, Dreidax D, Gartlgruber M, Menon R, et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature. 2015;526:700–4.
Ackermann S, Cartolano M, Hero B, Welte A, Kahlert Y, Roderwieser A, et al. A mechanistic classification of clinical phenotypes in neuroblastoma. Science. 2018;362:1165–70.
Fischer M, Moreno L, Ziegler DS, Marshall LV, Zwaan CM, Irwin MS, et al. Ceritinib in paediatric patients with anaplastic lymphoma kinase-positive malignancies: an open-label, multicentre, phase 1, dose-escalation and dose-expansion study. Lancet Oncol. 2021;22:1764–76.
Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008;455:967–70.
Lee CC, Jia Y, Li N, Sun X, Ng K, Ambing E, et al. Crystal structure of the ALK (anaplastic lymphoma kinase) catalytic domain. Biochem J. 2010;430:425–37.
Carpenter EL, Mosse YP. Targeting ALK in neuroblastoma-preclinical and clinical advancements. Nat Rev Clin Oncol. 2012;9:391–9.
Javanmardi N, Fransson S, Djos A, Sjoberg RM, Nilsson S, Truve K, et al. Low frequency ALK hotspots mutations in neuroblastoma tumours detected by ultra-deep sequencing: implications for ALK inhibitor treatment. Sci Rep. 2019;9:2199.
Bellini A, Bernard V, Leroy Q, Rio Frio T, Pierron G, Combaret V, et al. Deep sequencing reveals occurrence of subclonal ALK mutations in neuroblastoma at diagnosis. Clin Cancer Res. 2015;21:4913–21.
Schleiermacher G, Javanmardi N, Bernard V, Leroy Q, Cappo J, Rio Frio T, et al. Emergence of new ALK mutations at relapse of neuroblastoma. J Clin Oncol. 2014;32:2727–34.
De Brouwer S, De Preter K, Kumps C, Zabrocki P, Porcu M, Westerhout EM, et al. Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clin Cancer Res. 2010;16:4353–62.
Bresler SC, Weiser DA, Huwe PJ, Park JH, Krytska K, Ryles H, et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell. 2014;26:682–94.
Schulte JH, Bachmann HS, Brockmeyer B, Depreter K, Oberthur A, Ackermann S, et al. High ALK receptor tyrosine kinase expression supersedes ALK mutation as a determining factor of an unfavorable phenotype in primary neuroblastoma. Clin Cancer Res. 2011;17:5082–92.
O’Donohue T, Gulati N, Mauguen A, Kushner BH, Shukla N, Rodriguez-Sanchez MI, et al. Differential impact of ALK mutations in neuroblastoma. JCO Precis Oncol. 2021;5:492–500.
Bellini A, Potschger U, Bernard V, Lapouble E, Baulande S, Ambros PF, et al. Frequency and prognostic impact of ALK amplifications and mutations in the European Neuroblastoma Study Group (SIOPEN) High-Risk Neuroblastoma Trial (HR-NBL1). J Clin Oncol. 2021;39:3377–90.
Trigg RM, Turner SD. ALK in neuroblastoma: biological and therapeutic implications. Cancers. 2018;10:113.
Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455:930–5.
George RE, Sanda T, Hanna M, Frohling S, Luther W 2nd, Zhang J, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455:975–8.
Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008;455:971–4.
Fransson S, Hansson M, Ruuth K, Djos A, Berbegall A, Javanmardi N, et al. Intragenic anaplastic lymphoma kinase (ALK) rearrangements: translocations as a novel mechanism of ALK activation in neuroblastoma tumors. Genes Chromosomes Cancer. 2015;54:99–109.
Cazes A, Louis-Brennetot C, Mazot P, Dingli F, Lombard B, Boeva V, et al. Characterization of rearrangements involving the ALK gene reveals a novel truncated form associated with tumor aggressiveness in neuroblastoma. Cancer Res. 2013;73:195–204.
Mosse YP, Voss SD, Lim MS, Rolland D, Minard CG, Fox E, et al. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: a Children’s Oncology Group Study. J Clin Oncol. 2017;35:3215–21.
Mosse YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group Phase 1 Consortium Study. Lancet Oncol. 2013;14:472–80.
Brodeur GM, Pritchard J, Berthold F, Carlsen NL, Castel V, Castelberry RP, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol. 1993;11:1466–77.
Brodeur GM, Seeger RC, Barrett A, Berthold F, Castleberry RP, D’Angio G, et al. International criteria for diagnosis, staging, and response to treatment in patients with neuroblastoma. J Clin Oncol. 1988;6:1874–81.
Theissen J, Boensch M, Spitz R, Betts D, Stegmaier S, Christiansen H, et al. Heterogeneity of the MYCN oncogene in neuroblastoma. Clin Cancer Res. 2009;15:2085–90.
Ihle MA, Fassunke J, Konig K, Grunewald I, Schlaak M, Kreuzberg N, et al. Comparison of high resolution melting analysis, pyrosequencing, next generation sequencing and immunohistochemistry to conventional Sanger sequencing for the detection of p.V600E and non-p.V600E BRAF mutations. BMC Cancer. 2014;14:13.
Cun Y, Yang TP, Achter V, Lang U, Peifer M. Copy-number analysis and inference of subclonal populations in cancer genomes using Sclust. Nat Protoc. 2018;13:1488–501.
Herling CD, Abedpour N, Weiss J, Schmitt A, Jachimowicz RD, Merkel O, et al. Clonal dynamics towards the development of venetoclax resistance in chronic lymphocytic leukemia. Nat Commun. 2018;9:727.
Worst BC, van Tilburg CM, Balasubramanian GP, Fiesel P, Witt R, Freitag A, et al. Next-generation personalised medicine for high-risk paediatric cancer patients—the INFORM pilot study. Eur J Cancer. 2016;65:91–101.
van Tilburg CM, Pfaff E, Pajtler KW, Langenberg KPS, Fiesel P, Jones BC, et al. The pediatric precision oncology INFORM registry: clinical outcome and benefit for patients with very high-evidence targets. Cancer Discov. 2021;11:2764–79.
Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44:1104–10.
R Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria; 2021. https://www.R-project.org/.
Berlak M, Tucker E, Dorel M, Winkler A, McGearey A, Rodriguez-Fos E, et al. Mutations in ALK signaling pathways conferring resistance to ALK inhibitor treatment lead to collateral vulnerabilities in neuroblastoma cells. Mol Cancer. 2022;21:126.
Sausen M, Leary RJ, Jones S, Wu J, Reynolds CP, Liu X, et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet. 2013;45:12–7.
Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013;45:279–84.
Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. 2012;483:589–93.
Cheung NK, Zhang J, Lu C, Parker M, Bahrami A, Tickoo SK, et al. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA 2012;307:1062–71.
Ogawa S, Takita J, Sanada M, Hayashi Y. Oncogenic mutations of ALK in neuroblastoma. Cancer Sci. 2011;102:302–8.
Eleveld TF, Oldridge DA, Bernard V, Koster J, Colmet Daage L, Diskin SJ, et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet. 2015;47:864–71.
Padovan-Merhar OM, Raman P, Ostrovnaya I, Kalletla K, Rubnitz KR, Sanford EM, et al. Enrichment of targetable mutations in the relapsed neuroblastoma genome. PLoS Genet. 2016;12:e1006501.
Foster JH, Voss SD, Hall DC, Minard CG, Balis FM, Wilner K, et al. Activity of crizotinib in patients with ALK-aberrant relapsed/refractory neuroblastoma: a Children’s Oncology Group Study (ADVL0912). Clin Cancer Res. 2021;27:3543–8.
Kim C, Choi YB, Lee JW, Yoo KH, Sung KW, Koo HH. Excellent treatment outcomes in children younger than 18 months with stage 4 MYCN nonamplified neuroblastoma. Korean J Pediatr. 2018;61:53–8.
Berthold F, Rosswog C, Christiansen H, Fruhwald M, Hemstedt N, Klingebiel T, et al. Clinical and molecular characterization of patients with stage 4(M) neuroblastoma aged less than 18 months without MYCN amplification. Pediatr Blood Cancer. 2021;68:e29038.
Acknowledgements
We thank Nadine Hemstedt, Yvonne Kahlert, Witali Lorenz, and Anne Welte for their technical assistance and Martina Breuer for assistance with the clinical data. This work was funded through the Else Kröner-Fresenius Stiftung (2016-Kolleg-19 to CR) and supported by the Köln Fortune Program/Faculty of Medicine, University of Cologne (to CR). This work was further supported by the Deutsche Forschungsgemeinschaft (DFG; grant no. FI 1926/2-1 to MF), the Förderverein für krebskranke Kinder e.V. Köln (endowed chair to MF), the Fördergesellschaft Kinderkrebs-Neuroblastom-Forschung e.V. (to MF), the German Ministry of Science and Education (BMBF) as part of the e:Med initiative (grant no. 01ZX1303, 01ZX1603, 01ZX1307 and 01ZX1607 to MF), the Deutsche Krebshilfe (grant no. 70113130 to MF), and the DFG as part of the SFB1399 (to MF, MP, and RB). The INFORM program is financially supported by the German Cancer Research Center (DKFZ), the German Cancer Consortium (DKTK), the German Federal Ministry of Education and Research (BMBF), the German Federal Ministry of Health (BMG), the Ministry of Science, Research and the Arts of the State of Baden-Württemberg (MWK BW); the German Cancer Aid (DKH), the German Childhood Cancer Foundation (DKS), RTL television, the aid organization BILD hilft e.V. (Ein Herz für Kinder) and the generous private donation of the Scheu family.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
CR conceived of the study, participated in its design, coordination, and molecular genetic analysis, and drafted the manuscript. JF participated in molecular genetic analysis, carried out ddPCR analyses, and participated in drafting the manuscript. AE carried out statistical analysis. BS participated in formal analysis. SM provided resources for the study. CB participated in sequence alignment and subsequent analysis of WES and WGS data. MC carried out analyses on clonal evolution of longitudinal samples. SA participated in formal analysis. JT participated in formal analysis. MB provided resources for the study. BJ provided resources for the study. KS provided resources for the study. JA participated in sequencing analysis. PN participated in sequencing analysis. MO provided resources for the study. FB provided resources for the study. MP participated in sequence alignment and in interpretation of the results. RB provided resources for the study. FW provided resources for the study. JS provided resources for the study. TS participated in formal analysis and provided resources for the study. BH provided resources for the study and participated in formal and statistical analysis. MF conceived of the study, participated in its design and coordination, and helped to draft the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
All co-authors have read and approved the final version of the manuscript and its submission to this journal.
Ethics approval and consent to participate
Informed consent was given from all patients or their legal guardians. The Institutional Review Board of the Medical Faculty of the University of Cologne granted the ethical approval for the use of specimens.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rosswog, C., Fassunke, J., Ernst, A. et al. Genomic ALK alterations in primary and relapsed neuroblastoma. Br J Cancer 128, 1559–1571 (2023). https://doi.org/10.1038/s41416-023-02208-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-023-02208-y
