
Abstract
Background
Chemotherapy resistance is the major cause of recurrence in patients with colorectal cancer (CRC). A previous study found that Fusobacterium (F.) nucleatum promoted CRC chemoresistance. Additionally, metformin rescued F. nucleatum-induced tumorigenicity of CRC. Here, we aimed to investigate whether metformin could revert F. nucleatum-induced chemoresistance and explore the mechanism.
Methods
The role of metformin in F. nucleatum-infected CRC cells was confirmed using cell counting kit 8 assays and CRC xenograft mice. Stemness was identified by tumorsphere formation. Bioinformatic analyses were used to explore the regulatory molecules involved in metformin and F. nucleatum-mediated regulation of the sonic hedgehog pathway.
Results
We found that metformin abrogated F. nucleatum-promoted CRC resistance to chemotherapy. Furthermore, metformin attenuated F. nucleatum-stimulated stemness by inhibiting sonic hedgehog signaling. Mechanistically, metformin diminished sonic hedgehog signaling proteins by targeting the MYC/miR-361-5p cascade to reverse F. nucleatum-induced stemness, thereby rescuing F. nucleatum-triggered chemoresistance in CRC.
Conclusions
Metformin acts on F. nucleatum-infected CRC via the MYC/miR-361-5p/sonic hedgehog pathway cascade, subsequently reversing stemness and abolishing F. nucleatum-triggered chemoresistance. Our results identified metformin intervention as a potential clinical treatment for patients with chemoresistant CRC with high amounts of F. nucleatum.
Similar content being viewed by others


Background
Colorectal cancer (CRC) incidence and mortality remain high, constituting a major public health burden [1]. Chemotherapy is the most common treatment to reduce tumor growth and inhibit tumor metastasis. 5-fluorouracil (5-Fu) and oxaliplatin are the first-line chemotherapeutic drugs in the treatment of patients with advanced CRC [2, 3]. However, CRC cells usually become resistant to chemotherapeutic agents, leading to tumor recurrence. It has been reported that more than 30% patients receiving surgery for stage II and III colon cancer are resistant to 5-Fu-based adjuvant chemotherapy within 8 years of follow-up [4]. Furthermore, about 50% of patients who were diagnosed with metastatic CRC were resistant to 5-Fu-based chemotherapy and their 5-year survival rate was lower than 15% [5]. Thus, it is necessary to determine the molecular mechanisms of CRC chemoresistance and develop new and effective strategies to improve chemosensitivity.
Chemoresistance of CRC is a complex process resulting from the interplay between intrinsic and extrinsic factors, including gene regulation, epigenetic modification, hypoxia, and the gut microbiota [6, 7]. Accumulating evidence indicates that the gut microbiota profoundly influences the effect of chemotherapy with three main outcomes: facilitation of drug efficacy; restraint and compromise of anticancer effects; and mediation of toxicity effects [8]. Some bacteria, such as Lactobacillus johnsonii, L. murinus, and Bacteroidetes, promote chemotherapy efficacy [9, 10]. However, some bacteria, such as F. nucleatum, a well-known CRC pathogenic factor, drive high chemoresistance [11,12,13,14]. Our previous work demonstrated that an increased abundance of F. nucleatum contributes to post-chemotherapy recurrence in patients with CRC [15]. As such, this is an important clinical consideration when evaluating conventional chemotherapeutic treatment of patients with CRC with a high amount of F. nucleatum. Therefore, a strategy capable of modulating the effects of F. nucleatum in the management of CRC is highly desirable. However, to date, the problem of CRC chemoresistance caused by F. nucleatum has not been solved. Our previous work pinpointed that metformin could elicit an antitumor effect by restoring the disordered intestinal microbiota and alleviating F. nucleatum-induced colorectal tumorigenesis [16]. Metformin, which has been used widely in type 2 diabetes (T2D) for more than 60 years, has attracted interest in the prevention and treatment of various cancers recently [17,18,19,20]. A phase II clinical trial reported a modest efficacy profile of a combination of metformin and 5-Fu in patients with refractory CRC [21]. These raised the possibility that metformin might play a role in treating chemoresistance in patients with CRC with high abundance of F. nucleatum. In the current work, we aimed to determine whether and how metformin affects F. nucleatum-induced CRC chemoresistance. We found that metformin plays a critical role in abrogating F. nucleatum-induced CRC chemoresistance in response to 5-Fu and oxaliplatin through the MYC/miR-361-5p cascade, and subsequently suppression of sonic hedgehog signaling and CRC cell stemness. Our approach might inspire a future combined treatment comprising metformin with chemotherapeutic drugs to managing recurrence in patients with CRC with high amounts of F. nucleatum.
Methods
Cell culture and treatment
Human CRC cell lines with different degrees of differentiation (HT-29, well differentiated; HCT 116, moderately differentiated) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in McCoy’5 A medium (GIBCO, Carlsbad, CA, USA) with 10% (vol/vol) fetal bovine serum (FBS) (GIBCO) at 37 °C in a humidified 5% CO2 atmosphere. 1 mM metformin (Sigma-Aldrich, St. Louis, MO, USA) was used to treat CRC cells for 24–72 h. F. nucleatum at a multiplicity of infection (MOI) of 100 was used to treat CRC cells for 4 h. The miR-361-5p mimics and miR-361-5p inhibitor were transfected into CRC cells using the DharmaFECT transfection reagent (Thermo Fisher Scientific, Waltham, MA, USA). The lentivirus control and lentiviral vectors expressing miR-361-5p inhibitors were purchased from Shanghai OBiO medical biotechnology company (Shanghai, China).
F. nucleatum culture
The F. nucleatum strain (ATCC 25586) was obtained from the ATCC. F. nucleatum was cultured for 24 h at 37 °C under anaerobic conditions in brain heart infusion (BHI) broth containing hemin, Vitamin K1, K2HPO4, and L-Cysteine.
Mouse models
For the xenograft experiments, 4–5-week-old male Balb/c nude mice were purchased from the Shanghai Model Organisms Center (Shanghai, China) and housed in specific pathogen-free (SPF) conditions. HCT 116 cells (5 × 106 or 2.5 × 106) with different treatments were suspended in 100 μl of phosphate-buffered saline (PBS) and inoculated subcutaneously into the right axilla of the mice to establish the CRC xenograft model. Six days after subcutaneous injection, mice with xenograft tumors and no significant differences in tumor load were divided into different groups (n = 5). F. nucleatum (MOI = 100, 100 μl per mouse) were given by multipoint intratumoral injection, twice per week for 2 weeks. Metformin (125 mg/Kg) was administered via gavage every day for two weeks. Chemotherapeutic agents (FOX: 5-Fu 5 mg/Kg + Oxaliplatin 6 mg/Kg) were administered by intraperitoneal injection, twice per week for 2 weeks.
To explore the role of metformin in F. nucleatum-induced chemoresistance in vivo, we designed seven groups: (i) Control group; (ii) Metformin group; (iii) F. nucleatum group; (iv) FOX group; (v) FOX and metformin group; (vi) FOX and F. nucleatum group; (vii) FOX, F. nucleatum, and metformin group.
To explore whether metformin reversed F. nucleatum-mediated chemoresistance via miR-361-5p, we generated stable miR-361-5p knockdown HCT 116 cells by transducing lentiviruses expressing miR-361-5p inhibitors. We designed four groups: (i) FOX group; (ii) FOX and F. nucleatum group; (iii) FOX, F. nucleatum and metformin group; (iv) FOX, F. nucleatum, metformin, and miR-361-5p inhibitor group.
The length and width of the tumors were measured every 2–3 days. Tumor volume (mm3) was calculated as (length × width2) /2. If the tumor volume exceeded 2000 mm3, the mouse was killed for welfare reasons. After 2 weeks, the mice were killed humanely and subcutaneous tumors were collected for subsequent analysis of sonic hedgehog (SHH), GLI family zinc finger 1 (GLI1), SRY box transcription factor 2 (SOX2) and Nanog homeobox (NANOG) expression. Mouse experiments were conducted according to the guidelines approved by the Institutional Animal Care and Use Committee of Renji Hospital, School of Medicine, Shanghai Jiaotong University.
RNA extraction and quantitative real-time PCR
Total RNA was extracted from HT-29 and HCT 116 cells using the Trizol reagent (Takara, Shiga, Japan). For the analysis of mRNA and primary microRNA (pri-miRNA), 1 μg of total RNA was reverse transcribed into first-strand cDNA using a PrimeScript RT Reagent Kit (Takara). For the miRNAs, 500 ng of total RNA was reverse transcribed using Mir-X TM miRNA First-Strand Synthesis Kit (Takara). The quantitative real-time PCR step was conducted using TB Green Premix Ex Taq™ II (Takara) on an ABI StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The relative gene expression was calculated using 2−ΔΔCt method and expressed as the fold change [22]. ACTB (encoding β-actin) and U6 were used, respectively, as endogenous references for the mRNA, pri-miRNA, and miRNA assays. The mRNA primers used in real-time PCR are shown in Table 1. The miRNA primers and pri-miRNA primers were purchased from GeneCopoeia (Rockville, MD, USA).
Western blotting
Proteins (60 μg) were separated using 8% or 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), transferred onto polyvinylidene fluoride (PVDF) membranes (Bio-Rad, Hercules, CA, USA), and incubated with primary antibodies overnight. The membranes were then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (1:3000, KangChen, Shanghai, China) and the signals were visualized using an enhanced luminescence (ECL) Kit (Thermo Fisher Scientific). The following commercial antibodies were used: anti-SHH (Cell Signaling Technology, Danvers, MA, USA), anti-GLI1 (Cell Signaling Technology), anti-SOX2(Cell Signaling Technology), anti-NANOG (Cell Signaling Technology), anti-MYC antibody (Abcam, Cambridge, UK) and anti-β-actin (Cell Signaling Technology). All antibodies were used at a dilution of 1:1000.
Tumorsphere formation assay
CRC cells were exposed to F. nucleatum (MOI = 100) for 4 h. Then, the medium containing F. nucleatum was replaced with McCoy’5 A medium supplemented with 10% FBS. After 24 h, HT-29 and HCT 116 cells were seeded in low-adherent 96-well plates at a density of 400 cells per well in serum-free tumorsphere medium with or without 1 mM metformin. The tumorsphere medium consisted of Dulbecco’s Modified Eagle Medium/F12 (GIBCO) supplemented with necessary growth factors [23]. After a 4-day incubation, the tumorspheres were counted under a light microscope and their size were quantified using Image J software (NIH, Bethesda, MD, USA).
Cell Counting Kit-8 (CCK-8) assay
CRC cells were seeded at 2500 cells per well into 96-well plates with 100 μl of medium. After attachment, HT-29 and HCT 116 cells were treated with different interventions for 72 h. Then, 10 μl of CCK-8 solution (Dojindo, kumamoto, Japan) was added to each well at specific times and the absorbance value at 450 nm was measured after 2 h of incubation.
Data acquisition
For the analysis of cancer stem cell pathways, the data and sample information of GSE102573, GSE90944, and GSE67342 were downloaded from the Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/). GSE102573 comprises the expression profile of Caco-2 cells infected with F. nucleatum or not. GSE90944 comprises RNA sequencing (RNA-seq) data of HT-29 cells with or without F. nucleatum treatment. GSE67342 comprises expression data of LoVo cells treated by metformin. The single-sample gene set enrichment analysis (ssGSEA) score was calculated using Gene Set Variation Analysis (GSVA) in the R package with the chemical and genetic perturbations gene set collection (MSigDB C2 CGP; 3358 gene sets available) [24]. Differential expression analysis was conducted using the R package Limma and an adjusted (Bonferroni–Holm method) two-tailed P < 0.05 was considered statistically significant.
For the analysis of metformin-regulated genes, we used the dataset GSE67342, which includes gene expression of LoVo cells treated by metformin. An adjusted P < 0.1 was considered statistically significant.
Dual luciferase assay
HT-29 and HCT 116 cells were co-transfected with 100 ng of the reporter plasmids and 10 ng of the pRL-TK-Renilla-luciferase plasmids using the FuGene transfection reagent (Promega, Madison, WI). At 6 h after transfection, the medium was replaced and cells were treated with metformin and/or F. nucleatum. At 72 h after transfection, dual luciferase activities were estimated using a Dual-Luciferase Assay Kit (Promega) according to the manufacturer’s instructions. Each transfection was performed in four complex wells and repeated twice.
Statistical analysis
All data analyses were performed using GraphPad Prism version 7 (GraphPad Inc., La Jolla, CA, USA). Data are expressed as the mean ± standard deviation (SD) or the mean ± standard error of the mean (SEM). The differences between experimental groups were analyzed using an unpaired t test. In the figures, asterisks denote statistical significance (n.s., not significant, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
Results
Metformin abolishes F. nucleatum-induced chemoresistance in vivo and in vitro
To explore the role of metformin in F. nucleatum-induced chemoresistance, we initially verified metformin’s effect on CRC cell proliferation. After treating CRC cells with different concentration of metformin, we observed that 500 μM and 1 mM metformin did not affect the proliferation of HT-29 cells (Fig. S1a) or HCT 116 cells (Fig. S1b). However, 5 mM and 10 mM metformin inhibited the proliferation in both CRC cell lines (Fig. S1a, b). Importantly, 1 mM metformin is approximately the physiological concentration in the colon of patients taking this drug [25]. In addition, our previous work demonstrated that an MOI of 100 of F. nucleatum had no effect on CRC cell proliferation but could trigger CRC cell chemoresistance [15]. Therefore, we chose 100 MOI of F. nucleatum and 1 mM metformin for subsequent experiments. HT-29 and HCT 116 cells were pre-infected with F. nucleatum for 4 h and then treated with a gradient concentration of 5-Fu or oxaliplatin in the presence of 1 mM metformin. In line with our previous observations, F. nucleatum decreased the cytotoxicity of 5-Fu and oxaliplatin in HT-29 cells (Fig. 1a, b) and HCT 116 cells (Fig. 1c, d), respectively, after 72 h. These results suggest that F. nucleatum promotes chemoresistance of CRC cells. Interestingly, metformin markedly enhanced the cytotoxicity of 5-Fu and oxaliplatin in F. nucleatum-infected HT-29 cells (Fig. 1a, b) and HCT 116 cells (Fig. 1c, d), but not in F. nucleatum-free HT-29 cells and HCT 116 cells. Thus, metformin could abolish F. nucleatum-induced CRC chemoresistance in vitro.

a, b Cell proliferation was detected by CCK-8 assays in HT-29 cells. HT-29 cells were co-cultured with F. nucleatum or treated with 1 mM metformin, and different concentration of 5-Fu (a) and Oxaliplatin (b); unpaired t-test. c, d Cell proliferation was detected by CCK-8 assays in HCT 116 cells. HCT 116 cells were co-cultured with F. nucleatum or treated with 1 mM metformin, and different concentration of 5-Fu (c) and Oxaliplatin (d); unpaired t-test. e Representative data of tumors in mice under different conditions. f, g Statistical analysis of tumor volumes (f) and weights (g) in the different groups, n = 5/group; values indicate mean ± standard error of the mean (SEM); unpaired t-test. n.s., P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
To further verify this conclusion, HCT 116 cells were inoculated into Balb/c nude mice, followed by treatment with metformin (125 mg/Kg/d), F. nucleatum, chemotherapeutic agents (FOX: 5-Fu 5 mg/Kg + Oxaliplatin 6 mg/Kg) and other manipulations. We observed comparable tumor growth (Fig. 1e), tumor volumes (Fig. 1f), and tumor weights (Fig. 1g) in the control, metformin, and F. nucleatum groups. As expected, tumor growth (Fig. 1e), tumor volumes (Fig. 1f), and tumor weights (Fig. 1g) were significantly decreased by FOX treatment, and this decrease was blocked by F. nucleatum infection. These data further demonstrated that F. nucleatum participated in CRC chemoresistance. Remarkably, Balb/c nude mice treated with F. nucleatum plus 1 mM metformin exhibited a better response to FOX, with impaired tumor growth, reduced tumor volumes, and lower tumor weights, when compared with the F. nucleatum plus FOX group (Fig. 1e–g). Collectively, these data supported the view that metformin reverses F. nucleatum-induced CRC chemoresistance.
Metformin attenuates F. nucleatum-induced chemoresistance by inhibiting CRC cell stemness
We next explored the detailed mechanisms by which metformin abolished F. nucleatum-induced CRC chemoresistance. Given the crucial role of stemness in cancer chemoresistance [26], we hypothesized that metformin might rescue F. nucleatum-induced chemoresistance through inhibition of cancer stemness. To test this assumption, we performed bioinformatic analysis to predict the regulation of metformin and F. nucleatum in cancer stem cell pathways. We collected the gene expression data for CRC cells treated with metformin or F. nucleatum from GEO. Subsequently, ssGSEA showed that F. nucleatum upregulated cancer stem cell pathways, whereas metformin downregulated cancer stem cell pathways (Fig. 2a). Based on this data, we speculated that metformin might inhibit F. nucleatum-stimulated CRC stemness. Consistently, the tumorsphere formation assay showed that F. nucleatum increased the number and diameter of tumorspheres in both HT-29 cells (Fig. 2b) and HCT 116 cells (Fig. 2c). By contrast, metformin significantly inhibited the number and diameter of tumorspheres in F. nuleatum-stimulated HT-29 cells (Fig. 2b) and HCT 116 cells (Fig. 2c), but not in F. nuleatum-free cells.

a ssGSEA analysis was conducted to show the alteration of stemness-related pathways in colorectal cancer cells treated with F. nucleatum or metformin. b, c Tumorspheres were observed by light microscopy in HT-29 (b) and HCT 116 (c) cells co-cultured with F. nucleatum or treated with 1 mM metformin. The number and size of tumorspheres was quantified in the right panel. Scale bar, 400 μm. d, e Real-time PCR was performed on stemness marker expression in HT-29 (d) and HCT 116 (e) cells co-cultured with F. nucleatum or treated with 1 mM metformin for 24 h; unpaired t test. f Western blotting was performed on stemness marker expression in HT-29 and HCT 116 cells co-cultured with F. nucleatum or treated with 1 mM metformin for 24 h. g Western blotting was performed to detect stemness-related proteins in xenograft tumors after different treatments. n.s., P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
In support of this data, we detected the expression of stemness markers after metformin and F. nucleatum treatment. NANOG and SOX2 are two important markers of stemness. F. nucleatum enhanced the mRNA and protein expression levels of SOX2 and NANOG in HT-29 and HCT 116 cells (Fig. 2d–f). Metformin abolished F. nucleatum-induced upregulation of SOX2 and NANOG at both the mRNA (Fig. 2d, e) and protein (Fig. 2f) levels. In line with the above results, western blotting analysis also showed that metformin downregulated F. nucleatum-stimulated SOX2 and NANOG expression in FOX-treated subcutaneous xenograft mice (Fig. 2g). Taken together, our data indicated that metformin attenuates F. nucleatum-induced chemoresistance by inhibiting CRC cell stemness.
Metformin rescues F. nucleatum-induced stemness by inhibiting the sonic hedgehog pathway
We next examined how metformin rescued F. nucleatum-induced stemness in CRC cells. Notch, Wnt/β-catenin, and sonic hedgehog signaling pathways are known to play a decisive role in tumor stemness via regulating SOX2 and NANOG expression [27]. We hypothesized that Notch, Wnt/β-catenin, or sonic hedgehog signaling pathways were involved in the recovery effect of metformin in F. nucleatum-induced stemness. To test this hypothesis, we evaluated these three signaling pathways by testing the expression of critical transcription factors individually, including SHH (sonic hedgehog pathway), GLI1 (sonic hedgehog pathway), Transcription factor 1 (TCF1) (Wnt pathway), and Hes family BHLH transcription factor 1 (HES1) (Notch pathway). F. nucleatum enhanced the mRNA expression of SHH, GLI1, TCF, and HES1 in HT-29 cells (Fig. 3a and Fig. S2a) and HCT 116 cells (Fig. 3b and Fig. S2b). This indicated F. nucleatum activates these three pathways. To our surprise, metformin reduced the mRNA expression of SHH and GLI1 in F. nucleatum-infected HT-29 cells (Fig. 3a) and HCT 116 cells (Fig. 3b), but not in F. nucleatum-free CRC cells. Consistently, western blotting also showed metformin reduced SHH and GLI1 protein levels (Fig. 3c). By contrast, metformin could not downregulate the expression of TCF1 and HES1 in F. nucleatum-infected CRC cells (Fig. S2a, b). These data implied that the sonic hedgehog pathway might be involved in the metformin-mediated downregulation of F. nucleatum-induced stemness. In accordance with these results, CRC xenograft mice treated with FOX also showed that metformin diminished F. nucleatum-stimulated SHH and GLI1 protein levels (Fig. 3d). These data indicated that metformin inhibits F. nucleatum-induced stemness by inhibiting the sonic hedgehog pathway.

a, b Real-time PCR was performed to evaluate SHH and GLI1 expression in HT-29 (a) and HCT 116 (b) cells co-cultured with F. nucleatum or treated with 1 mM metformin for 24 h; unpaired t test. c SHH and GLI1 levels were detected by western blotting in HT-29 and HCT 116 cells. The cells were co-cultured with F. nucleatum or treated with 1 mM metformin for 24 h. d Western blotting was performed to detect sonic hedgehog pathway-related proteins in xenograft tumors after different treatments. n.s., P > 0.05; ***P < 0.001; ****P < 0.0001.
MiR-361-5p inhibits the sonic hedgehog signaling pathway and stemness
Next, we conducted luciferase assays to investigate the molecular mechanism by which metformin and F. nucleatum control sonic hedgehog signaling. When the SHH ligand binds to Patched 1 (PTCH1) at the cell membrane, GLI1 is accumulated and activated. GLI1 is the final transcriptional effector and its expression reflects the activation of sonic hedgehog signaling [28]. Thus, we constructed the recombinant luciferase reporter plasmid pGL3-GLI1, containing the promoter region of GLI1 ( − 979 to 33 nts) [29]. Luciferase assays showed that both F. nucleatum and metformin had no effect on the transcriptional activity of pGL3-GLI1 in HCT 116 cells (Fig. S3a). This result suggests that the regulation of GLI1 by metformin and F. nucleatum is not dependent on the direct transcriptional modulation of the GLI1 promoter. Therefore, we sought to determine if metformin and F. nucleatum regulated the GLI1 expression at the post-transcriptional level.
MicroRNAs (miRNAs) are important post-transcriptional regulators of gene expression [30]. We hypothesized that dysregulated miRNAs might be involved in metformin- and F. nucleatum-regulated GLI1. To test this hypothesis, we identified F. nucleatum-related miRNAs from two studies [15, 31]. F. nucleatum appeared to regulate 262 miRNAs significantly (Fig. 4a). Next, we used the bioinformatic tool, TargetScan (http://www.targetscan.org) to identify potential miRNAs that might regulate GLI1. Seventy-four potential GLI1-regulatory miRNAs were found (Fig. 4a). After overlapping these potential GLI1-regulatory miRNAs with the 262 F. nucleatum-related miRNAs, we identified six miRNAs (miR-361-5p, miR-509-3-5p, miR-509-5p, miR-103b, miR-4496, and miR-616-3p) that might regulate GLI1 (Fig. 4a).

a Schematic illustration of the target miRNA candidate screening process. b, c Expression of miR-361-5p was quantified by real-time PCR in HT-29 (b) and HCT 116 cells (c). The cells were co-cultured with F. nucleatum or treated with 1 mM metformin for 24 h; unpaired t test. d The predicted binding sequences for miR-361-5p within the human GLI1 3′ UTR. Seed sequences are highlighted. e Luciferase activity was measured in HCT 116 cells transfected with miR-316-5p mimics or control mimics, miR-316-5p inhibitors or control inhibitors for 72 h. The luciferase reporters expressing wild-type or mutant human GLI1 3′ UTRs were used; unpaired t test. f, g Real-time PCR (f) and western blotting (g) were performed in HT-29 cells to detect the expression levels of SHH, GLI1, and NANOG after transfection with miR-361-5p mimics or inhibitors for 48 h; unpaired t test. h, i Real-time PCR (h) and western blotting (i) were performed in HCT 116 cells to detect the expression levels of SHH, GLI1, and NANOG after transfection with miR-361-5p mimics or inhibitors for 48 h; unpaired t test. j, k Tumorspheres were observed by light microscopy in HT-29 (j) and HCT 116 (k) cells to detect the tumorsphere formation capability after transfection with miR-361-5p mimics; the number of the tumorspheres were quantified in the right panel. Scale bar, 400 μm; unpaired t test. n.s., P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
We validated whether these six miRNAs are regulated by metformin and F. nucleatum using real-time PCR. The results showed that 1 mM metformin alone had no regulatory effect on the six miRNAs (Fig. 4b, c and Fig. S3b, c). Interestingly, only miR-361-5p was significantly downregulated by F. nucleatum and this downregulation could be reverted by 1 mM metformin in both HT-29 (Fig. 4b) and HCT 116 cells (Fig. 4c). Although F. nucleatum inhibited miR-616-3p expression, the decreased miR-616-3p expression could not be rescued by 1 mM metformin in either CRC cell line (Fig. S3b, c). The other miRNAs were not significantly regulated by either F. nucleatum or metformin (Fig. S3b, c). Thus, these data implied that miR-361-5p might contribute to the downregulation of sonic hedgehog signaling in response to 1 mM metformin in F. nucleatum-infected CRC cells.
We next examined whether miR-361-5p could suppress sonic hedgehog signaling and stemness in CRC cells. Targetscan was used to predict the binding site of miR-361-5p in the 3′ untranslated region (UTR) of GLI1. Position163–184 of the GLI1 3′ UTR was identified (Fig. 4d). To verify whether the 3′ UTR of GLI1 mRNA was a functional target of miR-361-5p, we performed dual luciferase reporter gene assays. The 3′ UTR sequences of GLI1 (3′ UTR wild-type, WT) and the mutant 3′ UTR sequences of GLI1 (3′ UTR mutant, MUT) were cloned into a luciferase reporter vector, separately (Fig. 4d). The dual luciferase assay demonstrated that miR-361-5p mimics suppressed the luciferase activity and miR-361-5p inhibitors elevated the luciferase activity in HCT 116 cells transfected with the WT GLI1 reporter plasmid, but not with the MUT reporter plasmid (Fig. 4e). These findings indicated that GLI1 is the specific target of miR-361-5p.
Then, to assess the function of miR-361-5p on the sonic hedgehog pathway and stemness, we transfected miR-361-5p mimics and inhibitors into HT-29 and HCT 116 cells, separately. Overexpression of miR-361-5p decreased the expression levels of SHH, GLI1, and NANOG at both the mRNA and protein level. By contrast, inhibition of miR-361-5p increased the expression of SHH, GLI1, and NANOG in HT-29 cells (Fig. 4f, g) and HCT 116 cells (Fig. 4h, i). In addition, the number and size of tumorspheres were reduced in the miR-361-5p overexpressing HT-29 (Fig. 4j and Fig. S3d) and HCT 116 cells (Fig. 4k and Fig. S3e). Thus, these data support the view that miR-361-5p suppresses the sonic hedgehog pathway and subsequently inhibits CRC cell stemness.
Metformin reverts miR-361-5p in F. nucleatum-infected CRC cells via inhibiting MYC
We next investigated the potential mechanisms responsible for the upregulation of miR-361-5p by metformin in F. nucleatum-infected cells. MiRNA genes are transcribed into primary miRNA (pri-miRNA) transcripts; therefore, we first detected pri-miR-361 expression using real-time PCR. F. nucleatum inhibited the expression of pri-miR-361 in HT-29 and HCT 116 cells (Fig. 5a, b). Metformin increased the expression of pri-miR-361 in F. nucleatum-infected HT-29 and HCT 116 cells, but not in F. nucleatum-free cells (Fig. 5a, b). These data indicated that metformin and F. nucleatum might regulate the transcription of miR-361-5p. To identify potential transcription factors of miR-361-5p, we employed the TransmiR v2.0 database (http://www.cuilab.cn/transmirwas) and predicted 48 transcription factors that might govern the expression of miR-361-5p directly (Fig. 5c). To explore metformin-regulated transcription factors, we analyzed microarray data to compare gene expression profiles of metformin-treated LoVo cells and control. After application of the filtering criterion (adjusted P < 0.1), a total of 560 downregulated genes and 292 upregulated genes were detected (Fig. 5c). Four transcription factors (MYC, FOS, FOXF2, and JUN) were identified after overlapping the 48 potential transcription factors and the metformin-regulated genes (Fig. 5c). Among these four transcription factor genes, MYC was the most significantly downregulated gene by metformin and was well characterized to bind to miRNA promoters directly [32].

a, b Real-time PCR was performed in HT-29 cells (a) and HCT 116 (b) cells to detect the expression of pri-miR-361. The cells were co-cultured with F. nucleatum or treated with 1 mM metformin for 24 h; unpaired t test. c Venn diagram showing the candidate transcription factor screening process. d, e Real-time PCR was performed to evaluate MYC expression in HT-29 (d) and HCT 116 (e) cells treated with 1 mM metformin, F. nucleatum, and F. nucleatum plus 1 mM metformin for 24 h. f MYC levels were detected by western blotting in HT-29 and HCT 116 cells. The cells were co-cultured with F. nucleatum and/or treated with 1 mM metformin for 24 h. g, h Real-time PCR was performed to detect pri-miR-361 and miR-361-5p expression in HT-29 (g) and HCT 116 (h) cells co-cultured with F. nucleatum after MYC plasmid transfection, and subsequently treated with or without 1 mM metformin for 24 h. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
To determine whether MYC contributed to the recovery effect of metformin in F. nucleatum-inhibited miR-361-5p, we first detected the expression of MYC in CRC cells treated with metformin and F. nucleatum. The levels of the MYC transcript (Fig. 5d, e) and protein (Fig. 5f) were enhanced in response to F. nucleatum infection in HT-29 cells and HCT 116 cells. Metformin reduced the levels of MYC mRNA (Fig. 5d, e) and protein (Fig. 5f) in F. nucleatum-infected HT-29 cells and HCT 116 cells, but not in F. nucleatum-free CRC cells. Moreover, metformin failed to upregulate pri-miR-361 and miR-361-5p when MYC was overexpressed in F. nucleatum-infected HT-29 cells (Fig. 5g) and HCT 116 cells (Fig. 5h). Collectively, the data suggested that metformin decreases miR-361-5p transcription in F. nucleatum-infected cells by inhibiting MYC.
Metformin reverses F. nucleatum-induced stemness and chemoresistance via the miR-361-5p/ sonic hedgehog axis
To determine whether miR-361-5p regulates F. nucleatum-mediated sonic hedgehog pathway and CRC stemness, miR-361-5p mimics or inhibitors were transfected in F. nucleatum-treated CRC cells. Western blotting analysis revealed that miR-361-5p mimics decreased SHH, GLI1, and NANOG protein levels in HT-29 (Fig. 6a) and HCT 116 cells (Fig. 6b) cultured with F. nucleatum. Consistent with these results, a loss-of-function study showed miR-361-5p inhibitors increased the levels of SHH, GLI1, and NANOG in HT-29 (Fig. 6a) and HCT 116 cells (Fig. 6b), which presented the same effect as F. nucleatum. In addition, the number and size of tumorspheres stimulated by F. nucleatum were reduced in miR-361-5p overexpressing HT-29 cells (Fig. 6c) and HCT 116 cells (Fig. 6d). Thus, these data supported the view that F. nucleatum activates the sonic hedgehog signaling pathway and CRC stemness via the selective loss of miR-361-5p.

a, b HT-29 (a) and HCT 116 (b) cells were transfected with mimics or inhibitors of miR-361-5p. After culturing with F. nucleatum for 24 h, SHH, GLI1 and NANOG levels were detected by western blotting. c, d Tumorspheres were observed by light microscopy in HT-29 (c) and HCT 116 (d) cells transfected with miR-361-5p mimics, and then co-cultured with F. nucleatum. The number and size of the tumorspheres were quantified in the lower panel. Scale bar, 400 μm; unpaired t test. e, f Western blotting was performed to detect the levels of SHH, GLI1, and NANOG. After transfection with mimics or inhibitors of miR-361-5p, HT-29 (e) and HCT 116 (f) cells were treated with 1 mM metformin in the presence of F. nucleatum for 24 h. g, h After transfection with miR-361-5p mimics, HT-29 (g) and HCT 116 (h) cells were treated with 1 mM metformin in the presence of F. nucleatum. Tumorspheres were observed by light microscopy in HT-29 (g) and HCT 116 (h) cells. The number and size of the tumorspheres were quantified in the right panel. Scale bar, 400 μm; unpaired t test. i Representative data of the tumors in mice under different conditions. j, k Statistical analysis of tumor volumes (j) and weights (k) in the different groups, n = 5/group; unpaired t test. Values indicate mean ± standard error of the mean (SEM); unpaired t-test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
To address whether metformin blocked F. nucleatum-stimulated sonic hedgehog pathway and CRC stemness in a miR-361-5p-dependent manner, miR-361-5p mimics or 1 mM metformin were used to treat F. nucleatum-infected CRC cells. Similar to metformin, the miR-361-5p mimics decreased the SHH, GLI1, and NANOG levels stimulated by F. nucleatum in HT-29 cells (Fig. 6e) and HCT 116 cells (Fig. 6f). MiR-361-5p inhibitors abolished the inhibitory effects of metformin on SHH, GLI1, and NANOG levels in F. nucleatum-infected HT-29 cells (Fig. 6e) and HCT 116 cells (Fig. 6f). Consistently, tumorsphere formation assays showed that the miR-361-5p mimics repressed the number and size of tumorspheres in F. nucleatum-infected HT-29 cells (Fig. 6g) and HCT 116 cells (Fig. 6h). Besides, miR-361-5p inhibitors significantly blocked the inhibitory effects of metformin on tumorsphere numbers in F. nucleatum-stimulated cells (Fig. S3f).
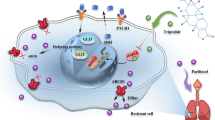
To further assess whether metformin blocked F. nucleatum-stimulated chemoresistance in a miR-361-5p-dependent manner, we established stable miR-361-5p knockdown HCT 116 cells by transducing lentiviruses expressing miR-361-5p inhibitors. We then created subcutaneous tumor models via inoculation of the stable miR-361-5p knockdown HCT 116 cells into nude mice, followed by treatment with chemotherapeutic agents, F. nucleatum, and metformin. As expected, metformin reversed F. nucleatum-induced chemoresistance in control HCT 116 cells (Fig. 6i–k). However, the rescue effect of metformin in F. nucleatum-stimulated chemoresistance was abrogated by the miR-361-5p inhibitors in vivo, as shown by the lack of decrease in tumor growth (Fig. 6i), tumor volumes (Fig. 6j), and tumor weights (Fig. 6k). Altogether, we reasoned that metformin acts on F. nucleatum-infected CRC via the MYC/miR-361-5p cascade, which downregulates the sonic hedgehog signaling pathway, subsequently reversing CRC stemness and abolishing F. nucleatum-triggered chemoresistance (Fig. S4).
Discussion
Chemotherapeutic agents, such as 5-Fu and oxaliplatin, remain the backbone of treatment for patients with CRC; however, the development of chemoresistance is the major cause for treatment failure. Cancer chemoresistance is a complex process and results from the interplay between intrinsic and extrinsic factors. Tumors with genetic and epigenetic alterations are critical for the CRC chemotherapeutic response [6]. Recent studies also showed that the gut microbiota controlled the response to chemotherapy by modulating the tumor microenvironment [33]. F. nucleatum abundance is associated with the properties of CRC, such as tumorigenesis, development, metastasis, and recurrence [34]. Through a combination of bioinformatic analyses, biological experiments, in vivo models, and clinical studies, we demonstrated that abnormal proliferation of F. nucleatum led to CRC chemoresistance and recurrence [15]. Given that broad spectrum antibiotics have a negative effect on the healthy intestinal microbiota, and no F. nucleatum-specific antimicrobial agent has been discovered, chemoresistance caused by F. nucleatum remains a thorny problem in the clinic [35]. Therefore, it is necessary to develop a safe and effective approach to eliminate chemoresistance caused by F. nucleatum.
Based on our previous work showing that metformin could attenuate F. nucleatum-induced tumorigenesis [16], we further explored the effect of metformin on F. nucleatum-induced chemoresistance. To our surprise, metformin abrogated F. nucleatum-induced CRC chemoresistance in CRC cells and xenograft mice. However, how metformin affects F. nucleatum-mediated chemoresistance was unknown. A distinct tumor cell subpopulation with stemness, known as the cancer stem cell population, exists in cancers, which mediates chemoresistance and metastatic progression [36, 37]. The use of cancer stemness inhibitors, such as napabucasin, can overcome chemoresistance in human cancers [38,39,40]. Emerging evidence supports the view that bacterial infections, such as Enterococcus faecalis, nonpathogenic E. coli, and Enterotoxigenic Bacteroides fragilis, might stimulate cancer cell stemness via upregulated expression of stemness markers [41,42,43]. Our bioinformatic studies demonstrated that a stemness-related pathway is enriched in the modulation of metformin and F. nucleatum in CRC cells. Indeed, the effect of metformin depends on the specific regulation of stemness in F. nucleatum-infected CRC cells. Accordingly, metformin decreased the expression of stemness-related proteins, SOX2 and NANOG, and inhibited tumorsphere formation in CRC cells co-cultured with F. nucleatum. Thus, we concluded that metformin attenuates F. nucleatum-induced chemoresistance by inhibiting stemness in CRC cells. These data might explain why metformin recovers chemosensitivity in F. nucleatum-infected CRC cells and xenograft mice.
We dissected the mechanisms by which metformin regulates F. nucleatum-induced stemness in CRC cells. Aberrant activity of the sonic hedgehog pathway has been linked to stem cell self-renewal and chemoresistance in variety of solid neoplasms [44]. We demonstrated that sonic hedgehog signaling is a bridge connecting metformin- and F. nucleatum-modulated stemness and chemoresistance. Metformin abolishes F. nucleatum-stimulated stemness and subsequently chemoresistance by selectively suppressing sonic hedgehog signaling. Furthermore, metformin and F. nucleatum do not affect the transcription of GLI1, which encodes the key effector of the sonic hedgehog pathway. Given that miRNAs are critical post transcriptional regulators [30], our bioinformatic and functional studies revealed miR-361-5p targets GLI1 and inhibits sonic hedgehog signaling and stemness. In addition, metformin reverses F. nucleatum-stimulated stemness, sonic hedgehog signaling and chemoresistance in a miR-361-5p-dependent manner. In line with this notion, we found that metformin decreases miR-361-5p transcripts in F. nucleatum-infected cells by downregulating MYC expression. Our data suggested that metformin reverses F. nucleatum-induced stemness by inhibiting the MYC/miR-361-5p/sonic hedgehog signaling axis, and then biologically and mechanistically reverting CRC chemoresistance induced by F. nucleatum.
In addition to its biological importance, our work might be relevant in the clinical management of patients with CRC. The abundance of F. nucleatum is associated with the risk of CRC chemoresistance and recurrence; therefore, combining conventional chemotherapeutic regimens with metformin might be an effective strategy to reverse CRC chemoresistance in patients with high amounts of F. nucleatum. Our results highlighted the need for a clinical trial of metformin as a potential treatment in Fusobacterium-associated CRC chemoresistance. Furthermore, an important question raised by our data is whether metformin directly affects F. nucleatum. It has been reported that metformin exerts its hypoglycemic benefits partly through alteration of the abundance of certain members of the gut microbiota, such as Bacteroides fragilis and Akkermansia muciniphila [45,46,47,48]. Researchers also found that metformin could regulate microbial folate and methionine metabolism, which is required for bacterial growth [46, 49]. These provide the possibility that patients with CRC might benefit from the direct modulation of metformin on F. nucleatum. In addition to the details of the mechanisms by which metformin affects F. nucleatum-induced host responses, we will explore the direct effect of metformin on F. nucleatum colonization and dissemination in a future study.
Our results provide a foundation for the metformin–gut microbiota–host response network in CRC. This network regulation mode might be a direction for meaningful basic and translational medicine research in the future.
Data availability
Previously published data sets were available in GEO under accession code GSE102573, GSE90944, and GSE67342.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30.
André T, Boni C, Mounedji-Boudiaf L, Navarro M, Tabernero J, Hickish T, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N. Engl J Med. 2004;350:2343–51.
Messersmith WA. Systemic management of colorectal cancer. J Natl Compr Canc Netw. 2017;15:699–702.
Sargent D, Sobrero A, Grothey A, O’Connell MJ, Buyse M, Andre T, et al. Evidence for cure by adjuvant therapy in colon cancer: observations based on individual patient data from 20,898 patients on 18 randomized trials. J Clin Oncol. 2009;27:872–7.
Douillard JY, Cunningham D, Roth AD, Navarro M, James RD, Karasek P, et al. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet. 2000;355:1041–7.
Vodenkova S, Buchler T, Cervena K, Veskrnova V, Vodicka P, Vymetalkova V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: Past, present and future. Pharm Ther. 2020;206:107447.
Roy S, Trinchieri G. Microbiota: a key orchestrator of cancer therapy. Nat Rev Cancer. 2017;17:271–85.
Alexander JL, Wilson ID, Teare J, Marchesi JR, Nicholson JK, Kinross JM. Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat Rev Gastroenterol Hepatol. 2017;14:356–65.
Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillère R, Hannani D, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013;342:971–6.
Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350:1079–84.
Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013;14:207–15.
Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013;14:195–206.
Wong SH, Yu J. Gut microbiota in colorectal cancer: mechanisms of action and clinical applications. Nat Rev Gastroenterol Hepatol. 2019;16:690–704.
Zhang S, Yang Y, Weng W, Guo B, Cai G, Ma Y, et al. Fusobacterium nucleatum promotes chemoresistance to 5-fluorouracil by upregulation of BIRC3 expression in colorectal cancer. J Exp Clin Cancer Res. 2019;38:14.
Yu T, Guo F, Yu Y, Sun T, Ma D, Han J, et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell. 2017;170:548–63.e16.
Huang X, Hong X, Wang J, Sun T, Yu T, Yu Y, et al. Metformin elicits antitumour effect by modulation of the gut microbiota and rescues Fusobacterium nucleatum-induced colorectal tumourigenesis. EBioMedicine. 2020;61:103037.
Ferrara A, Lewis JD, Quesenberry CP, Peng T, Strom BL, Van Den Eeden SK, et al. Cohort study of pioglitazone and cancer incidence in patients with diabetes. Diabetes Care. 2011;34:923–9.
Higurashi T, Hosono K, Takahashi H, Komiya Y, Umezawa S, Sakai E, et al. Metformin for chemoprevention of metachronous colorectal adenoma or polyps in post-polypectomy patients without diabetes: a multicentre double-blind, placebo-controlled, randomised phase 3 trial. Lancet Oncol. 2016;17:475–83.
Coyle C, Cafferty FH, Vale C, Langley RE. Metformin as an adjuvant treatment for cancer: a systematic review and meta-analysis. Ann Oncol. 2016;27:2184–95.
Li L, Jiang L, Wang Y, Zhao Y, Zhang XJ, Wu G, et al. Combination of metformin and gefitinib as first-line therapy for nondiabetic advanced NSCLC patients with EGFR mutations: a randomized, double-blind phase II trial. Clin Cancer Res. 2019;25:6967–75.
Miranda VC, Braghiroli MI, Faria LD, Bariani G, Alex A, Bezerra Neto JE, et al. Phase 2 trial of metformin combined with 5-fluorouracil in patients with refractory metastatic colorectal cancer. Clin Colorectal Cancer. 2016;15:321–8.e1.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–8.
Johnson S, Chen H, Lo P-K. In vitro tumorsphere formation assays. Bio Protoc. 2013;3:e325.
Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinforma. 2013;14:7.
Paleari L, Burhenne J, Weiss J, Foersch S, Roth W, Parodi A, et al. High accumulation of metformin in colonic tissue of subjects with diabetes or the metabolic syndrome. Gastroenterology. 2018;154:1543–5.
Lytle NK, Barber AG, Reya T. Stem cell fate in cancer growth, progression and therapy resistance. Nat Rev Cancer. 2018;18:669–80.
Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. 2011;8:97–106.
Machado MV, Diehl AM. Hedgehog signalling in liver pathophysiology. J Hepatol. 2018;68:550–62.
Huang J, Liu H, Sun T, Fang JY, Wang J, Xiong H. Omeprazole prevents CDX2 and SOX9 expression by inhibiting hedgehog signaling in Barrett’s esophagus cells. Clin Sci (Lond). 2019;133:483–95.
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33.
Yang Y, Weng W, Peng J, Hong L, Yang L, Toiyama Y, et al. Fusobacterium nucleatum increases proliferation of colorectal cancer cells and tumor development in mice by activating toll-like receptor 4 signaling to nuclear factor-κB, and up-regulating expression of microRNA-21. Gastroenterology. 2017;152:851–66.e24.
Psathas JN, Thomas-Tikhonenko A MYC and the art of microRNA maintenance. Cold Spring Harb Perspect Med. 2014;4. https://doi.org/10.1101/cshperspect.a014175.
Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342:967–70.
Brennan CA, Garrett WS. Fusobacterium nucleatum - symbiont, opportunist and oncobacterium. Nat Rev Microbiol. 2019;17:156–66.
Bullman S, Pedamallu CS, Sicinska E, Clancy TE, Zhang X, Cai D, et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science. 2017;358:1443–8.
Zhao J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharm Ther. 2016;160:145–58.
Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. 2015;16:225–38.
Li Y, Rogoff HA, Keates S, Gao Y, Murikipudi S, Mikule K, et al. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc Natl Acad Sci USA. 2015;112:1839–44.
MacDonagh L, Gray SG, Breen E, Cuffe S, Finn SP, O’Byrne KJ, et al. BBI608 inhibits cancer stemness and reverses cisplatin resistance in NSCLC. Cancer Lett. 2018;428:117–26.
Han D, Yu T, Dong N, Wang B, Sun F, Jiang D. Napabucasin, a novel STAT3 inhibitor suppresses proliferation, invasion and stemness of glioblastoma cells. J Exp Clin Cancer Res. 2019;38:289.
Wang X, Yang Y, Huycke MM. Commensal bacteria drive endogenous transformation and tumour stem cell marker expression through a bystander effect. Gut 2015;64:459–68.
Sahu U, Choudhury A, Parvez S, Biswas S, Kar S. Induction of intestinal stemness and tumorigenicity by aberrant internalization of commensal non-pathogenic E. coli. Cell Death Dis. 2017;8:e2667.
Liu QQ, Li CM, Fu LN, Wang HL, Tan J, Wang YQ, et al. Enterotoxigenic Bacteroides fragilis induces the stemness in colorectal cancer via upregulating histone demethylase JMJD2B. Gut Microbes. 2020;12:1788900.
Girardi D, Barrichello A, Fernandes G, Pereira A. Targeting the hedgehog pathway in cancer: current evidence and future perspectives. Cells. 2019;8. https://doi.org/10.1080/19490976.2020.1788900.
Sun L, Xie C, Wang G, Wu Y, Wu Q, Wang X, et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat Med. 2018;24:1919–29.
Wu H, Esteve E, Tremaroli V, Khan MT, Caesar R, Mannerås-Holm L, et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med. 2017;23:850–8.
Shin N-R, Lee J-C, Lee H-Y, Kim M-S, Whon TW, Lee M-S, et al. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut. 2014;63:727–35.
de la Cuesta-Zuluaga J, Mueller NT, Corrales-Agudelo V, Velásquez-Mejía EP, Carmona JA, Abad JM, et al. Metformin is associated with higher relative abundance of mucin-degrading akkermansia muciniphila and several short-chain fatty acid-producing microbiota in the gut. Diabetes Care. 2017;40:54–62.
Cabreiro F, Au C, Leung K-Y, Vergara-Irigaray N, Cochemé HM, Noori T, et al. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell. 2013;153:228–39.
Funding
This work was supported by grants from the Shanghai Municipal Education Commission—Gaoyuan and Gaofeng Clinical Medicine Grant (No: 20191907), the Major health science and technology projects in ZheJiang Province (No: WKJ-ZJ-2217), and medical research project of Science and Technology Innovation Action Plan in Shanghai (No: 21Y11912800).
Author information
Authors and Affiliations
Contributions
X-L Hong performed the experiments, analyzed the data, and wrote the manuscript; T-C Yu performed the experiments; X-W Huang performed the bioinformatics analyses; J-L Wang planned the project, and participated in data analysis, data interpretation; T-T Sun, T-T Yan, C-B Zhou, H-M Chen, and W-Y Su performed the experiments; H Xiong and W Du conceived, designed, and planned the project, and participated in experimental design, data analysis, data interpretation, and manuscript writing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Mouse experiments were conducted in accordance with the guidelines approved by the Institutional Animal Care and Use Committee of Renji Hospital, School of Medicine, Shanghai Jiaotong University.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hong, XL., Yu, TC., Huang, XW. et al. Metformin abrogates Fusobacterium nucleatum-induced chemoresistance in colorectal cancer by inhibiting miR-361-5p/sonic hedgehog signaling-regulated stemness. Br J Cancer 128, 363–374 (2023). https://doi.org/10.1038/s41416-022-02044-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-022-02044-6
This article is cited by
-
The roles and molecular mechanisms of non-coding RNA in cancer metabolic reprogramming
Cancer Cell International (2024)
-
miR-135b Aggravates Fusobacterium nucleatum-Induced Cisplatin Resistance in Colorectal Cancer by Targeting KLF13
Journal of Microbiology (2024)
-
Gut microbiota and microbiota-derived metabolites in colorectal cancer: enemy or friend
World Journal of Microbiology and Biotechnology (2023)
