
Abstract
Background
Overexpression of anti-apoptotic MCL-1 protein in oral squamous cell carcinoma (OSCC) is linked to disease progression, therapy resistance and poor outcome. Despite its characteristic short half-life owing to ubiquitin–proteasome-dependent degradation, oral tumours frequently show elevated MCL-1 protein expression. Hence, we investigated the role of deubiquitinase USP9X in stabilising MCL-1 protein and its contribution to oral tumorigenesis.
Methods
Expression of MCL-1 and USP9X was assessed by immunoblotting and immunohistochemistry in oral cancer cell lines and tissues. The association between MCL-1 and USP9X was confirmed by coimmunoprecipitation and immunofluorescence. Cell death assessment was performed by MTT, flow cytometry and clonogenic assays.
Results
Both USP9X and MCL-1 are significantly elevated in oral premalignant lesions and oral tumours versus normal mucosa. USP9X interacts with and deubiquitinates MCL-1, thereby stabilising it. Pharmacological inhibition of USP9X potently induced cell death in OSCC cells in vitro and in vivo. The elevated expression of USP9X and MCL-1 correlated with poor prognosis in OSCC patients.
Conclusion
We demonstrate the oncogenic role of USP9X in driving early-to-late stages of oral tumorigenesis via stabilisation of MCL-1, suggesting its potential as a prognostic biomarker and therapeutic target in oral cancers.
Similar content being viewed by others


Background
Oral squamous cell carcinoma (OSCC) or oral cancer, a major subset of head and neck cancers, is a leading cause of mortality among men in Southeast Asia primarily due to tobacco-chewing habit in combination with areca nut, betel quid and alcohol consumption.1 Apprehensively, tobacco-related cancers account for ~42% male and ~18% female deaths in India as evident from a nationwide representative survey.2 Although preventable to a large extent simply by changes in the habits and lifestyle, in most cases, delayed clinical presentation of oral cancer is associated with poor prognosis and high mortality.3 Consequently, late-stage oral cancers are largely refractory to therapeutics, leading to poor prognosis than early-stage cancers. Despite significant advances in anticancer therapeutics, the 5-year survival rate of OSCC patients remains dismal over the past several decades, primarily due to therapy resistance and disease recurrence.4,5 The 5-year survival rates for oral cancer patients drop significantly from stage I (75%) to stage IV (30%).6 Surgery still remains the primary treatment modality in a majority of cases with concurrent chemoradiotherapy being administered in the advanced-stage tumours.7 Thus, screening and treating the disease in the premalignant stages is clinically important.8 OSCC is almost invariably preceded by oral premalignant lesions and conditions, collectively known as oral potentially malignant disorders (OPMD).9 However, their malignant transformation is unpredictable due to the unavailability of reliable clinicopathological and molecular predicting factors.10,11 It is hence imperative to find potential biomarkers and therapeutic targets in oral cancers that can predict disease progression from the precancerous lesions to the tumour with high sensitivity and specificity.12
Apoptotic dysregulation owing to the overexpression of anti-apoptotic members of the B-Cell Lymphoma-2 (BCL-2) family is a critical factor underlying tumour progression in a variety of human cancers.13 Among the anti-apoptotic proteins of the BCL-2 family, predominant overexpression of myeloid cell leukaemia-1 (MCL-1) has been demonstrated in a variety of human cancer cell lines,14 diverse haematological malignancies15,16,17 and solid tumours,18,19,20 including oral cancers as reported by our group.21 Furthermore, earlier studies from our laboratory have attributed MCL-1 overexpression to the acquired chemo- and radioresistance in oral cancers and its correlation with reduced disease-free survival.22,23,24 In addition, targeted MCL-1 downregulation or its pharmacological inhibition was found to sensitise oral cancer cells to cell death in response to treatment with cisplatin and ionising radiation.23,24,25 All these reports suggest that MCL-1 is a critical pro-survival protein in oral cancers, which contributes to therapy resistance and poor prognosis.
In a mammalian cell, MCL-1 expression is tightly regulated at multiple levels, the most pivotal being its rapid turnover through the ubiquitin–proteasomal degradation pathway, which accounts for its characteristic short half-life.26 However, being a critical pro-survival protein, tumour cell adopts several strategies to maintain sustained elevated levels of MCL-1.27 Although our previous studies demonstrate overexpression of MCL-1 transcript in oral cancer cells and tumours as compared to the normal oral mucosa,21 elevated levels of MCL-1 protein in oral cancer cells suggest its plausible post-translational stabilisation. The deubiquitinase ubiquitin-specific peptidase 9 X-linked (USP9X) has been reported as an interacting partner of MCL-1, which may stabilise it.28 Therefore, in this study, we investigated the potential contribution of USP9X in MCL-1 protein stability in oral cancers and during the different stages of oral tumorigenesis. Here, we demonstrate for the first time that both USP9X and MCL-1 protein levels are elevated through early-to-late stages of oral tumorigenesis and exhibit a positive correlation. We also report that USP9X contributes to MCL-1 protein stability in oral cancers and its targeted downregulation or inhibition sensitises oral cancer cells to cell death through rapid degradation of MCL-1. Furthermore, we show that concomitant overexpression of both USP9X and MCL-1 in oral tumours correlates with disease recurrence and poor prognosis.
Methods
Cell culture
AW8507 and AW13516 cell lines were derived from epidermoid carcinoma and poorly differentiated squamous cell carcinoma of the tongue, respectively.29 SCC029B cell line was derived from poorly differentiated squamous cell carcinoma of the buccal mucosa.30 The three cell lines were maintained in IMDM (Gibco, Carlsbad, CA, USA) containing 10% foetal bovine serum. Cell line authentication was performed by short tandem repeat (STR) profiling of 21 markers (DNA labs, India).
Inhibitors and reagents
MTT, Cycloheximide, Chloroquine and E-64d were obtained from Sigma (St. Louis, MO, USA). WP1130 (Degrasyn) was procured from Selleck chemicals (Houston, TX, USA). All siRNAs were obtained from Dharmacon. siRNA transfections were performed using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA).
Antibodies
Antibodies against MCL-1 (sc-819, sc-20679), BCL-2 (sc-493), Actin (sc-1616R) and USP9X (sc-100628) were obtained from Santa Cruz Biotechnology (Dallas, TX, USA). Antibodies specific for BCL-XL (#2764), p62 (#5114) and PARP (#9542) were purchased from Cell Signaling Technologies (Danvers, MA, USA). An antibody against ubiquitin (U0508) was obtained from Sigma. For immunofluorescence, Alexa fluor 488 and Alexa fluor 568 secondary antibodies were used (Molecular Probes, Invitrogen).
Generation of USP9X-knockout clones by CRISPR/Cas9 approach
We used the USP9X target sequence CAGAGGAATCATCAGCCAG to design the guide RNA. The guide RNA oligonucleotides were annealed and cloned into the BbsI (BpiI) restriction site in the vector pSp-Cas9-BB-2A-Puro (PX459) V2.0 (a gift from Feng Zhang (Addgene plasmid # 62988); Addgene, MA, USA). The positive transformants were confirmed by colony PCR, restriction digestion and sequencing. AW8507 cells were transfected with the USP9X CRISPR construct using the Lipofectamine 2000 transfection reagent (Invitrogen). Twenty-four hours post transfection, the cells were selected using 1 µg/ml puromycin. Distinctly visible colonies were observed ~14 days post puromycin selection. The individual colonies were allowed to expand and were screened for the presence of USP9X protein by western blotting. The intended microdeletion in the USP9X gene was further confirmed by sequencing.
Protein extraction and western blotting
The cells were harvested by trypsinisation and lysed in mammalian cell lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl and 0.5% NP-40) containing protease inhibitor cocktail (Fermentas, Vilnius, Lithuania). The cell lysates were subjected to protein estimation by Bradford assay (BioRad, Hercules, CA, USA). Equal amounts of proteins were resolved on SDS-PAGE and electroblotted onto PVDF membranes (Pall Corporation, NY, USA). The membranes were blocked in 5% BSA followed by incubation with appropriate primary antibodies. The membranes were then probed with corresponding secondary antibodies. The proteins were visualised using an enhanced chemiluminescence kit (GE Healthcare, Chicago, Illinois, USA). Densitometric analysis was performed using Image J software (NIH, Bethesda, MD, USA).
Coimmunoprecipitation
The cells were harvested by trypsinisation and lysed in EBC lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA and 0.5% NP-40) containing protease inhibitors followed by centrifugation. The supernatants were precleared with protein G-sepharose beads (GE Healthcare) and subjected to overnight incubation with appropriate antibodies at 4 °C on a rocking platform. The next day, these cell lysates were incubated with protein G-sepharose bead slurry for 3 h at 4 °C on a rocker. The immunocomplexes were recovered by centrifugation, washed with EBC buffer at least five times, boiled in Laemelli buffer and subjected to SDS-PAGE followed by immunoblotting.
Real-time PCR
The total RNA was extracted from the cells by using TRIZol reagent (Invitrogen). In total, 500 ng of total RNA was reverse-transcribed to cDNA by High Capacity cDNA synthesis kit (Applied Biosystems, Foster City, CA, USA). The primers used for PCR amplification were MCL-1L F: AAGAGGCTGGGATGGGTTTG; MCL-1L R: CAGCAGCACATTCCTGATGC. GAPDH F: CTCTGCTCCTCCTGTTCGAC; GAPDH R: GCGCCCAATACGACCAAATC. Real-time PCR was performed on ABI QuantStudio 12 K Flex Sequence detection system (Applied Biosystems) using SYBR Green mastermix (Applied Biosystems). The results were analysed by relative quantitation and expressed as 2−ΔCt.
Cell viability assays
MTT assay
In total, 5000 cells per well were seeded in 96-well plates (Nunc, Roskilde, Denmark) and allowed to grow overnight. The next day, the cells were treated with indicated concentrations of WP1130 for the specified time points. At the end of the treatment period, 20 µl MTT reagent (from a stock of 5 mg/ml) was added to each well and the plates were again incubated in a CO2 incubator for 4 h. The formazan crystals were dissolved by the addition of DMSO and absorbance was recorded on a microplate reader (Spectrostar nano, BMG Labtech, Germany) at 540 nm with a reference wavelength of 690 nm.
Cell death quantification by flow cytometry
Cell death was quantified by using an FITC-Annexin V Apoptosis detection kit (BD Biosciences, Franklin Lakes, NJ, USA). Briefly, cells post treatment were harvested by trypsinisation, washed with cold PBS twice and resuspended in binding buffer followed by incubation with Annexin V-FITC and Propidium Iodide in the dark at room temperature for 30 min and subjected to acquisition on a FACS Calibur flow cytometer (BD Biosciences). Data analysis was performed by using Cell Quest software (BD Biosciences).
siRNA transfection
The USP9X siRNA was bought as a pool of four different siRNA oligos (ON-TARGETplus-SMARTpool from Dharmacon) targeting different regions of USP9X. Briefly, the siRNA was brought to the desired working concentration by appropriately diluting in serum-free medium, mixed with lipofectamine 2000 (Invitrogen), allowed to stand at room temperature for 20 min to ensure the complex formation and then added to the cells dropwise. The culture medium containing transfection mixture was replaced with fresh medium 6 h post transfection.
Radiation treatment and clonogenic assay
Twenty-four hours post transfection with siRNA, the cells were harvested, seeded in 35-mm plates and allowed to grow overnight. The next day, the cells were irradiated (γ-irradiation on a Cobalt-60 source) with their respective D0 doses (previously determined by standard clonogenic assays). The cells were allowed to grow for further 8–10 days until visible colonies appear. The colonies were fixed with 90% methanol, stained with 0.5% Crystal Violet, washed gently with water to remove excess stain, air-dried and scored manually.
Immunofluorescence and confocal microscopy
Cells were cultured on coverslips and at the end of the desired treatment periods, the cells were fixed with 4% PFA followed by permeabilisation with 0.5% Triton X-100. Blocking was done in 5% BSA for 1 h at room temperature (RT). The cells were then incubated with specific primary antibodies for 1 h at RT followed by incubation with appropriate fluorescent labelled secondary antibodies with 1-h incubation at RT in the dark. Nuclei were counterstained with DAPI. The coverslips were mounted with Vecta-shield (Vector Laboratories, Burlingame, CA, USA) on glass slides, sealed with nail polish and observed under a laser confocal microscope (LSM 510 Metaconfocal, Zeiss, Germany). Colocalisation analysis was performed by the Coloc2 plug-in of Image J and the extent of colocalisation was expressed in terms of Pearson’s correlation coefficient (R).
Oral tissue samples
The study was reviewed and duly approved by the Institutional Ethics Committee (IEC-III) of the Tata Memorial Centre (TMC) (project # 900211). Normal oral mucosa (NOM, n = 50) obtained during minor dental surgeries, Oral Leukoplakia (OL, n = 50) and Oral Submucous Fibrosis (OSMF, n = 50) tissue FFPE (Formalin Fixed Paraffin Embedded) blocks were obtained from the Department of Oral Pathology and Microbiology, KBH Dental College and Hospital, Nashik (after approval from the respective Institutional Ethics Committees and obtaining informed consent). Pre-treatment tissue FFPE blocks of OSCC patients (n = 100) and FFPE blocks of paired (n = 23) recurrent and primary oral tumours and nonrecurrent oral tumours (n = 25) were collected from the Department of Pathology, Tata Memorial Hospital (TMH) and Advanced Centre for Treatment, Research and Education in Cancer (ACTREC) (after approval from Institutional Ethics Committee-III of Tata Memorial Centre). The detailed clinicopathological parameters of the patients are indicated in Supplementary Tables 1, 2, 3 and 4. The paired oral tumour and adjacent normal samples were obtained from the Tumor Tissue Repository, Tata Memorial Centre, Mumbai, India.
Immunohistochemistry (IHC)
The IHC procedure was performed using a VectaStain ABC kit from Vector Laboratories. The tissue sections on glass slides were deparaffinised by passing through several grades of xylene and alcohol before being subjected to endogenous peroxidase blocking followed by heat-induced epitope retrieval in a microwave oven. The tissue sections were then incubated with goat or horse normal serum followed by overnight incubation with specific primary antibodies at 4 °C. The next day, the slides were washed with PBS, and the tissue sections were incubated with appropriate secondary antibodies conjugated with biotin. A conjugate of Avidin and HRP-tagged biotin was applied to the tissue sections followed by staining with DAB (3,3′-Diaminobenzidine tetrahydrochloride) chromogen (Sigma) and then counterstaining with haematoxylin. The tissue sections were again dehydrated by passing through different grades of alcohol and xylene and then mounted with DPX mountant. The immunostaining was evaluated in a double-blinded manner (by SNP & RW) by counting the percentage of cells expressing the proteins in at least 10–15 different microscopic fields and assessing the intensity of staining (grade): 0: negative, 1: weak, 2: moderate, 3: strong. The protein expression was determined by multiplication of percent positivity and grade (staining intensity) and expressed as H score.
In vivo studies
All the animal studies (Project no. 14/2013) were reviewed and approved by the Institutional Animal Ethics Committee (IAEC) of Tata Memorial Centre (TMC)-Advanced Centre for Treatment, Research and Education in Cancer (ACTREC) constituted under the guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Government of India. To assess the in vivo efficacy of WP1130 against oral cancer cells, we employed xenograft mouse models as described earlier.31,32 Six-to-eight-week-old female BALB/C nude mice were obtained from the in-house breeding facility of the Advanced Centre for Treatment, Research and Education in Cancer (ACTREC), Tata Memorial Centre (TMC). The animals were subcutaneously injected into the hind flank region with 1 × 106 SCC029B cells resuspended in 150 µl serum-free IMDM medium. A day after the cell inoculation, the animals were randomised into two groups (Vehicle control and 20 mg/kg WP1130) each containing six animals. Palpable tumours were observed at about 21 days post cell inoculation. WP1130 was formulated as DMSO:Polyethylene Glycol 300 1:1. The formulated drug was administered by intraperitoneal injection (100 µl per animal) every alternate day for 2 consecutive weeks. The tumour volume measurements were performed using a Vernier calliper every alternate day and calculated by the formula: length (mm) × (Width(mm))2/2. The tumour volume and weight of the animals was monitored every alternate day for 16–18 days post termination of drug treatment. The animals were euthanised by CO2 asphyxiation, and tumours and other vital organs were collected in 10% formalin (v/v in PBS) followed by mounting in paraffin blocks. Ten-micron sections of the tumour tissues were mounted on glass slides. Tumour volume regression was considered as a primary outcome. Alternatively, assessment of molecular markers of cell death in the tumour tissues was performed by TUNEL assay and cleaved Caspase-3 IHC. The tumour volumes were represented as mean + /− SEM of the 6 animals from the group and ANOVA was performed to assess the extent of tumour regression in response to drug treatment compared to vehicle control group. For evaluation of the molecular markers, t test was used. Statistical analyses were performed using GraphPad prism V 5.0 software.
TUNEL assay
The percentage of cells undergoing apoptosis in the tumours of the animals was determined by TUNEL assay using in situ Apoptosis Detection kit (Takara Bio Inc., Kusatsu, Shiga, Japan). Briefly, the formalin-fixed paraffin-embedded tissue sections were deparaffinised by passing through several grades of xylene and alcohol, followed by proteinase K-mediated epitope retrieval and blocking of endogenous peroxidase. The tissue sections were then incubated with labelling reaction mixture containing TdT enzyme for 90 min at 37 °C followed by washing with PBS. Finally, the tissue sections were exposed to anti-FITC HRP conjugate for 30 min at 37 °C and then washed with PBS. The tissue sections were stained with DAB chromogen and counterstained with haematoxylin.
Transmission electron microscopy (TEM)
The cells were harvested by trypsinisation and washed twice with PBS. The cells were then fixed with 3% glutaraldehyde at 4 °C for 2 h followed by washing twice with 0.1 M sodium cacodylate buffer (pH 7.4). The cell pellets were then fixed in 1% osmium tetroxide for 1 h at 4 °C in the dark, subjected to dehydration by passing through different grades of alcohol and then embedded with Araldite resin. Ultrathin sections (~60–70 nm) of the cells fixed within the Araldite resin were cut on an ultramicrotome (Leica, Wetzlar, Germany) and mounted on copper grids. These sections were stained with 10% uranyl acetate and counterstained with lead citrate. Electron micrographs were captured on a Jeol 100-CXII electron microscope (Jeol, Akishima, Tokyo, Japan) using Olympus camera and iTEM software.
Statistical analysis
For comparison of two datasets in an experiment, we used the two-tailed Student’s t test. The correlation between expression of MCL-1 and USP9X proteins was assessed by Spearman’s rank correlation coefficient. As recurrence-free survival (RFS) has the highest correlation with overall survival (OS), we considered the RFS as a primary endpoint. RFS corresponds to the time from the date of the first treatment of the patient to the date of the event (either loco-regional and/or distant metastasis). The median of each protein expression was considered as a cut-off to dichotomise the data. The values above the median were considered as high expression as opposed to those below the median, which were considered to be low expression. The survival rates between the categories of protein expression (high vs low) were compared by the Kaplan–Meier method of Log-rank test. The data are represented as mean ± standard error mean (SEM). The difference between mean was considered significant when p < 0.05. All the statistical analyses were performed using GraphPad Prism software version 5.01 and SPSS version 21 software.
Results
MCL-1 is primarily degraded by the ubiquitin–proteasomal pathway in oral cancer cells
MCL-1 is degraded through multiple pathways, including the ubiquitin–proteasomal pathway, caspase and granzyme B-mediated cleavage and ubiquitin-independent pathway.27 To identify the major pathway responsible for MCL-1 turnover in oral cancer cells, we treated AW8507 cells with protein synthesis inhibitor cycloheximide (CHX), lysosomal inhibitor chloroquine (CQ), pan-caspase inhibitor Z-VAD-FMK, calpain/cathepsin inhibitor E-64d and proteasome inhibitor MG132. We observed that CHX completely abolished the expression of MCL-1, suggesting that MCL-1 protein has rapid turnover. However, CQ, Z-VAD-FMK and E-64d did not significantly change the levels of MCL-1, indicating that lysosomal, caspase-mediated and proteolytic cleavage contribute minimally to MCL-1 turnover. Notably, MG132 significantly increased MCL-1 protein levels, suggesting that MCL-1 is primarily degraded through the proteasomal pathway in oral cancer cells (Supplementary Fig. 1A). To further confirm the contribution of proteasomal pathway specifically in MCL-1 degradation, we treated AW8507 and SCC029B cells with the proteasome inhibitors MG132 and Velcade (Bortezomib). Treatment with MG132 and Velcade significantly increased MCL-1 protein levels specifically, but the levels of both BCL-2 and BCL-XL proteins (fellow members of the anti-apoptotic BCL-2 subfamily) did not change significantly (Supplementary Fig. 1B, C). Further, immunoprecipitation of MCL-1 from MG132-treated AW8507 cells indicated that the increased level of MCL-1 protein represents its polyubiquitinated form (Supplementary Fig. 1D). All these results indicate that MCL-1 protein is primarily degraded by the ubiquitin–proteasomal pathway in OSCC cells.
MCL-1 expression correlates with USP9X in oral cell lines and tissues
As USP9X has been identified to be one of the several interacting partners of MCL-1, which may stabilise it,28,33 we first investigated the correlation between the expression of these two proteins in oral cell lines and tissues. In a set of three oral cancer cell lines screened, we observed a significant positive correlation (p < 0.05, Pearson’s correlation coefficient r = 0.9527) between MCL-1 and USP9X proteins (Fig. 1a). Next, we evaluated eight paired adjacent normal-tumour tissues of oral cancer patients for the expression of MCL-1 and USP9X proteins by western blotting. The expression of both USP9X and that of MCL-1 was significantly (p < 0.01) higher in the tumour versus the corresponding adjacent normal tissues (Fig. 1b). We further evaluated the expression of MCL-1 and USP9X proteins in oral tissues of various histopathological grades (normal oral mucosa (NOM, n = 50), oral leukoplakia with epithelial dysplasia (OL, n = 50), oral submucous fibrosis (OSMF, n = 50) and oral squamous cell carcinoma (OSCC, n = 100)) by immunohistochemistry. The expression of both MCL-1 and USP9X proteins is significantly (p < 0.0001) higher in OL, OSMF and OSCC as compared to NOM (Fig. 1c). Evidently, the expression of MCL-1 and USP9X correlates significantly in NOM (r = 0.3778, p = 0.0068), OL (r = 0.4762, p = 0.0005) and OSCC (r = 0.3066, p = 0.0019), except OSMF (r = 0.1097, p = ns) (Fig. 1d). Interestingly, among the OSCC cohort, the nonrecurrent oral tumours (n = 25, r = 0.4373, p = 0.0288), primary oral tumours (n = 23, r = 0.5046, p = 0.0141) and recurrent oral tumours (n = 23, r = 0.4332, p = 0.0389) exhibited a significant positive correlation between the expression of MCL-1 and USP9X proteins (Supplementary Fig. 2). These observations are suggestive of a probable role of USP9X in the stabilisation of MCL-1 protein in oral cells.

a The basal-level expression of USP9X, MCL-1, BCL-2, BCL-XL and actin was assessed by western blotting in three oral cancer cell lines. The blots are representative of three experiments. The data points in the graph are mean ± SEM of three independent repeats and represent a correlation between normalised expression of MCL-1 and USP9X proteins. b The expression of MCL-1 and USP9X proteins was assessed by western blotting in a set of eight adjacent normal-tumour paired samples of oral cancer patients. Densitometric analysis was performed, and the normalised expression of both proteins in paired normal and tumour tissues is represented as mean + SEM. c The expression of MCL-1 and USP9X proteins was assessed by IHC in the oral tissue samples of NOM, OL, OSMF and OSCC and represented in terms of H Score. d The expression of MCL-1 and USP9X proteins in oral tissue samples of NOM, OL, OSMF and OSCC was determined by IHC. The images are the best representatives of the oral histopathological type studied for the individual proteins (scale bar: 200 μm, inset scale bar: 50 μm). Correlation analysis was performed between MCL-1 and USP9X protein expression in terms of H Score. ns not significant, r Pearson’s correlation coefficient.
USP9X interacts with MCL-1 and stabilises it
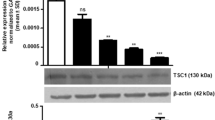
To bring about deubiquitination of its substrate protein, USP9X needs to physically interact with MCL-1. Here, we demonstrate a direct interaction between MCL-1 and USP9X proteins by coimmunoprecipitation and co-immunofluorescence experiments (Fig. 2a, b). We show that MCL-1 partly localises to the mitochondria, and also exhibits diffused cytoplasmic and punctate nuclear staining (Supplementary Fig. 3). Next, to ascertain the contribution of USP9X in stabilising MCL-1 protein in OSCC cells, we downregulated USP9X using siRNA. We first optimised the transfection efficiency and the extent of siRNA-mediated USP9X knockdown in a concentration titration experiment (Supplementary Fig. 4). USP9X knockdown significantly (p < 0.001) lowered MCL-1 protein levels in both AW8507 and SCC029B cells. However, there was no significant change in the levels of BCL-2 or BCL-XL proteins (Fig. 2c). The downregulation of MCL-1 protein in response to USP9X knockdown in this case, however, was not associated with any changes in MCL-1 transcript levels (Supplementary Fig. 5). To investigate whether USP9X contributes to the stabilisation of MCL-1 protein by increasing its half-life, we performed a cycloheximide chase assay. USP9X downregulation rendered a significant (p < 0.01) increase in the rate of MCL-1 degradation in OSCC cells, thereby contributing to its reduced levels (Fig. 2d). To further ascertain the importance of USP9X in stabilising MCL-1 protein in oral cancer cells, we generated USP9X-knockout AW8507 cell line by CRISPR–Cas9 approach. The USP9X-knockout AW8507 cells also exhibited a rapid turnover of MCL-1 protein as compared to the wild-type cells reiterating the fact that USP9X stabilises MCL-1 (Supplementary Fig. 6). Moreover, we observed that USP9X downregulation increased the extent of MCL-1 ubiquitination, suggesting that USP9X has an important role in MCL-1 deubiquitination (Fig. 2e). The reduction of MCL-1 protein levels upon USP9X downregulation in oral cancer cells was also confirmed by immunofluorescence (Fig. 2f and Supplementary Fig. 7). Previous studies from our lab demonstrated the upregulation of MCL-1 at the transcriptional and protein level upon exposure to ionising radiation that contributes to the radioresistance of oral cancer cells.22,23 Therefore, we investigated whether USP9X stabilises and thereby enhances MCL-1’s ability to confer radioresistance in these cells. We indeed observed that USP9X downregulation significantly (p < 0.05) increased the radiosensitivity of oral cancer cells, suggesting a critical role of USP9X in enhanced stabilisation of MCL-1, thereby contributing to radioresistance in oral cancer cells (Fig. 2g). All these observations are suggestive of a critical role of USP9X in MCL-1 stabilisation.

a AW8507 cell lysates were subjected to coimmunoprecipitation using an anti-MCL-1 antibody and an isotype control antibody. For the reverse IP, the cell lysates were incubated with the USP9X antibody. The immunoprecipitates and 5% total input were probed with USP9X and MCL-1 antibodies in western blot to confirm the interaction between them. b AW8507 cells were immunostained for endogenous MCL-1 and USP9X proteins and observed under a confocal microscope. A merged yellow-orange fluorescence indicates potential colocalisation of the two proteins. The images are representative of two independent experiments (scale bar: 10 µm). Colocalisation analysis was also performed by using Coloc2 plugin of Image J. c AW8507 and SCC029B cells were transfected with scramble siRNA (siControl) and USP9X siRNA (siUSP9X). Forty-eight hours post transfection, the cells were harvested and the whole-cell lysates were subjected to western blotting for the detection of USP9X, MCL-1, BCL-2, BCL-XL and actin proteins. The corresponding densitometric analyses are represented as mean + SEM of triplicate experiments. d Cycloheximide chase assay to determine the half-life of MCL-1 protein. AW8507 cells were transfected with 50 nM siControl or siUSP9X, and 48 h post transfection, the cells were treated with 100 µg/ml cycloheximide (CHX). The cell lysates corresponding to the indicated time points were subjected to western blotting for detection of MCL-1, USP9X and actin. The relative half-life of MCL-1 in the siControl vs siUSP9X-transfected cells were determined by densitometric analysis. The data are represented as mean ± SEM of three independent experiments. e siControl and siUSP9X-transfected AW8507 cells were subjected to immunoprecipitation using an anti-MCL-1 antibody. To assess the extent of ubiquitinated MCL-1, the immunoprecipitates were subjected to western blotting using an anti-ubiquitin antibody. f AW8507 cells were transfected with 50 nM of each of siControl or siUSP9X. Forty-eight hours post transfection, the cells were immunostained for MCL-1 and USP9X and observed under a confocal microscope (scale bar: 20 µm). The immunofluorescence images and western blots are representative of triplicate experiments. g AW8507 and SCC029B cells were transfected with 50 nM siUSP9X and harvested at the indicated time points post transfection and analysed for USP9X levels by western blotting. Alternatively, the cells were transfected with either siUSP9X or siControl, and 24 h post transfection, the cells were harvested and reseeded for clonogenic assay. The cells were either left untreated or irradiated with their respective D0 doses as indicated. The cells were allowed to grow for further 8–10 days until visible colonies appear and the colony-forming units (CFUs) were represented as percent survival (relative to unirradiated cells). Images are the best representatives of two experiments. Data are represented as mean ± SEM of two independent experiments.
Deubiquitinase inhibitor WP1130 potently induces apoptosis in OSCC cells
We next investigated the effect of USP9X inhibition by a small-molecule deubiquitinase inhibitor WP1130 (Degrasyn) in oral cancer cell lines. Although WP1130 is a selective deubiquitinase inhibitor, it inhibits several deubiquitinases, including USP5, UCH-L1, USP14, UCH37 and USP9X. Therefore, we first determined the extent of specificity of WP1130 against USP9X. We observed that the USP9X-knockout cells were significantly (p < 0.01) less sensitive to WP1130 than the wild-type cells, suggesting the specificity of WP1130 against USP9X (Supplementary Fig. 8). Treatment of OSCC cells with WP1130 induced a dose- and time-dependent decrease in their viability as evaluated by MTT assay (Fig. 3a, b). WP1130 rapidly induced apoptosis in oral cancer cells as evident by PARP cleavage, which was associated with a rapid decline of MCL-1 protein levels (Fig. 3c). The apoptosis induced by WP1130 was also confirmed by flow cytometry using the Annexin V/Propidium Iodide staining assay (Fig. 3d), by the apparent characteristic nuclear fragmentation (Fig. 3e) and by the caspase-3 cleavage (Supplementary Fig. 9). Next, we determined whether USP9X inhibition by low doses of WP1130, which does not compromise cell viability, can restrain the growth potential of OSCC cells. Treatment of OSCC cell lines AW8507 and SCC029B with sublethal concentration (2.5 μM) of WP1130 severely inhibited their clonogenic potential, suggesting that USP9X might be important for cell viability and proliferation (Fig. 3f). We demonstrate that WP1130-mediated deubiquitinase inhibition potently triggers aggresome formation in oral cancer cells. Immunofluorescence microscopy revealed that the p62/SQSTM1 bodies co-localised with ubiquitin foci, which probably represent polyubiquitinated protein aggregates accumulated upon deubiquitinase inhibition (Fig. 3g). We also observed that WP1130 treatment triggered an autophagic response in these cells as evident by western blotting and by the appearance of LC-3B foci that co-localise with ubiquitin foci. Interestingly, these p62 foci also co-localised with the lysosomal marker protein LAMP-1, indicating a possible turnover of these aggresomes through autophagy, termed as aggrephagy (Supplementary Fig. 10). Electron microscopy of WP1130-treated cells revealed numerous vesicles carrying electron-dense proteinaceous inclusions, possibly indicating aggrephagy (Fig. 3h). Finally, we evaluated the antitumour activity of WP1130 against oral cancer xenografts in immunocompromised mice. The animals that were administered with WP1130 (20 mg/kg) exhibited a significant (p < 0.05) reduction in the mean tumour volume as opposed to the vehicle control group, suggesting potent single-agent antitumour activity of WP1130. The regressing tumours of WP1130-administered animals showed marked induction of apoptosis in the tumour cells, which was evident by a significant (p < 0.0001) increase in TUNEL and caspase-3 staining. Notably, the tumour cells of WP1130-administered animals exhibited significantly (p < 0.01) lower expression of MCL-1 as compared to the vehicle control animals (Fig. 3I). These observations suggest that OSCC cells are dependent on MCL-1 for their survival, destabilisation of which through USP9X inhibition triggers apoptosis.

a AW8507 and SCC029B cells were treated with indicated concentrations of WP1130 for 24, 48 and 72 h. The percent cell viability was determined by MTT assay and expressed relative to the vehicle control. b AW8507 and SCC029B cells were treated with increasing concentrations of WP1130 for 24 h, and the percent cell viability was determined by MTT assay. c The cells were treated with 10 µM WP1130 for the indicated time points, and the cell lysates were subjected to western blotting for the detection of MCL-1, PARP, USP9X and actin proteins. (FL—full length, CL—cleaved PARP). d The cells were either treated as vehicle control (VC: 0.01% DMSO) or with 10 µM WP1130 for 1 h, and the percent cell death was measured by Annexin V/PI staining. e AW8507 cells were treated with 10 µM WP1130 for 1 h or as VC, and the nuclei were stained with DAPI. The characteristic nuclear fragmentation is indicative of cells undergoing apoptosis (scale bar: 20 µm). f AW8507 and SCC029B cells were exposed to 2.5 µM WP1130 or 0.01% DMSO (VC) for 24 h. The media was then replaced and cells were allowed to grow and form visible colonies that were then fixed and stained with crystal violet. g AW8507 cells were either treated as VC or with 10 µM WP1130 for 1 h and were immunostained for p62/SQSTM1 and ubiquitin followed by imaging with a confocal microscope (scale bar: 10 µm). The data are represented as mean ± SEM of triplicate experiments. The blots and images are representative of three independent experiments. h AW8507 cells were either treated as VC or with 10 µM WP1130 for 1 h and were processed for electron microscopy. Arrows indicate numerous cytoplasmic vesicles containing a variety of electron-dense inclusions in WP1130-treated cells. i In vivo antitumour activity of WP1130 was assessed against oral xenografts derived from SCC029B cells. The tissue sections of tumours of the individual animals were stained by haematoxylin and eosin stains or by antibodies specific for cleaved caspase-3 and MCL-1 by IHC and TUNEL as markers of apoptosis (scale bar: 50 μm). The data are represented as mean ± SEM for six animals per group.
Elevated MCL-1/USP9X expression correlates with aggressive oral tumours
Next, we aimed at elucidating the correlation between MCL-1 and USP9X proteins within the OSCC cohort. We first divided the cohort of OSCC patients into high- and low-expressing tumours based on a median expression (H score) of 100 for MCL-1 and 85 for USP9X. We did not find a significant association between MCL-1/USP9X expression with clinicodemographic/clinicopathological parameters such as age, gender, site of lesion, habits or differentiation status of the tumour (Supplementary Table 5). Next, we obtained a cohort of OSCC patients’ tumour samples comprising nonrecurrent oral tumours (n = 25), recurrent oral tumours (n = 23) and their corresponding primary tumours (n = 23) (Supplementary Table 6). The oral tumours exhibited a significant (p < 0.01) positive correlation between MCL-1 and USP9X protein expression as shown earlier (Fig. 1d and Supplementary Fig. 2). Interestingly, the recurrent oral tumours had a significantly (p < 0.0001) higher expression of both MCL-1 and USP9X proteins as compared to their nonrecurrent counterparts and the corresponding paired primary tumours (Fig. 4a). As our previous studies demonstrated the importance of MCL-1 overexpression in therapy resistance in oral cancers, we evaluated if the increased expression of MCL-1/USP9X proteins contributes to the occurrence of more resistant tumours. As expected, we observed that recurrent oral tumours show a significantly high co-expression of MCL-1 and USP9X as compared to the corresponding primary tumours as well as the nonrecurrent tumours (Fig. 4a). This suggests that elevated levels of USP9X in the recurrent tumours may enhance MCL-1 stability, increase its levels and thereby contribute to therapy resistance and thus disease relapse. Moreover, low-grade/early-stage (T1+T2) oral tumours exhibited significantly lower co-expression of MCL-1/USP9X as compared to the high-grade/late-stage (T3+T4) OSCC tumours. Interestingly, within the high-grade oral tumours (T3+T4), the recurrent (R) tumours had a significantly higher expression of MCL-1 and USP9X than the nonrecurrent (NR) ones (Fig. 4b). However, we did not find any association between nodal involvement and MCL-1/USP9X expression (Fig. 4c). These results indicate that elevated MCL-1 protein levels associated with elevated USP9X contribute to the development of more aggressive tumours, which coincides with therapy resistance and disease recurrence.

a The expression of MCL-1 and USP9X proteins was assessed in the primary tumours (PT, n = 23) and the corresponding recurrent tumours (R, n = 23) of OSCC patients with disease relapse and in the tumours of OSCC patients with no evidence of disease recurrence (NR, n = 25) by IHC. Representative IHC images of the two proteins are indicated (scale bar: 200 μm, inset scale bar: 50 μm). The protein expression was represented as H Scores. ***p < 0.0001. b The expression of MCL-1 and USP9X was assessed by IHC in oral tumours and were categorised based on their grades (T1+T2, T3+T4) in the nonrecurrent (NR) and recurrent (R) tissues. c The recurrent and nonrecurrent oral tumours were categorised based on the involvement of regional lymph nodes (N0: node negative, N+: node positive).
High co-expression of MCL-1/USP9X is associated with poor prognosis in OSCC patients
Finally, we aimed at predicting the overall survival (OS) as well as recurrence-free survival (RFS) outcomes of OSCC patients based on MCL-1/USP9X expression. As discussed above, the cohort of OSCC patients was dichotomised into high or low MCL-1/USP9X expression based on their median H scores (100 for MCL-1 and 85 for USP9X). There was a significant (p < 0.01) correlation between MCL-1 and USP9X expression (Fig. 5a). A high expression of MCL-1 alone (hazard ratio: 2.572, p < 0.05) or USP9X alone (hazard ratio: 2.281, p < 0.05) or their higher co-expression (hazard ratio: 3.241, p < 0.05) correlated with a poor OS of OSCC patients (Fig. 5b). This observation was true for RFS as well. Elevated expression of MCL-1 alone (hazard ratio: 2.323, p < 0.05) or USP9X alone (hazard ratio: 2.050, p < 0.05) or their increased co-expression correlated with a reduced RFS (hazard ratio: 3.022, p < 0.05) (Fig. 5c and Table 1). Cumulatively, these results suggest that elevated MCL-1 levels in conjunction with elevated USP9X contribute to therapy resistance and more aggressive tumours, which result in a poor prognosis in terms of recurrence-free survival as well as overall survival in oral cancer patients.

a The expression of MCL-1 and USP9X proteins was determined by IHC in the tumours of OSCC patients and was expressed as H score. The patient cohort was dichotomised into high and low expression based on the median values of H scores for MCL-1 (100) and USP9X (85) proteins. Representative OSCC tissues showing high and low expression of MCL-1 and USP9X proteins. Correlation analysis between high or low MCL-1/USP9X-expressing tumours. b Analysis of the overall survival (OS) and c recurrence-free survival (RFS) by Kaplan–Meier estimator. The survival curves were compared by Mantel–Cox Log Rank test and hazard ratios were calculated, which determine the survival estimates based on the high or low expression of MCL-1 and/or USP9X proteins either individually or in combination.
Discussion
Overexpression of anti-apoptotic proteins of the BCL-2 family, particularly MCL-1, has been demonstrated in many cancers, including OSCC, and is implicated in therapy resistance.8,14,21 Tumour cells maintain sustained elevated levels of pro-survival MCL-1 protein to ensure cell viability. Although increased transcription is known to contribute to its overexpression, enhanced stability of MCL-1 protein also appears to be instrumental for maintaining its sustained elevated levels in tumours,27 primarily because of its characteristic rapid turnover mediated by the ubiquitin–proteasomal degradation pathway.26 In the present study, we provide evidence that in OSCC cells, the deubiquitinase USP9X stabilises MCL-1 protein by deubiquitinating it and thereby contributes to oral tumorigenesis. Moreover, USP9X-mediated elevation of MCL-1 protein levels not only provides a survival advantage to the tumour cells but also contributes to more aggressive tumours, resistance to therapeutics and disease relapse, which eventually culminates in poor prognosis in OSCC patients.
We demonstrated that MCL-1 protein is primarily degraded by the ubiquitin–proteasomal pathway in OSCC cells. Further, we have demonstrated for the first time a striking positive correlation between MCL-1 and USP9X expression in OSCC cell lines and oral tissues. Interestingly, the expression of both MCL-1 and USP9X proteins was higher in oral premalignant lesions, oral carcinomas and recurrent tumours than normal oral mucosa, suggesting a probable role of USP9X in stabilising MCL-1 and thus contributing to oral tumorigenesis. Our data indicate a high co-expression of USP9X and MCL-1 proteins from early to late stages of oral tumorigenesis, suggesting a synergistic pro-survival and oncogenic role of these proteins. Indeed, of the ~90 DUBs encoded by the human genome, a tissue microarray screen identified USP9X to be one of the most significantly dysregulated deubiquitinases in multiple types of solid tumours.34 It is noteworthy that USP9X is found to be one of the most frequently altered genes in gingivo-buccal oral squamous cell carcinoma (GB-OSCC) among the Indian population.35 Owing to their critical role in regulating the stability of key proteins such as c-myc and p53, deubiquitinases (DUBs) are potential therapeutic targets.36
Our results show a direct interaction between MCL-1 and USP9X proteins, and that targeted downregulation of USP9X leads to decreased levels of MCL-1 protein by increasing its turnover. Evidently, USP9X has been shown to interact with and stabilise MCL-1 protein by deubiquitinating it.28 Consistent with our observations, Zhang et al. showed that gemcitabine-mediated disruption of MCL-1/USP9X interaction in solid tumour cell lines is critical for sensitising them to ABT-737, a BH3 mimetic to which MCL-1 is refractory.37 Our study also underscores the role of USP9X in radioresistance as its downregulation sensitised oral cancer cells to ionising radiation, predominantly through the destabilisation of MCL-1 protein. Indeed, earlier studies from our lab have shown the importance of MCL-1 protein in the radioresistance of oral cancer cells.22,23 Further, the targeted downregulation or pharmacological inhibition of MCL-1 protein has been shown to improve the efficacy of radiotherapy in OSCC cells.24,25
Consistent with its role in stabilising MCL-1 protein in OSCC cells, pharmacological inhibition of USP9X by WP1130 resulted in a potent induction of apoptosis owing to rapid degradation of MCL-1 protein. In accordance with our observation, it has been previously demonstrated that downregulation or inhibition of USP9X resulted in degradation of MCL-1 and subsequent sensitisation of solid tumour cell lines to BCL-XL inhibition.38,39 Interestingly, the cell death induced by WP1130 was associated with appearance of numerous aggresomes, polyubiquitinated protein aggregates that are refractory to proteasome-mediated destruction and are sequestered in the form of cytoplasmic inclusions. They probably accumulate in response to WP1130-mediated inhibition of deubiquitinases and elicit an autophagic response to ensure their bulk and safe disposal.40 The single agent in vivo potency of WP1130 against oral cancer xenografts was marked by apoptosis in the tumour cells and downregulation of MCL-1, suggesting that the oral xenografts are highly dependent on MCL-1 for their survival and growth, which in turn is stabilised by USP9X.
In contrast to its oncogenic role, USP9X is known to serve as a tumour suppressor protein in pancreatic ductal adenocarcinoma.41 USP9X not only deubiquitinates MCL-1 but also Beclin-1 and Smad4. Both Beclin-1 and MCL-1 compete for interaction with USP9X and thus negatively modulate the stability of each other in a reciprocal manner.42 USP9X also regulates TGFβ signalling pathway by deubiquitinating Smad4. Loss of USP9X prevents deubiquitination of Smad4, thereby enhancing tumour progression.43 Moreover, USP9X is also critical for many cellular processes and its deficiency or defect may cause certain disorders, including neurodegenerative diseases.44 These tumour suppressor functions of USP9X may correlate with its basal levels, whereas its overexpression may be associated with oncogenic functions, including MCL-1 stabilisation.
In summary, the expression of USP9X and MCL-1 is elevated and positively correlated through early-to-late stages of oral tumorigenesis implying their synergistic pro-survival and oncogenic role. The enhanced stabilisation of MCL-1 protein by the deubiquitinase USP9X might contribute to disease progression, therapy resistance and recurrence, resulting in poor outcome in oral cancers. Systemic targeting of MCL-1 by pan-BCL-2 inhibitors or MCL-1-specific inhibitors has seen setbacks in early phases of clinical trials primarily due to toxicity.45 Destabilising MCL-1 via USP9X inhibition may negotiate the systemic toxicity and the associated side effects. We believe that USP9X may serve as a surrogate biomarker as well as a potential therapeutic target in oral cancer, contributing to the improved management of oral cancer.
References
Mehrotra, R. & Yadav, S. Oral squamous cell carcinoma: etiology, pathogenesis and prognostic value of genomic alterations. Indian J. cancer 43, 60 (2006).
Dikshit, R., Gupta, P. C., Ramasundarahettige, C., Gajalakshmi, V., Aleksandrowicz, L., Badwe, R. et al. Cancer mortality in India: a nationally representative survey. Lancet 379, 1807–1816 (2012).
Warnakulasuriya, K., Harris, C., Scarrott, D., Watt, R., Gelbier, S., Peters, T. et al. Oral cancer: an alarming lack of public awareness towards oral cancer. Br. Dent. J. 187, 319 (1999).
Neville, B. W. & Day, T. A. Oral cancer and precancerous lesions. CA: A Cancer J. Clin. 52, 195–215 (2002).
Rapidis, A. D., Gullane, P., Langdon, J. D., Lefebvre, J. L., Scully, C. & Shah, J. P. Major advances in the knowledge and understanding of the epidemiology, aetiopathogenesis, diagnosis, management and prognosis of oral cancer. Oral Oncol. 45, 299–300 (2009).
Lo, W.-L., Kao, S.-Y., Chi, L.-Y., Wong, Y.-K. & Chang, R. C.-S. Outcomes of oral squamous cell carcinoma in Taiwan after surgical therapy: factors affecting survival. J. Oral Maxillofac. Surg. 61, 751–758 (2003).
Shah, J. P. & Gil, Z. Current concepts in management of oral cancer–surgery. Oral Oncol. 45, 394–401 (2009).
Napier, S. S. & Speight, P. M. Natural history of potentially malignant oral lesions and conditions: an overview of the literature. J. Oral Pathol. Med. 37, 1–10 (2008).
Warnakulasuriya, S., Johnson, N. W., Van & der Waal, I. Nomenclature and classification of potentially malignant disorders of the oral mucosa. J. Oral Pathol. Med. 36, 575–580 (2007).
Van der Waal, I. Oral potentially malignant disorders: is malignant transformation predictable and preventable? Med. Oral Patol.ía oral. y. cirugía bucal 19, e386 (2014).
Speight, P. M., Khurram, S. A. & Kujan, O. Oral potentially malignant disorders: risk of progression to malignancy. Oral Surg., Oral Med., Oral Pathol. Oral Radiol. 125, 612–627 (2018).
Warnakulasuriya, S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 45, 309–316 (2009).
Davids, M. S. & Letai, A. Targeting the B-cell lymphoma/leukemia 2 family in cancer. J. Clin. Oncol. 30, 3127 (2012).
Placzek, W., Wei, J., Kitada, S., Zhai, D., Reed, J. & Pellecchia, M. A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell Death Dis. 1, e40 (2010).
Derenne, S., Monia, B., Dean, N. M., Taylor, J. K., Rapp, M. J., Harousseau, J. L. et al. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-xL is an essential survival protein of human myeloma cells. Blood 100, 194–199 (2002).
Aichberger, K. J., Mayerhofer, M., Krauth, M.-T., Skvara, H., Florian, S., Sonneck, K. et al. Identification of mcl-1 as a BCR/ABL-dependent target in chronic myeloid leukemia (CML): evidence for cooperative antileukemic effects of imatinib and mcl-1 antisense oligonucleotides. Blood 105, 3303–3311 (2005).
Pepper, C., Lin, T. T., Pratt, G., Hewamana, S., Brennan, P., Hiller, L. et al. Mcl-1 expression has in vitro and in vivo significance in chronic lymphocytic leukemia and is associated with other poor prognostic markers. Blood 112, 3807–3817 (2008).
Thallinger, C., Wolschek, M. F., Maierhofer, H., Skvara, H., Pehamberger, H., Monia, B. P. et al. Mcl-1 is a novel therapeutic target for human sarcoma synergistic inhibition of human sarcoma xenotransplants by a combination of Mcl-1 antisense oligonucleotides with low-dose cyclophosphamide. Clin. Cancer Res. 10, 4185–4191 (2004).
Shigemasa, K., Katoh, O., Shiroyama, Y., Mihara, S., Mukai, K., Nagai, N. et al. Increased MCL-1 expression is associated with poor prognosis in ovarian carcinomas. Cancer Sci. 93, 542–550 (2002).
Sieghart, W., Losert, D., Strommer, S., Cejka, D., Schmid, K., Rasoul-Rockenschaub, S. et al. Mcl-1 overexpression in hepatocellular carcinoma: a potential target for antisense therapy. J. Hepatol. 44, 151–157 (2006).
Mallick, S., Patil, R., Gyanchandani, R., Pawar, S., Palve, V., Kannan, S. et al. Human oral cancers have altered expression of Bcl‐2 family members and increased expression of the anti‐apoptotic splice variant of Mcl‐1. J. Pathol. 217, 398–407 (2009).
Mallick, S., Agarwal, J., Kannan, S., Pawar, S., Kane, S. & Teni, T. PCNA and anti-apoptotic Mcl-1 proteins predict disease-free survival in oral cancer patients treated with definitive radiotherapy. Oral Oncol. 46, 688–693 (2010).
Palve, V. C. & Teni, T. R. Association of anti-apoptotic Mcl-1L isoform expression with radioresistance of oral squamous carcinoma cells. Radiat. Oncol. 7, 135 (2012).
Palve, V., Mallick, S., Ghaisas, G., Kannan, S. & Teni, T. Overexpression of Mcl-1L splice variant is associated with poor prognosis and chemoresistance in oral cancers. PLoS ONE 9, e111927 (2014).
Sulkshane, P. & Teni, T. BH3 mimetic Obatoclax (GX15-070) mediates mitochondrial stress predominantly via MCL-1 inhibition and induces autophagy-dependent necroptosis in human oral cancer cells. Oncotarget 8, 60060 (2017).
Ertel F., Nguyen M., Roulston A. & Shore G. C. Programming cancer cells for high expression levels of Mcl1. EMBO Rep. 14, 328–336 (2013).
Thomas, L. W., Lam, C. & Edwards, S. W. Mcl-1; the molecular regulation of protein function. FEBS Lett. 584, 2981–2989 (2010).
Schwickart, M., Huang, X., Lill, J. R., Liu, J., Ferrando, R., French, D. M. et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 463, 103 (2010).
Tatake, R. J., Rajaram, N., Damle, R., Balsara, B., Bhisey, A. & Gangal, S. G. Establishment and characterization of four new squamous cell carcinoma cell lines derived from oral tumors. J. Cancer Res. Clin. Oncol. 116, 179–186 (1990).
Martin, C. L., Reshmi, S. C., Ried, T., Gottberg, W., Wilson, J. W., Reddy, J. K. et al. Chromosomal imbalances in oral squamous cell carcinoma: examination of 31 cell lines and review of the literature. Oral Oncol. 44, 369–382 (2008).
Bartholomeusz, G., Talpaz, M., Bornmann, W., Kong, L.-Y. & Donato, N. J. Degrasyn activates proteasomal-dependent degradation of c-Myc. Cancer Res. 67, 3912–3918 (2007).
Bartholomeusz, G. A., Talpaz, M., Kapuria, V., Kong, L. Y., Wang, S., Estrov, Z. et al. Activation of a novel Bcr/Abl destruction pathway by WP1130 induces apoptosis of chronic myelogenous leukemia cells. Blood 109, 3470–3478 (2007).
Gomez-Bougie, P., Ménoret, E., Juin, P., Dousset, C., Pellat-Deceunynck, C. & Amiot, M. Noxa controls Mule-dependent Mcl-1 ubiquitination through the regulation of the Mcl-1/USP9X interaction. Biochem. Biophys. Res. Commun. 413, 460–464 (2011).
Luise, C., Capra, M., Donzelli, M., Mazzarol, G., Jodice, M. G., Nuciforo, P. et al. An atlas of altered expression of deubiquitinating enzymes in human cancer. PLoS ONE 6, e15891 (2011).
of the International IPT, Maitra, A., Biswas, N. K., Amin, K., Kowtal, P., Kumar, S. et al. Mutational landscape of gingivo-buccal oral squamous cell carcinoma reveals new recurrently-mutated genes and molecular subgroups. Nat. Commun. 4, 2873 (2013).
Turcu, F. E. R., Ventii, K. H. & Wilkinson, K. D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 78, 363 (2009).
Zhang, C., Cai, T.-Y., Zhu, H., Yang, L., Jiang, H., Dong, X.-W. et al. Synergistic anti-tumor activity of gemcitabine and ABT-737 in vitro and in vivo through disrupting the interaction of USP9X and Mcl-1. Mol. Cancer Ther. 1091, 2010 (2011).
Peddaboina, C., Jupiter, D., Fletcher, S., Yap, J. L., Rai, A., Tobin, R. P. et al. The downregulation of Mcl-1 via USP9X inhibition sensitizes solid tumors to Bcl-xl inhibition. BMC Cancer 12, 541 (2012).
Kapuria, V., Peterson, L. F., Fang, D., Bornmann, W. G., Talpaz, M. & Donato, N. J. Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res. 70, 9265–9276 (2010).
Lamark, T. & Johansen, T. Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int. J. Cell Biol. 2012 (2012).
Pérez-Mancera, P. A., Rust, A. G., Van Der Weyden, L., Kristiansen, G., Li, A., Sarver, A. L. et al. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature 486, 266–270 (2012).
Elgendy, M., Ciro, M., Abdel-Aziz, A. K., Belmonte, G., Dal Zuffo, R., Mercurio, C. et al. Beclin 1 restrains tumorigenesis through Mcl-1 destabilization in an autophagy-independent reciprocal manner. Nat. Commun. 5, 1–11 (2014).
Dupont, S., Mamidi, A., Cordenonsi, M., Montagner, M., Zacchigna, L., Adorno, M. et al. FAM/USP9x, a deubiquitinating enzyme essential for TGFβ signaling, controls Smad4 monoubiquitination. Cell 136, 123–135 (2009).
Murtaza, M., Jolly, L. A., Gecz, J. & Wood, S. A. La FAM fatale: USP9X in development and disease. Cell. Mol. Life Sci. 72, 2075–2089 (2015).
Xiang, W., Yang, C.-Y. & Bai, L. MCL-1 inhibition in cancer treatment. OncoTargets Ther. 11, 7301 (2018).
Acknowledgements
We thank Dr S. Gollins, the University of Pittsburg for kindly providing the SCC029B cell line. P.S. was supported by Senior Research Fellowship from the Department of Biotechnology, Government of India. We sincerely thank members of the light microscopy, electron microscopy, flow cytometry, histology and animal house facility at ACTREC. We extend our thanks to all the members of the Teni laboratory for their technical help, suggestions and critical comments while preparing the paper.
Author information
Authors and Affiliations
Contributions
Conceptualisation: P.S., S.S.P. and T.T.; data curation: P.S., S.N.P., R.W., S.S.P., A.U., S.O., P. Rajput and T.T.; formal analysis: P.S., R.W., S.S.P., P. Rane, R.P. and S.N.; methodology: P.S., S.N.P., R.W., S.S.P., A.U., S.O., P. Rajput, P.K. and H.W.; funding acquisition: P.S., S.N.P., R.P., S.N. and T.T.; writing original draft: P.S.; review and editing: P.S., R.P., S.N., P. Rane and T.T.; resources: S.N.P., R.W., S.S.P., R.P., S.N. and T.T.; supervision: S.N.P., R.P., S.N. and T.T.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This work was approved by the Institutional Ethics Committee (IEC-III) of TMC-ACTREC (Project No. 900211) and was performed in accordance with the Declaration of Helsinki. All patients provided written informed consent. The animal studies were reviewed and approved by the Institutional Animal Ethics Committee (IAEC) of TMC-ACTREC constituted under the guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Government of India.
Consent to publish
Not applicable.
Data availability
All the data generated or analysed in this study are included in this published article and supplementary information file. If required, the clinical outcomes/follow-up and biomarker data are readily available. Supplementary information is available at the British Journal of Cancer’s website.
Competing interests
The authors declare no competing interests.
Funding information
This project was supported by an Intramural grant from Tata Memorial Centre.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Sulkshane, P., Pawar, S.N., Waghole, R. et al. Elevated USP9X drives early-to-late-stage oral tumorigenesis via stabilisation of anti-apoptotic MCL-1 protein and impacts outcome in oral cancers. Br J Cancer 125, 547–560 (2021). https://doi.org/10.1038/s41416-021-01421-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-021-01421-x
This article is cited by
-
Protein degradation: expanding the toolbox to restrain cancer drug resistance
Journal of Hematology & Oncology (2023)
-
USP32 deubiquitinase: cellular functions, regulatory mechanisms, and potential as a cancer therapy target
Cell Death Discovery (2023)
-
Identification of bicyclic compounds that act as dual inhibitors of Bcl-2 and Mcl-1
Molecular Diversity (2023)
-
Myeloid cell leukemia-1 expression in cancers of the oral cavity: a scoping review
Cancer Cell International (2022)
-
Stabilization of MCL-1 by E3 ligase TRAF4 confers radioresistance
Cell Death & Disease (2022)
