
Abstract
Background
This Phase 1 dose-escalation/expansion study assessed safety/tolerability of sapanisertib, an oral, highly selective inhibitor of mTORC1/mTORC2, in advanced solid tumours.
Methods
Eligible patients received increasing sapanisertib doses once daily (QD; 31 patients), once weekly (QW; 30 patients), QD for 3 days on/4 days off QW (QD × 3dQW; 33 patients) or QD for 5 days on/2 days off QW (QD × 5dQW; 22 patients). In expansion cohorts, 82 patients with renal cell carcinoma (RCC), endometrial or bladder cancer received sapanisertib 5 mg QD (39 patients), 40 mg QW (26 patients) or 30 mg QW (17 patients).
Results
Maximum tolerated doses of sapanisertib were 6 mg QD, 40 mg QW, 9 mg QD × 3dQW and 7 mg QD × 5dQW. Frequent dose-limiting toxicities (DLTs) included hyperglycaemia, maculo-papular rash (QD), asthenia and stomatitis (QD × 3dQW/QD × 5dQW); expansion phase doses of 5 mg QD and 30 mg QW were selected based on tolerability beyond the DLT evaluation period. One patient with RCC achieved complete response; nine experienced partial responses (RCC: seven patients; carcinoid tumour/endometrial cancer: one patient each). Sapanisertib pharmacokinetics were time-linear and supported multiple dosing. Pharmacodynamic findings demonstrated treatment-related reductions in TORC1/2 biomarkers.
Conclusions
Sapanisertib demonstrated a manageable safety profile, with preliminary antitumour activity observed in RCC and endometrial cancer.
Clinical trial registration
ClinicalTrials.gov, NCT01058707.
Similar content being viewed by others
Background
The phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) signalling pathway is a central regulator of cellular growth, proliferation and survival.1 Dysregulation of PI3K/AKT/mTOR activity is frequently observed in human cancers.2 As part of the mammalian target of rapamycin complex 1 (mTORC1) and 2 (mTORC2), mTOR is a key intracellular point of convergence for several pathways, thus representing an important therapeutic target. Inhibition of mTOR may decrease protein translation and prevent abnormal cell proliferation and tumour angiogenesis.3,4 Accordingly, rapamycin analogues (“rapalogs”), such as temsirolimus and everolimus, have been approved by the US Food and Drug Administration for the treatment of advanced renal cell cancer (RCC) and several other cancers.1,5,6,7,8,9,10 Rapalogs exert their effect mainly on mTORC1, with only a mild inhibitory effect on mTORC2.11 Inhibition of mTORC1, without mTORC2 inhibition, can result in the activation of AKT through a negative feedback mechanism, which may limit rapalog efficacy by accelerating tumour progression.12
Preclinical data have demonstrated that inhibition of AKT activity, through mTORC2 inhibition, may block tumour progression.13 Subsequently, a new generation of mTORC1/2 inhibitors have been developed, as dual inhibition may offer an advantage over mTORC1 inhibitors by targeting at least three key enzymes (PI3K, AKT and mTOR).14 Proof of this concept was demonstrated in preclinical models for several epithelial malignancies.15,16,17,18
Sapanisertib is an investigational, oral and highly selective adenosine triphosphate-competitive mTOR kinase inhibitor that suppresses both mTORC1 and mTORC2. Preclinical models of efficacy, safety and pharmacokinetics (PK) of sapanisertib have largely relied on daily dosing.19,20,21,22,23 Investigation of different dosing schedules in preclinical efficacy models has demonstrated commensurate tumour growth inhibition with administration of sapanisertib in daily or intermittent dosing schedules.19,20,21,22,23 These preclinical data generally indicated that efficacy was related to total exposure (i.e. area under the curve) and independent of dosing schedule. However, the clinical tolerability was expected to be different for different schedules. Therefore, we evaluated a range of dosing schedules in this first-in-human Phase 1 study (NCT01058707) of sapanisertib in patients with advanced solid tumours, with an expansion phase including patients with RCC, endometrial or bladder cancer.
Methods
Study design
This Phase 1, open-label, dose-escalation and expansion study aimed to determine the safety, tolerability and preliminary efficacy of sapanisertib in patients with advanced solid tumours. During dose-escalation, patients received one of four sapanisertib dosing schedules: once daily (QD), once weekly (QW; days 1, 8, 15 and 22), QD for 3 days on/4 days off each week (QD × 3dQW; days 1–3, 8–10, 15–17 and 22–24) and QD for 5 days on/2 days off each week (QD × 5dQW; days 1–5, 8–12, 15–19 and 22–26) in a 28-day cycle with dose escalation based on a modified Fibonacci schema.
Definitions for dose-limiting toxicities (DLTs), as predetermined in the study protocol, were: any grade ≥3 non-haematologic toxicity (except inadequately treated grade 3 nausea and/or vomiting and grade 3 diarrhoea [all patients should have received optimal antiemetic and/or antidiarrhoeal prophylaxis treatment], grade 3 hyperglycaemia lasting ≤14 days [all patients should have received optimal anti-glycaemic treatment, including insulin) and grade 3 rash lasting ≤3 days [all patients should have received topical steroid treatment, oral antihistamines and pulse oral steroids, if necessary]); grade 4 neutropenia lasting >7 days in the absence of growth factor support; grade 4 neutropenia of any duration accompanied with fever ≥38.5 °C and/or systemic infection; any other grade ≥4 haematologic toxicity.
Sapanisertib dosing was withheld for Grade ≥ 3 treatment-related toxicities. If the event resolved to Grade ≤ 1 or baseline values within 28 days of interrupting therapy, the patient could resume study treatment at a ≥25% dose reduction or, for patients in the dose-escalation phase, at the next lower dose level with the sponsor’s approval. If dose modification was required for subjects receiving ≤4 mg QD, then the dosing frequency was decreased to 5 days per week, instead of decreasing the daily dose administered. If sapanisertib dosing was delayed for >28 consecutive days for treatment-related toxicity, despite supportive treatment per standard clinical practice, or more than 2 dose reductions of sapanisertib were required in a patient, sapanisertib therapy was stopped, the patient was discontinued from the study and the follow-up visit was completed within 30 days of the last administration of sapanisertib.
The maximum tolerated dose (MTD) was to be determined in the QD schedule before enrolment of initial single-patient cohorts for the QW and QDx3d QW schedules. If a grade ≥ 2 AE was observed in any single-patient cohort, an additional 2–5 patients were assigned to that cohort. Subsequent dose cohorts for that schedule included 3–6 patients. Dose cohorts for the QDx5dQW schedule enrolled three patients with an additional three patients enrolled if a DLT was observed per a standard 3 + 3 design. Once the MTD was identified for each of the dosing schedules, an additional six patients were enrolled to obtain further PK and safety data prior to expansion.
Based on data collected during dose-escalation, the expansion phase evaluated the safety and efficacy of QD and QW sapanisertib in patients with RCC, endometrial or bladder cancer. Patients could continue receiving sapanisertib for ≤1 year (or beyond, if the investigator and sponsor agreed) in the absence of disease progression or unacceptable toxicity.
The primary objective was to determine the MTD and DLTs for each sapanisertib dosing schedule and evaluate the safety and tolerability of sapanisertib in both phases. Secondary objectives were to evaluate preliminary antitumour activity, PK in peripheral blood, and pharmacodynamics (PD) of sapanisertib, as measured by modulation in phosphorylation of S6, 4EBP1 and NDRG1 in surrogate tissue (skin) and tumour.
The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. Institutional review boards approved all aspects of the study. All participants provided written informed consent.
Patients
Eligible patients were aged ≥18 years with locally advanced or metastatic solid tumours who had failed standard-of-care therapy, had an Eastern Cooperative Oncology Group performance status of 0–1, and had adequate bone marrow, hepatic, renal and metabolic (fasting serum glucose ≤ 130 mg/dL and fasting triglycerides ≤ 300 mg/dL) function.
Patients with locally advanced or metastatic brain tumours were eligible if their brain metastases had been treated (without evidence of progression or haemorrhage post-treatment), and if they had not taken dexamethasone 4 weeks prior to the first study drug administration and with no ongoing requirement for dexamethasone or antiepileptic drugs. In the expansion phase, eligible patients had measurable disease per the Response Evaluation Criteria in Solid Tumors (RECIST), v1.1.
Patients could be considered for enrolment in one of three disease-specific cohorts: (1) advanced or recurrent endometrial adenocarcinoma with disease progression following ≥1 prior chemotherapy regimen; (2) advanced/metastatic urothelial cancer (carcinoma of the bladder, ureter and/or renal pelvis), progressive after ≥1 prior therapy in the metastatic/unresectable setting; (3) advanced RCC after failure of ≥1 prior antivascular endothelial growth factor therapy with no prior TORC1 inhibitor therapy, or progressed on treatment with TORC1 inhibitor therapy.
Patients who had received prior cancer therapy within 2 weeks, systemic corticosteroid therapy within 1 week prior to the first dose of study drug, or bisphosphonates within 30 days prior to the first sapanisertib dose were not eligible. Additionally, patients with impaired cardiac function or significant active cardiovascular disease were also excluded. In the expansion phase, patients who had received prior AKT, PI3K, dual PI3K/TORC1/2 or TORC1/2 inhibitors were excluded.
Assessments
Patients who received ≥1 dose of sapanisertib were included in the safety population. The response-evaluable population included patients who received ≥1 dose of sapanisertib, had measurable disease at baseline, and had ≥1 postbaseline assessment. In the dose-escalation phase, the evaluable population included patients who received ≥75% of the planned sapanisertib doses in cycle 1 or experienced a DLT. Response was assessed according to the RECIST v1.124 after every two treatment cycles. Adverse events (AEs) were assessed using the National Cancer Institute Common Terminology Criteria for Adverse Events, v4.0. Patients were provided with a home blood-glucose meter to monitor their fasting predose blood-glucose measurements to assess hyperglycaemia as an on-target AE and PD marker.
Peripheral blood was collected serially to quantify plasma levels of sapanisertib for PK analysis via validated liquid chromatography tandem mass spectrometry with an assay range of 1–1000 ng/mL (MicroConstants, San Diego, CA), during both dose-escalation and expansion phases. Blood samples were collected before each dose and 0.5, 1, 2, 4, 8 and 24 h after dosing on cycle 1, day 1 and cycle 2, day 1, in the dose-escalation phase. In the expansion phase, samples were collected predose and 2, 4 and 6 h postdose on cycle 1, day 1, and then predose and 2 h postdose on day 8, 15 or 22 of cycle 1.
The tissue PK and PD of sapanisertib were investigated in skin and tumour biopsies. During dose escalation, 3-mm core skin biopsies were collected predose and on any day from 8–15 (3 h postdose) of cycle 1 for immunohistochemistry (IHC) assessment of mTORC1 downstream effects (p4EBP1, pS6) and pNDRG1 (Mosaic Laboratories, Lake Forest, CA). IHC for p-AKT did not pass validation and is not reported. Archival tumour tissues for assessment of prognostic markers were collected at baseline.
Statistical analyses were primarily descriptive and graphical in nature, with no formal statistical hypothesis testing.
Results
Patients
From December 2009 to January 2013, 116 patients were enrolled to the dose-escalation phase and received single-agent sapanisertib in the following schedules: QD (n = 31), QW (n = 30), QD × 3dQW (n = 33) and QD × 5dQW (n = 22). From March 2013 to April 2014, an additional 82 patients with RCC, endothelial or bladder cancer were enrolled in the expansion phase to receive sapanisertib 5 mg QD (n = 39), 40 mg QW (n = 26) or 30 mg QW (n = 17). Baseline demographics and characteristics are shown in Table 1.
DLTs and MTD determination
Dose escalation, DLTs and MTDs are summarised in Table 2. The MTD was determined to be 6 mg for the QD schedule based on 4/10 patients reporting a DLT in cycle 1 (grade 3 maculo-papular rash [n = 1], grade 3 diarrhoea [n = 1], grade 3 asthenia [n = 1], grade 5 ventricular fibrillation/cardiac arrest [n = 1]) and 40 mg for the QW schedule based on 2/12 patients reporting a DLT (grade 3 dry mouth and fatigue [n = 1], grade 3 asthenia [n = 1]), 9 mg for the QD × 3dQW schedule based on 1/6 evaluable patients reporting a DLT (grade 3 hypophosphatemia) and 7 mg for the QD × 5dQW schedule based on all three patients receiving 13 mg reporting DLTs (grade 3 fatigue, asthenia and stomatitis [n = 1 each]), 4/13 patients receiving 10 mg reporting DLTs (grade 2 stomatitis [n = 1], grade 3 stomatitis [n = 1], grade 3 asthenia [n = 1], grade 3 stomatitis and fatigue [n = 1]) and no patients reported DLTs at 7 mg.
Based on biochemical, PK and tolerability data, and early signs of antitumour activity, the QD and QW schedules were selected for expansion. Sapanisertib 5 mg QD was selected rather than the MTD of 6 mg, because the 6 mg dose was poorly tolerated beyond the DLT evaluation period in 10 evaluable patients. The QW 40 mg dose was determined as the MTD based on DLT criteria and was initially selected for expansion. However, follow-up of the first 21 patients enrolled at this dose revealed that several patients required dose modifications or discontinued treatment due to AEs; the recommended expansion dose was thereafter reduced to 30 mg QW.
Treatment exposure and safety
During dose escalation, patients received a median of two treatment cycles (range, 1–58; Supplementary Table 1). Across all doses, 97% of patients experienced treatment-related AEs (TRAEs), and 11% experienced at least one treatment-related grade ≥ 3 AE (Supplementary Table 1). The most common TRAEs were hyperglycaemia, nausea, stomatitis, vomiting, decreased appetite and diarrhoea (Table 3). Hyperglycaemia was the most common grade ≥ 3 TRAE (Supplementary Table 2), which was generally well controlled with metformin and home glucose monitoring. Serious AEs occurred in 43% of patients; 24% discontinued treatment due to an AE (Supplementary Table 1).
During expansion, patients received a median of two treatment cycles (range, 1–26; Supplementary Table 1). All patients in this phase experienced at least one TRAE; 46% experienced at least one grade ≥ 3 TRAE (Supplementary Table 1). TRAEs and all-cause AEs in the expansion phase are summarised in Table 3 and Supplementary Table 3, respectively. The most common TRAEs according to schedule were fatigue (56%) and nausea (51%) at 5 mg QD, fatigue (88%), nausea and hyperglycaemia (77% each) at 40 mg QW, and hyperglycaemia (71%), nausea and fatigue (65% each) at 30 mg QW (Table 3). Supplementary Table 2 and Supplementary Table 4 summarise grade ≥ 3 TRAEs and all-cause AEs, respectively, in the expansion phase. Hyperglycaemia was also the most common grade ≥ 3 TRAE in the expansion phase. Serious AEs occurred in 41% of patients and 15% discontinued due to an AE.
AEs resulting in treatment discontinuation during the expansion phase occurred in 18% of patients receiving 5 mg QD or 30 mg QW dosing and 8% of patients receiving 40 mg QW dosing. Fewer patients in the 30 mg QW dose group had their dose modified or reduced than in the other dose groups (47% for 30 mg QW vs 69% for 5 mg QD and 77% for 40 mg QW) (Supplementary Table 1).
There were 7 on-study deaths within 30 days of last dose. Four patients died during dose-escalation: 1 due to ventricular fibrillation and cardiac arrest (6 mg QD considered possibly related to sapanisertib by the investigator, in a patient with pre-existing cardiovascular risk factors), 3 from progressive cancer; 2 gastric (10 mg QW [n = 1], 9 mg QD × 3dQW [n = 1]) and 1 breast (12 mg QD × 3dQW). Three patients died during expansion due to disease progression (30 mg QW [n = 2], 40 mg QW [n = 1]).
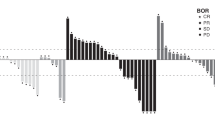
Antitumour activity
At data cut-off (02 October 2015), two patients, one with RCC receiving 15 mg QW (dose escalation; 58 cycles) and one with TORC1 inhibitor−naïve RCC receiving 30 mg QW (expansion; 26 cycles), were still receiving study drug. Tumour response across both study phases is summarised in Table 4 and Supplementary Fig. 1. Supplementary Fig. 1 also shows study duration and best percentage change for the expansion phase. During dose-escalation, two patients with RCC receiving 15 mg QW and 40 mg QW achieved a partial response (PR) maintained for >32 months for an objective response rate (ORR) of 8%, and one with a carcinoid tumour receiving 10 mg QD × 5dQW achieved a PR maintained for > 32 months for an ORR of 6%. Five patients in dose-escalation maintained stable disease (SD) for ≥ 6 months. In the expansion phase, one patient with TORC1 inhibitor−naïve RCC receiving 40 mg QW achieved a complete response (CR) maintained for 16 months. Five additional patients with RCC achieved PRs: three TORC1 inhibitor–naïve (5 mg QD, 30 mg QW, and 40 mg QW) for an ORR of 22%, and two with TORC1 inhibitor failure (both 5 mg QD) for an ORR of 9%. There was also one PR in a patient with endometrial cancer receiving 5 mg QD (ORR of 6%). Four of the seven objective responses reported in the expansion phase persisted for > 16 months: three patients with TORC1 inhibitor–naïve RCC (30 mg QW [n = 1], 40 mg QW [n = 2]) and one with TORC1 inhibitor failure RCC (5 mg QD). Six patients with RCC maintained SD for ≥6 months. Among patients with bladder cancer (n = 13), no objective responses were achieved, and 1/5 patients with SD maintained SD for ≥ 6 months.
Pharmacokinetics
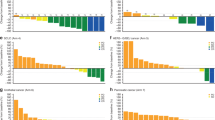
Cycle 1, day 1, PK data were available from 112 patients and cycle 2, day 1, PK data were available from 70 patients. Single-dose (cycle 1, day 1) and multiple-dose (cycle 2, day 1) PK data are summarised in Fig. 1, Supplementary Fig. 2 and Supplementary Table 5. Sapanisertib exhibited rapid oral absorption, with a median Tmax ranging from 1.0 − 2.8 h across all doses (Supplementary Table 5). Sapanisertib plasma concentrations generally increased in a dose-dependent manner within the 2–40 mg dose range; mean plasma half-life ranged from 5.9 to 9.4 h, with PK variability percentage coefficient of variation (%CV) values for area under the curve from time zero to 24 h postdose (AUC0–24h) ranging from ~26 to 87%. Sapanisertib did not accumulate in plasma to any appreciable extent with repeat dosing in any of the schedules (Fig. 1). Sapanisertib PK were generally consistent during both assessment periods (cycle 1, day 1, and cycle 2, day 1), indicating a lack of a time-dependent accumulation with repeat dosing.

Mean (SD) plasma concentration–time profiles of multiple-dose sapanisertib (cycle 2, day 1) on the a QD, b QW, c QD × 3dQW and d QD × 5dQW dosing schedules. Error bars indicate SD. QD once daily, QD × 3dQW QD for 3 days on/4 days off QW, QD × 5dQW QD for 5 days on/2 days off QW, QW once weekly, SD standard deviation.
Pharmacodynamics
During dose-escalation, the PD effect of sapanisertib on downstream effectors of TORC1 (p4EBP1 and pS6) and TORC2 (pPRAS40 and pNDRG1) was measured in skin-tissue biopsies from 88 patients collected at baseline and days 8–15 (3 h postdose) of cycle 1. Treatment-related decreases in p4EBP1, pS6, pPRAS40 and pNDRG1 were consistent with dual TORC1/2 inhibition at sapanisertib doses of ≥4 mg (Supplementary Fig. 3). There was a trend toward a dose-dependent PD effect between 2–6 mg sapanisertib; however, no noteworthy differences in PD marker trends were observed at doses ≥6 mg or across the different dosing schedules and dose levels. PD data in tumour tissue were limited due to the small number of samples. For the five patients with paired tumour samples available for review (biopsies at baseline and cycle 1, week 2), the levels of p4EBP1, pS6, pPRAS40 and pNDRG1 decreased after sapanisertib treatment (Supplementary Fig. 4). An integrated PK/PD analysis correlated the plasma concentration of sapanisertib at a single time point with PD findings in skin and demonstrated a concentration-related change in pS6, p4EBP1, pNDRG1 and pPRAS40 (Supplementary Fig. 5).
Discussion
In this open-label, Phase 1, first-in-human study, the safety profile of sapanisertib was characterised and shown to be manageable and consistent with the toxicity profiles of other mTOR inhibitors. Sapanisertib showed preliminary antitumour activity in patients with RCC (TORC1 inhibitor–naïve and exposed) and in endometrial cancer.
The sapanisertib MTDs were determined as 6 mg QD, 40 mg QW, 9 mg QD × 3dQW and 7 mg QD × 5dQW. In a previous study in advanced haematological malignancies, the sapanisertib MTD for QD dosing was slightly lower (4 mg) and the MTD for QD × 3dQW was the same (9 mg).25 Consistent with the lower MTD determined for QD dosing, 5 mg QD (expansion phase) was selected for further investigation, as 6 mg QD was poorly tolerated beyond the DLT evaluation period. DLTs varied across the dosing schedules: maculo-papular rash and hyperglycaemia, considered on-target toxicities for this class of agents, were the most frequently reported DLTs with QD dosing, and stomatitis and asthenia with the QD × 3dQW and QD × 5dQW schedules. Sapanisertib DLTs previously reported in patients with haematologic malignancies, were stomatitis, urticaria, blood creatinine elevation, fatigue, nausea and vomiting with QD dosing, and erythematous rash, fatigue, asthenia, mucosal inflammation and thrombocytopenia with QD × 3dQW dosing.25
The safety profile of sapanisertib in this Phase 1 study was generally manageable across all schedules, and tolerability was greater with increased intermittence of dosing. Common AEs related to sapanisertib across all schedules in both phases included hyperglycaemia, nausea and vomiting. Hyperglycaemia is a known side effect of PI3K pathway inhibition reported in other studies investigating dual mTORC inhibitors.26,27 Hyperglycaemia was found to be dose-dependent in the QD dosing schedule, occurring more frequently at the higher QW dose. Hyperglycaemia in both phases was generally grade 1–2 in severity, consistent with episodes previously reported,25 and easily controlled with metformin therapy. Otherwise, there were no consistent dose-dependent trends in AEs related to sapanisertib among the schedules, and AEs reported for sapanisertib 5 mg QD and 30 mg QW in the expansion phase were generally manageable. In the expansion phase, the sapanisertib dose for QW dosing was reduced from 40 mg (MTD determined during dose escalation) to 30 mg due to high rates of nausea and vomiting.
Preliminary antitumour activity was observed with sapanisertib across different schedules in patients with RCC and endometrial cancer. One patient receiving sapanisertib 40 mg QW had a CR and nine patients achieved PR, with response maintained in two patients for >32 months and four for >16 months. This is consistent with efficacy reported in other phase 1/2 studies of dual TORC1/2 kinase inhibitors in patients with advanced malignancies,25,27,28,29 and the ORR of 22% reported in patients with RCC naïve to a TORC1 inhibitor compares favourably to ORRs observed with rapalogs in patients with RCC (ORR 8.6% with temsirolimus8 and 1–5% with everolimus).9,7,30,31 However, a recent Phase 2 study of sapanisertib 30 mg QW versus everolimus 10 mg QD in patients with refractory clear-cell RCC reported no responses with single-agent sapanisertib versus an ORR of 13% with everolimus (NCT02724020).32
The PK profile showed that single doses of oral sapanisertib were rapidly absorbed and concentrations increased in a dose-dependent manner between 2 and 40 mg; the PK of sapanisertib was time-linear and supported the use of multiple dosing. Following repeat dosing, sapanisertib did not accumulate in plasma to any appreciable extent in any of the dosing schedules. Ghobrial et al.25 reported a similar PK profile, supporting the administration of both daily and intermittent schedules. Therefore, QW and QD dosing schedules were selected for expansion based on their apparent similar preliminary antitumour activity and better tolerability. PD findings demonstrated treatment-related reductions in TORC1/2 biomarkers (p4EBP1, pS6, pPRAS40 and pNDRG1), which supports dual TORC1/2 inhibition of sapanisertib in doses ≥4 mg.
In conclusion, sapanisertib had a manageable safety profile across the various schedules studied. Recommended Phase 2 doses, based on the data presented here, include 30 mg QW and 5 mg QD. Preliminary antitumour activity was observed in RCC and endometrial cancer. Phase 2 studies of sapanisertib (QW dosing) in patients with previously treated, metastatic clear-cell RCC (NCT03097328) and endometrial cancer (NCT02725268) have completed enrolment. Findings from these studies and those presented here contribute to sapanisertib dosing decisions in ongoing Phase 1/2 studies as a single agent or in combination with standard-of-care therapy in multiple malignancies.
References
Porta, C., Paglino, C. & Mosca, A. Targeting PI3K/Akt/mTOR signaling in cancer. Front. Oncol. 4, 64 (2014).
Zoncu, R., Efeyan, A. & Sabatini, D. M. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 12, 21–35 (2011).
Duran, I., Lambea, J., Maroto, P., Gonzalez-Larriba, J. L., Flores, L., Granados-Principal, S. et al. Resistance to targeted therapies in renal cancer: the importance of changing the mechanism of action. Target. Oncol. 12, 19–35 (2017).
Moschetta, M., Reale, A., Marasco, C., Vacca, A. & Carratu, M. R. Therapeutic targeting of the mTOR-signalling pathway in cancer: benefits and limitations. Br. J. Pharm. 171, 3801–3813 (2014).
Yardley, D. A., Noguchi, S., Pritchard, K. I., Burris, H. A. 3rd, Baselga, J., Gnant, M. et al. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Adv. Ther. 30, 870–884 (2013).
Yao, J. C., Fazio, N., Singh, S., Buzzoni, R., Carnaghi, C., Wolin, E. et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet 387, 968–977 (2016).
Motzer, R. J., Escudier, B., Oudard, S., Hutson, T. E., Porta, C., Bracarda, S. et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 372, 449–456 (2008).
Hudes, G., Carducci, M., Tomczak, P., Dutcher, J., Figlin, R., Kapoor, A. et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 356, 2271–2281 (2007).
AFINITOR® (everolimus) Prescribing Information. April 2018 revision.
Torisel® (temsorolimus) Prescribing Information. February 2015 revision.
Kajiwara, M., Masuda, S. Role of mTOR inhibitors in kidney disease. Int. J. Mol. Sci. 17, 1–12 https://doi.org/10.3390/ijms17060975 (2016).
O’Reilly, K. E., Rojo, F., She, Q. B., Solit, D., Mills, G. B., Smith, D. et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 66, 1500–1508 (2006).
Guertin, D. A., Stevens, D. M., Saitoh, M., Kinkel, S., Crosby, K., Sheen, J. H. et al. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 15, 148–159 (2009).
Zhou, H., Luo, Y. & Huang, S. Updates of mTOR inhibitors. Anticancer Agents Med. Chem. 10, 571–581 (2010).
Bhagwat, S. V., Gokhale, P. C., Crew, A. P., Cooke, A., Yao, Y., Mantis, C. et al. Preclinical characterization of OSI-027, a potent and selective inhibitor of mTORC1 and mTORC2: distinct from rapamycin. Mol. Cancer Ther. 10, 1394–1406 (2011).
Janes, M. R., Vu, C., Mallya, S., Shieh, M. P., Limon, J. J., Li, L. S. et al. Efficacy of the investigational mTOR kinase inhibitor MLN0128/INK128 in models of B-cell acute lymphoblastic leukemia. Leukemia 27, 586–594 (2013).
Korets, S. B., Musa, F., Curtin, J., Blank, S. V. & Schneider, R. J. Dual mTORC1/2 inhibition in a preclinical xenograft tumor model of endometrial cancer. Gynecol. Oncol. 132, 468–473 (2014).
Zheng, B., Mao, J. H., Qian, L., Zhu, H., Gu, D. H., Pan, X. D. et al. Pre-clinical evaluation of AZD-2014, a novel mTORC1/2 dual inhibitor, against renal cell carcinoma. Cancer Lett. 357, 468–475 (2015).
Garcia-Garcia, C., Ibrahim, Y. H., Serra, V., Calvo, M. T., Guzman, M., Grueso, J. et al. Dual mTORC1/2 and HER2 blockade results in antitumor activity in preclinical models of breast cancer resistant to anti-HER2 therapy. Clin. Cancer Res. 18, 2603–2612 (2012).
Gokmen-Polar, Y., Liu, Y., Toroni, R. A., Sanders, K. L., Mehta, R., Badve, S. et al. Investigational drug MLN0128, a novel TORC1/2 inhibitor, demonstrates potent oral antitumor activity in human breast cancer xenograft models. Breast Cancer Res. Treat. 136, 673–682 (2012).
Hernandez-Prat, A., Rodriguez-Vida, A., Juanpere-Rodero, N., Arpi, O., Menendez, S., Soria-Jimenez, L. et al. Novel oral mTORC1/2 inhibitor TAK-228 has synergistic antitumor effects when combined with paclitaxel or PI3Kalpha inhibitor TAK-117 in preclinical bladder cancer models. Mol. Cancer Res. 17, 1931–1944 (2019).
Ingels, A., Zhao, H., Thong, A. E., Saar, M., Valta, M. P., Nolley, R. et al. Preclinical trial of a new dual mTOR inhibitor, MLN0128, using renal cell carcinoma tumorgrafts. Int. J. Cancer 134, 2322–2329 (2014).
Kang, M. H., Reynolds, C. P., Maris, J. M., Gorlick, R., Kolb, E. A., Lock, R. et al. Initial testing (stage 1) of the investigational mTOR kinase inhibitor MLN0128 by the pediatric preclinical testing program. Pediatr. Blood Cancer 61, 1486–1489 (2014).
Eisenhauer, E. A., Therasse, P., Bogaerts, J., Schwartz, L. H., Sargent, D., Ford, R. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247 (2009).
Ghobrial, I. M., Siegel, D. S., Vij, R., Berdeja, J. G., Richardson, P. G., Neuwirth, R. et al. TAK-228 (formerly MLN0128), an investigational oral dual TORC1/2 inhibitor: a phase I dose escalation study in patients with relapsed or refractory multiple myeloma, non‐Hodgkin lymphoma, or Waldenström’s macroglobulinemia. Am. J. Hematol. 91, 400–405 (2016).
Bauer, T. M., Patel, M. R. & Infante, J. R. Targeting PI3 kinase in cancer. Pharm. Ther. 146, 53–60 (2015).
Bendell, J. C., Kelley, R. K., Shih, K. C., Grabowsky, J. A., Bergsland, E., Jones, S. et al. A phase I dose-escalation study to assess safety, tolerability, pharmacokinetics, and preliminary efficacy of the dual mTORC1/mTORC2 kinase inhibitor CC-223 in patients with advanced solid tumors or multiple myeloma. Cancer 121, 3481–3490 (2015).
Basu, B., Dean, E., Puglisi, M., Greystoke, A., Ong, M., Burke, W. et al. First-in-human pharmacokinetic and pharmacodynamic study of the dual m-TORC 1/2 inhibitor AZD2014. Clin. Cancer Res. 21, 3412–3419 (2015).
Powles, T., Wheater, M., Din, O., Geldart, T., Boleti, E., Stockdale, A. et al. A randomised phase 2 study of AZD2014 versus everolimus in patients with VEGF-refractory metastatic clear cell renal cancer. Eur. Urol. 69, 450–456 (2016).
Choueiri, T. K., Escudier, B., Powles, T., Tannir, N. M., Mainwaring, P. N., Rini, B. I. et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): final results from a randomised, open-label, phase 3 trial. Lancet Oncol. 17, 917–927 (2016).
Motzer, R. J., Escudier, B., McDermott, D. F., George, S., Hammers, H. J., Srinivas, S. et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 373, 1803–1813 (2015).
Choueiri, T. K., Porta, C., Suárez, C., Hainsworth, J., Voog, E., Duran, I. et al. Randomised phase 2 study of sapanisertib (TAK‑228/MLN0128) ± TAK-117 versus everolimus in patients with VEGF-targeted therapy-refractory metastatic clear cell renal cell carcinoma. (Oral presentation at EIKCS, Dubrovnik, 2019).
Acknowledgements
We thank all the patients who participated in these studies and their families, as well as all the investigators and site staff who made these studies possible. We also acknowledge writing support from Helen Wilkinson, PhD (FireKite, an Ashfield company, part of UDG Healthcare plc, which was funded by Millennium Pharmaceuticals, Inc.) and editorial support from Janice Y. Ahn, PhD (Millennium Pharmaceuticals, Inc.) in compliance with Good Publication Practice 3 ethical guidelines (Battisti et al., Ann Intern Med 2015;163:461–4).
Author information
Authors and Affiliations
Contributions
Conception and design: M.H.V., H.A.B., R.I.N., A.E., Y.S., F.S. and D.V.F. Collection and assembly of data: All authors. Data analysis and interpretation: M.H.V., H.A.B., R.I.B., A.E., Y.S., F.S. and D.V.F. Drafting the manuscript: All authors. Final approval of manuscript: All authors. Agree to be accountable for all aspects of the work, which includes ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved: All authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. Institutional review boards (listed in the Supplementary Information) approved all aspects of the study. All participants provided written informed consent.
Data availability
The datasets, including the redacted study protocol, redacted statistical analysis plan and individual participants data supporting the results reported in this article, will be made available within three months from initial request, to researchers who provide a methodologically sound proposal. The data will be provided after its de-identification, in compliance with applicable privacy laws, data protection and requirements for consent and anonymisation.
Competing interests
M.H.V. has received honoraria from Novartis and consultancy fees from Alexion Pharmaceuticals, Bayer, Calithera Biosciences, Corvus Pharmaceuticals, Exelixis, Eisai, GlaxoSmithKline, Natera, Novartis, Ontarget therapeutics and Pfizer. He has received research funding from Bristol-Myers Squibb, Genentech and Pfizer, and travel grants from Takeda and Eisai. M.S.G. has ownership of stock/shares with Medelis, CARE Mission and Beckon Call, and received consulting fees from Agenus, Deciphera and Salarius. B.R. is an advisor for and has received consultancy fees and research funding from Pfizer, Merck, Bristol-Myers Squibb and Roche. He has also received researching funding from AstraZeneca and Peloton. V.M. is an advisor for Eisai, Merck and Karyopharm, and has received honoraria from Eisai and Merck. He has received research funding from Eisai, Merck, Lilly, Karyopharm, Bristol-Myers Squibb, AstraZeneca, Takeda and Genentech. T.M. is an advisor for Roche, Amgen, Novartis, Sanofi, Shire, Incyte, Celgene and Baxter, and is a speaker for Celgene, Shire, Sanofi, Amgen and Servier. She has received research funding from Beigene, Novartis and Celgene. D.C.S. has received research funding from Agensys, Bayer, Exelixis, Incyte, Lilly, MedImmune, Novartis, OncoMed, Seattle Genetics, Bristol-Myers Squibb/Medarex, Essa, Genentech, Medivation/Astellas, Merck, Astellas and Takeda. A.C. is an advisor for Merck Serono, Roche, Beigene, Bayer, Servier, Lilly, Novartis, Takeda, Astelas, Pierre Fabre, Amgen and Foundation Medicine. He has received grants from Merck Serono and Roche, and research funding from Genentech, Merck Serono, Roche, Beigene, Bayer, Servier, Lilly, Novartis, Takeda, Astelas, Fibrogen, Amcure, Sierra Oncology, AstraZeneca, MedImmune, Bristol-Myers Squibb and MSD. I.P. is an advisor for and has received honoraria and consultancy fees from Amgen. He has received research funding from Nektar, Amgen, Rgenix and ADC. S.J. has received grants from AstraZeneca, Tesaro and Astex. S.P. has received honoraria from UD Pharma and Tyme Inc. R.I.N., A.E., Y.S., F.S. and D.V.F. are employed by Millennium Pharmaceuticals Inc., a wholly owned subsidiary of Takeda Pharmaceutical Co. Ltd. H.B. is an advisor for Mersana, AstraZeneca, FORMA Therapeutics, Janssen, Novartis, Roche/Genentech, MedImmune, Bristol-Myers Squibb, Celgene, Incyte, Boehringer Ingelheim, Eisai and Tolero Pharmaceuticals. He has received research funding from Roche/Genentech, Bristol-Myers Squibb, Incyte, AstraZeneca, MedImmune, Macrogenics, Novartis, Boehringer Ingelheim, Lilly, Seattle Genetics, Merck, Celgene, Agios, Jounce Therapeutics, Morderna Therapeutics, CytomX Therapeutics, GlaxoSmithKline, Verastem, Tesaro, Immunocore, Takeda, BioMed Valley Discoveries, TG Therapeutics, Loxo, Vertex, eFFECTOR Therapeutics, Janssen, Gilead Sciences, BioAtla, CicioMed, Harpoon Therapeutics, Jiangsu Hengrui Medicine, Arch, Kyocera, Arvinas and Revolution Medicines. He has provided expert testimony for Novartis. All remaining authors have declared no conflicts of interest.
Funding information
This study was supported by Millennium Pharmaceuticals Inc., Cambridge, MA, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. NIH/NCI Cancer Center Support Grant (Memorial Sloan Kettering Cancer Center, P30 CA008748).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Voss, M.H., Gordon, M.S., Mita, M. et al. Phase 1 study of mTORC1/2 inhibitor sapanisertib (TAK-228) in advanced solid tumours, with an expansion phase in renal, endometrial or bladder cancer. Br J Cancer 123, 1590–1598 (2020). https://doi.org/10.1038/s41416-020-01041-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-020-01041-x
This article is cited by
-
Modulating the PI3K Signalling Pathway in Activated PI3K Delta Syndrome: a Clinical Perspective
Journal of Clinical Immunology (2024)
-
Phase I dose escalation study and pilot efficacy analysis of LXI-15029, a novel mTOR dual inhibitor, in Chinese subjects with advanced malignant solid tumors
BMC Cancer (2023)
-
Identification and subsequent validation of transcriptomic signature associated with metabolic status in endometrial cancer
Scientific Reports (2023)
-
Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease
Signal Transduction and Targeted Therapy (2023)
-
Phase I study of sapanisertib with carboplatin and paclitaxel in mTOR pathway altered solid malignancies
npj Precision Oncology (2023)
