
Abstract
Background
Tumour cells with BRCA1/2 gene mutations demonstrate increased sensitivity to platinum and poly (ADP-ribose) polymerase (PARP) inhibitors. 6-mercaptopurine (6MP) was found to selectively kill BRCA-defective cells in a xenograft model as effectively as the PARP inhibitor AG014699, even after these cells acquired resistance to a PARP inhibitor or cisplatin.
Methods
This phase II single-arm trial investigated the activity of 6MP 55–75 mg/m2 per day, and methotrexate 15–20 mg/m2 per week in advanced breast or platinum-resistant ovarian cancer patients with a BRCA1/2 germline mutation, who had progressed after ≥1 previous line of chemotherapy. The primary outcome was objective response including stable disease (SD) as an assessment of clinical benefit rate (CBR), at 8 weeks, by RECIST v1.1. Secondary outcomes included overall survival (OS) and progression-free survival (PFS).
Results
In total, 67 evaluable patients were recruited; 55 ovarian and 11 breast cancer patients. In total, 21 patients had SD (31%), one had a partial response (1.5%); CBR was 33% at 8 weeks. In total, 12/67 patients (18%) had SD at 16 weeks. In total, five ovarian cancer patients had SD for over 200 days. Median OS was 10.3 months (95% CI 6.9–14.5), median PFS 1.9 months (1.7–2.8).
Conclusions
The overall activity of 6MP and methotrexate in these patients was low; however, there was a small group of patients who appeared to derive longer-term clinical benefit.
Trial registration
NCT01432145 http://www.ClinicalTrials.gov.
Similar content being viewed by others

Background
BRCA1 and BRCA2 genes play an important role in homologous recombination DNA repair and have been implicated in familial breast and ovarian cancer syndromes. Ovarian cancer is the fifth commonest cancer in women,1 with 46% 5-year survival rate.2 Over 15% of women who are diagnosed with high-grade serous ovarian carcinoma will have a germline BRCA mutation present.3,4 Breast cancer is the most common cancer in women and accounts for between 18 and 25% of all female malignancies worldwide.5 There is a familial component in 5–10% of all breast cancer cases, with most commonly, mutations in the BRCA1/2 genes and p53, ATM or PTEN genes.6,7 The triple-receptor negative breast cancer phenotype, i.e. negative for oestrogen receptor, progesterone receptor and HER2, who also carries an adverse prognosis, accounts for 80–90% of BRCA1-associated breast cancers.8 For patients with metastatic cancer, the challenge is to develop more effective therapies that maximise tumour cell killing (efficacy) and minimise toxicity.
In patients with BRCA1/2-deficient cancers, the use of molecular targeted therapy by using poly (ADP-ribose) polymerase (PARP) inhibitors, has demonstrated a clear benefit. The molecular mechanisms that underlie the selective killing of homologous recombination-deficient BRCA mutant cells by PARPi were initially thought to be solely due to inhibition of base excision repair (BER), with PARPi causing an increase in DNA single-strand breaks (SSBs) that led to toxic double-strand breaks at replication forks.9,10 However, other mechanisms, such as PARP trapping on DNA at sites of unrepaired SSB causing physical obstruction,11 and PARPi enhancing non-homologous end joining in some tumour cells,12 also may play a significant role in cell death.
PARP inhibitors have revolutionised the treatment of high-grade serous ovarian cancer and have shown particular efficacy in women with a BRCA mutation. Between 2014 and 2017, three PARP inhibitors, olaparib (LYNPARZA®, AstraZeneca Pharmaceuticals LP13), niraparib14 and rucaparib15 have been licensed in the treatment of recurrent high-grade ovarian cancer. Olaparib has recently shown efficacy in the front-line setting, with an improvement in disease-free survival when used as a maintenance therapy trial in women with newly diagnosed ovarian cancer, which may result in a new treatment option in the near future.16
Among patients with HER2-negative metastatic breast cancer and a germline BRCA mutation, olaparib monotherapy provided a significant benefit over standard therapy; median progression-free survival was 2.8 months longer and the risk of disease progression or death was 42% lower with olaparib monotherapy than with standard therapy.17
There are multiple mechanisms of PARP inhibitor resistance, including restoration of the homologous recombination pathway through secondary BRCA reversion mutations,18 hyperactivation of non-homologous end joining19 and increased stabilisation of replication forks independent of BRCA1/2 reversion mutations.20 Given the expanding clinical use of PARP inhibitors and the high likelihood of acquired resistance, there is a significant need for new treatment strategies to manage PARP inhibitor-resistant disease.
In a screen for novel drugs that selectively kill BRCA2-defective cells, Helleday and colleagues identified 6-thioguanine (6TG)21 and demonstrated that 6TG induces DNA double-strand breaks that are repaired by homologous recombination. They found that 6TG was as efficient as the PARP inhibitor, AG014699, in selectively killing BRCA2-defective tumours in a xenograft model, and that 6TG also kills cisplatin-resistant or PARP inhibitor-resistant (PIR) BRCA2-defective cells.21 Although homologous recombination is reactivated in some PIR cells in response to PARP inhibitors, it is not fully restored for the repair of 6TG-induced lesions. This is likely to be due to the repair of 6TG defects also being dependent on mismatch repair (MMR), in contrast to the MMR-independent replication defects produced by PARP inhibitors. This suggested that 6TG may be effective in the treatment of tumours that have developed resistance to PARP inhibitors or cisplatin chemotherapy.21
6-Mercaptopurine (6MP) is a prodrug that is converted to the same cytotoxic moiety as 6TG, i.e. 6-thioguanine nucleotides (6TGN), but with fewer toxic effects.22 Low-dose methotrexate is used in combination with 6MP as it promotes the formation of 6TGN. 6MP is metabolised to 6-thioinosine monophosphate and subsequently to 6TGN by hypoxanthine guanine phosphoribosyl transferase and can then be incorporated into nucleic acids. See Supplementary Fig. 1. The cytotoxic effects of 6MP are predominantly due to this incorporation of 6TGN into the DNA, as they are structurally similar to endogenous purine-based guanine. Thiopurine toxicity is delayed as TGNs require passage through one S phase of the cell cycle to incorporate into DNA in place of guanine. The incorporated 6TGN (about 1 in 104 bases) is methylated to 6-meTG by endogenous S-adenosylmethionine, which becomes a substrate for mismatch repair in the second replication round to mediate its toxicity.23,24,25,26
Inactivation of 6MP to 6-thiouric acid occurs via xanthine oxidase and also by thiopurine methyltransferase (TPMT). TPMT methylates 6MP to 6-methylmercaptopurine (6MMP) at the expense of TGNs and 6MMP is a strong inhibitor of purine de novo synthesis and can also result in immunosuppression.27 Quantitation of 6TG (activation of 6MP) and 6MMP (inactivation of 6MP) may give an indication of TPMT activity and hence the potential efficacy of 6MP in a patient.28
It can take 2–3 months (depending on TPMT status) to see therapeutic benefits from 6MP in acute lymphoblastic leukaemia (ALL) and inflammatory bowel disease.29
Methotrexate is also an antimetabolite antineoplastic agent. It inhibits tetrahydrofolate dehydrogenase and prevents formation of tetrahydrofolate, which is required for synthesis of thymidylate, an essential component of DNA. It is routinely used in combination with 6MP in patients with ALL as it has been shown to reduce de novo purine synthesis and thus enhance the cytotoxicity of 6MP by promoting its conversion to 6TGNs.30 Methotrexate has also been widely used in a number of solid tumours such as breast, ovarian, lung and cervical cancers and shown to induce regression. Furthermore, a synthetic lethality screen has identified methotrexate as an agent that has activity in DNA MMR-defective cancers, as evidenced by its selective toxicity in cells lacking functional mutS homologue 2 (MSH2) gene mutations.31
Based on these preclinical findings, 6MP, combined with low-dose methotrexate, was used to test a new therapeutic option for women with known BRCA mutations and relapsed breast or ovarian cancer. This is the first trial to assess the efficacy and toxicity of 6MP and weekly methotrexate in women with BRCA-mutated platinum-resistant ovarian cancer or relapsed BRCA-mutated breast cancer.
Methods
Study design
The 6MP study was a single-arm, two-stage Phase II trial, which recruited from 14 centres across the United Kingdom. Conduct of the trial complied with the Declaration of Helsinki, and ethical approval was obtained prior to the study opening.
Further details of the study design, patient selection, interventions and outcome measures are provided in the previously published protocol paper.32
Participants and treatment
To be eligible for inclusion in the 6MP study, patients must have been aged 18 or older, have proven BRCA1 or BRCA2 germline mutations and measurable disease as defined by Response Evaluation Criteria in Solid Tumours (RECIST) v1.1 criteria. Ovarian cancer patients had to have disease that was either platinum-resistant or be those in whom further platinum-based therapy was inappropriate. Breast cancer patients must have had locally advanced or metastatic breast cancer and may have received up to three previous lines of chemotherapy in the locally advanced or metastatic setting. All patients must have had an Eastern Cooperative Oncology Group (ECOG) performance score between 0 and 2, a life expectancy of greater than 12 weeks and adequate haematological and biochemical function. Patients were excluded from participating in the study if they had a Low/Low genotype on TPMT testing.
Registered patients received 6MP once daily and methotrexate once weekly in 28-day cycles until they developed disease progression, unacceptable toxicity or patient/clinician decision to stop treatment. Starting doses were modified from 75 mg/m2 of 6MP and 20 mg/m2 of methotrexate to 55 and 15 mg/m2, respectively, because many of the first 26 registered patients required dose reductions or treatment delays due to myelosuppression.
Outcome measures
Primary outcome was objective response including stable disease (SD), as an assessment of clinical benefit (CB), according to RECIST v1.1, defined as complete response (CR), partial response (PR) and stable disease confirmed by computerised tomography or magnetic resonance imaging. Tumour assessments were carried out at baseline and every 8 weeks until the end of treatment. Quality assurance of tumour assessments was conducted by an independent radiographer for at least one patient at each site. Two time points of interest were 8 and 16 weeks after the first treatment. Long-term stable disease patients were defined as those having SD for over 200 days. Secondary endpoints included overall survival (OS), progression-free survival (PFS), pharmacokinetics (PK) and quality of life (QoL). Outcome definitions are provided in the protocol paper. Safety was evaluated by using National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.0. Feasibility of a multicentre trial was evaluated by at least three patients per year per site. A CA125 response according to the Gynaecological Cancer Intergroup (GCIG) criteria occurred if there was at least a 50% reduction in CA125 levels from a pre-treatment sample and this was maintained for at least 28 days.
Sample size determination
The Simon compromise/admissible two-stage design33,34 was implemented to provide enough power for an estimated 65 evaluable patients to detect a 10% change from 10% in the absolute response rate including SD with a power of 90% and a significance level of 20%.
Statistical analysis and patient groups
The proportions of clinical benefit and of CR, PR and SD at 8 and 16 weeks were described. Association of objective response at 8 weeks with baseline characteristics (prior PARP inhibitor treatment, BRCA status, TPMT status and volume of disease) was assessed by using a chi-squared test or Fisher’s exact test, as appropriate.
OS and PFS were presented as median and quartiles overall and separately for patients previously treated with PARP inhibitors. A Tarone–Ware test rather than a log-rank test was applied to assess the association of OS and PFS with prior PARP inhibitor treatment because of the indication of non-proportional hazards.
Objective response rate (ORR), stable disease rate, OS and PFS analyses were conducted on a modified intention-to-treat population, which included evaluable patients who had received at least one dose of treatment and who had a response assessment, irrespective of their compliance to the planned course of treatment.
The safety analysis population comprised all registered patients.
6MMP (inactivation of 6MP) and 6TG (activation of 6MP) were measured in red blood cells for pharmacokinetic analysis, by using a validated high-performance liquid chromatography method (HPLC) with absorbance detection using similar methods as described.35 Red blood cell (RBC) 6TG and 6MMP was assessed at Cycle 1 days 1 and 8, Cycle 2 day 1 and Cycle 3 day 1. Acid hydrolysis is used to convert 6MMP and 6TGN to AMTCI and 6TG, respectively, this is detectable at wavelengths of 310 and 342 nm, respectively. The ratio of 6TG and 6MMP could give an indication of TPMT activity and hence the efficacy of 6MP in a patient. The inter-patient variability in 6MP and 6TG levels was assessed graphically as well as their relationship with response over time. TPMT status was assessed in all patients.
Results
Patient demographics and recruitment feasibility
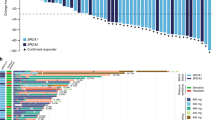
In total, 133 patients with advanced ovarian or breast cancer were screened for eligibility from 14 UK sites between May 2011 and October 2014; 74 patients were consented and registered, and 67 of these registered patients were found to be evaluable (Fig. 1). This is larger than the planned sample size of 65 patients, to compensate for unevaluable patients. On average, 1.4 patients were recruited per site per year.

CONSORT flow diagram.
Baseline characteristics are shown in Table 1. Of the 67 evaluable, 57 (85%) were ovarian cancer patients and 10 (15%) had breast cancer. In total, 40 (60%) patients had a BRCA1 mutation and 27 (40%) had a BRCA2 mutation. Of all the patients, 26 (39%) had received a prior PARP inhibitor. These patients were heavily pre-treated, and the mean number of prior therapies received for ovarian cancer was 4.7 (standard deviation (STD) 2.2), and 4.2 (STD 2.3) for breast cancer. The median time between finishing previous therapy and entering the 6MP trial was just 1.9 (interquartile range (IQR) 1.1–4.6) months across all patients.
Treatment exposure
Median total duration of trial treatment was 55 days overall; 55 days for ovarian cancer patients and 91 days for breast cancer patients. In total, 18 patients (27%) remained on the trial for over 100 days, and 7 patients (10%) for over 200 days. In total, 5 patients (7%) discontinued treatment within 4 weeks due to disease progression. Among the 24 patients who started with 75 mg/m2 of 6MP and 20 mg/m2 of methotrexate, 11 (46%) required dose modifications. Their mean daily 6MP dose intensity was 94.4 mg/day (STD 29.9) and their median time on trial was 63 days (IQR 30–135). After modification of the starting dose, 10 of the remaining 43 patients (23%) required dose modifications, their mean daily 6MP dose intensity was 82.6 mg/day (STD 23.6) and their median time on trial was 55 days (IQR 40–61).
Objective response (clinical benefit)
At stage 1 of the trial, the first milestone of at least 3 out of 30 evaluable patients having SD, PR or CR at 8 weeks was surpassed; 10/30 patients (33%) had SD at 8 weeks, and hence the trial continued to recruit a further 35 patients.
Of 67 evaluable patients at the second stage, 22 (CBR: 33% with 95% CI: 22–45%) patients had an objective response or stable disease at 8 weeks. Only one patient had a partial response; hence the stable disease rate was 31% (Table 2). At 16 weeks, 12 (18%) patients had SD. No statistically significant correlation between 8- or 16-week response/disease stabilisation and any baseline characteristics, such as BRCA status, ECOG performance status or volume of disease, was found.
Biochemical response rate
In total, 6/57 (11%) ovarian cancer patients had a CA125 response by GCIG criteria over the course of the trial.
Progression-free and overall survival
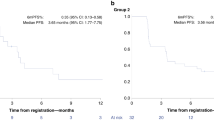
Median OS was 10.3 months (95% CI 6.9–14.5 months). Median PFS was 1.9 months (95% CI 1.7–2.8) and the proportion of patients who were progression free at 6 months was 16%. OS and PFS Kaplan–Meier curves are shown in Fig. 2a, b, respectively.

Overall survival (a) and progression-free survival (b).
Response by prior PARPi exposure
There were no statistically significant differences in OS or PFS in those 26 patients (39%) who had received prior PARP inhibitor treatment compared with those 41 patients (61%) who had not (Tarone–Ware test, p = 0.84 for OS, p = 0.93 for PFS). The median OS for patients exposed and unexposed to prior PARP were 9.9 (5.9–17.7 months) and 11.9 months (7.1–14.5 months). The median PFS values were 2.1 (1.7–3.4) and 1.8 (1.6–3.7).
Longer-term stable disease patients
In total, 5 ovarian cancer patients had SD for more than 200 days; their clinical characteristics are summarised in Table 3. All but one of these patients had visceral disease. Two patients also had a biochemical response according to GCIG criteria. One patient had just one Grade 3 or 4 AE during the course of the trial, and the other four patients had between three and five Grade 3 or 4 AEs.
Tolerability
A pre-planned safety analysis was performed after the first 12 patients had received 3 months of therapy. The initial starting dose was 75 mg/m2 of 6MP and 20 mg/m2 of methotrexate. In total, 11 of the first 24 patients (46%) required a dose reduction, and therefore the starting dose was reduced to 55 mg/m2 of 6MP and 15 mg/m2 of methotrexate. Subsequently 11 of 43 patients (26%) required a dose reduction.
In total, 67 (91%) of 74 registered patients had at least one AE, of which 45 patients (61%) had 106 AEs of CTCAE grade 3 or above. The most frequently occurring grade 3 or above AEs were abdominal pain, anaemia and neutropenia. In total, 23 episodes of grade 3–4 neutropenia were reported in the trial, experienced by 18/74 (24%) patients (Table 4). There were also 167 CTCAE grade 2 adverse drug reactions, experienced by 48/74 (65%) patients, most commonly anaemia, fatigue, neutropenia and nausea. SAEs were reported by 33 (49%) patients; 47 SAEs in total, of which 12 were Grade 3 or above. There were no treatment-related deaths; all deaths were disease related.
Over the course of the trial, 12 patients (18%) were withdrawn from study treatment due to toxicity. Other causes of treatment discontinuation, without PFS, were investigator decision (2 patients, 3%), patient request (1 patient, 2%) and death (1 patient, 2%). All other patients (76%) had a PFS event.
Pharmacokinetics
Measurement of 6TG and 6MMP
Our analysis demonstrated considerable inter-patient variability in the levels of RBC 6TG and 6MMP levels (Supplementary Fig. 2). There was no apparent relationship between the levels of the active moiety (6TGN measured as 6TG) and response to 6MP or duration of response or PFS. The wide inter-patient variability in 6TGN levels observed is thought likely to be as a result of genetic polymorphisms of TPMT.
Pharmacogenomics (TPMT status)
The TPMT status in the subset of patients who derived the greatest clinical benefit, extending beyond 200 days, demonstrated that all of the five long-term stable disease patients had high/high TPMT status.
Quality of life (QoL)
Quality of life could not be analysed due to the low questionnaire completion rate; only two patients (3%) completed the baseline and 12-month follow-up QoL questionnaires.
Discussion
The aim of this trial was to determine the activity and efficacy of 6MP in combination with weekly methotrexate in women with relapsed BRCA-mutated breast and ovarian cancer.
Despite the fact that 6MP 55 mg/kg and MTX 15 mg/m2 was a tolerable combination, there was only one partial response to treatment. However, disease stabilisation (8 weeks) was seen in 33% (95% CI 22–45%) of patients and a small proportion of patients (n = 5, 7%) derived disease stabilisation and clinical benefit (>200 days) with this combination. The response rates and stable disease rates observed are lower than the rates observed in a single-arm phase II trial of the PARP inhibitor, olaparib, that demonstrated a 31% response rate by RECIST v1.1 and a 40% disease stabilisation rate in women with BRCA-mutated platinum-resistant ovarian cancer, and a 12.9% (95% CI, 5.7–23.9) response rate and 47% (95% CI, 34.0–59.9) stable disease rate in advanced breast cancer.36
In comparison with standard chemotherapy in this setting, i.e. women with advanced platinum-resistant ovarian cancer, the SaPPrOC trial demonstrated a 43% response rate (combined CA125 and RECIST response) and PFS of 5.3 months in women treated with weekly paclitaxel.37 Retrospective data indicate similar response rates to weekly paclitaxel in women with BRCA- mutated ovarian cancer.38 Furthermore, a trial of caelyx chemotherapy vs olaparib (200 vs 400 mg) in women with BRCA- mutated ovarian cancer that had recurred within 12 months of prior platinum chemotherapy demonstrated a PFS of 7.1 months in the group receiving caelyx.39
Despite the use of cross-trial comparisons, it is clear that the clinical efficacy of 6MP with MTX is lower in comparison with standard chemotherapeutic agents.
TPMT status and TGN levels
We hypothesised that higher levels of intracellular 6TGN may result in increased efficacy of 6MP due to the long half-life of these metabolites.40 Our study demonstrated significant inter-patient and intra-patient variability in the levels of RBC 6TGN (measured as 6TG). The main determinants of 6MP activity have been found to be BSA and TPMT mutational status.41,42 We assessed TPMT status and excluded those patients homozygous for TPMT, due to the high chance of life-threatening myelosuppression.43 In this study, we did not demonstrate a correlation between dose intensity and TPMT status. We also did not alter 6MP dose based on TPMT status, white blood cell (WBC) counts or RBC TGN levels as is recommended in the treatment of paediatric ALL.44 In fact, following the pre-planned safety review in the first 12 patients, a dose reduction from 75 to 55 mg/m2 was required due to the high rates of myelosuppression and dose reductions required. It is likely that this cohort of heavily pre-treated patients, with an average of 4.7 (ovarian) and 2.8 (breast) prior lines of therapy, had significantly reduced bone marrow capacity compared with paediatric ALL patients.
The five long-term SD ( > 200 days) all had high/high TPMT status and experienced similar toxicity to non-long-term responders. TPMT is the predominant enzyme involved in methylation and formation of inactive metabolites of 6MP, and it could therefore be expected that these patients may have lower levels of RBC TGN and higher levels of 6MMP and therefore reduced activity of 6MP. This is contrary to the results seen in paediatric ALL patients, where the greatest efficacy is seen in the heterozygote TPMT group and where dose escalation of 6MP has been shown to be of benefit.27 However, the main methylation product, 6MMP, has been shown to be a strong inhibitor of purine de novo synthesis and also causes immunosuppression,27 and therefore this group (high/high TPMT status) may have thus had greater anticancer effects from treatment. Studies in ALL patients have demonstrated that the greater the inhibition of de novo purine synthesis, the greater the anti-leukaemic effects observed.45 We might also have expected decreased toxicity in this subgroup of patients, due to the increased formation of inactive metabolites due to their high TPMT status.
It can take 2–3 months (depending on TPMT status) to see therapeutic benefits from 6MP in ALL and inflammatory bowel disease.29 In our study, five patients (7%) discontinued treatment within 4 weeks and 27 patients (40%) discontinued within 8 weeks due to disease progression, suggesting that there was inadequate initial disease control with 6MP with MTX. This is in comparison with olaparib, where therapeutic effects are seen within a few weeks. It may therefore have been better to consider the use of this combination as a maintenance therapy post chemotherapy, rather than upfront treatment, due to the increased time 6MP takes to reach therapeutic levels compared with agents such as olaparib or chemotherapy.
PARPi resistance
Preclinical data demonstrated activity of 6MP in PARP inhibitor-resistant cells and we therefore included patients who had previously received PARP inhibitors. Planned subgroup analysis of the 26 patients (39%) who had received prior PARP inhibitor therapy was performed and compared with the 41 patients (61%) who were PARP inhibitor naive. No difference in efficacy was demonstrated in the two patient groups to support the preclinical findings. However, this might be due to the lack of statistical power in the subgroup analyses.
Study limitations
This was a group with heavily pre-treated patients, who had 4.7 (ovarian) and 4.2 (breast) prior lines of therapy, and therefore there was a limited chance of seeing benefit in this group. Furthermore, the time taken for 6MP to reach therapeutic levels may also have contributed to the low response rates seen at 8 weeks.
We used 6TGN incorporation into RBCs as a surrogate for tumour and WBC levels of incorporation of active cytotoxic metabolites. Whilst there is a good correlation between RBC TGN levels and lymphocyte TGN levels, there is less evidence with respect to tumour levels of active metabolites. As patients did not have biopsies whilst on trial, we were unable to determine intra-tumoral levels of active TGNs.
Final conclusions
This was the first trial to investigate the use of a novel, cost-effective combination of 6MP and MTX in patients with multiple relapsed breast and ovarian cancer with a known BRCA mutation. Although overall the activity of this combination was low, there was a small subset of patients who derived longer-term clinical benefit. There may have been enhanced activity if we had used 6MP/MTX as maintenance therapy or individually dose escalated based on TPMT status, as in the paediatric ALL practice. However, dose escalation is more problematic in patients who have had multiple lines of prior treatment compared with the paediatric ALL setting, where this approach is standard, and we therefore chose a flat dosing schedule.
There are now a number of ongoing trials in this setting with PARP inhibitors both as a single agent and in combination with agents such as cediranib (e.g. the OCTOVA trial), and immune checkpoint inhibitors. Epithelial ovarian cancers that develop in patients with BRCA (germline and somatic) mutations have been shown to have a higher mutational load, leading to an increase in the number of neoantigens and to higher levels in tumour-infiltrating lymphocytes, CD3+ and CD8+ counts and higher levels of PD-1 and PD-L1.46,47 This group of patients may therefore derive increased benefit from immune checkpoint inhibitors, either as single-agent therapy or in combination with PARP inhibitors or chemotherapy. Similarly, combination approaches of cisplatin-based chemotherapy and immunotherapy may also be beneficial in women with BRCA-mutated triple-negative breast cancer.48 In view of these developments, it is unlikely that further investigation of this combination will be feasible.
References
Ferlay, J. S. I, Ervik, M, Dikshit, R, Eser, S, Mathers, C., Rebelo, M. et al. Rebelo M et al. GLOBOCAN2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11, Lyon, France: International Agency for Research on Cancer. http://globocan.iarc.fr (2013).
Eisenhauer, E. A. Real-world evidence in the treatment of ovarian cancer. Ann. Oncol. 28, viii61–viii65 (2017).
Risch, H. A., McLaughlin, J. R., Cole, D. E., Rosen, B., Bradley, L., Kwan, E. et al. Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am. J. Hum. Genet. 68, 700–710 (2001).
Alsop, K., Fereday, S., Meldrum, C., deFazio, A., Emmanuel, C., George, J. et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 30, 2654–2663 (2012).
McPherson, K., Steel, C. M. & Dixon, J. M. ABC of breast diseases. Breast cancer-epidemiology, risk factors, and genetics. BMJ. 321, 624–628 (2000).
Easton, D., Ford, D. & Peto, J. Inherited susceptibility to breast cancer. Cancer Surv. 18, 95–113 (1993).
Oesterreich, S. & Fuqua, S. A. Tumor suppressor genes in breast cancer. Endocr. Relat. Cancer. 6, 405–419 (1999).
Lakhani, S. R., Reis-Filho, J. S., Fulford, L., Penault-Llorca, F., van der Vijver, M., Parry, S. et al. Prediction of BRCA1 status in patients with breast cancer using estrogen receptor and basal phenotype. Clin. Cancer Res. 11, 5175–5180 (2005).
Bryant, H. E., Schultz, N., Thomas, H. D., Parker, K. M., Flower, D., Lopez, E. et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 (2005).
Farmer, H., McCabe, N., Lord, C. J., Tutt, A. N. J., Johnson, D. A., Richardson, T. B. et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 (2005).
Murai, J., Huang S-yN, Das, B. B., Renaud, A., Zhang, Y., Doroshow, J. H. et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 72, 5588–5599 (2012).
Polak, P., Kim, J., Braunstein, L. Z., Karlic, R., Haradhavala, N. J., Tiao, G. et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 49, 1476 (2017).
FDA. Lynparza (olaparib) tablets, for oral use: prescribing information. https://www.fda.gov/drugs/fda-approved-olaparib-lynparza-astrazeneca-pharmaceuticals-lp-maintenance-treatment-adult-patients (2018).
Mirza, M. R., Monk, B. J., Herrstedt, J., Oza, A. M., Mahner, S., Redondo, A. et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N. Engl. J. Med. 375, 2154–2164 (2016).
Coleman, R. L., Oza, A. M., Lorusso, D., Aghajanian, C., Oaknin, A., Dean, A. et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet 390, 1949–1961 (2017).
Moore, K., Colombo, N., Scambia, G., Kim, B.-G., Oaknin, A., Friedlander, M. et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N. Engl. J. Med. 379, 2495–2505 (2018).
Robson, M., Im, S.-A., Senkus, E., Xu, B., Domchek, S. M., Masuda, N. et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N. Engl. J. Med. 377, 523–533 (2017).
Barber, L. J., Sandhu, S., Chen, L., Campbell, J., Kozarewa, I., Fenwick, K. et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J. Pathol. 229, 422–429 (2013).
Makvandi, M., Xu, K., Lieberman, B. P., Anderson, R.-C., Effron, S. S., Winters, H. D. et al. A radiotracer strategy to quantify PARP-1 expression in vivo provides a biomarker that can enable patient selection for PARP Inhibitor Therapy. Cancer Res. 76, 4516 (2016).
Ray Chaudhuri, A., Callen, E., Ding, X., Gogola, E., Duarte, A. A., Lee, J.-E. et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535, 382 (2016).
Issaeva, N., Thomas, H. D., Djureinovic, T., Jaspers, J. E., Stoimenov, I., Kyle, S. et al. 6-thioguanine selectively kills BRCA2-defective tumors and overcomes PARP inhibitor resistance. Cancer Res. 70, 6268–6276 (2010).
Coulthard, S. & Hogarth, L. The thiopurines: An update. Investigational New Drugs. 23, 523–532 (2005).
Swann, P. F., Waters, T. R., Moulton, D. C., Xu, Y. Z., Zheng, Q., Edwards, M. et al. Role of postreplicative DNA mismatch repair in the cytotoxic action of thioguanine. Science 273, 1109–1111 (1996).
Griffin, S., Branch, P., Xu, Y. Z. & Karran, P. DNA mismatch binding and incision at modified guanine bases by extracts of mammalian cells: implications for tolerance to DNA methylation damage. Biochemistry 33, 4787–4793 (1994).
Waters, T. R. & Swann, P. F. Cytotoxic mechanism of 6-thioguanine: hMutSalpha, the human mismatch binding heterodimer, binds to DNA containing S6-methylthioguanine. Biochemistry. 36, 2501–2506 (1997).
Yan, T., Berry, S. E., Desai, A. B. & Kinsella, T. J. DNA mismatch repair (MMR) mediates 6-thioguanine genotoxicity by introducing single-strand breaks to signal a G2-M arrest in MMR-proficient RKO cells. Clin. Cancer Res. 9, 2327–2334 (2003).
Lennard, L. The clinical pharmacology of 6-mercaptopurine. Eur. J. Clin. Pharmacol. 43, 329–339 (1992).
Relling, M. V., Hancock, M. L., Rivera, G. K., Sandlund, J. T., Ribeiro, R. C., Krynetski, E. Y. et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst. 91, 2001–2008 (1999).
Dean L. Mercaptopurine Therapy and TPMT Genotype (eds Pratt V., McLeod H., Dean L., Malheiro A. & Rubinstein W.). (Medical Genetics Summaries, Bethesda, MD, 2012).
Bokkerink, J. P., De Abreu, R. A., Bakker, M. A., Hulscher, T. W., van Baal, J. M., Schretlen, E. D. et al. Effects of methotrexate on purine and pyrimidine metabolism and cell-kinetic parameters in human malignant lymphoblasts of different lineages. Biochem. Pharmacol. 37, 2329–2338 (1988).
Martin, S. A., McCarthy, A., Barber, L. J., Burgess, D. J., Parry, S., Lord, C. J. et al. Methotrexate induces oxidative DNA damage and is selectively lethal to tumour cells with defects in the DNA mismatch repair gene MSH2. EMBO Mol. Med. 1, 323–337 (2009).
Nicum, S., Roberts, C., Boyle, L., Kopijasz, S., Gourley, C., Hall, M. et al. A phase II clinical trial of 6-mercaptopurine (6MP) and methotrexate in patients with BRCA defective tumours: a study protocol. BMC Cancer 14, 983 (2014).
Jung, S. H., Carey, M. & Kim, K. M. Graphical search for two-stage designs for phase II clinical trials. Control Clin. Trials. 22, 367–372 (2001).
Jung, S. H., Lee, T., Kim, K. & George, S. L. Admissible two-stage designs for phase II cancer clinical trials. Stat Med. 23, 561–569 (2004).
Hawwa, A. F., Millership, J. S., Collier, P. S. & McElnay, J. C. Development and validation of an HPLC method for the rapid and simultaneous determination of 6-mercaptopurine and four of its metabolites in plasma and red blood cells. J. Pharm. Biomed. Anal. 49, 401–409 (2009).
Kaufman, B., Shapira-Frommer, R., Schmutzler, R. K., Audeh, M. W., Friedlander, M., Balmana, J. et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 33, 244–250 (2015).
McNeish, I. A., Ledermann, J. A., Webber, L., James, L., Kaye, S. B., Hall, M. et al. A randomised, placebo-controlled trial of weekly paclitaxel and saracatinib (AZD0530) in platinum-resistant ovarian, fallopian tube or primary peritoneal cancerdagger. Ann. Oncol. 25, 1988–1995 (2014).
Tan, D. S., Yap, T. A., Hutka, M., Roxburgh, P., Ang, J., Banerjee, S. et al. Implications of BRCA1 and BRCA2 mutations for the efficacy of paclitaxel monotherapy in advanced ovarian cancer. Eu.r J. Cancer 49, 1246–1253 (2013).
Kaye, S. B., Lubinski, J., Matulonis, U., Ang, J. E., Gourley, C., Karlan, B. Y. et al. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J. Clin. Oncol. 30, 372–379 (2012).
Lilleyman, J. S. & Lennard, L. Mercaptopurine metabolism and risk of relapse in childhood lymphoblastic leukaemia. Lancet. 343, 1188–1190 (1994).
Hawwa, A. F., Collier, P. S., Millership, J. S., McCarthy, A., Dempsey, S., Cairns, C. et al. Population pharmacokinetic and pharmacogenetic analysis of 6-mercaptopurine in paediatric patients with acute lymphoblastic leukaemia. Br. J. Clin. Pharmacol. 66, 826–837 (2008).
Cheok, M. H. & Evans, W. E. Acute lymphoblastic leukaemia: a model for the pharmacogenomics of cancer therapy. Nat Rev Cancer. 6, 117–129 (2006).
Colombel, J. F., Ferrari, N., Debuysere, H., Marteau, P., Gendre, J. P., Bonaz, B. et al. Genotypic analysis of thiopurine S-methyltransferase in patients with Crohn’s disease and severe myelosuppression during azathioprine therapy. Gastroenterology. 118, 1025–1030 (2000).
Lennard, L., Welch, J. & Lilleyman, J. S. Mercaptopurine in childhood leukaemia: the effects of dose escalation on thioguanine nucleotide metabolites. Br. J. Clin. Pharmacol. 42, 525–527 (1996).
Dervieux, T., Brenner, T. L., Hon, Y. Y., Zhou, Y., Hancock, M. L., Sandlund, J. T. et al. De novo purine synthesis inhibition and antileukemic effects of mercaptopurine alone or in combination with methotrexate in vivo. Blood. 100, 1240–1247 (2002).
McAlpine, J. N., Porter, H., Kobel, M., Nelson, B. H., Prentice, L. M., Kalloger, S. E. et al. BRCA1 and BRCA2 mutations correlate with TP53 abnormalities and presence of immune cell infiltrates in ovarian high-grade serous carcinoma. Mod. Pathol. 25, 740–750 (2012).
Strickland, K. C., Howitt, B. E., Shukla, S. A., Rodig, S., Ritterhouse, L. L., Liu, J. F. et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 7, 13587–13598 (2016).
Nolan, E., Savas, P., Policheni, A. N., Darcy, P. K., Vaillant, F., Mintoff, C. P., et al. Combined immune checkpoint blockade as a therapeutic strategy for BRCA1-mutated breast cancer. Sci. Transl. Med. 9, pii: eaal4922 (2017).
Acknowledgements
The views expressed in this publication are those of the authors and not necessarily those of the Department of Health. We thank present and former staff at Oncology Clinical Trials Office and Centre for Statistics in Medicine (Oxford University, Oxford, United Kingdom) who have been involved with the 6MP trial, particularly senior staff for trial management advice and guidance, and members of the Independent Early Phase Trials Oversight Committee for their support and guidance. We thank Professor Thomas Helleday for his help with the development of this trial. We also thank all the patients involved in the study and the nursing and administrative staff involved in the running of this trial. The 14 sites across the United Kingdom who recruited for this study were Royal United Hospital, Bath; Belfast City Hospital, Belfast; Velindre Hospital, Cardiff; University Hospital, Coventry and Warwickshire; Western General Hospital, Edinburgh; Guy’s Hospital, London; Royal Marsden Hospital, London; University College London Hospital, London; Christie Hospital, Manchester; Freeman Hospital, Newcastle; Mount Vernon Hospital, Middlesex; Churchill Hospital, Oxford; Royal Marsden Hospital, Sutton; Clatterbridge, Wirral.
Author information
Authors and Affiliations
Contributions
S.N. was largely involved in the conception and design of the 6MP trial and made substantial contributions to the protocol. S.N., C.G., M.H., A.M., J.A., A.C., R.K. and S.B. provided patients for the study. C.G., M.H. and A.M. were members of the 6MP Trial Management Group overseeing the trial. S.K. was the trial coordinator, with responsibility for the day-to-day running of the trial. C.R. and V.S. were involved in data analysis and interpretation. L.F. and M.S. carried out PK, C.R. and SN drafted this paper and it was revised critically by V.S. All authors read and approved the final paper
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The appropriate ethical guidelines and legislation were observed in conducting the 6MP study; namely, the study complied with the ethical principles of the Declaration of Helsinki (1996) and the regulatory requirements for clinical trials of an IMP under the European Union Clinical Trials Directive. Subjects provided their informed consent to participate in the study. The study was approved by the National Research Ethics Committee South Central–Oxford B (REC reference 10/H0605/79).
Consent to publish
All authors consent for publication.
Data availability
The datasets used and/or analysed during this study are available from the corresponding author on reasonable request with permission of 6MP trial management group committee.
Competing interests
S.N. has received honoraria from AstraZeneca, Tesaro, Clovis and Roche and has provided consultancy for AstraZeneca, Clovis, Tesaro and Roch, and has received research funding from AstraZeneca. C.G. has received honoraria from AstraZeneca, Tesaro, Cor2Ed and Medscape; has provided consultancy for AstraZeneca, Clovis, Nucana, Tesaro, Roche, Foundation One and Cor2Ed; his institution receives research funding from AstraZeneca, Aprea AB, Nucana and Tesaro. R.K. receives payment as medical director for Almac Diagnostics. J.A. has received honorarium in the past 24 months, which has included travel expenses for advisory roles from Novartis and Genomic Health. S.B. has received research funding from Astrazeneca and has served on advisory boards for Astrazeneca, Tesaro and Clovis. All other authors jointly declare that they have no competing interests.
Funding information
The trial was sponsored by the University of Oxford, with funding from Cancer Research UK’s Clinical Trials Award and Advisory Committee (CTAAC) who approved a full funding application on 26th October 2010 (grant no: C1488/A12447), and this work was supported by the Oxford Partnership Comprehensive Biomedical Research Centre with funding from the Department of Health’s National Institute of Health Research (NIHR) Biomedical Research Centre funding scheme. VS was funded by Cancer Research UK (grant no. C5527/A16895).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Roberts, C., Strauss, V.Y., Kopijasz, S. et al. Results of a phase II clinical trial of 6-mercaptopurine (6MP) and methotrexate in patients with BRCA-defective tumours. Br J Cancer 122, 483–490 (2020). https://doi.org/10.1038/s41416-019-0674-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-019-0674-4
This article is cited by
-
Targeting purine metabolism in ovarian cancer
Journal of Ovarian Research (2022)
-
Cytotoxic and targeted therapy for BRCA1/2-driven cancers
Hereditary Cancer in Clinical Practice (2021)
-
Application of pharmacogenetics in oncology
Biomarker Research (2020)
