
Abstract
Background
Testicular germ cell cancer (TGCC), being the most frequent malignancy in young Caucasian males, is initiated from an embryonic germ cell. This study determines intratumour heterogeneity to unravel tumour progression from initiation until metastasis.
Methods
In total, 42 purified samples of four treatment-resistant nonseminomatous (NS) TGCC were investigated, including the precursor germ cell neoplasia in situ (GCNIS) and metastatic specimens, using whole-genome and targeted sequencing. Their evolution was reconstructed.
Results
Intratumour molecular heterogeneity did not correspond to the supposed primary tumour histological evolution. Metastases after systemic treatment could be derived from cancer stem cells not identified in the primary cancer. GCNIS mostly lacked the molecular marks of the primary NS and comprised dominant clones that failed to progress. A BRCA-like mutational signature was observed without evidence for direct involvement of BRCA1 and BRCA2 genes.
Conclusions
Our data strongly support the hypothesis that NS is initiated by whole-genome duplication, followed by chromosome copy number alterations in the cancer stem cell population, and accumulation of low numbers of somatic mutations, even in therapy-resistant cases. These observations of heterogeneity at all stages of tumourigenesis should be considered when treating patients with GCNIS-only disease, or with clinically overt NS.
Similar content being viewed by others

Background
Malignant germ cell tumours of the adult testis, referred to as type II TGCTs of testicular germ cell cancer (TGCC), are the most frequent cancer in young Caucasian males.1 TGCC are thought to be initiated during early embryogenesis affecting an embryonic germ cell, and become clinically manifest during young adulthood with an annual frequency of approximately 5–12 per 100,000 men in the western world and may require “aggressive” medical treatment. These cancers are clinically and histologically classified into two variants, being seminoma (SE) and nonseminoma (NS). Both arise from a common cancer stem cell, currently referred to as germ cell neoplasia in situ (GCNIS),2,3 which resembles totipotent primordial germ cells (PGCs)/gonocytes. Patients with proven GCNIS have a 70% chance of progression to TGCC (both SE and NS) within 7 years. SE consists of a homogeneous population of cells with similarity to GCNIS and PGC/gonocytes. About 50% of the TGCC patients present with a NS that can be composed of different histological elements, embryonal carcinoma (EC), teratoma (TE), yolk sac tumour (YST), and choriocarcinoma, either pure or mixed. The EC is the pluripotent stem cell component of NS, which can mimic normal early embryogenesis including the formation of so-called embryonal bodies (EBs), and thereby give rise to all differentiated components.3,4,5
Although all TGCC, including mature TE, are in principle capable to metastasise, about 80–85% of the SE patients and 55–60% of the NS patients present with localised (stage I) disease. Patients with metastatic TGCC are generally cured by standard treatment regimens involving platinum compounds and additional surgery for residual TE, while only few patients show resistance to treatment.6 So far, detailed studies into the molecular profile of TGCCs and their progression stages were focused on specific genes, like KIT7 and TP53,8,9,10 and on the chromosomal constitution. Analyses have revealed many changes in the (relative) number of individual chromosomes in the different tumour components.5,11,12,13,14,15,16 Gain of the short arm of chromosome 12 is a hallmark of (invasive) TGCCs,17 but as yet no causative gene(s) have been identified. Information on driver mutations underlying the development of these NS using exome sequencing is scarce.11,12,13,14,18,19,20
In order to unravel the molecular heterogeneity of NS, we extensively investigated four rare cases of primary therapy-resistant NS and performed WGS on the primary cancer and targeted sequencing analyses on 42 enriched histological components, precursor cell populations, and metastatic lesions after treatment (Figs. 1a, b). Focus was on the early events of tumour formation, the molecular heterogeneity within the primary lesion and the retention of molecular markers in the metastatic recurrences. Additionally, data from RNA expression (RNAseq) and copy number alterations (CNA) from high-throughput DNA methylation profiling and DEPArrayTM/LowPass WGS were interrogated to decipher the evolution of the disease.

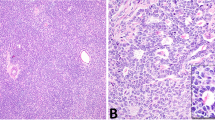
Dissection of nonseminoma (NS). a NS may consist of multiple histological elements (examples case T1382, TE and EC; case T618, germ cell neoplasia in situ (GCNIS), OCT3/4 stained nuclei), which were each enriched by laser capture micro dissection. The primary NS was subjected to WGS and the purified components to targeted sequencing using Ion Torrent technology. b The results of the whole genome sequencing (WGS) are shown per case. Complete genomics (CG) somatic DNA variant (SNV) were retrieved from CG output files, and putative candidates were selected after visual inspection of the reads. SNV validation was performed by mutation-specific PCR, targeted DNA sequencing and/or RNA sequencing (Supplementary Table S4). Structural variants (SVs) were confirmed for selected cases using targeted sequencing. Mutations occurring near exon boundaries were evaluated for potential effect on splicing using Alamut Software. Details are provided in Supplementary Tables S4-S6. The bottom panel shows the number of purified histological components isolated and analysed for these cases. PBL peripheral blood leukocytes, NAP nonmalignant adjacent parenchyma. c The mutational profiles of these four NS were compared with the 30 COSMIC signatures described. The mutational signatures contributing significantly (>5%) are presented. In addition, the SNV of NS and SE cases from the Taylor-Weiner study (*)14 were pooled for tumour subtype and similarly analysed. The number of SNV used for the analysis is indicated below each bar. d, e Profiles of lesser allele frequencies (LAF) of heterozygote SNP and relative read frequencies of SNV derived from targeted sequencing of primary NS (red) and dissected EC components (green) are shown. SNV are presented as filled symbols (NS: yellow diamonds; EC: green squares). Positions are provided on chromosomes scaled according to size
Materials and methods
A brief description is provided here. Further details are provided in the Supplementary Methods.
Patient samples
NS samples of patients with established intrinsic resistance to standard first-line chemotherapy (detailed in Supplementary Methods) were included in this study. Use of tissue samples remaining after diagnosis for scientific reasons was approved by Medical Ethical Committee of the Erasmus MC Rotterdam (The Netherlands), permission 02.981. This included the permission to use the secondary tissue without further consent. Samples were used according to the “Code for Proper Secondary Use of Human Tissue in The Netherlands” developed by the Dutch Federation of Medical Scientific Societies (Version 2002, update 2011).
Omics analyses of patient samples
Purified tumour components (Supplementary Table S1), as defined by an experienced pathologist (J.W.O.), were isolated from frozen tissue slices after staining for alkaline phosphatase enzyme reactivity,21 using laser capture micro dissection (Zeiss). Tumour and paired normal DNA samples were whole-genome sequenced (40 times coverage) and analysed at Complete Genomics Inc. (CG) (Mountain View, CA, USA) using NCBI build 36.3 as human reference genome and pipeline software version 2.0.2.22.22 Lists of putative somatic DNA variants (SNVs) were established from the WGS data as described in the Supplementary Methods. SNV were verified using mutation-specific quantitative real-time PCR, targeted sequencing (Supplementary Tables S2 and S3), and RNAseq. Structural variants were evaluated for gene fusions with iFuse.23 Characterisation of the mutational signature was done by comparison of the trinucleotide context of each SNV to the established COSMIC signatures using the MutationalPatterns R package (v1.0).24
Targeted sequencing was performed by semiconductor sequencing with the Ion Torrent Personal Genome Machine with supplier’s materials and protocols (ThermoFisher Scientific) as previously described.25 Amplicons were designed to cover tumour-specific SNV, structural variants and heterozygote positions (Supplementary Table S3). Median sequencing depth was at least 250 reads. Allele frequencies were established for the heterozygous single-nucleotide polymorphisms (SNPs) in the matched normal samples present on the amplicons. Details for calling of SNP, SNV and structural variants in the targeted sequencing experiments are provided in the Supplementary Methods. Evolutionary trees of different samples of a specific tumour were drawn based on the lesser allele frequencies (LAF) and SNV profiles and supported by the TargetClone tool.26 TargetClone was designed to reconstruct evolutionary trees for multiple samples of a cancer using allele frequencies and SNV (Supplementary Methods).
RNA samples of T6107 and T3209 were rRNA-reduced and Ion Proton sequenced (90 bases, 50 million mapped reads) using the supplier’s protocols and reagents (ThermoFisher Scientific). Generation of methylation profiles of primary tumour DNA was performed as previously described27 or at the Microarray unit of the Genomics and Proteomics Core Facility of the German Cancer Research Centre (DKFZ, Heidelberg) strictly adhering to the Illumina EPIC protocols for the T6107-YSTmeta. CNA based on methylation intensities were resolved using the Conumee package.28 DEPArrayTM experiments on a T6107 metastatic sample, and GCNIS and YST samples of T618 were performed by Menarini Silicon Biosystems (Castel Maggiore, Italy), essentially as described.29
Results
Primary NS characteristics
To address tumour heterogeneity and progression, WGS data from four primary chemo-naive NS were exploited. Comparison of the primary tumours with the matched normal provided a set of 1239 somatic putative DNA variants (SNV) for these cases (Fig. 1b). RNAseq, mutation-specific PCR and targeted sequencing experiments validated 150 out 158 SNV and 9 out of 15 structural variants (Fig. 1b, Supplementary Tables S4 and S5). The identified mutations causing protein changes have been listed in Table 1. Only four SNV resulted in protein truncation and another 13 were predicted to be damaging (Supplementary Table S6). In addition, detailed information regarding structural variants, LAF and chromosome CNAs were obtained from the WGS and methylation profiling (details in Supplementary Figs. S1 and S2). The trinucleotide profile of single base SNV identified by WGS of the four NS was determined and compared with the established set of COSMIC mutational signatures.30 In all cases (Fig. 1c), signature 3 contributed significantly or was the predominant signature. Analysis of the pooled SNV (from whole-exome sequencing) of independent cases of NS (N = 18) and SE (N = 18),14 also revealed this signature to be the most prominent (Fig. 1c and further details in Supplementary Fig. S3). This signature 3 is strongly associated with mutations in BRCA1 or BRCA2, genomic deletion and insertion events smaller than 100 kb, and a deficiency in homologous recombination repair in breast cancer.31 In the absence of substantial numbers of indels and genomic deletion and insertion events in our WGS data, additional support for recombination repair deficiency is lacking. Furthermore, pathogenic somatic mutations in BRCA1 or BRCA2 were not observed in the four included TGCC, although case T1382 carried a predicted, non-pathogenic BRCA1 missense variant (Supplementary Table S6), nor in the cases of the Taylor-Weiner study.14 In addition, promoter hypomethylation and RNAseq reads observed for both genes did not support loss of BRCA function (Supplementary Fig. S4). Furthermore, we did not observe pathogenic mutations in other genes responsible for homologous recombination repair defects,32 and transcripts of these genes were detected by RNAseq (data not shown).
Molecular heterogeneity and evolution
For the study of the molecular heterogeneity within these histologically complex primary NS (containing EC, EB, TE and YST components, Fig. 1a), matched metastases and precursor lesions, DNA was prepared from various micro- and macro-dissected components (N = 42, Figs. 1a, b and Supplementary Table S1). The histological identity was determined by an experienced pathologist, and using direct alkaline phosphatase-staining for EC, EB and GCNIS in frozen tissue (examples in Supplementary Fig. S5).21 To evaluate the allelic imbalances and the presence of SNV in these enriched specimens, amplicons were designed across the genome containing a tumour-specific SNV and additional heterozygous SNPs (Supplementary Fig. S1 and Supplementary Table S3). For the primary tumour DNA samples, an excellent agreement between the LAF profiles of the WGS and targeted analyses was observed (Supplementary Fig. S6). Analyses of the enriched samples were focused on the LAF of germ line heterozygote SNPs, the read frequencies of the SNV and the presence of specific breakpoints. Furthermore, evolutionary trees based on the LAF and the presence of SNV, and supported by TargetClone, were generated for each case. Results of these analyses will be discussed per case below.
T6107
The majority of the allelic imbalances and SNV found in the primary cancer were present and more easily detected in the enriched malignant histologies due to their increased purity and the removal of contaminating normal cells (example in Fig. 1d). Loss of heterozygosity (LOH) was clearly resolved for chromosome (arm) 4q, 10, 11, 13 and 16q for the purified EC component. The false colour plot showed extensive overlap in regional allelic imbalances and SNV among the enriched histological components of T6107 and compared with the primary NS (Figs. 2a, b and Supplementary Fig. S7A). LOH on the chromosomes mentioned above was preserved in all histological elements (homogeneously red coloured blocks indicating LAF < 0.1, Fig. 2a). Sample EC21 displayed additional LOH resulting from copy losses of chromosomes arms 9q and 22q. In the GCNIS preparations (CIS30 and FCIS31, Fig. 2a), very little overlap in LAF patterns with the primary NS was observed and the majority of SNV were completely absent (Fig. 2b). The YST metastasis in the lung (YSTmeta) showed minor overlap with the primary NS and its histological components with regard to LAF pattern and presence of SNV. The shared LOH of chromosome arm 22q between EC21 and this metastasis represented independent events based on the different parental alleles retained (Supplementary Fig. S8). Presence of chromosome arm 12p gain (Supplementary Fig. S9), and two SNV (Fig. 2b) in this metastasis demonstrated a shared origin with the primary NS. The copy number profiles of this lung metastasis and a prior retroperitoneal lymph node metastasis with the histology of mature TE displayed many novel alterations, including amplification of the MDM2 gene region (details in Supplementary Fig. S9). Immunohistochemistry (IHC) and fluorescent in situ hybridisation (FISH) analysis confirmed the amplification of MDM2, and targeted sequencing did not reveal TP53 mutation in the lung metastasis (data not shown). An evolutionary tree for this case (Fig. 3) was based on the general profiles of the LAF and SNV (Figs. 2a, c), and required two unidentified EC precursors (ECx1 and ECx2) to explain the variance between the primary NS components and the YST lung metastasis. EC21 represented a separate progression line with additional chromosome losses. Furthermore, the early ECx1 precursor containing few aberrations was the founder of the late appearing lung metastasis (YSTmeta) and likely of the mature TE in the lymph node metastasis (Supplementary Fig. S9), which both lacked many of genomic marks of the primary tumour.

False colour plots of allelic imbalances, somatic DNA variant (SNV) and structural variants in primary testicular germ cell cancer (TGCC) (nonseminoma (NS)) and various purified tumour components of two cases (T6107 (panels a, b) and T618 (panels c, d)). Data from (tumour) samples analysed more than once were averaged. Top panels a, c Each line represents the amplicon averaged lesser allele frequencies (LAF) of heterozygote SNPs using the LAF colour scheme in panel e (LAF). Blue indicates heterozygosity and red refers to loss of heterozygosity (LOH). Ordering is based on chromosomal position (indicated on the right). For comparison, the 100 kb interval WGS LAF (CG-LAF) is also shown. In addition, the WGS relative read coverage (CGrelCov) data of the primary NS are provided for the specific chromosomal regions using the colour scheme (Rel Cov) in panel e. Missing data are white. Sample types have been marked by coloured dashed lines (germ cell neoplasia in situ (GCNIS): orange, histological components: red and metastases: purple). (*) Formalin-fixed paraffin-embedded (FFPE) tissue blocks derived DNA samples. Bottom panels b, d Occurrence of tumour-specific SNV and structural variants in the different tumour samples (grey indicates absence, black indicates >3% of the reads carrying the variant, missing data in white). Tumour-specific structural variants are indicated with green arrow heads. Genes with a mutation resulting in amino-acid change (T618: only those observed in GCNIS) have been indicated (see Table 1). A red arrow marks a SNV present in all samples (except PBL) from case T6107 (chr19:56131557), residing in a 2-kb region between two Zn-finger genes. The number at the bottom indicates the months after surgery of the primary tumour for the removal of the metastasis. e Colour keys for the different categories. Sample information and targeted sequence data details are provided in Supplementary Table S1 and Table S8

Evolution of nonseminoma (NS). Evolutionary trees for the different histological specimens for all cases are shown on this developmental model. Colouring of boxes is according to sample type: germ cell neoplasia in situ (GCNIS; yellow), precursor embryonal carcinoma (EC; light orange), primary tumour (dark orange) and metastasis (purple). The order and grouping of the samples is based on the similarities in allele and somatic DNA variant (SNV) profiles (as shown in Fig. 2 and Supplementary Fig. S7) and supported by TargetClone (Materials and methods section), and implementing the biological constraint for NS development that the differentiated components (teratoma (TE) and yolk sac tumour (YST)) originated from an EC-type precursor. Samples with comparable profiles are grouped together. In specific cases of samples with partially non-overlapping features (i.e. SNV and/or loss of heterozygosity (LOH)) without an immediate precursor, a non-identified precursor (GCNISx or ECx) was inserted at the branch point in the tree (T6107, T618). Similarly, to comply with the evidence that an EC is the precursor of TE and YST, a non-identified precursor ECx was introduced (T6107, T1382). Specific gains of LOH or SNV compared with their immediate precursor are indicated. *) indicates SNV present at very low read frequencies, suggesting polyclonality for GCNIS or the presence of a minor subclone in some of the primary tumour components of T3209. For case T1382, a minimal tree is presented with ordering of samples based on SNV only due to sequencing noise
T3209
Detection of LOH (chromosomes 4, 14, 15 and 22) was markedly improved for the enriched EC sample, with increased read frequencies for most SNV (Fig. 1e). Absence of specific SNV in this EC sample and present in low read frequencies (10–20%) in the primary tumour indicated clonal variation. Major overlap in LAF patterns (LOH on the above mentioned chromosomes) and most SNV was observed for the enriched histological components and the primary NS (Supplementary Fig. S7B). A single EB (EB23) showed additional regions of LOH (involving chromosomes 1 and 5). The GCNIS preparations essentially lacked allelic imbalances and SNV. The evolutionary tree for this case suggested separate developmental lineages for EB23 and four samples of EC, TE and YST (Fig. 3). The occurrence of very low frequency SNV (<5% of the reads, details Supplementary Fig. S7B) in the GCNIS preparation (which were abundant in the histological components), suggested the presence of a minor population of further progressed GCNIS.
T618
Laser capture of the histological components in the primary NS was not successful due to the presence of excess TE of cartilage differentiation. Purification of YST cells was achieved from FFPE sections using the DEPArray and provided copy number profiles comparable to the primary NS (Supplementary Figs. S1C, S2 and S10). Abundant numbers of GCNIS in the “normal” adjacent parenchyma allowed for the preparation of these cancer stem cells (Supplementary Fig. S5). Subtypes located isolated (CIS29), basal (BCIS28) or floating (FCIS27) and probably reflecting their progression state, were obtained.17 Inspection of the LAF profiles showed increasing allelic imbalances up to LOH for chromosomes 4 and 5 in these GCNIS stages (Figs. 2c, d). In addition, alterations on chromosome arms 11q and 22qter, and seven SNV were observed in these GCNIS. Copy number analysis of DEPArray purified GCNIS did not reveal gain of chromosome arm 12p (Supplementary Fig, S10). The evolutionary tree of this case (Fig. 3) required the insertion of an unidentified precursor (GCNISx) as the progressed state GCNIS (BCIS28 and FCIS27) lacked four SNV (Fig. 2d) and showed loss of alleles on chromosome arms 11q and 22qter, which were retained by the primary tumour (Supplementary Fig. S8). These data strongly suggest that the abundant basal and floating GCNIS adjacent to the tumour mass represent a precursor clone that did not progress to a full malignant state and was not the founder of the primary NS.
T1382
The enriched samples were all derived from old FFPE tissue blocks and showed more amplicon drop out and noisy LAF data (Supplementary Fig. S7D). In spite of this limitation, the enriched TE and EC histologies showed good overlap for LAF, SNV, and breakpoints with the primary NS. A set of SNV, breakpoints and regions of imbalance appeared preserved in some of the metastases, while novel alterations were also observed (details in Supplementary Fig. S7D). The evolutionary tree based primarily on the SNV required the insertion of a non-identified EC precursor to explain the differences between the primary NS components and the metastases (Fig. 3).
Discussion
Our studies on multiple samples of four cases of therapy-resistant NS provided insight in the complexity of tumourigenesis (Fig. 4). NS carried low numbers of SNV (~0.1 per Mb, Fig. 1b), somewhat lower than reported and comparable to some of the paediatric cancers and spermatocytic tumours.12,13,14,20,33 The total number of SNV with a predicted impact on the encoded protein (3–18, Table 1) was similar to other reports.11,12,13 Overlap of mutated genes belonging to specific pathways was not observed within this small series of TGCC cases and was limited with genes reported for primary untreated TGCC (ASAP1, BRCA1, CCKBR, CROCC, CSE1L, KAT6A, KEAP1, MTOR, MYOM3 and TAF7).11,12,13,14,20,34,35

Nonseminoma (NS) evolution. A model of the different steps in NS development from normal primordial germ cell (PGC)/gonocytes to metastasised NS is shown. The model is based on the available literature regarding early genome duplication, acquisition of extra copies of the short arm of chromosome 12, the pluripotent capacity of the embryonal carcinoma (EC),4 and the results from this study. Following initial whole-genome doubling, during puberty chromosome loss may be the predominant way to change the copy numbers in the formation of germ cell neoplasia in situ (GCNIS) cancer stem cell (yellow). GCNIS represent a polyclonal mixture of cells, some may remain dormant and others may progress to malignancy. The gain of copies of 12p (12p++) is a hallmark of the precursor with invasive potential (light orange). Further losses and gains of chromosomes or chromosome fragments may contribute to the formation of the primary tumour with its distinct histological components (orange) and the typical metastases (purple). Somatic mutation appears to be limited and occurring at later stages. The metastases may also originate from precursor EC not detected in the primary tumour
NS exhibited clonal heterogeneity, which did not correlate with the histological subtypes (Fig. 3). This was expected because of EC being the stem cell component of all differentiated NS elements in the primary NS and different EC precursors providing independent lineages of differentiated cells.5,36 A typical example is provided by the T3209 EB23, which resembles an early developing embryo derived from a single EC with a different genomic make-up (Supplementary Fig. S7B). Extensive intratumour heterogeneity was also reported for non-small-cell lung cancer.37 Our analyses also showed that metastatic clones can be derived from very early cancer stem cells that are underrepresented or even absent in the primary lesion, and not detected with the current approaches (Fig. 4). Early disseminated cells have been shown to seed metastases in models of breast, pancreatic, bladder and melanoma,38,39,40,41 but appeared less predominant in breast cancer patients.42
The comparison of the primary tumour and the highly enriched tumour histologies revealed that the read frequencies of SNV on the autosomes (even in regions with LOH) hardly ever exceeded the 50% level (examples in Supplementary Figs. S1, S6 and S7), indicating that always a wild-type copy of the particular gene was retained within the cancer genome. Similarly, the relative read frequencies of the SNV present in the GCNIS cells were low (<0.34 for case T618, Fig. 2d) and therefore likely limited to a single allele copy. These results indicate that whole-genome duplication preceded the gain of most somatic mutations.20,37,42 The single SNV identified in the GCNIS of T6107 was present in high relative read frequencies (up to 50%), suggesting that this mutational event preceded genome duplication (Fig. 2b). Alternatively, this specific variant may represent mosaicism resulting from somatic mutation in early embryonic cells.43,44 Genome doubling may also underlie development of oesophageal cancer following early TP53 inactivation.45 The status of the overrepresentation of the 12p region in the purified cancer stem cells GCNIS remains uncertain, but the observed balanced allele frequencies are in line with absence of 12p gain (Figs. 2a, c and Supplementary Fig. S8). Furthermore, LowPass WGS on DEPArray purified T618 GCNIS, revealed no gain of 12p and absence of the majority of CNA present in the primary tumour (Supplementary Fig. S10). These results are in agreement with the notion that accumulation of chr12p copies coincides with acquirement of invasive behaviour (Fig. 4).17,46,47
Our data and the lack of recurrent driver mutations support the hypothesis that whole-genome duplication is the primary event in NS development (Fig. 4), to be followed by overall net chromosome copy losses.4,48 Proof for early whole-genome duplication may be obtained using digital NGS based on Barcode-In-Genome technology on many individual GCNIS to determine actual chromosome copy numbers.49 Subsequently, gain of 12p copies (which may be dynamic in subclones, details Supplementary Fig. S7), gain of limited numbers of somatic mutations, and additional CNA will trigger the development of the primary NS (Fig. 4). This model is in agreement with the conclusions of Shen et al.20 for the majority of the TGCTs. Only a fraction of KIT-mutated SE may have acquired the mutation before whole-genome duplication. In order to prevent accumulation of genomic mutations in germ cells, active surveillance and removal of PGCs with an aberrant genome is very efficient.50,51 All PGCs are completely de-methylated and considered to be prone to aneuploidy,52 but incidence of TGCC is low in the male population. Removal of aberrant GCNIS may require functional TP53, which could be interrupted by gene mutation or by defined miRNAs.53,54 In agreement with their extreme sensitivity to cisplatin-based therapies, TP53 mutations are extremely rare in primary TGCC indicating no selective pressure.10,14,54 The presence of an amplified MDM2 locus in two metastases of case T6107 (Supplementary Fig. S9) may have provided an alternative route for inactivation of TP53 and therapy resistance.10,34,55 The BRCA-like base substitution signature of this cancer hints at inefficient homologous recombination repair.56 This BRCA-like signature was not reported in a recent exome sequencing of TGCC.20 There is currently no evidence for the direct involvement of the BRCA genes (i.e., absent pathogenic gene mutation and no rearrangement signature,31 and no association of TGCC with familiar BRCA1/2 mutations), although increased methylation of the BRCA1 gene promoter was observed in some NS without EC.20 It is tempting to speculate that other components of this homologous recombination repair pathway may be affected and responsible for the specific base substitution signature in the absence of direct involvement of BRCA genes.31,56 Of other genes linked to this repair pathway,32 ATM, CHEK1 and MRE11 show aberrations (predominantly deep deletions) in approximately 10% of the NS,57 but complete gene deletions were not observed in our cases (Figs S1 and S2) and normal levels of transcripts were detected, as well. Alternatively, BRCA-related repair functions may be low or turned off intrinsically in the embryonic PGC/gonocytes58 and during early TGCC development, and thus contribute to the accumulation of this specific pattern of base substitutions. Therefore, TGCC development may be the result of the properties of the PGC (de-methylated and reduced repair pathway), which allows the tetraploid GCNIS to evade apoptosis and survive till puberty and subsequently progress to malignancy due to genomic destabilisation.59 The (epi)-genetic triggers for destabilisation of the tetraploid genome and initiation of the development of malignant clones remain as yet largely unknown, although the induction of mitogenic signalling by testosterone may contribute.
The observed heterogeneity in the primary tumour, metastases and precursor lesions of NS may impact on clinical decisions and treatment strategies. The occurrence of metastatic tumours with little molecular overlap with the primary lesion indicates that treatment of therapy-resistant recurrences should be targeted at their molecular properties according to the concepts of personalised medicine. The identification of abundant cancer stem cells GCNIS, which did not contribute to the development of the NS further complicates the clinical advice to patients with GCNIS-only disease.60 In view of its tendency to progress in 70% of the patients within 7 years, novel predictive markers for the GCNIS progression are needed. The BRCA-like mutational signature in TGCC indicative for reduced homologous recombination repair, may be suggestive for combined use of poly ADP ribose polymerase (PARP) inhibitors and platinum-based therapy20,61 but requires additional support. Further studies into NS and SE cases, with GCNIS adjacent to the primary tumour and different histologies or localisation, are needed to identify reliable markers for GCNIS progression, to unravel the critical steps for malignancy and therapy resistance, and to decipher the origin of the BRCA-like mutational signature of TGCC.
References
Ulbright, T.M. et al. Germ cell tumours. (eds H. M., Humphrey, P.A., Ulbright, T.M., Reuter, V.E.) in WHO Classification of Tumours of the Urinary System and Male Genital Organs. 4th ed. pp. 189–226 (IARC Press, Lyon, 2016).
Berney, D. M. et al. Germ cell neoplasia in situ (GCNIS): evolution of the current nomenclature for testicular pre-invasive germ cell malignancy. Histopathology 69, 7–10 (2016).
Spiller, C. M. & Bowles, J. Germ cell neoplasia in situ: the precursor cell for invasive germ cell tumors of the testis. Int. J. Biochem. Cell. Biol. 86, 22–25 (2017).
Oosterhuis, J. W. & Looijenga, L. H. Testicular germ-cell tumours in a broader perspective. Nat. Rev. Cancer 5, 210–222 (2005).
Killian, J. K. et al. Imprints and DPPA3 are bypassed during pluripotency- and differentiation-coupled methylation reprogramming in testicular germ cell tumors. Genome Res. 26, 1490–1504 (2016).
Beyer, J. et al. Maintaining success, reducing treatment burden, focusing on survivorship: highlights from the third European consensus conference on diagnosis and treatment of germ-cell cancer. Ann. Oncol. 2, 878–888 (2013).
McIntyre, A. et al. Amplification and overexpression of the KIT gene is associated with progression in the seminoma subtype of testicular germ cell tumors of adolescents and adults. Cancer Res. 65, 8085–8089 (2005).
Chaganti, R. S. & Houldsworth, J. Genetics and biology of adult human male germ cell tumors. Cancer Res. 60, 1475–1482 (2000).
Oosterhuis, J. W. et al. Patient with two secondary somatic-type malignancies in a late recurrence of a testicular non-seminoma: illustration of potential and flaw of the cancer stem cell therapy concept. Int. J. Dev. Biol. 57, 153–157 (2013).
Kersemaekers, A. M. et al. Role of P53 and MDM2 in treatment response of human germ cell tumors. J. Clin. Oncol. 20, 1551–1561 (2002).
Brabrand, S. et al. Exome sequencing of bilateral testicular germ cell tumors suggests independent development lineages. Neoplasia 17, 167–174 (2015).
Cutcutache, I. et al. Exome-wide sequencing shows low mutation rates and iidentifies novel mutated genes in seminomas. Eur. Urol. 68, 77–83 (2015).
Litchfield, K. et al. Whole-exome sequencing reveals the mutational spectrum of testicular germ cell tumours. Nat. Commun. 6, 5973 (2015).
Taylor-Weiner, A. et al. Genomic evolution and chemoresistance in germ-cell tumours. Nature 540, 114–118 (2016).
de Jong, B., Oosterhuis, J. W., Castedo, S. M., Vos, A. & te Meerman, G. J. Pathogenesis of adult testicular germ cell tumors. A cytogenetic model. Cancer Genet. Cytogenet. 48, 143–167 (1990).
Oosterhuis, J. W. et al. Ploidy of primary germ cell tumors of the testis. Pathogenetic and clinical relevance. Lab. Invest. 60, 14–21 (1989).
Rosenberg, C., Van Gurp, R. J., Geelen, E., Oosterhuis, J. W. & Looijenga, L. H. Overrepresentation of the short arm of chromosome 12 is related to invasive growth of human testicular seminomas and nonseminomas. Oncogene 19, 5858–5862 (2000).
Bilen, M. A. et al. Intratumoral heterogeneity and chemoresistance in nonseminomatous germ cell tumor of the testis. Oncotarget 7, 86280–86289 (2016).
Ichimura, K. et al. Recurrent neomorphic mutations of MTOR in central nervous system and testicular germ cell tumors may be targeted for therapy. Acta Neuropathol. 131, 889–901 (2016).
Shen, H. et al. Integrated molecular characterization of testicular ggerm cell tumors. Cell Rep. 23, 3392–3406 (2018).
Stoop, H. et al. Diagnosis of testicular carcinoma in situ ‘(intratubular and microinvasive)’ seminoma and embryonal carcinoma using direct enzymatic alkaline phosphatase reactivity on frozen histological sections. Histopathology 58, 440–446 (2011).
Drmanac, R. et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327, 78–81 (2010).
Hiltemann, S. et al. iFUSE: integrated fusion gene explorer. Bioinformatics 29, 1700–1701 (2013).
Blokzijl F, Janssen R, Van Boxtel R, Cuppen E. MutationalPatterns: An Integrative R Package for Studying Patterns in Base Substitution Catalogues. http://www.biorxivorg/content/early/2016/10/17/071761 (2016).
Dubbink, H. J. et al. Molecular classification of anaplastic oligodendroglioma using next-generation sequencing: a report of the prospective randomized EORTC Brain Tumor Group 26951 phase III trial. Neuro. Oncol. 18, 388–400 (2016).
Nieboer, M. M., Dorssers, L. C. J., Straver, R., Looijenga, L. H. J. & de Ridder, J. TargetClone: a multi-sample approach for reconstructing subclonal evolution of tumors. PLoS ONE 13, e0208002 (2018).
Rijlaarsdam, M. A. et al. Genome wide DNA methylation profiles provide clues to the origin and pathogenesis of germ cell tumors. PLoS One 10, e0122146 (2015).
Hovestadt V, Zapatka M. Conumee: Enhanced Copy-Number Variation Analysis Using Illumina DNA Methylation Arrays. R package version 1.6.0. http://www.packages/packages/conumee/ (2015).
Bolognesi, C. et al. Digital sorting of pure cell populations enables unambiguous genetic analysis of heterogeneous formalin-fixed paraffin-embedded tumors by next generation sequencing. Sci. Rep. 6, 20944 (2016).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Nik-Zainal, S. et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534, 47–54 (2016).
Lord, C. J. & Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 16, 110–120 (2016).
Giannoulatou, E. et al. Whole-genome sequencing of spermatocytic tumors provides insights into the mutational processes operating in the male germline. PLoS ONE 12, e0178169 (2017).
Bagrodia A. et al. Genetic determinants of cisplatin resistance in patients with advanced germ cell tumors. J. Clin. Oncol. 34, 4000–4007. https://doi.org/10.1200/JCO.2016.68.7798 (2016).
TCGA-TGCT. Testicular Germ Cell Cancer (TCGA, Provisional). http://www.cbioportalorg/study?id=tgct_tcga#summary (2016).
Stevens, L. C. Experimental production of testicular teratomas in mice of strains 129, A/He, and their F1 hybrids. J. Natl. Cancer Inst. 44, 923–929 (1970).
Jamal-Hanjani, M. et al. Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med. 376, 2109–2121 (2017).
Hosseini, H. et al. Early dissemination seeds metastasis in breast cancer. Nature 50, 552–558 (2016).
Rhim, A. D. et al. EMT and dissemination precede pancreatic tumor formation. Cell 148, 349–361 (2012).
Eyles, J. et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J. Clin. Invest. 120, 2030–2039 (2010).
Shin, K. et al. Cellular origin of bladder neoplasia and tissue dynamics of its progression to invasive carcinoma. Nat. Cell Biol. 16, 469–478 (2014).
Yates, L. R. et al. Genomic evolution of breast cancer metastasis and relapse. Cancer Cell. 32, 169–84e7 (2017).
Lodato, M. A. et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 350, 94–98 (2015).
Ju, Y. S. et al. Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. Nature 543, 714–718 (2017).
Stachler, M. D. et al. Paired exome analysis of Barrett’s esophagus and adenocarcinoma. Nat. Genet. 47, 1047–1055 (2015).
Summersgill, B., Osin, P., Lu, Y. J., Huddart, R. & Shipley, J. Chromosomal imbalances associated with carcinoma in situ and associated testicular germ cell tumours of adolescents and adults. Br. J. Cancer 85, 213–220 (2001).
Ottesen, A. M. et al. High-resolution comparative genomic hybridization detects extra chromosome arm 12p material in most cases of carcinoma in situ adjacent to overt germ cell tumors, but not before the invasive tumor development. Genes Chromosomes Cancer 38, 117–125 (2003).
van Echten, J. et al. No recurrent structural abnormalities apart from i(12p) in primary germ cell tumors of the adult testis. Genes Chromosomes Cancer 14, 133–144 (1995).
Xi, L. et al. New library construction method for single-cell genomes. PLoS ONE 12, e0181163 (2017).
Salmina, K. et al. Up-regulation of the embryonic self-renewal network through reversible polyploidy in irradiated p53-mutant tumour cells. Exp. Cell Res. 316, 2099–2112 (2010).
Fotovati, A., Nakayama, K. & Nakayama, K. I. Impaired germ cell development due to compromised cell cycle progression in Skp2-deficient mice. Cell. Div. 1, 4 (2006).
Hoffman, R. M. Is DNA methylation the new guardian of the genome? Mol. Cytogenet. 10, 11 (2017).
Lane, D. P. Cancer. p53, guardian of the genome. Nature 358, 15–16 (1992).
Voorhoeve, P. M. et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell 124, 1169–1181 (2006).
Bauer, S. et al. Therapeutic potential of Mdm2 inhibition in malignant germ cell tumours. Eur. Urol. 57, 679–687 (2010).
Polak, P. et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 49, 1476–1486 (2017).
TCGA-testis. TCGA-Testicular Germ Cell Cancers. http://wwwcbioportalorg/ (2018).
Ahmed, E. A. et al. Differences in DNA double strand breaks repair in male germ cell types: lessons learned from a differential expression of Mdc1 and 53BP1. Dna. Repair. (Amst.). 6, 1243–1254 (2007).
van der Zwan, Y. G., Biermann, K., Wolffenbuttel, K. P., Cools, M. & Looijenga, L. H. Gonadal maldevelopment as risk factor for germ cell cancer: towards a clinical decision model. Eur. Urol. 67, 692–701 (2015).
Elzinga-Tinke, J. E., Dohle, G. R. & Looijenga, L. H. Etiology and early pathogenesis of malignant testicular germ cell tumors: towards possibilities for preinvasive diagnosis. Asian J. Androl. 17, 381–393 (2015).
Petljak, M. & Alexandrov, L. B. Understanding mutagenesis through delineation of mutational signatures in human cancer. Carcinogenesis 37, 531–540 (2016).
Acknowledgements
We greatly acknowledge Lieke Dons and Dragana Bertovic for assistance in sample purification. We thank Remko Hersmus for performing FISH experiments; Daphne Heijsman, Arne S. IJpma and Peter van de Spek for help with WGS data; Saskia D. Hiltemann, and Andrew P. Stubbs for support with Galaxy and analysis of structural variants (IFuse); Ans van den Ouweland for functional evaluation of SNV; Erik-Jan Dubbink for targeted sequencing options, and Alex Nigg for the plug-in to enhance FISH images. We thank the microarray unit of the DKFZ Genomics and Proteomics Core Facility for providing the Illumina Human Methylation arrays and related services, and Volker Hovestadt and colleagues for help with the copy number analyses of methylation intensity data. We also acknowledge the Erasmus MC Cancer Computational Biology Centre for giving access to their IT-infrastructure and software that was used for computations and/or data analysis in this study. This study was supported in part by a WGS grant from Complete Genomics, Inc. Mountain View, CA 94043, USA. We appreciate the support from Rosanna Lanzellotto, Alberto Ferrarini, Bechir Boughaba, Raimo Tanzi and Francesca Fontana from Menarini Silicon Biosystems S.p.a. Castel Maggiore (Italy) for the DEPArray experiments. Whole-genome sequencing was provided by Complete Genomics, Inc. Mountain View, CA 94043, USA, and DEPArray experiments were performed by Menarini Silicon Biosystems S.p.a. (Castel Maggiore, Italy).
Author contributions
L.C.J.D. and L.H.J.L.: design of experiments and writing the manuscript. A.J.M.G., H.S., R.v.M. and L.C.J.D.: execution of experiments, J.v.R., H.J.G.v.W, M.M.N., J.d.R. and L.C.J.D. for bioinformatics analyses, J.W.O.: pathology review. All authors approved the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Data availability
WGS, ENA PRJEB20644, accession numbers ERX2100523–530; targeted sequencing, ENA PRJEB20644, accession numbers: ERX2019898-958; RNAseq, ArrayExpress, accession number: E-MTAB-5746; DNA methylation, GEO (GSE58538, GSM1413103–GSM1413106) or ArrayExpress (E-MTAB-5842, sample T6107-YSTmeta).
Ethics approval
Use of tissue samples remaining after diagnosis for scientific reasons was approved by Medical Ethical Committee of the Erasmus MC Rotterdam (The Netherlands), permission 02.981. This included the permission to use the secondary tissue without further consent. Samples were used according to the “Code for Proper Secondary Use of Human Tissue in The Netherlands” developed by the Dutch Federation of Medical Scientific Societies.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dorssers, L.C.J., Gillis, A.J.M., Stoop, H. et al. Molecular heterogeneity and early metastatic clone selection in testicular germ cell cancer development. Br J Cancer 120, 444–452 (2019). https://doi.org/10.1038/s41416-019-0381-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-019-0381-1
This article is cited by
-
Impact of tumor markers on diagnosis, treatment and prognosis in CNS germ cell tumors: correlations with clinical practice and histopathology
Brain Tumor Pathology (2023)
-
Integrated genomic analysis reveals aberrations in WNT signaling in germ cell tumors of childhood and adolescence
Nature Communications (2023)
-
ZFP281 drives a mesenchymal-like dormancy program in early disseminated breast cancer cells that prevents metastatic outgrowth in the lung
Nature Cancer (2022)
-
Atypical cartilage in type II germ cell tumors of the mediastinum show significantly different patterns of IDH1/2 mutations from conventional chondrosarcoma
Modern Pathology (2022)
-
Clonal diversification and histogenesis of malignant germ cell tumours
Nature Communications (2022)
