
Abstract
Background
Liquid biopsy offers a minimally invasive alternative to tissue-based evaluation of mutational status in cancer. The goal of the present study was to evaluate the aggregate performance of OncoBEAM RAS mutation analysis in plasma of colorectal cancer (CRC) patients at 10 hospital laboratories in Spain where this technology is routinely implemented.
Methods
Circulating cell-free DNA from plasma was examined for RAS mutations using the OncoBEAM platform at each hospital laboratory. Results were then compared to those obtained from DNA extracted from tumour tissue from the same patient.
Results
The overall percentage agreement between plasma-based and tissue-based RAS mutation testing of the 236 participants was 89% (210/236; kappa, 0.770 (95% CI: 0.689–0.852)). Re-analysis of tissue from all discordant cases by BEAMing revealed two false negative and five false positive tumour tissue RAS results, with a final concordance of 92%. Plasma false negative results were found more frequently in patients with exclusive lung metastatic disease.
Conclusions
In this first prospective real-world RAS mutation performance comparison study, a high overall agreement was observed between results obtained from plasma and tissue samples. Overall, these findings indicate that the plasma-based BEAMing assay is a viable solution for rapid delivery of RAS mutation status to determine mCRC patient eligibility for anti-EGFR therapy.
Similar content being viewed by others


Introduction
Colorectal cancer (CRC) remains one of the most common cancers worldwide, and accounts for 12% of all cancer-related deaths in Europe.1 The epidermal growth factor receptor (EGFR) has become an important therapeutic target in CRC,2 but ~40% of patients with metastatic colorectal cancer (mCRC) have tumours with mutations in KRAS and are not expected to respond to treatment with the anti-EGFR monoclonal antibodies cetuximab and panitumumab.3,4 Several studies have shown that an extended analysis of RAS mutations (including KRAS exons 2, 3, and 4 and NRAS exons 2, 3, and 4) may optimise the identification of patients most likely to benefit from anti-EGFR therapy,5,6,7,8,9 and clinical practice guidelines in the US and Europe include the indication for expanded RAS testing before the use of anti-EGFR agents.10,11,12
Typically, the evaluation of RAS mutation status requires the acquisition of tumour tissue, subsequent processing to formalin-fixed, paraffin-embedded (FFPE) specimens and molecular testing with various techniques. As an alternative, the analysis of circulating tumour DNA (ctDNA) can provide a rapid genotype result with a streamlined clinical workflow and minimal disturbance to the patient. The recent approval of the OncoBEAM RAS CRC liquid biopsy assay by the European Commission as an in vitro diagnostic tool allows a practical and sensible approach for determination of RAS mutations in ctDNA.13
In this study, which included 10 hospital centres across Spain certified to run OncoBEAM RAS in routine practice, we evaluated the concordance between RAS status determined by OncoBEAM in plasma and the reference test performed on tissue at each centre from a large cohort of mCRC patients. We also examined the characteristics of discordant cases and the mutant allele fraction (MAF) in RAS mutated patients.
Patients and methods
Study design and patients
This was a multicenter, prospective, real-world study performed in 10 Spanish centres from November 2015 to October 2016. The study was approved by the Institutional Review Board at each hospital and was conducted in accordance with the principles of the Declaration of Helsinki. Prior to participation, all patients signed the inform consent form. Newly-diagnosed mCRC patients or presenting with recurrent disease after resection and/or chemotherapy were eligible. Patients having surgery with total disease removal or that received the last cycle of chemotherapy <6 months prior to blood draw were excluded.
Procedures
Plasma was obtained from 10 ml of blood collected in Streck cell-free DNA BCT® or EDTA tubes before any therapeutic intervention. All patients had FFPE tissue (either primary tumour or metastasis) available for mutation analysis. OncoBEAM™ RAS CRC assay, which detects 34 mutations in KRAS/NRAS codons 12, 13, 59, 61, 117, and 146, was used to analyse RAS mutations and determine MAF in plasma. The mutation profile in tissue samples was determined by standard-of-care (SoC) procedures validated by each hospital (Supplementary Table S1). Tissue RAS testing by BEAMing (1% mutant allele cut-off) was centrally performed by the Service Laboratory of Sysmex Inostics. The commercially available mutation testing service using the RAS OncoBEAM panel (33 single mutations, covering the same base exchanges like the IVD kit product OncoBEAM™ RAS CRC assay) was used in the laboratory of Sysmex Inostics GmbH, Hamburg (Germany) on FFPE samples from every patient case where the SoC RAS result was discordant with the plasma RAS result. The same tissue block was used for the central re-analysis by BEAMing.
Statistical analysis
Categorical variables were summarised in numbers and percentages, continuous variables were presented as medians, minima and maxima. Concordance between plasma and tissue RAS testing was determined using a Kappa statistic (kappa) with 95% confidence interval (CI). Positive percent agreement (PPA), negative percent agreement (NPA) and overall percent agreement (OPA) were also calculated. For MAF levels correlations with clinical variables, we performed non-parametric statistics (Mann–Whitney U test for dichotomous and Kruskal–Wallis test for polychotomous variables). All statistical tests were considered significant when P < 0.05. Statistical analyses were performed using the SAS version 9.4 statistical software.
Results
Patient characteristics and RAS mutation status analysis from plasma and tissue
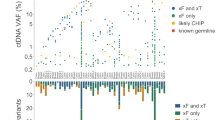
A total of 239 mCRC patients were initially included, 3 of which were excluded because total disease removal during primary surgery. The remaining 236 participants, 144 men and 92 women, comprised the study population (see their baseline characteristics in Table 1). The majority of patients (95.4%) had colorectal adenocarcinoma with distant metastases at diagnosis (85.1%), 50.4% underwent surgery to remove the primary tumour or some portion of metastasis (16.9%) before blood sample collection. The most frequent site of metastasis was the liver (71.2%) followed by the lung (29.3%).
RAS mutation status was evaluable in both plasma and tissue of all 236 patients. Overall, RAS mutations were detected in 55.5% of tumour-tissue samples and in 51.3% of plasma samples (Table 1). The OPA of RAS results between ctDNA and SoC for tissue analysis was 89% (210/236 patients), with a kappa index of 0.770 (95% CI: 0.689–0.852) (Fig. 1a). To clarify the 26 discrepant RAS status results, all FFPE samples except one (not available) were centrally re-analysed by BEAMing technology (Table 2). Of the 18 plasma WT/RAS+ cases, five were finally concordant (Plasma WT/Tissue SoC Mutated/Tissue BEAMing WT); of the 8 plasma RAS+/tissue WT cases, two were concordant (Plasma BEAMing Mutated/Tissue SoC WT/Tissue BEAMing Mutated). This final analysis resulted in a total 217 concordant patients from the 236, representing a 92% overall concordance (κ: 0.853, 95% CI: 0.786–0.920) (Fig. 1b).

Concordance between plasma and tissue results obtained by SOC (a) or SOC + BEAMing (b). SOC standard of care
Discordant samples description and factors affecting concordance
Among the samples with discordant RAS status between ctDNA and tissue BEAMing (n = 19), we observed that 5 of 13 Plasma WT/RAS+ Tissue cases involved patients that had exclusively lung metastases (Table 2). Among the group with RAS+ Plasma/Tissue WT discordance, the sites of metastases in these 6 patients were widely distributed, including locations such as ovary and bone.
Table 3 shows concordant/discordant paired samples according to different clinical and pathological factors. Concordance was lower (87.4% vs 95.7%; P = 0.033) in cases where primary tumour surgery was initially performed. Those with metastatic disease at diagnosis had a higher agreement than patients without metastasis (94.5% vs 78.1%; P = 0.006). A higher concordance of plasma and tissue RAS results was observed in patients having liver metastases (94.5–94.8%) versus those not having liver metastases (83.8%; P = 0.040), whereas the lowest concordance rate was associated with the presence of lung metastases only (68.8%).
MAF analysis in mutated plasma samples
For the 121 patients with detectable plasma RAS mutations, the median MAF [range] was 2.9% [0–68]. No differences in mutational load were observed in relation to age, tumour histology, metastasis at diagnosis, or primary site of disease (Table 3). The median MAF in ctDNA according to the number of metastatic sites was 2% [0–68] in those with one metastatic site, 3.9% [0–35.7] in those with two metastatic sites, and 13% [0.3–51.8] in those with three or more (P = 0.074). The median MAF for those with at least one liver metastasis was 6.7% [0–51.8], whereas the patients with metastases only in the lung showed a median of 0.7% [0.1–24.7].
Discussion
A timely assessment of the current RAS mutation profile of mCRC patients provides the opportunity to deliver the most optimal therapy regimen matched to tumour molecular status.14 With a liquid biopsy we can determine the presence of circulating tumour cells (CTCs), cancer‐derived exosomes, and ctDNA. Despite the technological advances in identifying and characterising CTCs, there are still significant biological challenges that limit their clinical application.15 As ctDNA is released from primary tumours, CTCs, micrometastasis or overt metastases, this might better reflect the molecular changes that occur during disease progression.16 Mutations in ctDNA may be detected in blood using several techniques, such as digital PCR (dPCR) assays such as droplet-digital PCR (ddPCR) and BEAMing, or next-generation sequencing (NGS). dPCR platforms offer easy workflow and better allele-specific sensitivity and reproducibility than standard quantitative PCR, but are limited in its multiplexing capability.17 When multiple targets have to be analysed, NGS technology reduces the cost of screening compared to analysis with a lower throughput technology.18 The principles and different characteristics of these techniques have been reviewed elsewhere.13,19
To the best of our knowledge, this is the first prospective real-world study in which a RAS mutational analysis was compared between plasma OncoBEAM RAS CRC assay and tissue-based techniques in a network of hospital laboratories certified to perform OncoBEAM testing in routine clinical practice. Overall, a high concordance rate of RAS status was observed between plasma and tissue analysis performed by SoC procedures (89%). This rate was even better when certain tissue specimens which were seemingly mischaracterised by the local SoC technique were re-evaluated with BEAMing (92%). These results support the use of plasma testing with the OncoBEAM platform as a valuable alternative to tissue SoC to identify patients eligible for anti-EGFR therapy in routine clinical practice. Moreover, concordance rates are comparable to those obtained in previous retrospective and prospective studies,20,21,22 which corroborate the consistency of the technique among different mCRC patient populations. In fact, the frequency of RAS mutations in patients evaluated in this study was in agreement with the results of other groups performing expanded RAS analysis (plasma 51.3%; tissue 55.5%).21,22,23
Some reports have demonstrated that testing of DNA from a single colorectal tumour tissue block will wrongly assign KRAS wild-type status in 8–11.6% of patients.24,25 Thus, the sole reliance on RAS mutation results obtained from a primary tumour sample might misinform effective treatment of residual systemic disease, imposing significant costs both clinically and financially. This sampling bias could largely be avoided by determining the RAS mutational status on multiple tumour blocks, but this is neither practical nor feasible. Studies evaluating inter-tumour heterogeneity between primary tumours and metastases have also revealed mutational discordance in a significant proportion of cases26,27,28 with high levels of inter-tumour heterogeneity observed between primary tumour and matched lung metastases (32.4%).27 Thus, mCRC patients eligible for surgical resection can have primary tumours with a RAS mutation and metastases without RAS mutations, and vice-versa. Though there is no definitive guideline for determining which sample should be tested for RAS mutations, it is often the case in metastatic disease that the primary site is surgically resected while distant metastases are treated with systemic therapy.
Previous studies have shown that BEAMing is an accurate technique for the mutational analysis of archival FFPE tumour tissue.6,8,29 In the present study, we found seven cases in which BEAMing identified the same RAS mutation in tissue that was identified in plasma, contrary to the original SoC result. Differences in tissue RAS mutation detection capabilities ranging between 3 and 20% among diverse routine methodologies have been reported,30,31,32 possibly associated with different sensitivity thresholds.20 Accordingly, the agreement between plasma and tissue RAS testing results will likely improve when both the methods of plasma and FFPE preparation are standardised, underlying the importance of selecting a reliable laboratory for routine testing.33 In our population, 13 patients had mutations in tissue that could not be detected in plasma, which may be attributed to tumour heterogeneity, low ctDNA shedding or low tumour burden. In fact, five of these discordant cases had archival primary tumour mutated and excised, maybe their metastases were WT or low-shedding lesions. Other authors found similar results in patients with RAS mutant on tissue and WT on liquid biopsy that had recurrence of the disease after surgical resection of the primary and a lower tumour burden, with metastatic lesions often localised in the lung and lymph nodes.34 This finding is consistent with the significantly lower MAF found in our cohort of patients subjected to primary tumour resection. In line with this, our results also showed that the degree of RAS mutational concordance varied according to the metastatic site, with more discrepancies in patients with lung only metastases and a higher agreement in liver metastases; similarly, Thierry et al found higher specificity of plasma mutation analysis in patients with at least one liver metastasis,35 whereas Kim et al.,27 reported higher discordance rates when compared paired primary tumour and lung samples, so it is possible that lung metastases more frequently have a different RAS status than other metastatic sites. Another explanation may be that tumour budding in the metastatic lesions triggers different levels of ctDNA release. Metastatic deposits in the liver are likely highly vascularised as compared to the lungs, and this may contribute to greater levels of ctDNA released in the bloodstream.
Here, the median MAF obtained by plasma BEAMing was 2.9%, higher than in Vidal et al. (1.84%)22 but lower than Schmiegel et al. (6.82%).21 In the first study, 8 of the 59 RAS+ patients had received previous treatment with chemotherapy ± anti-VEGF within a month prior ctDNA blood extraction and showed significantly lower RAS plasma MAF as compared to treatment-naive patients, whereas our population had at least 6 treatment-free months before plasma collection. It has been reported that changes in ctDNA may occur during the course of the chemotherapy, with significant reductions in ctDNA levels observed even after the first cycle.29 Thus, mutational load in patients exposed to therapy may decrease in parallel to radiological response.36,37 Indeed, Schmiegel et al.21 included a cohort of stage IV newly diagnosed patients with intact primary CRC whose MAF was 6.5-fold higher (9.63%) compared with those patients who presented with recurrent disease after removal of their primary tumours (1.49%). These findings highlight the significance of determining and monitoring the MAF of RAS+ mCRC patients throughout the course of the disease management and the impact of any surgical procedure and/or systemic treatment on it. Moreover, based on our results, the rate of release of tumour DNA into circulation may serve as an important clinical observation to consider, as highly vascularised metastatic sites (i.e. liver) and an elevated number of metastases were associated with higher MAF values.
In conclusion, ctDNA analysis by OncoBEAM RAS CRC assay is comparable to SoC tissue testing techniques. It represents a minimally invasive method easily implemented in routine clinical practice to rapidly determine mCRC patient eligibility for anti-EGFR therapy. This technique likely avoids the potential pitfalls of selecting a targeted therapy strategy based on the molecular profile of a single lesion. A unique feature of ctDNA genotyping is its ability to evaluate the extent of an individual patient’s tumour burden, eliminating sampling issues related to tissue molecular heterogeneity and the development of mutations during the metastatic process.
References
Malvezzi, M. et al. European cancer mortality predictions for the year 2017, with focus on lung cancer. Ann. Oncol. 28, 1117–1123 (2017).
Lockhart, A. C. & Berlin, J. D. The epidermal growth factor receptor as a target for colorectal cancer therapy. Semin Oncol. 32, 52–60 (2005).
Di Fiore, F. et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br. J. Cancer 96, 1166–1169 (2007).
Karapetis, C. S. et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 359, 1757–1765 (2008).
Douillard, J.-Y. et al. Panitumumab–FOLFOX4 treatment and RAS mutations in colorectal cancer. New Engl. J. Med. 369, 1023–1034 (2013).
Bokemeyer, C. et al. FOLFOX4 plus cetuximab treatment and RAS mutations in colorectal cancer. Eur. J. Cancer 51, 1243–1252 (2015).
Peeters, M. et al. Analysis of KRAS/NRAS mutations in a phase III study of panitumumab with FOLFIRI compared with FOLFIRI alone as second-line treatment for metastatic colorectal cancer. Clin. Cancer Res. 21, 5469–5479 (2015).
Van Cutsem, E. et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J. Clin. Oncol. 33, 692–700 (2015).
Stintzing, S. et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab for metastatic colorectal cancer (FIRE-3): a post-hoc analysis of tumour dynamics in the final RAS wild-type subgroup of this randomised open-label phase 3 trial. Lancet Oncol. 17, 1426–1434 (2016).
Benson, A. B. et al. Colon cancer, version 3. 2014. J. Natl Compr. Cancer Netw. 12, 1028–1059 (2014).
Allegra C. J. et al. Extended RAS gene mutation testing in metastatic colorectal carcinoma to predict response to anti–epidermal growth factor receptor monoclonal antibody therapy: American Society of Clinical Oncology Provisional Clinical Opinion Update 2015. J. Clin. Oncol. 34, 179–185 (2016).
Van Cutsem, E. et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 27, 1386–1422 (2016).
Garcia-Foncillas, J. et al. Incorporating BEAMing technology as a liquid biopsy into clinical practice for the management of colorectal cancer patients: an expert taskforce review. Ann. Oncol. 28, 2943–2949 (2017).
Atreya, C. E., Corcoran, R. B. & Kopetz, S. Expanded RAS: refining the patient population. J. Clin. Oncol. 33, 682–685 (2015).
Jeong, K. Y., Kim, E. K., Park, M. H. & Kim, H. M. Perspective on cancer therapeutics utilizing analysis of circulating tumor cells. Diagnostics 8, E23 (2018).
Spindler, K. G. et al. Cell-free DNA in metastatic colorectal cancer: a systematic review and meta-analysis. Oncologist 22, 1049–1055 (2017).
Janku, F. et al. Multiplex KRASG12/G13 mutation testing of unamplified cell-free DNA from the plasma of patients with advanced cancers using droplet digital polymerase chain reaction. Ann. Oncol. 28, 642–650 (2017).
Luthra, R., Chen, H., Roy-Chowdhuri, S. & Singh, R. R. Next-generation sequencing in clinical molecular diagnostics of cancer: advantages and challenges. Cancers 7, 2023–2036 (2015).
Denis, J. A., Guillerm, E., Coulet, F., Larsen, A. K. & Lacorte, J. M. The Role of BEAMing and Digital PCR for Multiplexed Analysis in Molecular Oncology in the Era of Next-Generation Sequencing. Mol. Diagn. Ther. 21, 587–600 (2017).
Grasselli, J. et al. Concordance of blood- and tumor-based detection of RAS mutations to guide anti-EGFR therapy in metastatic colorectal cancer. Ann. Oncol. 28, 1294–1301 (2017).
Schmiegel, W. et al. Blood-based detection of RAS mutations to guide anti-EGFR therapy in colorectal cancer patients: concordance of results from circulating tumor DNA and tissue-based RAS testing. Mol. Oncol. 11, 208–219 (2017).
Vidal, J. et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Ann. Oncol. 28, 1325–1332 (2017).
Sorich, M. J. et al. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of random, controlled trials. Ann. Oncol. 26, 13–21 (2015).
Baldus, S. E. et al. Prevalence and heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin. Cancer Res. 16, 790–799 (2010).
Watanabe, T. et al. Heterogeneity of KRAS status may explain the subset of discordant KRAS status between primary and metastatic colorectal cancer. Dis. Colon Rectum 54, 1170–1178 (2011).
Artale, S. et al. Mutations of KRAS and BRAF in primary and matched metastatic sites of colorectal cancer. J. Clin. Oncol. 26, 4217–4219 (2008).
Kim, M. J. et al. Different metastatic pattern according to the KRAS mutational status and site-specific discordance of KRAS status in patients with colorectal cancer. BMC Cancer 12, 347 (2012).
Mostert, B. et al. KRAS and BRAF mutation status in circulating colorectal tumor cells and their correlation with primary and metastatic tumor tissue. Int J. Cancer 133, 130–141 (2013).
Morelli, M. P. et al. Character the patterns of clonal selection in circulating tumor DNA from patients with colorectal cancer refractory to anti-EGFR treatment. Ann. Oncol. 26, 731–736 (2015).
Laurent-Puig, P. et al. Clinical relevance of KRAS-mutated subclones detected with picodroplet digital PCR in advanced colorectal cancer treated with anti-EGFR therapy. Clin. Cancer Res. 21, 1087–1097 (2015).
de Castro, D. G. et al. A comparison of three methods for detecting KRAS mutations in formalin-fixed colorectal cancer specimens. Br. J. Cancer 107, 345–351 (2012).
Azuara, D. et al. Nanofluidic digital PCR and extended genotyping of RAS and BRAF for improved selection of metastatic colorectal cancer patients for anti-EGFR therapies. Mol. Cancer Ther. 15, 1106–1112 (2016).
Tack, V. et al. External quality assessment unravels interlaboratory differences in quality of RAS testing for anti-EGFR therapy in colorectal cancer. Oncologist 20, 257–262 (2015).
Normanno, N. et al. RAS testing of liquid biopsy correlates with the outcome of metastatic colorectal cancer patients treated with first-line FOLFIRI plus cetuximab in the CAPRI-GOIM trial. Ann. Oncol. 29, 112–118 (2018).
Thierry, A. R. et al. Clinical utility of circulating DNA analysis for rapid detection of actionable mutations to select metastatic colorectal patients for anti-EGFR treatment. Ann. Oncol. 28, 2149–2159 (2017).
Tie, J. et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann. Oncol. 26, 1715–1722 (2015).
Garlan, F. et al. Early evaluation of circulating tumor DNA as marker of therapeutic efficacy in metastatic colorectal cancer patients (PLACOL Study). Clin. Cancer Res. 23, 5416–5425 (2017).
Acknowledgements
We would like to acknowledge Anabel Herrero, who provided writing support on behalf of Springer Healthcare, with funding from Sysmex Inostics, Inc.
Author information
Authors and Affiliations
Contributions
Design or conceptualisation of the study: Jesús García-Foncillas; data acquisition: all the authors; analysis or interpretation of the data: all the authors; manuscript preparation and editing: Anabel Herrero; manuscript review: all the authors.
Corresponding author
Ethics declarations
Competing interests
J.G.F.: Advisory role for Amgen, Astellas, AstraZeneca, Bayer, Boehringer Ingelheim, BMS, Celgene, Gilead, GSK, Janssen, Lilly, Merck Serono, MSD, Novartis, Pharmamar, Pfizer, Roche, Sanofi, and Sysmex-Inostics; J.T.: Has had advisory role activities for Bayer, Boehringer Ingelheim, Genentech/Roche, Lilly, MSD, Merck Serono, Merrimack, Novartis, Peptomyc, Sanofi, Symphogen, and Taiho; E.A.: Consultant or advisory role for Amgen, Bayer, Celgene, Merck Serono, Roche, and Sanofi; C.M.: Funding (equipment) to the Institution; Advisory role for Sysmex; E.D.R.: Consulting: Amgem, Bayer, Genomica, Servier, Merck Serono, Speaker Bureau: Servier, MSD. Research Funding: Roche, Merck Serono, Amgem, Astra Zeneca, Sysmex; A.V.: Consultant role for Sysmex Inostics; M.B.: Funding (equipment) to the Institution. The remaining authors declare no competing interests.
Availability of data and material
All necessary data are included in the manuscript. The authors have no supplementary data to share.
Ethics approval and consent to participate
The study was approved by the Institutional Review Board at each hospital and was performed in accordance with the Declaration of Helsinki. All participants signed the informed consent form.
Funding
This work was supported by Sysmex Inostics, Inc as well as the AES Programme [Grant Numbers PI15/00934, PT17/0015/0006]. The funders had no involvement in the writing of this manuscript but did review for medical accuracy prior to submission.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
García-Foncillas, J., Tabernero, J., Élez, E. et al. Prospective multicenter real-world RAS mutation comparison between OncoBEAM-based liquid biopsy and tissue analysis in metastatic colorectal cancer. Br J Cancer 119, 1464–1470 (2018). https://doi.org/10.1038/s41416-018-0293-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-018-0293-5
This article is cited by
-
The current state of molecular profiling in gastrointestinal malignancies
Biology Direct (2022)
-
KRAS and NRAS mutational analysis in plasma ctDNA from patients with metastatic colorectal cancer by real-time PCR and digital PCR
International Journal of Colorectal Disease (2022)
-
Update of the recommendations for the determination of biomarkers in colorectal carcinoma: National Consensus of the Spanish Society of Medical Oncology and the Spanish Society of Pathology
Clinical and Translational Oncology (2020)
-
Comparison of the Clinical Sensitivity of the Idylla Platform and the OncoBEAM RAS CRC Assay for KRAS Mutation Detection in Liquid Biopsy Samples
Scientific Reports (2019)
-
A multicentre, prospective study of plasma circulating tumour DNA test for detecting RAS mutation in patients with metastatic colorectal cancer
British Journal of Cancer (2019)
